

Abstract
AIMS
The specific TP receptor antagonist terutroban improves endothelial function after a single dose in patients with coronary artery disease. Our aim was to evaluate the effects and dose dependency of repeated-dose terutroban on endothelial function and platelet aggregation in high-cardiovascular-risk patients with carotid atherosclerosis.
METHODS
We randomly allocated 48 patients taking 300 mg aspirin per day to placebo or to one of three terutroban dosages (2.5, 5 or 10 mg) for 15 days in a double-blind study. Flow-mediated vasodilatation was evaluated before and 2 h after the first oral dose on day 0 and 2 h after the last oral dose on day 14.
RESULTS
On day 0 and day 14, all three terutroban dosages improved flow-mediated vasodilatation and abolished platelet aggregation induced by the TP receptor agonist U46619, without changing the aggregation response to ADP or collagen.
CONCLUSION
Terutroban, by chronically improving endothelium-dependent vasodilatation and inhibiting platelet aggregation, may prove useful for preventing cardiovascular events in high-risk patients.
Keywords: antiplatelet therapy, atherosclerosis, endothelium, terutroban, thromboxane receptor
WHAT IS ALREADY KNOWN ABOUT THIS SUBJECT
Terutroban is a selective TP receptor antagonist, i.e. a specific antagonist of the thromboxane A2 and prostaglandin endoperoxide receptors, shown to improve endothelial function after a single administration in patients with coronary artery disease.
WHAT THIS STUDY ADDS
This randomized, double-blind, placebo-controlled trial demonstrates that repeated-dose terutroban for 15 days improves endothelial function and inhibits thromboxane A2-induced platelet aggregation in high-cardiovascular-risk patients taking 300 mg of aspirin per day. Terutroban may prove useful for preventing cardiovascular events in such patients.
Introduction
Atherosclerosis is a chronic vascular disease in which inflammation, endothelial dysfunction and platelet hyperactivity play crucial roles. Endothelial dysfunction, an early phenomenon in atherosclerosis that often antedates the structural changes and clinical manifestations [1], is characterized by deficient release by the endothelial cells of vasodilating substances such as nitric oxide, prostacyclin and endothelium-derived hyperpolarizing factor [2]. One of the current hypotheses is that impairment of endothelium-dependent relaxation in atherosclerosis may be accompanied by a propensity for releasing endothelium-contracting factors, such as thromboxane A2 (TXA2), superoxide anions and the peptide endothelin [3]. One consequence of endothelial dysfunction is enhancement of platelet–endothelial cell interactions, which leads to increased TXA2 levels.
Thromboxane A2 is a vasoconstricting substance produced from arachidonic acid via the cyclo-oxygenase pathway. It binds to a specific membrane-bound thromboxane prostaglandin (TP) receptor, thereby promoting vasoconstriction, platelet aggregation and cellular proliferation. However, TP receptors can bind to other vasoconstrictor prostanoids, such as isoprostanes [4], which are non-enzymatic products of cell membrane phospholipids. Isoprostanes are released in response to oxidative stress [5] in diseases such as hypercholesterolaemia [6, 7], diabetes mellitus [8] and acute coronary syndromes [9].
Aspirin, which decreases the risk of myocardial infarction and stroke by inhibiting TXA2 release from platelets, inhibits neither the release of TXA2 from nonplatelet sources nor the formation of isoprostanes. Therefore, an important question is whether vasoconstrictor prostanoids (TXA2 and isoprostanes) contribute to endothelial dysfunction and affect vascular tone even in patients treated with aspirin.
