

Abstract
The study of genetic variation has the potential to aid understanding of the mechanisms underlying the observed inter-individual variation in drug response and by which idiosyncratic adverse effects occur. In this review, we outline current progress in pharmacogenetics using examples to highlight both mechanisms of influence of polymorphisms and research strategies for their detection. In the final sections we discuss contemporary challenges for both researchers and clinicians.
Keywords: adverse drug effects, genome-wide association, pharmacogenetics
Introduction
What is pharmacogenetics?
Each prescription written constitutes an excursion into the unknown that is fundamental to the balance of risk and benefit in medicine. Will the patient respond fully, partially or not at all? Will there be unacceptable or even life-threatening adverse effects? Decision making can sometimes be informed by past experience with the drug in that patient as treatment responses are more similar within than between individuals. It is also true that responses aggregate within families and are most similar between monozygotic twins[1]. This genetic component to drug response has been documented and investigated for over 50 years, largely through a time of paucity of genetic markers with which to associate observations. However, the advent of high throughput genetic platforms, such as those used in genome wide association studies (GWAS) and in deep sequencing approaches for the identification of intermediate frequency or rare polymorphisms, has provided the field with a timely opportunity which allows much more extensive exploration of the effects of genetic variation on treatment response. Pharmacogenetics is the study of the clinically relevant inherited differences in drug response that can be in part explained by genetic variations. In this review we will consider pharmacogenetics also to include pharmacogenomics, a term increasingly applied when a whole genome approach is used to investigate or predict drug responses, or to relate the application of these technologies to drug discovery.
The increasing role of pharmacogenetics
Pharmacogenetics has clear potential to influence the practice of medicine. The selection of drugs based upon a one-off acquisition of genetic information has the potential to facilitate the selection of the most efficacious drugs at the optimal dose, and avoid many severe adverse effects. Translational research in pharmacogenetics is a priority for major institutions such as the FDA and NIH [2]. Furthermore, as prices for dedicated genetic testing and whole-genome screens continue to fall below that of a private whole body CT scan, there will be an increasing pressure arising from patients presenting with genetic results that we must then interpret. However, at present there are relatively few clear examples where pharmacogenetic information has led to a change in the recommended guidance in the use of individual drugs, although examples are increasing in number. A table of drugs where regulatory guidance suggests the use of genetic testing to help guide prescribing behaviour is given in Table 1.
Table 1.
Genomic markers mentioned on FDA approved drug labels (tumour expression profiles excluded)
| Gene | Example drugs | Effect of polymorphism | FDA label recommendation |
|---|---|---|---|
| CYP2C19 | Clopidogrel | Reduced metabolism of clopidogrel, lower exposure to active metabolite and, thus, higher cardiovascular risk | Warning of potential effect; consider alternative treatment |
| Voriconazole | Reduced metabolism of voriconazole, thus increased drug exposure | Warning of potential effect | |
| CYP2C9 | Warfarin | Reduced metabolism resulting in higher bleeding risk | Warning of potential effect; reduced dose may be required |
| CYP2D6 | Codeine | Leads to ultra-rapid metabolism to active metabolite | Warning of potential effect; use lowest effective dose |
| Tamoxifen | Poor metabolizers have higher plasma concentrations | Warning of potential effect | |
| G6PD | Rasburicase | G6PD deficiency leads to severe haemolysis with several drugs | Screen patients in high risk ethnic groups to exclude risk genotype before commencement |
| HLA-B*1502 | Carbamazepine | Greatly increased risk of severe dermatological hypersensitivity reaction | Screen patients in high ethnic risk groups to exclude genotype before commencement |
| Abacavir | Greatly increased risk of generalized hypersensitivity reaction | Screen all patients prior to drug commencement | |
| LDLR | Atorvastatin | LDL receptor deficiency or absence leads to familial hypercholesterolaemia | Dose adjustments in genetic risk groups |
| NAT | Rifampicin | Slow metabolism and greater drug exposure | Warning of potential effect |
| TPMT | Azathioprine | Slower metabolism and greater resultant risk of myelotoxicity | Screen all patients prior to drug commencement |
| UGT1A1 | Irinotecan | Reduced metabolism and increased neutropenia risk | Reduce initial dose in those known to be homozygous for UGT1A*28 |
| Nilotinib | Exacerbation of jaundice caused by drug | Warning of potential effects | |
| Urea cycle disorders | Valproate | Six potential culprit genes | Evaluate for UCD before commencement |
| VKORC1 | Warfarin | SNPs associated with lower dose requirements | Warning of potential effects |
Data taken from FDA website 25 August 2010.
In this review we outline the progress in pharmacogenetics from early successes, increasing functional data, and initial stratified clinical trials, through to the genomic era and end with a discussion of future clinical and research challenges.
Disease-related polymorphisms and pharmacotherapy
Current treatments
Although this article is not concerned primarily with genetic predisposition to disease, there are clearly a number of instances in which genetic polymorphisms have been identified because they lead to an intermediate phenotype and that subsequently influences the choice or duration of therapy.
