

Abstract
The New York Consortium on Membrane Protein Structure (NYCOMPS) was formed to accelerate the acquisition of structural information on membrane proteins by applying a structural genomics approach. NY-COMPS comprises a bioinformatics group, a centralized facility operating a high-throughput cloning and screening pipeline, a set of associated wet labs that perform high-level protein production and structure determination by x-ray crystallography and NMR, and a set of investigators focused on methods development. In the first three years of operation, the NYCOMPS pipeline has so far produced and screened 7,250 expression constructs for 8,045 target proteins. Approximately 600 of these verified targets were scaled up to levels required for structural studies, so far yielding 24 membrane protein crystals. Here we describe the overall structure of NYCOMPS and provide details on the high-throughput pipeline.
Keywords: Membrane proteins, Structural genomics, High throughput, NMR, X-ray
Introduction
Lipid-bilayer membranes produce water-impermeable barriers that define the boundaries of biological cells and of specialized compartments within these cells. Protein molecules embedded into or intimately associated with the lipid bilayer control communication and transport across these biological membranes. Such activity is intrinsically directional with respect to inside and out, and membrane proteins are necessarily oriented relative to this polarity. Both external surfaces of a lipid bilayer are hydrophilic, but the bilayer interior is hydrophobic as it is composed of aliphatic chains. Accordingly, protein molecules embedded into a membrane have hydrophobic surfaces in association with the lipids and, typically, hydrophilic portions protrude from the membrane surface. Such integral membrane proteins (IMPs) are not directly soluble in aqueous media but require detergents to cover the hydrophobic surfaces for extraction and solubilization [17]. These properties make biochemical manipulation of IMPS significantly more complex than for soluble proteins.
Because of biochemical complexities, integral membrane proteins present formidable, but not insurmountable problems for structural analysis. There have been striking successes starting with the first result in three dimensions, by electron crystallography at 7 Å resolution, on bacteriorhodopsin [20] and the first atomic-level structure, at 3 Å resolution by X-ray crystallography, on a photosynthetic reaction center [10]. Membrane protein structures have been determined at an accelerated pace in recent years, and many of these new structures have had dramatic impact as in the cases of cytochrome c oxidases [21, 42], potassium channels [13, 22], aquaporins [32, 39] and G-protein coupled receptors (for a review see [19]) Nevertheless, the structural output on membrane proteins is a very small fraction of that for soluble macromolecules. Through February 2010, White had recorded 231 unique membrane protein structures and 596Protein Data Bank (PDB) depositions on his website (http://blanco.biomol.uci.edu/Membrane_Proteins_xtal.html) whereas there were over 60,000 PDB entries determined by diffraction methods at the same time (www.rcsb.org/pdb). Thus, while membrane proteins comprise 20–30% of all proteins in both prokaryotic and eukaryotic organisms [43], they comprise at most one percent of those with known structure.
Challenges that complicate structural analysis of membrane proteins arise at almost every stage. Only the initial cloning for recombinant expression is no more difficult than for soluble proteins. However, difficulties in recombinant expression specific to membrane proteins arise at all other stages. Although there have been recent successes in producing recombinant bacterial proteins for structure analysis, eukaryotic membrane proteins have been strikingly recalcitrant to expression at the scale needed for such studies. Although there are structures of important eukaryotic membrane proteins, all but a few have come from natural sources.
Biochemical purification and characterization is intrinsically much more challenging for membrane proteins than for naturally soluble counterparts. One must isolate them in bilayers, either as naturally enriched or reconstituted, or make them water soluble in detergent micelles. Two-dimensional membrane protein arrays can be used for electron crystallography, and there are now at least seven such atomic-level (sub-4 Å in the best dimension) structures [37], or for solid-state NMR experiments now just coming of age [31]. Soluble detergent micelles can be used for solution NMR experiments or for x-ray crystallography, which has dominated the field until now. The added size due to adherent detergent complicates NMR analysis, but TROSY and selective labeling techniques offer promising solutions [31].
The crystallization of proteins in detergent micelles has its own special difficulties. These are at least threefold: (1) the protein may not be stable outside the lipid bilayer [5], (2) detergent interactions that occur during crystallization are important, providing another variable that must be screened [18], and (3) the detergent-covered lipophilic surfaces can be highly mobile and thus unsuitable for lattice contacts [34], which theoretically reduces the probability of crystallization by a high power of the fractional surface area [27]. Initial crystals, once obtained, typically require substantial effort in optimization to reach diffraction for atomic-level resolution. While the ultimately-achieved resolution can be stunning [24, 15], commonly, and much more often than for soluble proteins, resolution is limited to a level that frustrates model building and refinement [6].