Terutroban, previously called S18886, is a selective TP receptor antagonist, i.e. a specific antagonist of the TXA2 and prostaglandin endoperoxide receptors [10]. Terutroban is a potent antithrombotic agent and also possesses antiatherosclerotic [11, 12] and antivasoconstrictor properties [13]. The antithrombotic properties of terutroban have been investigated in animal models [14, 15], including ex vivo models of thrombosis in a Badimon chamber [16]. Ex vivo experiments have also been performed in patients previously treated with aspirin for the primary or secondary prevention of ischaemic stroke. In this setting, the antithrombotic effect of terutroban was greater than that of aspirin and similar to that of the clopidogrel–aspirin combination [17]. The antivasoconstrictor properties of a single oral 10 mg dose of terutroban were investigated in 20 patients with coronary artery disease (CAD) who were on aspirin at a dosage of 100 mg day−1[10]. Endothelial function was assessed based on flow-mediated vasodilatation (FMD) and acetylcholine (ACh)-induced vasodilatation. The single terutroban dose was followed by an FMD increase and by significant potentiation of the response to ACh compared with the placebo, with no change in the effect of sodium nitroprusside. These results suggest that the release of endogenous TP receptor agonists may contribute to induce endothelial dysfunction despite aspirin treatment in patients with CAD. However, this study did not determine whether the effects of terutroban persisted after repeated dosing, nor did it identify the lowest effective dose. Moreover, aspirin was used in the previous study only at a dosage of 100 mg day−1, raising the possibility that terutroban might not be effective in patients given higher aspirin dosages of up to 300 mg day−1, which are widely prescribed.
The present study was designed to investigate the following three hypotheses in high-cardiovascular-risk patients with atherosclerosis: (i) terutroban in repeated doses is effective in improving endothelium-dependent vasodilatation; (ii) terutroban is effective in low doses; and (iii) terutroban improves endothelium-dependent vasodilatation in patients taking larger aspirin doses (300 mg) than in the previous study (100 mg). We consequently designed a randomized, double-blind, placebo-controlled trial of terutroban (2.5, 5 or 10 mg day−1) in patients with carotid atherosclerosis (elevated carotid intima–media thickness) who were already taking 300 mg day−1 aspirin. Endothelial function was evaluated based on FMD, and blood samples were collected for measurement of ex vivo platelet aggregation.
Patients and methods
Inclusion and exclusion criteria
Men aged 40–80 years and postmenopausal women aged 55–80 years were included if they were ambulatory and had both carotid atherosclerosis, defined as intima–media thickness ≥0.75 mm at the common carotid artery examined using high-resolution B-mode ultrasonography within the past 3 months, and proven forearm endothelial dysfunction, defined as FMD <4.1% during a hyperaemia test. In addition, patients were selected based on a previous history of cardiovascular disease (myocardial infarction, coronary artery disease, angina, ischaemic stroke, haemorrhagic stroke, transient ischaemic attack and/or peripheral artery disease), or a Framingham score greater than 15 for men or 18 for women (defining a 10 year risk of cardiovascular disease greater than 20%) [18, 19]. We did not include patients with any of the following conditions: severe carotid artery stenosis (≥70% reduction in diameter of the common or internal carotid artery), history of intracerebral haemorrhage, myocardial infarction in the past 3 months, polycythaemia, heart failure, cardiac arrhythmias, uncontrolled hypertension, bleeding disorders, hypersensitivity to aspirin, or type 1 diabetes, platelet count <100 × 109 l−1 or >600 × 109 l−1, prothrombin time <70%, haemoglobin <10 g dl−1, or activated partial thromboplastin time >8 s longer than the control value. No anti-inflammatory drugs other than aspirin were allowed. Among the 48 included patients, 32 had a previous history of cardiovascular disease and the other 16 had a Framingham score indicating a high cardiovascular risk. All patients gave written informed consent before study inclusion, and the study protocol was approved by the independent ethics committee of the Henri Mondor Teaching Hospital (Créteil, France). The study was conducted in accordance with the ethical principles of the Declaration of Helsinki.
Study design and treatment
This was a randomized, double-blind, placebo-controlled, single-centre trial. All patients received prophylactic aspirin at a higher dosage (300 mg day−1) than usual for at least 10 days prior to inclusion and throughout the study period. Patients were randomly allocated to one of four parallel groups treated with terutroban 2.5, 5 or 10 mg or a placebo orally, once daily for 15 consecutive days, which allowed the terutroban levels to reach the steady state. The number of patients was set at 12 completed patients per group, based on previous studies [10]. Terutroban and placebo capsules were provided by Institut de Recherches Internationales Servier (Courbevoie, France).