A common clinical example is the single nucleotide polymorphism rs6025 in the gene FV known as Factor V Leiden. It was identified as the cause of activated protein C deficiency in 1994, and occurs in around 5% of the general population but 20% of those with thrombo-embolic events [3]. People homozygous for the polymorphism are 50 times more likely to have a venous thrombosis than those in the general population, and the presence of other relatively common mutations (e.g. Prothrombin 20210A) greatly increases this risk in a fashion that departs from what would be expected statistically (epistasis). The presence of these polymorphisms leads to individual tailoring of standard thromboprophylaxis interventions or course of anticoagulation for a thrombotic event.
Aside from thrombophilia, SNPs can also lead to other intermediate phenotypes which confer risk and influence the choice of medication. The genetic basis for the long QT syndrome has been extensively investigated, with culprit polymorphisms in a number of genes including KCNH2. Individuals with mutations in such genes risk ventricular dysrhythmia with common drugs such as macrolide antibiotics [4].
Another important aspect is that understanding the genetic basis for disease can contribute to diagnosis (by defining a subgroup of patients with a specific phenotype) and treatment. One excellent example of this is the identification of the genetic defect underlying some patients presenting with neonatal diabetes. A proportion of these patients have a mutation in the KCNJ11 gene, which codes for the ATP-sensitive K+ channel. This channel regulates insulin secretion, but in patients with activating mutations, the channel remains open despite elevation in intracellular ATP concentrations, thus resulting in reduced insulin secretion. These patients were usually treated with insulin in the past. However, because sulphonylureas such as glibenclamide act through an ATP-independent mechanism to close the channel, patients can be converted from insulin to a sulphonylurea, with improvement in glycaemic control compared with that achieved previously with insulin [5]. Hence, in this example, not only has understanding of the genetic defect in this subgroup of patients with diabetes led to redefining disease phenotypes based on genetic information, but in addition this understanding has led to altered treatment approaches with improved clinical outcomes.
Drug discovery
One consequence of the use of GWAS approaches for the study of common diseases or subphenotypes of disease is that a large number of previously unsuspected genes and pathways are being identified which appear to contribute to the pathophysiology of disease. For example, in two recent meta-analyses of GWAS data performed to identify genetic factors predicting lung function in the general population, several novel associations were identified: four were common to both analyses, namely HHP, AGER, HTR4 and GSTCD[6, 7]. While it does not automatically follow that targeting these genes will prove an effective therapeutic intervention, it is likely that at least some of these genes or associated pathways may provide novel strategies for therapy. Clearly it is easier to study this when the function of the proteins coded for by the genes identified (such as the serotonin receptor HTR4 in the above studies) are well understood, and where small molecule agonists or antagonists already exist, than for those genes such as GSTCD for which little is known in terms of function.
Polymorphisms influencing drug response
Drug disposition
Early research efforts focused on the genetics of drug metabolism as several drugs exhibit marked toxic adverse events that aggregate within families or along racial groups. The commonest clinical example of a single gene variation with a major clinical effect is thiopurine S-methyl transferase (TPMT) in the context of therapy with azathioprine or mercaptopurine. Several single nucleotide polymorphisms (SNPs) can each lead to an inactive form of this enzyme and thus active thioguanine nucleotides accumulate in tissues with resultant haemapoetic toxicity. However, subsequent research has delineated a more complex relationship between TPMT genotype and phenotype, highlighting the need to consider both clinical and additional genetic influences (such as polymorphisms in inosine triphosphate pyrophosphatase [8] in this instance).
Family studies of the observed variation in response to the antihypertensive debrisoquine led to the identification of SNPs in CY2D6 leading to its inactivity in around 10% of the population. These variants also influence the metabolism of codeine (reduce conversion to the active metabolite) and antidepressant medications (increase toxicity). Recent papers have also highlighted the effect of CYP2D6 genotype on the efficacy of tamoxifen for metastatic breast cancer [9], and brought to the fore concerns over the co-prescription of medications sharing this metabolic pathway (e.g. serotonergic antidepressants and tamoxifen). Genetic variation can affect drug transport as well as metabolism, with the best studied example being that of MDR1 which codes for an ATP-binding cassette membrane transporter, P-glycoprotein. Two SNPs within this gene influence its action in the excretion of xenobiotics and metabolites, and have demonstrable effects on plasma concentrations of common medications such as digoxin and fexofenadine [10].
Influential variants need not lie in coding regions of the genes nor be single base changes. For example, a common version of the promoter region tandem repeat polymorphism in the gene encoding the glucoronosyltransferase that conjugates bilirubin (UGT1A1) leads to Gilbert's syndrome. The protein is also responsible for the conjugation of several drugs, so this polymorphism can also lead to the toxic accumulation of active metabolites such as for the chemotherapy agent irinotecan. A commercial test is available for this polymorphism and is referred to in the product literature for irinotecan, though the clinical utility of this test is still debated [11].