To help circumvent these problems, and to thereby advance the field of membrane protein research, we founded the NYCOMPS consortium under the aegis of the NIH Protein Structure Initiative (PSI) in July 2005. The overall objective of the consortium is to accelerate the acquisition of structural information about membrane proteins by applying a structural genomics approach informed by experience gained in studies driven by biological and biochemical problems. A pipeline for structure determination has been established (Fig. 1), which begins with a bioinformatics analysis of all known sequences, moving onto recombinant cloning, protein expression and automated screening, protein purification at moderate throughput, and then into structure determination by X-ray crystallography and NMR spectroscopy. A centralized Protein Production Facility has been established in laboratories of the New York Structural Biology Center (NYSBC) to implement cloning and screening activities. Large scale protein production and structure determination of targets verified by the central facility are carried out in the laboratories of NYCOMPS participants. NYCOMPS members are also engaged in the development of new techniques for membrane proteins, relating to their purification, stabilization, crystallization, phasing, and structure determination by NMR spectroscopy. NYCOMPS has also recruited many members of the local structural community as ‘adjunct’ members who may request target processing via the NYCOMPS pipeline. This process has been popular and rewarding for participants.
Fig. 1.

Flow chart depicting the pipeline workflow at NYCOMPS
Results and discussion
NYCOMPS approach to target selection
The protocol that we follow to select targets for membrane proteins differs from that exploited typically for structural genomics (SG) of non-membrane proteins. Given a large set of valid targets, SG usually performs exhaustive clustering of all valid sequences, with the resulting clusters representing cloning families [29]. At NYCOMPS, we instead create cloning families dynamically. That is, whenever a protein of interest (a “seed”) becomes available we expand it into a cluster of valid targets. The expansion entails identification of proteins that are likely to have similar structures as the seed. Seeds are selected based on criteria such as novelty, feasibility of production, and biological/biomedical relevance. New seeds can become available at any time during the project from newly published data, from nominations by participating groups, and through feedback from our central experimental pipeline. While target selection for non-membrane proteins typically follows a top-down approach, that for NYCOMPS employs a bottom-up strategy. When clustering is based predominantly on simple sequence similarity thresholds, the two approaches may generate quite different sets of cloning families. In particular, our seed-centered cloning families are likely to contain more targets featuring characteristics similar to the ones of the seed (for example, in terms of function or feasibility) with respect to families that contain the seed but are created starting from a different target. Effectively our strategy aims at maximizing the number of proteins most similar to a given seed.
Creating the NYCOMPS98 set of valid targets
All NYCOMPS targets (with the exception of biological theme targets) are selected from 96 fully sequenced prokaryotic genomes for which genomic DNA is available from ATCC®. Protein sequences for these genomes are obtained following the annotation provided by RefSeq [35]. From this initial pool of protein sequences (310,357 in total), we remove proteins that meet any of the following four conditions (Filter 1, Fig. 2): (1) those predicted to have fewer than two transmembrane (TM) helices [26], (2) those with over 98% pairwise sequence identity to others in the set (i.e. we keep only one representative. Sequence identity is established through CD-HIT [28], (3) those predicted with two TM helices with the most N-terminal TM helix overlapping with a predicted signal peptide [14], and (4) those predicted to have over 15 consecutive disordered residues [12]. All remaining sequences (39,037 total) represent our set of valid targets, referred to as NYCOMPS98, where “98” stands for “98% redundancy reduced” (Fig. 2).
Fig. 2.

Target selection at NYCOMPS. We start from 96 prokaryotic genomes and we create (Filter 1) a set of valid targets (NYCOMPS98, see text). We then select proteins of interest (seeds) and expand them into the set of valid targets to create seed families (we use sequence similarity in the predicted TM region as a criterion for family membership). All members in a selected seed family are subjected to additional filtering steps (Filter 2) to ensure novelty or increase feasibility. Finally, remaining targets are sent to cloning
Seed selection and expansion
Seeds can enter our pipeline either by central selection or by nomination. Central seed selection has so far utilized sequences from a list of 613 Escherichia coli proteins that were successfully over-expressed in a previous genome-wide study [9]. Nominated seeds are submitted by individual groups. All seeds are expanded into families by selecting those proteins within the NYCOMPS98 dataset that align [1] to the seed with E-value < 10−3 and such that the alignment covers at least 50% of the predicted transmembrane region of both seed and target. This step serves to avoid association between integral membrane proteins simply based on the fact that they share one or more soluble domains, while having unrelated TM regions. Finally, all retrieved targets are further subjected to additional filters to ensure novelty or to increase feasibility (Filter 2, Fig. 2). These steps include discarding sequences that have significant similarity to a PDB entry within its TM regions (E-value < 1, alignment covering at least 25% of the predicted TM region of the target protein) and families for which there is evidence that one or more members represent individual subunits of heteromeric complexes [36]. Filter 2 criteria are not enforced on nominated targets, leaving the decision of whether or not to pursue a given family of proteins to the nominating group.
Results-based refinement of target selection procedures
Our experimental pipeline produces large amounts of informative data at different stages, including results of cloning, expression, crystallization, and structure determination stages. These data are periodically and systematically analyzed to help formulate “rules” that may improve our target selection protocol. These investigations have already led to the retirement of 19 genomes that showed low cloning and/or expression success. We continue to refine our analysis and plan to use cloning and expression data to develop methods that will enhance the success at every stage of the pipeline. All targets are deposited in a LIMS system (Sesame, a public domain LIMS from University of Wisconsin), and data from all the downstream process are collected here. This data is deposited on a regular basis at the TargetDB and PepCDB sites.