Evaluation of flow-mediated vasodilatation
Flow-mediated vasodilatation was measured before and 2 h after the first study-drug dose on day 0 (D0) and 2 h after the last study-drug dose on day 14 (D14). The method used to measure FMD has been described elsewhere [10, 20]. All measurements were performed after a 30 min rest in the supine position in a temperature-controlled room at 22°C with continuous blood-pressure monitoring. A high-resolution ultrasound Wall Track system (Pie Medical, Maastricht, The Netherlands) with a 7 MHz linear probe was used to measure the internal diastolic diameter (with a precision of 30 µm) of the distal brachial artery after 5 min ischaemia of the ipsilateral hand induced by inflating a wrist cuff at 200 mmHg (hyperaemia test). With this test, cuff deflation causes a transient increase in shear stress, which induces endothelial NO release and therefore flow-dependent vasodilatation. The maximal diameter (MD) was defined as the greatest diastolic diameter measured after cuff deflation. Measurements were made at deflation over five or six cardiac cycles, then at 30 s intervals for 3 min. The diameter at deflation was taken as the basal (minimal) diameter (BD). The FMD (%) was defined as [(MD – BD)/BD] × 100.
Ex vivo platelet aggregation tests
Blood (15 ml) was collected into a standard lithium heparin tube maintained at 4°C in 0.129 mol l−1 sodium citrate (3.8% w/v). Blood samples were drawn before and 2 h after the first dose on D0 and 2 h after the last dose on D14. The first few drops of each sample were discarded. After centrifugation (1500g, 10 min, 25°C), the platelet-rich plasma preparation was adjusted to 300 × 109 platelets l−1. If the platelet count was <150 × 109 l−1, the sample was discarded. Aggregation was assessed within 3 h of sample collection using an aggregometer (Beckman Coulter, Brea, CA, USA) by operators blinded to the study treatment. Platelet aggregation was induced by the selective TP receptor agonist U46619 (7 µmol l−1; Calbiochem), collagen (2 µg ml−1) or ADP (5 µmol l−1). Results are expressed as percentage of maximal platelet aggregation.
Safety data
Bleeding time was determined 4 h after the last study drug dose on D14. If the value on D14 was >12 min, it was determined again on day 35. We used the Ivy Nelson method with a Simplate® 1R device (Organon Technika, Eppelheim, Germany) at the forearm, with a blood pressure cuff on the upper arm inflated at 40 mmHg throughout the procedure. We collected the following data at baseline, on D14, and at follow-up completion on day 35: physical findings, adverse events, cardiovascular parameters (supine blood pressure, heart rate and electrocardiogram), blood biochemical parameters and haematological parameters.
Statistical analysis
Statistical analyses of FMD and platelet aggregation 2 h after the first dose (D0) and last dose (D14) were conducted on the per-protocol populations. The treatment effect was estimated using analysis of covariance with the baseline value as a covariate, followed by Dunnett's test to compare each terutroban dose vs. placebo. In each treatment group, the mean and SD were computed for each variable. Associated P values were determined for each terutroban–placebo comparison. To assess change over time in each treatment group, Student's paired t-test was used. For all analyses, P < 0.05 was considered significant. Safety analyses were done on the patients exposed to the study treatment.
Results
Study population
Of 51 patients who were screened and selected for the study, 48 patients were randomized (Figure 1). The per-protocol population included 47 patients on D0 (one patient in the 2.5 mg terutroban group was excluded due to noncompliance with aspirin treatment) and 46 patients on D14 (one patient in the 10 mg terutroban group was excluded due to nonperformance of the evaluation planned for D14). All 48 patients were included in the safety analysis.
Figure 1.
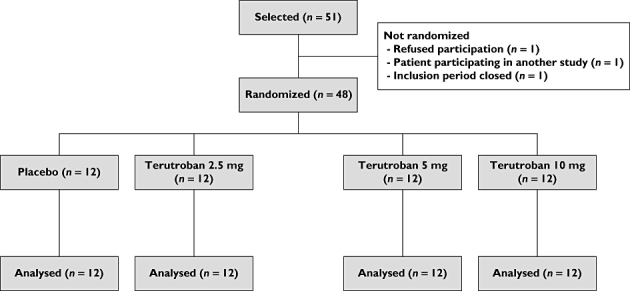
Patient flow chart
There were no differences in baseline characteristics across the four treatment groups (n = 12 in each group; Table 1). Mean age was 63.5 ± 8.7 years overall and mean intima–media thickness was 0.91 ± 0.09 mm.