Drug response
While the magnitude of effect of polymorphisms influencing drug metabolism can be large, in general, the work to date examining markers for efficacy rather than adverse drug reactions has not led to major changes in prescribing activity. A good example is the extensive work undertaken examining responses to β2-adrenoceptor agonists. The human β2-adrenoceptor is the target for the most commonly used medications in obstructive airways diseases, and studies on the gene ADRB2 have highlighted many of the difficulties in assessing the influence of genetic variants and translating this to clinically relevant outcomes. The region of ADRB2 is highly polymorphic and includes three non-synonymous coding region polymorphisms with functional effects on receptor down-regulation, ligand-binding and adenyl cyclase. However, study of these polymorphisms in vitro does not account for the influence of neighbouring SNPs, and commonly seen combinations of genotypes across the gene region (haplotypes) vary between racial groups, casting doubt on the applicability of such data. Several small studies investigated the effect of ADRB2 coding polymorphisms on bronchodilator response, and this has been followed by retrospective genotyping of a large clinical trial of a short-acting β-adrenoceptor agonist, then a small genotype-stratified prospective crossover trial [12]. Overall, individuals who were homozygous for the polymorphism leading to an arginine at position 16 (Arg16) had lower FEV1 and PEFR, and higher symptom scores compared with those possessing glycine (Gly16) in response to frequent or regular inhaled short-acting beta-agonists [13]. However, while these data seem robust, they are not relevant to the currently accepted guidance for the use of β-adrenoceptor agonists in asthmatic patients and scope for meta-analysis is notably limited by significant heterogeneity in the outcomes considered across studies. Debate has therefore ensued over whether these results can be extended to long-acting β-adrenoceptor agonists (LABAs), especially used in the clinically relevant fashion alongside inhaled corticosteroids. Retrospective analysis of patients treated in clinical trials with LABAs has failed to identify genotypic specific effects on efficacy [14]. Prospective, randomized, trials of the addition of salmeterol or placebo to inhaled corticosteroid and the use of salmeterol with or without corticosteroid found benefit from LABA use for all genotype groups (retrospectively genotyped), and no significant difference between them [15, 16]. Persisting questions remain as to the aetiology of the excess mortality seen particularly in African Americans in response to LABA therapy. Whether or not genetic markers may predict treatment response to the new group of ‘ultra-LABAs’ such as the recently launched indacaterol [17] remains to be determined.
Although not the focus of this review, perhaps the best evidence for the use of genetic approaches to predict efficacy comes from the oncology arena. Here it is the tumour genetic abnormality which can be used to guide therapy. Perhaps the best example is in the use of trastuzumab to treat breast cancer in individuals with tumours which are HER-2 positive [18].
In the above sections we have mentioned notable examples of ‘low-hanging fruit’ where single SNPs have a major impact on phenotype. The story of research into the influence of ADRB2 polymorphisms serves as a reminder that the situation is usually much more complex. Most drugs act through, and are metabolized by, complex pathways which include partial redundancy and that have components that are not readily inactivated in the presence of a single SNP (canalization). In addition, these pathways are often not fully elucidated and gene–environmental interaction may have a profound effect on drug effectiveness. In recent years, the plethora of genetic marker data that has become available and the falling cost of genotyping have enabled a hypothesis-free approach to seek a bank of polymorphisms that can explain part of the variability in observed drug response, and shed light on previously unrecognized aspects of the drug's mode of action. Several genome-wide association studies (GWAS) have now been published for drug response, generating hundreds of thousands of genotypes for participants of clinical trials [19] or for in vitro model systems such as transformed cells with expression data [20].
As with GWAS approaches to the study of disease susceptibility, these studies have both raised questions as to our understanding of the basic mechanisms at work by highlighting SNPs in genes not thought to be relevant previously, and have so far been able to explain less than expected of the observed inter-individual variability. This said, encouragement comes from results of studies of the pharmacogenomics of nicotine. Candidate gene and GWAS have identified SNPs robustly associated with smoking-related phenotypes. Although these polymorphisms explain little of the observed variation in isolation, a panel of up to 60 SNPs (alongside age and gender) in a multivariate predictive model derived using Bayesian networks predicts nicotine dependence with 75% accuracy [21].
Drug toxicity
In common with the previous sections, there have been a small number of successes in identifying polymorphisms underlying the genetic predisposition to some uncommon adverse drug effects. However, more recently studies of more common adverse outcomes have been facilitated by astute use of both integrated association and expression data in cell lines [22], and GWAS of patient populations. Promising examples of the latter approach have been the investigation of flucloxacillin-induced liver injury where replicated association has been seen with HLA type B*5701 with an observed odds ratio of the order of 80 [23], and of statin-induced myopathy where variants in the SLCO1B1 gene explain 60% of cases [24]. Further investigation into mechanisms underlying focal adverse reactions remain necessary, although it could be argued that the mode of action of significantly predictive genotypes need not be fully understood before their clinical application. This is the case for the antiviral drug, abacavir, whose potential to cause a widespread hypersensitivity reaction (that may relatively spare the liver) is also predicted by the HLA-B*5701 genotype. In a double-blind, prospective, randomized study involving almost 2000 individuals, genotype screening prior to commencement of abacavir completely eliminated immunologically confirmed hypersensitivity reactions, and greatly reduced clinically suggestive episodes [25].