Expression vectors and automated cloning
The core facility of NYCOMPS utilizes pET-derived, kanamycin resistant, IPTG inducible expression vectors (Fig. 3) for the production of integral membrane proteins. The vectors are available from the PSI materials repository. These vectors have been modified to introduce sites for ligation independent cloning [2] into both C- and N-terminal tag encoding versions. Accessibility of the tag and expression levels of the fusion protein can vary depending on the location of the tag in an often unpredictable way, thus warranting testing of both orientations. A ‘death gene’ (ccdB; [4]) has also been inserted into the subcloning site to minimize background in bacterial transformations. In both versions of this expression vector, proteins are produced with a FLAG tag for immuno-detection or purification, a deca-histidine tag for metal affinity chromatography, and a tobacco etch virus protease (TEV; [33]) recognition site for tag cleavage. The deca-hisitidine tag enables tight binding to metal-containing resins allowing for the use of high imidazole levels in wash buffers, resulting in higher purity over the more commonly used hexa-histidine version. We have chosen the TEV protease as its activity has proven to be relatively insensitive to detergent [30].
Fig. 3.

N-terminal (upper panel) and C-terminal (lower panel) fusion expression vectors for the production of His/FLAG-tagged membrane proteins in E. coli
PCR amplification primers are designed using an automated procedure to amplify the full length coding sequence. PCR reactions are set up robotically on a 384 well format by a Beckman Biomeck FX robot, utilizing a genomic DNA as template. After amplification, the inserts are purified using Agencourt Solid Phase Reversible Immobilization magnetic resins (Agencourt, Inc.) and adapted for LIC into the expression vectors. Vectors are transformed into a DH10B phage-resistant strain of E. coli and plated robotically. After overnight growth, single colonies are manually picked and grown for automated plasmid purification. These plasmids are sequenced to confirm the integrity and identity of each insert.
Small scale expression tests
The expression constructs are transformed into BL21 (DE3) pLysS phage resistant cells and grown in 96 well deep well blocks overnight. Overnight growths are diluted 100 fold into fresh media and grown in shakers at 700 rpm with a 2 mm orbit. This enables fermentation-like growth conditions to be achieved in deep well block format. Cultures are induced with 0.4 mM IPTG and further grown for 4 h at 37°C to express the membrane protein before harvest. Cell densities at harvest are typically above 10 OD units at 600 nm (OD600).
To test for expression and purification, induced cells are lysed by sonication in deep well blocks, using a robotic system. This custom apparatus is designed to ensure complete cell lysis without excessive sample heating. Membrane fractions may optionally be isolated by robotically transferring the lysate to 96 ultracentrifuge tubes and pelleting with a high-speed spin.
Typically the membrane isolation step is omitted and the crude lysate is solubilized by addition of dodecylmaltoside (DDM) to 2% w/v final concentration. Insoluble matter is removed by centrifugation and the lysate is mixed with 25 μl metal affinity resin overnight at 4°C, transferred to a 96 well filter plate, washed with 500 μl of wash buffer containing 75 mM imidazole. The protein of interest is then eluted with 35 μl of 0.5 M imidazole. Purified membrane proteins are detected on Coommassie Blue stained SDS–PAGE gels (see Fig. 4). Alternatively, and less frequently, purification results can be evaluated by western blot using anti-FLAG antibodies.
Fig. 4.

Coommassie blue stained SDS–PAGE showing expression and purification results of 22 different membrane proteins. Cells were grown in 0.6 ml of media in a deep well block, and metal affinity purified and eluted in a buffer containing Dodecyl-maltoside detergent. Well-expressed proteins can clearly be identified (without western blot or GFP labeling methods). Clones producing membrane proteins of approximately the correct molecular weight are re-grown at a larger scale prior to detergent stability analysis. For membrane proteins, molecular weights judged by electrophoretic mobility are often underestimated by ~10%. Also, SDS-resistant multimers are frequently observed, as is the case for samples in lanes 1, 2, 14, 15, 17 and 22
Samples yielding a band of approximately correct molecular weight and minimal proteolytic breakdown are re-arrayed into new plates. These targets are transferred to a mid-scale expression and purification platform to produce sufficient protein for detergent selection and stability analysis.
Mid-scale protein production and detergent stability assay
Target-expressing clones are grown to high cell densities 65 ml culture volume reaching a final optical density of 18 OD units (600 nm) final, at 37°C in an Airlift fermenter (GNF systems, San Diego, CA). Cell pellets are harvested and lysed robotically. Whole cell lysates are solubilized with 2% DDM and incubated with metal affinity resin before being transferred to 96 10 ml drip columns held in a purpose built rack. The columns are washed and the proteins eluted as described above. Using this protocol, 96 proteins can be purified every 2 days by a single laboratory worker.