Table 1.
Main baseline characteristics and medical history of randomized patients
| Placebo (n = 12) | Terutroban 2.5 mg day−1(n = 12) | Terutroban 5 mg day−1(n = 12) | Terutroban 10 mg day−1(n = 12) | |
|---|---|---|---|---|
| Age (years) | 63 ± 9 | 60 ± 8 | 64 ± 9 | 67 ± 8 |
| Male, n | 12 | 12 | 10 | 10 |
| Body mass index (kg m−2) | 28 ± 3 | 27 ± 3 | 28 ± 3 | 26 ± 3 |
| Heart rate (beats min−1) | 65 ± 4 | 63 ± 6 | 60 ± 8 | 65 ± 7 |
| Systolic blood pressure (mmHg) | 140 ± 17 | 135 ± 12 | 142 ± 15 | 142 ± 15 |
| Diastolic blood pressure (mmHg) | 83 ± 9 | 77 ± 8 | 83 ± 7 | 80 ± 13 |
| Blood glucose (mmol l−1) | 8 ± 4 | 7 ± 2 | 7 ± 3 | 6 ± 1 |
| Intima–media thickness (mm) | 0.95 ± 0.10 | 0.90 ± 0.10 | 0.92 ± 0.08 | 0.87 ± 0.08 |
| Disorders of lipid metabolism, n | 7 | 9 | 4 | 7 |
| History of essential hypertension, n | 9 | 4 | 8 | 5 |
| History of myocardial infarction, n | 3 | 3 | 2 | 2 |
| History of type 2 diabetes, n | 4 | 2 | 4 | – |
| History of angina pectoris, n | 4 | 1 | 2 | 2 |
| Concomitant treatment | ||||
| β-Blockers, n | 2 (16.7) | 7 | 9 | 7 |
| HMG CoA reductase inhibitors, n | 9 (75.0) | 8 | 6 | 6 |
| Angiotensin-converting enzyme inhibitors, n | 6 (50.0) | 1 | 4 | 3 |
| Calcium channel blockers, n | 2 (16.7) | – | 2 | 4 |
Unless otherwise stated, values are means ± SD.
Six patients had documented carotid stenoses between 30 and 67%. Most patients (97.9%) had a history of previous disease such as dyslipidaemia (56.3%), essential hypertension (54.2%), coronary atherosclerosis (29.2%), acute myocardial infarction (20.8%), type 2 diabetes (20.8%) and angina pectoris (18.8%). At inclusion, all patients were receiving concomitant treatments, including medications used to treat atherosclerosis (statins, 60.4%; β-blockers, 52.1%; and/or angiotensin-converting enzyme inhibitors, 29.2%).
The mean treatment duration was 14.9 ± 1.1 days. No patient withdrew from the study or was lost to follow-up. Mean overall treatment compliance (as estimated by counting unused tablets) was 99.9%.
Flow-dependent vasodilatation
The hyperaemia test at baseline induced comparable arterial vasodilatation in all groups, with a mean FMD change ranging across groups from 2.16 to 2.70%, indicating the presence of endothelial dysfunction. Two hours after the first 2.5 mg terutroban dose (D0), mean FMD significantly increased by 92%, to 4.14 ± 1.25% [95% confidence interval (CI) of the difference, 1.23–2.73; P < 0.001 vs. baseline; Table 2 and Figure 2]. This increase was sustained over the next 2 weeks, with the mean FMD on D14 being 4.42 ± 1.22% (95% CI of the difference, 1.60–2.91; P < 0.001 vs. baseline). Both postdrug values were significantly different from those recorded at the same time points in the placebo group (both P < 0.001).
Table 2.