Studies investigating the adverse effects of medications have also attempted to address the great majority of inter-individual variability that cannot be explained by whole-genome SNP results (‘missing variation’). Kalari and colleagues, for example, sought association between toxicity of cytidine analogues and copy number variants (CNVs) [26]. CNVs are a relatively recently described class of genomic structural variation with clear potential to influence therapeutic response. They consist of relatively large sections of DNA (1 kilobase to several megabases) that include segmental duplications, deletions or inversions. An accurate estimate of the frequency of these variants is still awaited, although they account for approximately 15% of the genome [27]. The technology available to detect these variants is improving rapidly, with several new generation arrays having been used to show association between gene-dose and disease risk [28]. Pharmacogenetic studies of drug toxicity have also begun to examine pathways [29], although not yet by GWA pathway analyses. This promising approach seeks association between variation in components of a molecular process and a clinical endpoint on the premise that our true target in the discovery phase of genetics is a pathway [30], and that a number or perturbations in a pathway is likely to be required for an effect on phenotype to occur [31]. This appealing concept is currently tempered by statistical challenges, although a good deal of work is ongoing to overcome these issues [32].
Challenges
Research challenges
The challenges facing pharmacogenetics are largely common to all population genetic research. In the first instance detailed phenotyping is required on subjects, who are preferably in receipt of a standardized intervention. Although genotyping in clinical trials is now common, trials are powered to detect a change in a common clinical outcome and so may not be informative with regard to uncommon adverse effects or therapeutic response determined by (relatively uncommon) multilocus genotypes. Trials may also exclude individuals at risk of adverse drug effects through multiple medical conditions or polypharmacy, and full datasets may not be released for academic scrutiny. The need to record accurately and subsequently account for variation in administered medication and potential confounders will continue to be an important feature of such studies.
Researchers face difficult questions as to the design of the genetic analysis in any pharmacogenetic study. The candidate-gene approach, analysis systematically informed by existing metabolic data [33], and hypothesis-free genome-wide analysis all have their place when used appropriately. Increasingly, methods addressing the ‘missing variation’ mentioned above will incorporate studies of copy number variants, sequencing for rare variants in key genes and attempts to address the biologically plausible but statistically challenging field of gene–gene and gene–environment interaction [34] will be used. In the near future, further layers of complexity will be introduced with the increasing study of epigenetics, changes in the genomic environment that lead to altered gene expression can be inherited, or altered by environmental factors or even medications themselves [35].
In all cases, a clear investigation strategy prior to the study is essential to temper enthusiasm for results produced by multiple testing or subgroup analysis in a single population. A major issue is that of replication of results as the aforementioned well-phenotyped and genome-wide genotyped populations are rare commodities. Replication, however, is essential to avoid being misled by initial extreme results [36]. Furthermore, because of the linkage structure and frequencies in the polymorphic background across racial and ethnic groups, a finding uncovering a genetic marker of drug response must also be replicated in diverse populations if it is to be widely clinically employed. A suggested method of investigating adverse drug reactions with genome-wide analysis is shown in Figure 1.
Figure 1.
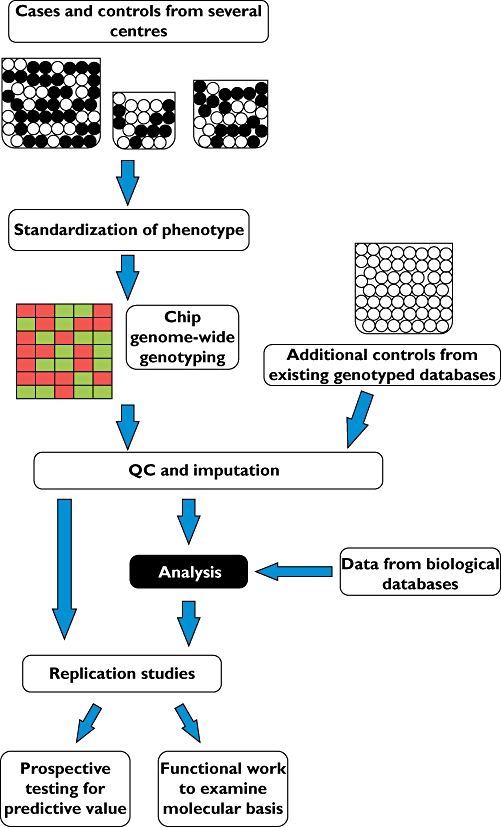
An idealized pathway for the use of genome-wide association data in the investigation of the genetic basis for adverse drug reactions
Although the genome era of pharmacogenetics has promise to influence widely clinical practice, it is worth considering an adage in medicine that has been attributed to William Osler: ‘Common sense in matters medical is rare, and is usually in inverse ratio to the degree of education’. We should not be so taken with the statistical significance of findings that we do not strive to undertake prospective clinical trials to determine predictive values and/or further functional biology research to elucidate basic mechanisms. For example, in our experience the majority of problems with warfarin dosing and side effects on medical wards would seem to relate to suboptimal judgement, communication or concordance rather than the marked effect of common polymorphisms.
Pharmacogenetic approaches have the potential to make existing medications safer and possibly more efficacious, but it should also be noted that they may play a key role in the rediscovery of discarded drugs. For example, it is common practice for compounds that are metabolized by CYP2D6 not to be pursued in drug development in light of the potential problems with common existing therapies such as codeine, fluoxetine, metoprolol and tamoxifen. However, a genetic test could identify those at greatest risk of side effects from these medications and thus allow potentially efficacious drugs to the market with an increased safety profile. Initial drug company concerns over the release of drugs that require pharmacogenetic testing have been at least partially assuaged by the increased prescription of abacavir since testing became available [37]. It may be that the first drug to be reappraised in this manner is lumiracoxib, the cyclo-oxygenase 2 inhibitor that was either not approved or withdrawn because of concerns over hepatotoxicity. A recent GWAS and fine mapping effort has identified a HLA type strongly associated with risk of adverse outcome [38].