DDM is the only detergent employed for solubilization and purification. DDM is considered a relatively mild detergent, however successful crystallization often requires the use of shorter chain detergents. To assess the tolerance of target proteins to shorter chain detergents, the purified proteins are subject to an ad hoc stability assay. Samples are split into aliquots and incubated with a large excess of a second, short chain detergent for 2 h at room temperature. Subsequently, the samples are clarified by centrifugation, and loaded on a size exclusion chromatography column equilibrated in DDM (Fig. 5). Proteins that show a single, symmetrical elution profile after treatment with one or more short chain detergents are prioritized for scale up and crystallization experiments. Targets suitable for NMR experiments are also screened by size exclusion chromatography, but using a panel of detergents tailored to this method, (e.g. DM, FC12, LysoFC14, and DHPC).
Fig. 5.

UV absorbance monitored elution profiles from a size exclusion column for two membrane proteins post detergent stability testing. Metal affinity elution’s of membrane proteins in DDM containing buffer are treated at an elevated temperature with a large excess of various short chain ‘harsh’ detergents and a DDM control. After a time period, the reactions are clarified to remove large aggregates and the samples are subjected to size exclusion chromatography in a mobile phase containing DDM. a Shows a membrane protein that is largely detergent insensitive, as the peak shape and height (as measured by mAU) are not significantly altered by the detergent treatment. b Shows a detergent sensitive membrane protein, where the detergent stability treatment has resulted in the peak shifting to the void or being absent, in the short chain detergents, but not the more mild DDM control. Blue, DDM; Green, C8E4; Red, LDAO; Pink β-OG
Scale-up to production scale
For production of proteins at a scale suitable for structural studies, expression-verified detergent-screened clones are distributed from the Center to the participating research groups. This arrangement recognizes that optimization of protein production, quality, and subsequent steps including crystallization and NMR sample evaluation require an individualized, often time consuming approach. This is at odds with the high-throughput model employed at the Center, and integrates well with the “classical” structural biology approach for which most individual labs are equipped. Expression and purification are based on a standard set of protocols, which can be modified as needed. Typically, proteins are expressed on a scale of more than 1 liter, depending on expression levels. Cells are grown in baffled shake flasks using rich media (800 ml per 2 liter flask) at 37°C to an OD600 of ~0.6, at which time the temperature is reduced 20°C and expression is induced by addition of IPTG to a final concentration of 0.2 mM. Expression is then allowed to continue for 18 h. Cells are harvested by centrifugation, and resuspended and solubilized in standard buffer conditions containing 1% DDM at a ratio of 1 g DDM per 10 g of cells. After solubilization the solution is clarified by ultracentrifugation, and the fusion protein is purified by metal affinity chromatography, with a washing step with 50–60 mM imidazole, and elution of the fusion protein with 250 mM imidazole. The affinity tag can then optionally be proteolytically removed with TEV protease, in which case a second pass of the dialyzed sample over metal affinity resin removes the protease, uncleaved fusions, and most contaminants. The protein is concentrated using a Centricon with a YM-50 membrane, and applied to a gel filtration column for further purification. The gel filtration step also serves the purpose of detergent exchange when desired.
Crystallization and crystallography
Crystallization screening is carried out robotically in 96-well plates. Robotic crystallization allows for rapid parallel screening of multiple parameters, including different substrates, additives, or detergents. NYCOMPS invested in a Mosquito crystallization robot, whose positive displacement mode of action is well suited to working with detergent solubilized membrane proteins. Once leads are discovered using commercial sparse matrix screens, optimization of crystallization conditions is carried out with the vapor diffusion technique in 24-well plates. Following the protein, the detergent is one of the most important parameters determining the success of crystallization. Therefore, we carry out crystallization experiments in as many short chain detergents validated by the stability assay. In one particular case, the switching of detergent greatly improved the quality of the crystals, extending the diffraction limit from 20 Å in DDM to 2.2 Å in β-octyl glucoside (β-OG) (Fig. 6). NYCOMPS has also designed and built an economical lipidic cubic phase dispensing robot that is available for setting up crystal trials in meso. We have also been utilizing the excellent crystallization service provided by the Center for High-Throughput Structural Biology (CHTSB) which conducts crystallization trials on a 1,536 experiment scale, under oil in batch mode [25].
Fig. 6.