Flow-mediated vasodilatation in the per-protocol population at baseline and 2 h after the first study drug dose on day 0 and 2 h after last study drug dose on day 14
| Flow-mediated vasodilatation (%) | |||||||
|---|---|---|---|---|---|---|---|
| Baseline | 2 h after intake on day 0 | 2 h after intake on day 14 | Day 0 vs. placebo‡P value | Day 0 vs. baseline§P value | Day 14 vs.placebo‡P value | Day 14 vs. baseline§P value | |
| Placebo (n = 12) | 2.30 ± 0.70 | 2.57 ± 0.65* | 2.39 ± 0.47 | ||||
| Terutroban 2.5 mg day−1 (n = 11) | 2.16 ± 0.51 | 4.14 ± 1.25 | 4.42 ± 1.22 | <0.001 | <0.001 | <0.001 | <0.001 |
| Terutroban 5 mg day−1 (n = 12) | 2.51 ± 0.72* | 3.88 ± 0.96* | 4.00 ± 1.23 | 0.001 | <0.001 | 0.001 | 0.001 |
| Terutroban 10 mg day−1 (n = 12) | 2.66 ± 0.81 | 4.07 ± 0.77† | 4.17 ± 0.72† | <0.005 | <0.001 | <0.001 | <0.001 |
Values are means ± SD.
Data available for n = 11 patients
data available for n = 10 patients
Dunnett's test
Student's paired t-test.
Figure 2.
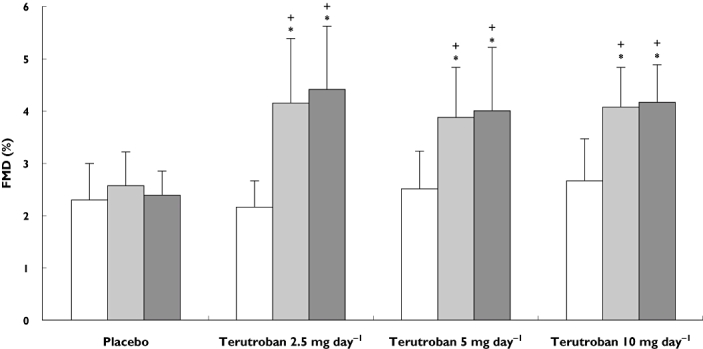
Flow-mediated vasodilatation (FMD) at baseline, 2 h after the first study drug dose on day 0 (D0) and 2 h after the last study drug dose on day 14 (D14) in patients receiving placebo or terutroban at a dosage of 2.5, 5 or 10 mg day−1. *P ≤ 0.001 vs. placebo; +P ≤ 0.001 vs. baseline. Baseline (□); D0 ( ); D14 (
); D14 ( )
)
The higher terutroban dosages of 5 and 10 mg day−1 produced similar results, with FMD increases to 3.88 ± 0.96% (95% CI of the difference, 0.95–1.79) and 4.07 ± 0.77% (95% CI of the difference, 0.92–1.66), respectively, 2 h after the first dose on D0, and sustained increases on D14 [4.00 ± 1.23% (95% CI of the difference, 10.60–1.98) and 4.17 ± 0.72% (95% CI of the difference, 1.03–2.05), respectively]. Flow-mediated dilatation values after each terutroban dose were always significantly higher than the values at baseline or in the placebo group (Table 2 and Figure 1). No dose–response relation was found within the range of terutroban doses tested. There was no significant difference between FMD on D14 and FMD 2 h postdose on D0.
Platelet aggregation
The results of the ex vivo platelet aggregation tests using the selective TP receptor agonist U46619 are summarized in Table 3. U46619-induced platelet aggregation was almost completely inhibited (<20%) within 2 h after the first dose on D0 in all patients receiving terutroban at any dosage. The differences were highly significant vs. baseline and vs. placebo (all P < 0.001). The ex vivo platelet aggregation results 2 h after the last dose on D14 were similar to those measured 2 h after the first dose on D0 (data not shown). Platelet aggregation induced by ADP or collagen was not significantly altered.
Table 3.