Clinical challenges
Presently there are many instances when the dosing of common drugs is influenced by existing patient data such as renal function and age [39]. Problems encountered in achieving compliance with these adjustments (e.g. [40]) hint at the difficulties that may be seen when incorporating genetic information (which has to be specifically requested). Clearly, as the range of medications influenced by genetic testing becomes greater, there will be an increasing onus on interested parties across specialities to disseminate clear information as to the availability of specific tests, when they should be employed and to guide their interpretation. This latter issue is perhaps the most challenging of all, as most genetic tests will influence risk prediction rather than give categorical reassurance of efficacy or freedom from adverse effects. It is likely that pharmacogenetic guidance will need to be increasingly included in national and society guidelines for common conditions and that medical professionals and pharmacists will need further education and support in interpreting these risk estimates [41].
Finally and most importantly, challenges lie in explaining the rationale for and results of pharmacogenetic testing to patients. Experience from dealing with the results of existing complex genetic diseases would suggest that well-informed practitioners will need additional time and supportive resources to discuss genetic results [42]. However, it is as yet unclear how such extended consultations will be accommodated and reimbursed in a pressured NHS, and the likely overall morbidity and cost benefit is yet to be elucidated. In the near future, falling costs and increasing availability will lead to an increase in the number of individuals who have purchased their own genome-wide SNP genotype profile. Such tests are already commercially available for as little as £250 and will soon place a further burden on general practitioners, although company records may well become a resource for future pharmacogenetic study [43].
Ethical challenges
The major current ethical issues arising from pharmacogenetic study are common to many aspects of medicine in that they relate to justice in the allocation of the resources and the autonomy of patients involved in research. It is evident that genetics has consumed a huge quantity of research capital, much of it from the public purse, and although research technologies and information processing move on apace, there is a notable discrepancy in the speed at which findings have been translated into direct clinical benefit. This concerning divergence must also be taken in the context of tremendous worldwide morbidity for want of appropriate medical therapy, and the frequency of adverse drug reactions that are preventable with existing clinical information [39]. Researchers and funding bodies must therefore be vigilant that pharmacogenetics is not an expensive pursuit of knowledge for its own sake, but that it complements existing practice and education in drug prescribing, administration and monitoring.
It has become increasingly clear that pharmacogenetic research will rarely be able to provide clear binary signals describing subpopulations at risk of adverse drug reactions or increased drug benefit. The more common scenarios will be of genetically defined populations with substantially overlapping distributions of drug response. The summary statistics of such subpopulations are unlikely to describe adequately those who are most refractory or responsive to a medication. In such scenarios, genetic testing has greatest benefit for those in the tails of the distributions (‘edge’ effects [44]), especially in the case of rare drug effects. Hence, few individuals who will undergo genetic testing are likely to directly benefit from it, so detailed evaluation of the cost of testing and the severity of the complication prevented (or treatment benefit gained) will be necessary in each case. There is therefore clear scope for adopted and future pharmacogenetic strategies to increase further existing disparities between optimal and universal healthcare provision, creating difficult priority selection issues for national healthcare providers and insurers.
The balance of risk and reward for prospective participants in pharmacogenetic trials also differs from other drug trials. Here, the risks of the drug administered are not diminished in the testing cohort when compared with standard practice and there is no scope for additional benefit as there is no new drug on trial. They additionally carry the risk of misinterpreting the inevitably more complex study design, and investigators must consider concerns over the subsequent handling of their DNA samples. Indeed, the issue of informed consent for medical research has taken on a new dimension with the drawing of samples for genetic analysis within drug trials. Often broad consent allows samples to be stored for protracted periods and used for studies not explicitly detailed. There have also been issues with the mechanism of withdrawal of consent and location of DNA samples as commercial entities merge or relocate. Such issues with commercial and university biobanking are discussed in detail elsewhere, for example, by Corrigan and colleagues [45].
Conclusion
In summary, we have given examples of the promise, increasing translational value and varied challenges in pharmacogenetics. It is to be expected that the current research focus and political will in this area facilitate potential increases in patient safety and drug effectiveness through genetic testing. It is to be hoped that the educated interpretation of results realizes this potential, although the timescale for this major change in the practice of medicine to become commonplace is very difficult to predict.
Acknowledgments
Work in IPH's laboratory is supported by the MRC, Wellcome Trust and Asthma UK. JDB is an NIHR-funded Clinical Lecturer.
Competing Interests
There are no competing interests to declare.