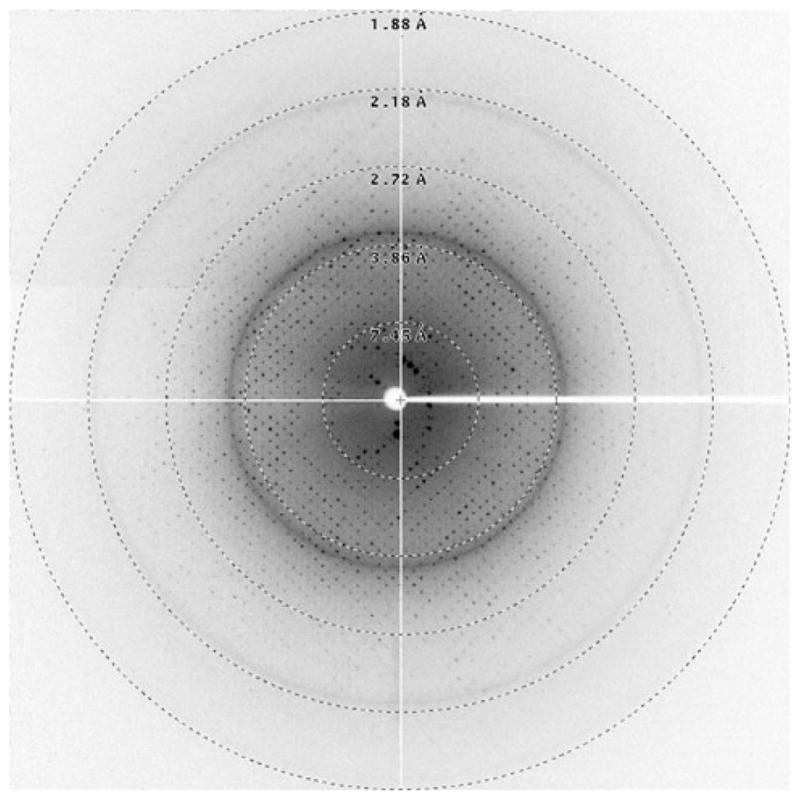
X-ray diffraction pattern of a membrane protein crystal. The highest resolution spots are visible to 2.0 Å. The resolution of the edge of the screen is indicated
NMR
NMR is used on a specifically-selected set of small (under 20 kDa) target proteins, as well as on slightly larger proteins that behave favorably through the stage of detergent screening, but fail to crystallize. The first NMR-specific step in the pipeline is to test for acceptable expression levels (~2 mg protein per liter of culture) in the defined minimal media that will be used for the 15N13C or 2H15N13C isotopic labeling ultimately required for triple resonance NMR methods. 15N-labeled protein is produced and exchanged into detergents that appear promising in the gel filtration screen, and two-dimensional 1H15N NMR spectral quality—the count, intensity, and line widths of the amide cross-peaks—are evaluated as a function of temperature and time. Additional optimizations, if needed and warranted, include pH (ranging from 5.0 to 8.0), ionic strength (ranging from 0 to 300 mM), and limited variations in detergent chain length and head group.
If one or more sets of conditions for a given target yield a good 1H15N HSQC or TROSY spectrum in less than 20 min, uniformly 13C15N-labeled samples are produced, and a fairly traditional triple resonance NMR strategy [23] is used for protein structure determination. Because of the added mass of the detergent micelle, membrane proteins larger than ~15 kD, like their larger water-soluble counterparts, typically require deuteration of alpha-carbon and side chain proton positions to achieve good sensitivity and resolution in NMR experiments. The uniformly 2H13C15N labeled samples are prepared by expression using a deuterated carbon source and D2O, and backbone resonances are assigned using TROSY-based triple resonance methods [38]. Two complications arise from perdeuteration—the absence of long range NOEs from side chains needed for structural constraints, and the potential loss of signals from residues in the transmembrane α-helices resulting from poor exchange of amide deuterons incorporated during expression with solvent protons over the course of protein purification. Long range side chain distance constraints are partially recovered by the reintroduction of protons in side chain methyl and aromatic groups [23], which can be supplemented by other constraint types. Signals from slowly exchanging amides can sometimes be recovered by an extended incubation in a harsher detergent during the first purification steps. If complete back exchange is not possible due to protein instability or hyper-stability, separate samples are prepared to selectively examine the slowly exchanging and more rapidly exchanging regions of the protein [7].
Functional analysis of solved structures
Our major focus has been on using structures of membrane proteins to obtain mechanistic insights. Since, to date, most solved membrane protein structures have been of bacterial proteins, it has been necessary to use homology modeling to obtain structures for and infer function for human proteins. To determine the validity of modeling methods, we established a database (HOMEP) of homologous pairs of structures of integral membrane proteins [16]. HOMEP is particularly useful resource for testing structure prediction methods for membrane proteins since one member can be used as a template for predicting the structure of the other member, and vice versa. We used HOMEP to compared various sequence alignment approaches for membrane proteins and observed that high-level profile-based sequence alignment methods offer significant improvements over existing methods that have been applied to membrane proteins [16]. We also showed that the prediction of secondary structures in membrane proteins can be accomplished with similar accuracy as for water-soluble proteins.
Experimental methods research at NYCOMPS
Although the majority of NYCOMPS resources are devoted to pipeline operations, several research projects aimed at membrane protein methods development have been initiated. These include collaborative projects on membrane nanodiscs, which may provide an alternative method for membrane protein solubilization ([3, 8, 11, 40]; a method for “single protein production” (SPP) in bacterial cells that uses the activity of the MazF toxin to degrade all cellular mRNA that contains the codon ACA, whereby a synthetic “ACA-less” gene encoding the protein of interest is the only protein produced after MazF induction [41]; evaluation of numerous eukaryotic expression systems for the recombinant production of membrane proteins; and development of novel G-matrix FT methods to massively reduce data collection in NMR which is absolutely necessary for the multidimensional experiments conducted on membrane proteins [44]. Results from these initiatives will be published elsewhere.