Platelet aggregation (percentage of maximal aggregation) induced by U46619 at baseline, 2 h after the first study drug dose on day 0 and 2 h after the last study drug dose on day 14
| Aggregation induced by U46619 (%) | |||
|---|---|---|---|
| Baseline | Day 0 | Day 14 | |
| Placebo (n = 12) | 95.3 ± 5.8 | 91.6 ± 10.4 | 88.3 ± 11.8 |
| Terutroban 2.5 mg day−1 (n = 11) | 99.4 ± 1.6 | 2.0 ± 2.0*† | 2.6 ± 1.8*† |
| Terutroban 5 mg day−1 (n = 12) | 90.3 ± 11.1 | 4.7 ± 2.2*† | 4.0 ± 3.4*† |
| Terutroban 10 mg day−1 (n = 12) | 87.8 ± 14.2 | 4.6 ± 3.5*† | 6.2 ± 4.2*† |
Values are means ± SD.
P < 0.001 vs. baseline (Dunnett's test)
P < 0.001 vs. placebo (Student's paired t-test).
Safety
All three terutroban dosages administered in addition to aspirin 300 mg day−1 were well tolerated. No serious adverse events or adverse events leading to discontinuation of treatment were observed. Treatment-emergent adverse events occurred in three patients, as follows: one patient in the placebo group experienced a mild erythematous rash and mild leg cramps on D14; one patient in the 5 mg day−1 terutroban group had a bleeding-time increase to 15 min on D14, with a normal value of 5 min on day 35; and one patient in the 10 mg day−1 terutroban group experienced mild pharyngitis on D14. Additionally, one patient in the 2.5 mg day−1 terutroban group experienced mild pruritus 1 day after treatment completion. Bleeding time on D14 was similar in all treatment groups (Table 4). No changes were observed in blood pressure, other vital signs, or other biological or electrocardiogram parameters.
Table 4.
Bleeding time at treatment completion (day 14) in the populations receiving placebo or terutroban at 2.5, 5 or 10 mg day−1
| Placebo (n = 12) | Terutroban 2.5 mg day−1(n = 11) | Terutroban 5 mg day−1(n = 12) | Terutroban 10 mg day−1(n = 12) | |
|---|---|---|---|---|
| Bleeding time (min) | 6.63 ± 1.86 | 5.55 ± 1.42 | 7.25 ± 3.75 | 7.21 ± 2.04 |
Values are means ± SD.
Discussion
Treatment with 2.5, 5 or 10 mg day−1 terutroban added to 300 mg day−1 aspirin improved endothelium-dependent vasodilatation in the peripheral arteries of high-cardiovascular-risk patients with carotid atherosclerosis. The beneficial effect of terutroban on FMD was detectable after the first intake and persisted up to the end of the 2 week treatment period. The FMD increases recorded with terutroban were significant compared with baseline values and with values in the placebo group, and they were similar with all three dosages tested (2.5, 5 and 10 mg day−1). These results are in line with the FMD increase observed after a single 10 mg terutroban dose in CAD patients treated with 100 mg day−1 of aspirin [10], and they establish that the effect persists during longer term treatment added to low-dose aspirin (300 mg day−1). These findings provide further confirmation of the vascular properties of terutroban in a population already treated with aspirin. Terutroban was safe and well tolerated in our study.
Measurement of FMD at the brachial artery is widely used to assess peripheral and coronary endothelial function [21, 22]. This method is non-invasive and can therefore be used for repeated measurements. Flow-mediated vasodilatation reflects the ability of blood vessels to adjust their vascular tone and blood flow in response to changes in the local environment and, more specifically, to dilate in response to an increase in shear stress. The absolute terutroban-induced FMD changes in our study of 1.4–2.3% are in line with the guidelines for using FMD measurement in clinical trials [21]. In a previous study, terutroban improved acetylcholine-induced vasodilatation without changing the response to sodium nitroprusside, indicating that one major effect of terutroban was to improve endothelium-dependent vasodilatation.