REFERENCES
- 1.Vessel ES. Pharmacogenetic perspectives gained from twin and family studies. Pharmacol Ther. 1989;41:535–52. doi: 10.1016/0163-7258(89)90130-7. [DOI] [PubMed] [Google Scholar]
- 2.Hamburg MA, Collins FS. The path to personalized medicine. N Engl J Med. 2010;363:301–4. doi: 10.1056/NEJMp1006304. [DOI] [PubMed] [Google Scholar]
- 3.Rosendaal FR. Venous thrombosis: a multicausal disease. Lancet. 1999;353:1167–73. doi: 10.1016/s0140-6736(98)10266-0. [DOI] [PubMed] [Google Scholar]
- 4.Aerssens J, Paulussen AD. Pharmacogenomics and the long QT syndrome. Pharmacogenomics. 2008;6:259–70. doi: 10.1517/14622416.6.3.259. [DOI] [PubMed] [Google Scholar]
- 5.Flechtner I, de Lonlay P, Polak M. Diabetes and hypoglycaemia in young children and mutations in the Kir6.2 subunit of the potassium channel: therapeutic consequences. Diabetes Metab. 2006;32:569–80. doi: 10.1016/S1262-3636(07)70311-7. [DOI] [PubMed] [Google Scholar]
- 6.Hancock DB, Eijgelsheim M, Wilk JB, Gharib SA, Loehr LR, Marciante KD, Franceschini N, van Durme YM, Chen TH, Barr RG, Schabath MB, Couper DJ, Brusselle GG, Psaty BM, van Duijn CM, Rotter JI, Uitterlinden AG, Hofman A, Punjabi NM, Rivadeneira F, Morrison AC, Enright PL, North KE, Heckbert SR, Lumley T, Stricker BH, O'Connor GT, London SJ. Meta-analyses of genome-wide association studies identify multiple loci associated with pulmonary function. Nat Genet. 2010;42:45–52. doi: 10.1038/ng.500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Repapi E, Sayers I, Wain LV, Burton PR, Johnson T, Obeidat M, Zhao JH, Ramasamy A, Zhai G, Vitart V, Huffman JE, Igl W, Albrecht E, Deloukas P, Henderson J, Granell R, McArdle WL, Rudnicka AR, Wellcome Trust Case Control Consortium. Barroso I, Loos RJ, Wareham NJ, Mustelin L, Rantanen T, Surakka I, Imboden M, Wichmann HE, Grkovic I, Jankovic S, Zgaga L, Hartikainen AL, Peltonen L, Gyllensten U, Johansson A, Zaboli G, Campbell H, Wild SH, Wilson JF, Gläser S, Homuth G, Völzke H, Mangino M, Soranzo N, Spector TD, Polasek O, Rudan I, Wright AF, Heliövaara M, Ripatti S, Pouta A, Naluai AT, Olin AC, Torén K, Cooper MN, James AL, Palmer LJ, Hingorani AD, Wannamethee SG, Whincup PH, Smith GD, Ebrahim S, McKeever TM, Pavord ID, MacLeod AK, Morris AD, Porteous DJ, Cooper C, Dennison E, Shaheen S, Karrasch S, Schnabel E, Schulz H, Grallert H, Bouatia-Naji N, Delplanque J, Froguel P, Blakey JD, NSHD Respiratory Study Team. Britton JR, Morris RW, Holloway JW, Lawlor DA, Hui J, Nyberg F, Jarvelin MR, Jackson C, Kähönen M, Kaprio J, Probst-Hensch NM, Koch B, Hayward C, Evans DM, Elliott P, Strachan DP, Hall IP, Tobin MD. Genome-wide association study identifies five loci associated with lung function. Nat Genet. 2010;42:36–44. doi: 10.1038/ng.501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stocco G, Cheok MH, Crews KR, Dervieux T, French D, Pei D, Yang W, Cheng C, Pui CH, Relling MV, Evans WE. Genetic polymorphism of inosine triphosphate pyrophosphatase is a determinant of mercaptopurine metabolism and toxicity during treatment for acute lymphoblastic leukemia. Clin Pharmacol Ther. 2009;85:164–72. doi: 10.1038/clpt.2008.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lammers LA, Mathijssen RH, van Gelder T, Bijl MJ, de Graan AJ, Seynaeve C, van Fessem MA, Berns EM, Vulto AG, van Schaik RH. The impact of CYP2D6-predicted phenotype on tamoxifen treatment outcome in patients with metastatic breast cancer. Br J Cancer. 2010;103:765–71. doi: 10.1038/sj.bjc.6605800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brinkmann U, Roots I, Eichbaum M. Pharmacogenetics of the human drug transporter gene MDR1. Drug Discov Today. 2001;6:835–9. doi: 10.1016/s1359-6446(01)01892-x. [DOI] [PubMed] [Google Scholar]
- 11.Marsh S, Hopkins JM. Irinotecan pharmacogenomics. Pharmacogenomics. 2010;11:1003–10. doi: 10.2217/pgs.10.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Israel E, Chinchilli VM, Ford JG, Boushey HA, Cherniack R, Craig TJ, Deykin A, Fagan JK, Fahy JV, Fish J, Kraft M, Kunselman SJ, Lazarus SC, Lemanske RF, Jr, Liggett SB, Martin RJ, Mitra N, Peters SP, Silverman E, Sorkness CA, Szefler SJ, Wechsler ME, Weiss ST, Drazen JM. Use of regularly scheduled albuterol treatment in asthma: genotype-stratified, randomised, placebo-controlled cross-over trial. Lancet. 2004;364:1505–12. doi: 10.1016/S0140-6736(04)17273-5. [DOI] [PubMed] [Google Scholar]