Conclusion
Structural studies of membrane proteins present formidable challenges. The NYCOMPS initiative addresses several of these, with the goal of bringing structural studies of membrane proteins close to parity with studies of their soluble counterparts. The NYCOMPS process provides a high-throughput platform where targets generated from bioinformatics can be screened to select those with the highest probability of success in structural studies.
Acknowledgments
We thank past and present colleagues who have participated in the development of the Protein Production Facility: Brandon Hillerich, Brian Kloss, Renato Bruni, Arianne Morrison, Patricia Rodriguez, Amanda Meyer, Jeff Bonanno, Zsolt Zolnai, Michael Weiner, Reinhard Grisshammer, and Gunnar von Heijne. This work was supported in part by a Cooperative Agreement from the NIGMS Protein Structure Initiative, U54GM075026.
Contributor Information
James Love, NYCOMPS Core Laboratory, New York Structural Biology Center, New York, NY 10027, USA.
Filippo Mancia, Department of Physiology and Cellular Biophysics, Columbia University, New York, NY 10032, USA.
Lawrence Shapiro, Department of Biochemistry and Molecular Biophysics, Columbia University, New York, NY 10032, USA.
Marco Punta, Department of Biochemistry and Molecular Biophysics, Columbia University, New York, NY 10032, USA.
Burkhard Rost, Department of Biochemistry and Molecular Biophysics, Columbia University, New York, NY 10032, USA.
Mark Girvin, Department of Biochemistry, Albert Einstein College of Medicine, Bronx, New York, NY 10461, USA.
Da-Neng Wang, Skirball Institute of Biomolecular Medicine, New York University Medical Center, New York, NY 10016, USA.
Ming Zhou, Department of Physiology and Cellular Biophysics, Columbia University, New York, NY 10032, USA.
John F. Hunt, Department of Biological Sciences, Columbia University, New York, NY 10027, USA
Thomas Szyperski, Department of Chemistry, University of Buffalo, Buffalo, NY 14260, USA.
Eric Gouaux, Howard Hughes Medical Institute, The Vollum Institute, Oregon Health & Science University, Portland, OR 97239, USA.
Roderick MacKinnon, Howard Hughes Medical Institute, The Rockefeller University, New York, NY 10021, USA.
Ann McDermott, Department of Chemistry, Columbia University, New York, NY 10027, USA.
Barry Honig, Howard Hughes Medical Institute, Department of Biochemistry and Molecular Biophysics, Columbia University, New York, NY 10032, USA.
Masayori Inouye, Department of Molecular Biology and Biochemistry, Rutgers, The State University of New Jersey, Piscataway, NJ 08854, USA.
Gaetano Montelione, Department of Biochemistry, UMDNJ, Piscataway, NJ 08854, USA.
Wayne A. Hendrickson, Email: wayne@convex.hhmi.columbia.edu, Howard Hughes Medical Institute, Department of Biochemistry and Molecular Biophysics, Columbia University, New York, NY 10032, USA
References
- 1.Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Aslanidis C, de Jong PJ. Ligation-independent cloning of PCR products (LIC-PCR) Nucleic Acids Res. 1990;18:6069–6074. doi: 10.1093/nar/18.20.6069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Baas BJ, Denisov IG, Sligar SG. Homotropic cooperativity of monomeric cytochrome P450 3A4 in a nanoscale native bilayer environment. Arch Biochem Biophys. 2004;430:218–228. doi: 10.1016/j.abb.2004.07.003. [DOI] [PubMed] [Google Scholar]
- 4.Bernard P, Couturier M. Cell killing by the F plasmid CcdB protein involves poisoning of DNA-topoisomerase II complexes. J Mol Biol. 1992;226:735–745. doi: 10.1016/0022-2836(92)90629-x. [DOI] [PubMed] [Google Scholar]
- 5.Bowie JU. Stabilizing membrane proteins. Curr Opin Struct Biol. 2001;11:397–402. doi: 10.1016/s0959-440x(00)00223-2. [DOI] [PubMed] [Google Scholar]
- 6.Chang G, Roth CB, Reyes CL, Pornillos O, Chen YJ, Chen AP. Retraction. Science. 2006;314:1875. doi: 10.1126/science.314.5807.1875b. [DOI] [PubMed] [Google Scholar]
- 7.Chill JH, Louis JM, Miller C, Bax A. NMR study of the tetrameric KcsA potassium channel in detergent micelles. Protein Sci. 2006;15:684–698. doi: 10.1110/ps.051954706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Civjan NR, Bayburt TH, Schuler MA, Sligar SG. Direct solubilization of heterologously expressed membrane proteins by incorporation into nanoscale lipid bilayers. Biotechniques. 2003;35(3):556–563. doi: 10.2144/03353rr02. [DOI] [PubMed] [Google Scholar]