The beneficial effect of terutroban on FMD was found in patients receiving concomitant aspirin, 300 mg day−1, which is within the recommended dosage range for decreasing ischaemic events in high-risk patients. The absence of an arm without aspirin treatment may constitute a limitation of the study. However, stopping aspirin in these high-risk patients would have been unethical, and one of the main goals of the study was to test terutroban in patients taking the largest widely used dosage of prophylactic aspirin. The additional effect of terutroban may be related to the different mechanisms of action of terutroban and aspirin. Aspirin reduces platelet aggregation by inhibiting the production of TXA2 by platelets (i.e. by inhibiting cyclo-oxygenase 1) but does not inhibit the production of TXA2 by other sources and has no effect on the activity of other TP receptor ligands with platelet-activating or vasoconstricting effects such as prostanoids and isoprostanes. The TP receptors participate in thromboxane- and prostanoid endoperoxide-dependent regulation of vascular tone and vasoconstriction. This may explain why up to 20% of patients with atherosclerosis or CAD experience recurrent vascular events despite aspirin therapy and why some patients exhibit aspirin resistance [23]. By specifically antagonizing the TP receptor, terutroban inhibits the activity of all TP receptor ligands (both enzymatic and non-enzymatic in origin), not only on platelet TP receptors, but also on endothelial TP receptors, thereby limiting the vasoconstricting effects of prostanoids and isoprostanes. Furthermore, TP receptor antagonism does not interfere with the synthesis of endogenous vasodilator prostanoids, such as prostaglandin. These broader effects of terutroban compared with aspirin were shown in apoplipoprotein E knockout mice, in which atherogenesis was delayed by terutroban but not by aspirin [12].
As expected, all three terutroban dosages completely inhibited U46619-induced platelet aggregation, in keeping with the specific receptor-inhibiting effect of terutroban and with available data. Our platelet aggregation results are consistent with those reported by Gaussem et al. in patients with peripheral arterial disease [24]. Those authors showed a direct relation between pharmacokinetics and pharmacodynamics and predicted that complete inhibition of platelet aggregation would be achieved with 10 or 30 mg terutroban for at least 24 h in patients with stable peripheral artery disease. However, they found no dose dependency of the effect on platelet aggregation 2 h after terutroban administration [22].
We found no evidence that the FMD response to terutroban was dose dependent within the range tested (2.5–10 mg day−1). It should be noted that FMD was measured 2 h postdose, that is, at the time of the peak effect, when the plasma concentration was probably effective with all three dosages. This fact may explain why we found no dose–effect relation. Measuring FMD before terutroban administration on day 14 would have been useful, especially with the lowest dosage. The absence of such a measurement is a limitation of this study.
Terutroban was well tolerated in the dosages used in our study. Despite co-administration with aspirin, which is known to increase the risk of bleeding [25, 26], no increase in bleeding was observed in the patients receiving terutroban. A single patient had a moderate bleeding time increase, which resolved spontaneously.
Conclusions
This study demonstrated that once-daily administration of terutroban at a dosage of 2.5, 5 or 10 mg day−1 for 2 weeks significantly improved endothelium-dependent FMD and inhibited platelet aggregation in high-risk patients with atherosclerosis receiving prophylactic aspirin therapy. These results constitute useful information for further drug development, because both platelet inhibition and improved vascular function are essential for prevention of cardiovascular events in patients with atherosclerosis.
Competing Interests
There are no competing interests to declare. The authors of this study do not have a financial relationship with a commercial entity that has an interest in the subject of this manuscript. The drug was supplied by Institut de Recherches Internationales Servier (IRIS), which funded the study. IRIS had no role in conducting the study, interpreting the study data, deciding to publish the study results, or writing the manuscript. None of the authors received fees from the company.