- 13.Sayers I, Hall IP. Pharmacogenetic approaches in the treatment of asthma. Curr Allergy Asthma Rep. 2005;5:101–8. doi: 10.1007/s11882-005-0082-0. [DOI] [PubMed] [Google Scholar]
- 14.Bleecker ER, Postma DS, Lawrance RM, Meyers DA, Ambrose HJ, Goldman M. Effect of ADRB2 polymorphisms on response to longacting β2-agonist therapy: a pharmacogenetic analysis of two randomised studies. Lancet. 2007;370:2118–25. doi: 10.1016/S0140-6736(07)61906-0. [DOI] [PubMed] [Google Scholar]
- 15.Wechsler ME, Kunselman SJ, Chinchilli VM, Bleecker E, Boushey HA, Calhoun WJ, Ameredes BT, Castro M, Craig TJ, Denlinger L, Fahy JV, Jarjour N, Kazani S, Kim S, Kraft M, Lazarus SC, Lemanske RF, Jr, Markezich A, Martin RJ, Permaul P, Peters SP, Ramsdell J, Sorkness CA, Sutherland ER, Szefler SJ, Walter MJ, Wasserman SI, Israel E. Effect of β2 adrenergic polymorphism on response to long-acting β2 agonist in asthma (LARGE trial): a genotype-stratified, randomised, placebo-controlled, crossover trial. Lancet. 2009;374:1754–64. doi: 10.1016/S0140-6736(09)61492-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bleecker ER, Nelson HS, Kraft M, Corren J, Meyers DA, Yancey SW, Anderson WH, Emmett AH, Ortega HG. β2-receptor polymorphisms in patients receiving salmeterol with or without fluticasone propionate. Am J Respir Crit Care Med. 2010;181:676–87. doi: 10.1164/200809-1511OC. [DOI] [PubMed] [Google Scholar]
- 17.Dahl R, Chung KF, Buhl R, Magnussen H, Nonikov V, Jack D, Bleasdale P, Owen R, Higgins M, Kramer B. Efficacy of a new once daily long-acting beta agonist indacaterol versus twice daily formoterol in COPD. Thorax. 2010;65:473–9. doi: 10.1136/thx.2009.125435. [DOI] [PubMed] [Google Scholar]
- 18.Mackey J, McLeod D, Ragaz J, Gelmon K, Verma S, Pritchard K, Laing K, Provencher L, Charbonneau LF. Adjuvant targeted therapy in early breast cancer. Cancer. 2009;115:1154–68. doi: 10.1002/cncr.24114. [DOI] [PubMed] [Google Scholar]
- 19.Barber MJ, Mangravite LM, Hyde CL, Chasman DI, Smith JD, McCarty CA, Li X, Wilke RA, Rieder MJ, Williams PT, Ridker PM, Chatterjee A, Rotter JI, Nickerson DA, Stephens M, Krauss RM. Genome-wide association of lipid-lowering response to statins in combined study populations. PLoS ONE. 2010;5:e9763. doi: 10.1371/journal.pone.0009763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li L, Fridley BL, Kalari K, Jenkins G, Batzler A, Weinshilboum RM, Wang L. Gemcitabine and arabinosylcytosine pharmacogenomics: genome-wide association and drug response biomarkers. PLoS ONE. 2009;4:e7765. doi: 10.1371/journal.pone.0007765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ramoni RB, Saccone NL, Hatsukami DK, Bierut LJ, Ramoni MF. A testable model of nicotine dependence. J Neurogenet. 2009;23:283–92. doi: 10.1080/01677060802572911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Huang RS, Duan S, Bleibel WK, Kistner EO, Zhang W, Clark TA, Chen TX, Schweitzer AC, Blume JE, Cox NJ, Dolan ME. A genome-wide approach to identify genetic variants that contribute to etoposide-induced cytotoxicity. Proc Natl Acad Sci USA. 2007;104:9758–63. doi: 10.1073/pnas.0703736104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Daly AK, Donaldson PT, Bhatnagar P, Shen Y, Pe'er I, Floratos A, Daly MJ, Goldstein DB, John S, Nelson MR, Graham J, Park BK, Dillon JF, Bernal W, Cordell HJ, Pirmohamed M, Aithal GP, Day CP. HLA-B*5701 genotype is a major determinant of drug-induced liver injury due to flucloxacillin. Nat Genet. 2009;41:816–9. doi: 10.1038/ng.379. [DOI] [PubMed] [Google Scholar]
- 24.The SEARCH Collaborative Group. SLCO1B1 variants and statin-induced myopathy – a genome-wide study. N Engl J Med. 2008;359:789–99. doi: 10.1056/NEJMoa0801936. [DOI] [PubMed] [Google Scholar]
- 25.Mallal S, Phillips E, Carosi G, Molina JM, Workman C, Tomazic J, Jägel-Guedes E, Rugina S, Kozyrev O, Cid JF, Hay P, Nolan D, Hughes S, Hughes A, Ryan S, Fitch N, Thorborn D, Benbow A. HLA-B*5701 screening for hypersensitivity to abacavir. N Engl J Med. 2008;358:568–79. doi: 10.1056/NEJMoa0706135. [DOI] [PubMed] [Google Scholar]