- 9.Daley DO, Rapp M, Granseth E, Melen K, Drew D, von Heijne G. Global topology analysis of the Escherichia coli inner membrane proteome. Science. 2005;308:1321–1323. doi: 10.1126/science.1109730. [DOI] [PubMed] [Google Scholar]
- 10.Deisenhofer J, Epp O, Miki K, Huber R, Michel H. X-ray structure analysis of a membrane protein complex. Electron density map at 3 A resolution and a model of the chromophores of the photosynthetic reaction center from Rhodopseudomonas viridis. J Mol Biol. 1984;180:385–398. doi: 10.1016/s0022-2836(84)80011-x. [DOI] [PubMed] [Google Scholar]
- 11.Denisov IG, Grinkova YV, Lazarides AA, Sligar SG. Directed self-assembly of monodisperse phospholipid bilayer Nanodiscs with controlled size. J Am Chem Soc. 2004;126:3477–3487. doi: 10.1021/ja0393574. [DOI] [PubMed] [Google Scholar]
- 12.Dosztanyi Z, Csizmok V, Tompa P, Simon I. The pairwise energy content estimated from amino acid composition discriminates between folded and intrinsically unstructured proteins. J Mol Biol. 2005;347:827–839. doi: 10.1016/j.jmb.2005.01.071. [DOI] [PubMed] [Google Scholar]
- 13.Doyle DA, Morais Cabral J, Pfuetzner RA, Kuo A, Gulbis JM, Cohen SL, Chait BT, MacKinnon R. The structure of the potassium channel: molecular basis of K+ conduction and selectivity. Science. 1998;280:69–77. doi: 10.1126/science.280.5360.69. [DOI] [PubMed] [Google Scholar]
- 14.Emanuelsson O, Brunak S, von Heijne G, Nielsen H. Locating proteins in the cell using TargetP, SignalP and related tools. Nat Protoc. 2007;2:953–971. doi: 10.1038/nprot.2007.131. [DOI] [PubMed] [Google Scholar]
- 15.Fischer G, Kosinska-Eriksson U, Aponte-Santamaría C, Palmgren M, Geijer C, Hedfalk K, Hohmann S, de Groot BL, Neutze R, Lindkvist-Petersson K. Crystal structure of a yeast aquaporin at 1.15 Ångstrom reveals a novel gating mechanism. PLoS Biol. 2009;7(6):e1000130. doi: 10.1371/journal.pbio.1000130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Forrest LR, Tang CL, Honig B. On the accuracy of homology modeling and sequence alignment methods applied to membrane proteins. Biophys J. 2006;91:508–517. doi: 10.1529/biophysj.106.082313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Garavito RM, Ferguson-Miller S. Detergents as tools in membrane biochemistry. J Biol Chem. 2001;276:32403–32406. doi: 10.1074/jbc.R100031200. [DOI] [PubMed] [Google Scholar]
- 18.Garavito RM, Picot D, Loll PJ. Strategies for crystallizing membrane proteins. J Bioenerg Biomembr. 1996;28:13–27. [PubMed] [Google Scholar]
- 19.Hanson MA, Stevens RC. Discovery of new GPCR biology: one receptor structure at a time. Structure. 2009;17:8–14. doi: 10.1016/j.str.2008.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Henderson R, Unwin PN. Three-dimensional model of purple membrane obtained by electron microscopy. Nature. 1975;257:28–32. doi: 10.1038/257028a0. [DOI] [PubMed] [Google Scholar]
- 21.Iwata S, Ostermeier C, Ludwig B, Michel H. Structure at 2.8 A resolution of cytochrome c oxidase from Paracoccus denitrificans. Nature. 1995;376:660–669. doi: 10.1038/376660a0. [DOI] [PubMed] [Google Scholar]
- 22.Jiang Y, Lee A, Chen J, Ruta V, Cadene M, Chait BT, MacK-innon R. X-ray structure of a voltage-dependent K+ channel. Nature. 2003;423:33–41. doi: 10.1038/nature01580. [DOI] [PubMed] [Google Scholar]
- 23.Kanelis V, Forman-Kay JD, Kay LE. Multidimensional NMR methods for protein structure determination. IUBMB life. 2001;52:291–302. doi: 10.1080/152165401317291147. [DOI] [PubMed] [Google Scholar]
- 24.Khademi S, O’Connell J, III, Remis J, Robles-Colmenares Y, Miercke LJ, Stroud RM. Mechanism of ammonia transport by Amt/MEP/Rh: structure of AmtB at 1.35 A. Science. 2004;305:1587–1594. doi: 10.1126/science.1101952. [DOI] [PubMed] [Google Scholar]
- 25.Koszelak-Rosenblum M, Krol A, Mozumdar N, Wunsch K, Ferin A, Cook E, Veatch CK, Nagel R, Luft JR, Detitta GT, Malkowski MG. Determination and application of empirically derived detergent phase boundaries to effectively crystallize membrane proteins. Protein Sci. 2009;18:1828–1839. doi: 10.1002/pro.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Krogh A, Larsson B, von Heijne G, Sonnhammer EL. Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J Mol Biol. 2001;305:567–580. doi: 10.1006/jmbi.2000.4315. [DOI] [PubMed] [Google Scholar]
- 27.Kwong PD, Wyatt R, Desjardins E, Robinson J, Culp JS, Hellmig BD, Sweet RW, Sodroski J, Hendrickson WA. Probability analysis of variational crystallization and its application to gp120, the exterior envelope glycoprotein of type 1 human immunodeficiency virus (HIV-1) J Biol Chem. 1999;274:4115–4123. doi: 10.1074/jbc.274.7.4115. [DOI] [PubMed] [Google Scholar]
- 28.Li W, Godzik A. Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics. 2006;22:1658–1659. doi: 10.1093/bioinformatics/btl158. [DOI] [PubMed] [Google Scholar]
- 29.Liu J, Hegyi H, Acton TB, Montelione GT, Rost B. Automatic target selection for structural genomics on eukaryotes. Proteins. 2004;56:188–200. doi: 10.1002/prot.20012. [DOI] [PubMed] [Google Scholar]
- 30.Lundback AK, van den Berg S, Hebert H, Berglund H, Eshaghi S. Exploring the activity of tobacco etch virus protease in detergent solutions. Anal Biochem. 2008;382:69–71. doi: 10.1016/j.ab.2008.07.018. [DOI] [PubMed] [Google Scholar]
- 31.McDermott A. Structure and dynamics of membrane proteins by magic angle spinning solid-state NMR. Ann Rev Biophys. 2009;38:385–403. doi: 10.1146/annurev.biophys.050708.133719. [DOI] [PubMed] [Google Scholar]
- 32.Murata K, Mitsuoka K, Hirai T, Walz T, Agre P, Heymann JB, Engel A, Fujiyoshi Y. Structural determinants of water permeation through aquaporin-1. Nature. 2000;407:599–605. doi: 10.1038/35036519. [DOI] [PubMed] [Google Scholar]
- 33.Nallamsetty S, Kapust RB, Tozser J, Cherry S, Tropea JE, Copeland TD, Waugh DS. Efficient site-specific processing of fusion proteins by tobacco vein mottling virus protease in vivo and in vitro. Protein Expr Purif. 2004;38:108–115. doi: 10.1016/j.pep.2004.08.016. [DOI] [PubMed] [Google Scholar]
- 34.Ostermeier C, Michel H. Crystallization of membrane proteins. Curr Opin Struct Biol. 1997;7:697–701. doi: 10.1016/s0959-440x(97)80080-2. [DOI] [PubMed] [Google Scholar]
- 35.Pruitt KD, Tatusova T, Maglott DR. NCBI reference sequences (RefSeq): a curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic Acids Res. 2007;35:D61–D65. doi: 10.1093/nar/gkl842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Punta M, Love J, Handelman S, Hunt JF, Shapiro L, Hendrickson WA, Rost B. Structural genomics target selection for the New York consortium on membrane protein structure. J Struct Funct Genomics. 2009;10:255–268. doi: 10.1007/s10969-009-9071-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Raunser S, Walz T. Electron crystallography as a technique to study the structure on membrane proteins in a lipidic environment. Ann Rev Biophys. 2009;38:89–105. doi: 10.1146/annurev.biophys.050708.133649. [DOI] [PubMed] [Google Scholar]
- 38.Salzmann M, Pervushin K, Wider G, Senn H, Wuthrich K. TROSY in triple-resonance experiments: new perspectives for sequential NMR assignment of large proteins. Proc Natl Acad Sci USA. 1998;95:13585–13590. doi: 10.1073/pnas.95.23.13585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Savage DF, Egea PF, Robles-Colmenares Y, O’Connell JD, 3rd, Stroud RM. Architecture and selectivity in aquaporins: 2.5 a X-ray structure of aquaporin Z. PLoS biology. 2003;1:E72. doi: 10.1371/journal.pbio.0000072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shaw AW, McLean MA, Sligar SG. Phospholipid phase transitions in homogeneous nanometer scale bilayer discs. FEBS Lett. 2004;556:260–264. doi: 10.1016/s0014-5793(03)01400-5. [DOI] [PubMed] [Google Scholar]
- 41.Suzuki M, Mao L, Inouye M. Single protein production (SPP) system in Escherichia coli. Nat Protoc. 2007;2:1802–1810. doi: 10.1038/nprot.2007.252. [DOI] [PubMed] [Google Scholar]
- 42.Tsukihara T, Aoyama H, Yamashita E, Tomizaki T, Yamaguchi H, Shinzawa-Itoh K, Nakashima R, Yaono R, Yoshikawa S. The whole structure of the 13-subunit oxidized cytochrome c oxidase at 2.8 A. Science. 1996;272:1136–1144. doi: 10.1126/science.272.5265.1136. [DOI] [PubMed] [Google Scholar]
- 43.Wallin E, von Heijne G. Genome-wide analysis of integral membrane proteins from eubacterial, archaean, and eukaryotic organisms. Protein Sci. 1998;7:1029–1038. doi: 10.1002/pro.5560070420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhang Q, Atreya HS, Kamen DE, Girvin ME, Szyperski T. GFT projection NMR based resonance assignment of membrane proteins: application to subunit C of E. coli F(1)F (0) ATP synthase in LPPG micelles. J Biomol NMR. 2008;40:157–163. doi: 10.1007/s10858-008-9224-8. [DOI] [PubMed] [Google Scholar]