- 26.Kalari KR, Hebbring SJ, Chai HS, Li L, Kocher JP, Wang L, Weinshilboum RM. Copy number variation and cytidine analogue cytotoxicity: a genome-wide association approach. BMC Genomics. 2010;11:357. doi: 10.1186/1471-2164-11-357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Estivill X, Armengol L. Copy number variants and common disorders. PLoS Genet. 2007;3:e190. doi: 10.1371/journal.pgen.0030190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ibáñez P, Bonnet AM, Débarges B, Lohmann E, Tison F, Pollak P, Agid Y, Dürr A, Brice A. Causal relation between alpha synuclein gene duplication and familial Parkinsons disease. Lancet. 2004;364:1167–69. doi: 10.1016/S0140-6736(04)17104-3. [DOI] [PubMed] [Google Scholar]
- 29.van Erp NP, Eechoute K, van der Veldt AA, Haanen JB, Reyners AK, Mathijssen RH, Boven E, van der Straaten T, Baak-Pablo RF, Wessels JA, Guchelaar HJ, Gelderblom H. Pharmacogenetic pathway analysis for determination of sunitinib-induced toxicity. J Clin Oncol. 2009;27:4406–12. doi: 10.1200/JCO.2008.21.7679. [DOI] [PubMed] [Google Scholar]
- 30.Hirschhorn JN. Genomewide association studies – illuminating biologic pathways. N Engl J Med. 2009;360:1699–701. doi: 10.1056/NEJMp0808934. [DOI] [PubMed] [Google Scholar]
- 31.Moore JH. A global view of epistasis. Nat Genet. 2005;37:13–4. doi: 10.1038/ng0105-13. [DOI] [PubMed] [Google Scholar]
- 32.Cantor RM. Prioritizing GWAS results: a review of the statistical methods and recommendations for their application. Am J Hum Gen. 2010;86:6–22. doi: 10.1016/j.ajhg.2009.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Saccone SF, Bolze R, Thomas P, Quan J, Mehta G, Deelman E, Tischfield JA, Rice JP. SPOT: a web-based tool for using biological databases to prioritize SNPs after a genome-wide association study. Nucleic Acids Res. 2010;38:W201–9. doi: 10.1093/nar/gkq513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cordell HJ. Detecting gene-gene interactions that underlie human diseases. Nat Rev Genet. 2009;10:392–404. doi: 10.1038/nrg2579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rodenhiser D, Mann M. Epigenetics and human disease: translating basic biology into clinical applications. CMAJ. 2006;174:341. doi: 10.1503/cmaj.050774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ioannidis JP, Trikalinos TA. Early extreme contradictory research estimates may appear in published research: The Proteus phenomenon in molecular genetics research and randomised controlled trials. J Clin Epidemiol. 2005;58:543–9. doi: 10.1016/j.jclinepi.2004.10.019. [DOI] [PubMed] [Google Scholar]
- 37.Ingelman-Sundberg M. Pharmacogenomic biomarkers for prediction of severe adverse drug reactions. N Engl J Med. 2008;358:637–9. doi: 10.1056/NEJMe0708842. [DOI] [PubMed] [Google Scholar]
- 38.Singer JB, Lewitzky S, Leroy E, Yang F, Zhao X, Klickstein L, Wright TM, Meyer J, Paulding CA. A genome-wide study identifies HLA alleles associated with lumiracoxib-related liver injury. Nat Genet. 2010;42:711–4. doi: 10.1038/ng.632. [DOI] [PubMed] [Google Scholar]
- 39.Pirmohamed M, James S, Meakin SGreen C, Scott AK, Walley TJ, Farrar K, Park BK, Breckenridge AM. Adverse drug reactions as cause of admission to hospital: prospective analysis of 18 820 patients. BMJ. 2004;329:15–9. doi: 10.1136/bmj.329.7456.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pillans PI, Landsberg PG, Fleming AM, Fanning M, Sturtevant JM. Evaluation of dosage adjustment in patients with renal impairment. Intern Med J. 2003;33:10–3. doi: 10.1046/j.1445-5994.2003.00330.x. [DOI] [PubMed] [Google Scholar]
- 41.Frueh FW, Goodsaid F, Rudman A, Huang SM, Lesko LJ. The need for education in pharmacogenomics: a regulatory perspective. Pharmacogenomics J. 2005;5:218–20. doi: 10.1038/sj.tpj.6500316. [DOI] [PubMed] [Google Scholar]
- 42.Meilleur KG, Littleton-Kearney MT. Interventions to improve patient education regarding multifactorial genetic conditions: a systematic review. Am J Med Genet A. 2009;149A:819–30. doi: 10.1002/ajmg.a.32723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Prainsack B, Wolinsky H. Direct-to-consumer genome testing: opportunities for pharmacogenomics research? Pharmacogenomics. 2010;11:651–5. doi: 10.2217/pgs.10.33. [DOI] [PubMed] [Google Scholar]
- 44.Kalow W. Both populations and individuals are evolutionary targets. Pharmacogenomics J. 2002;2:12–4. doi: 10.1038/sj.tpj.6500083. [DOI] [PubMed] [Google Scholar]
- 45.Tutton R, Corrigan OP. Genetic Databases: Socio-Ethical Issues in the Collection and Use of DNA. London: Routledge; 2004. [Google Scholar]