REFERENCES
- 1.Ludmer PL, Selwyn AP, Shook TL, Wayne RR, Mudge GH, Alexander RW, Ganz P. Paradoxical vasoconstriction induced by acetylcholine in atherosclerotic coronary arteries. N Engl J Med. 1986;315:1046–51. doi: 10.1056/NEJM198610233151702. [DOI] [PubMed] [Google Scholar]
- 2.Vanhoutte PM. Endothelial dysfunction and atherosclerosis. Eur Heart J. 1997;18(Suppl. E):E19–29. doi: 10.1016/s0195-668x(97)90005-1. [DOI] [PubMed] [Google Scholar]
- 3.Luscher TF, Boulanger CM, Dohi Y, Yang ZH. Endothelium-derived contracting factors. Hypertension. 1992;19:117–30. doi: 10.1161/01.hyp.19.2.117. [DOI] [PubMed] [Google Scholar]
- 4.Takahashi K, Nammour TM, Fukunaga M, Ebert J, Morrow JD, Roberts LJ, II, Hoover RL, Badr KF. Glomerular actions of a free radical-generated novel prostaglandin, 8-epi-prostaglandin F2 alpha, in the rat. Evidence for interaction with thromboxane A2 receptors. J Clin Invest. 1992;90:136–41. doi: 10.1172/JCI115826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Morrow JD, Hill KE, Burk RF, Nammour TM, Badr KF, Roberts LJ., II A series of prostaglandin F2-like compounds are produced in vivo in humans by a non-cyclooxygenase, free radical-catalyzed mechanism. Proc Natl Acad Sci U S A. 1990;87:9383–7. doi: 10.1073/pnas.87.23.9383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Davi G, Alessandrini P, Mezzetti A, Minotti G, Bucciarelli T, Costantini F, Cipollone F, Bon GB, Ciabattoni G, Patrono C. In vivo formation of 8-Epi-prostaglandin F2 alpha is increased in hypercholesterolemia. Arterioscler Thromb Vasc Biol. 1997;17:3230–5. doi: 10.1161/01.atv.17.11.3230. [DOI] [PubMed] [Google Scholar]
- 7.Reilly MP, Pratico D, Delanty N, DiMinno G, Tremoli E, Rader D, Kapoor S, Rokach J, Lawson J, FitzGerald GA. Increased formation of distinct F2 isoprostanes in hypercholesterolemia. Circulation. 1998;98:2822–8. doi: 10.1161/01.cir.98.25.2822. [DOI] [PubMed] [Google Scholar]
- 8.Davi G, Ciabattoni G, Consoli A, Mezzetti A, Falco A, Santarone S, Pennese E, Vitacolonna E, Bucciarelli T, Costantini F, Capani F, Patrono C. In vivo formation of 8-iso-prostaglandin f2alpha and platelet activation in diabetes mellitus: effects of improved metabolic control and vitamin E supplementation. Circulation. 1999;99:224–9. doi: 10.1161/01.cir.99.2.224. [DOI] [PubMed] [Google Scholar]
- 9.Cipollone F, Ciabattoni G, Patrignani P, Pasquale M, Di Gregorio D, Bucciarelli T, Davi G, Cuccurullo F, Patrono C. Oxidant stress and aspirin-insensitive thromboxane biosynthesis in severe unstable angina. Circulation. 2000;102:1007–13. doi: 10.1161/01.cir.102.9.1007. [DOI] [PubMed] [Google Scholar]
- 10.Belhassen L, Pelle G, Dubois-Rande JL, Adnot S. Improved endothelial function by the thromboxane A2 receptor antagonist S 18886 in patients with coronary artery disease treated with aspirin. J Am Coll Cardiol. 2003;41:1198–204. doi: 10.1016/s0735-1097(03)00048-2. [DOI] [PubMed] [Google Scholar]
- 11.Cayatte AJ, Du Y, Oliver-Krasinski J, Lavielle G, Verbeuren TJ, Cohen RA. The thromboxane receptor antagonist S18886 but not aspirin inhibits atherogenesis in apo E-deficient mice: evidence that eicosanoids other than thromboxane contribute to atherosclerosis. Arterioscler Thromb Vasc Biol. 2000;20:1724–8. doi: 10.1161/01.atv.20.7.1724. [DOI] [PubMed] [Google Scholar]
- 12.Viles-Gonzalez JF, Fuster V, Corti R, Valdiviezo C, Hutter R, Corda S, Anand SX, Badimon JJ. Atherosclerosis regression and TP receptor inhibition: effect of S18886 on plaque size and composition – a magnetic resonance imaging study. Eur Heart J. 2005;26:1557–61. doi: 10.1093/eurheartj/ehi175. [DOI] [PubMed] [Google Scholar]
- 13.Michel F, Simonet S, Vayssettes-Courchay C, Bertin F, Sansilvestri-Morel P, Bernhardt F, Paysant J, Silvestre JS, Levy BI, Feletou M, Verbeuren TJ. Altered TP receptor function in isolated, perfused kidneys of nondiabetic and diabetic ApoE-deficient mice. Am J Physiol Renal Physiol. 2008;294:F120–9. doi: 10.1152/ajprenal.00111.2007. [DOI] [PubMed] [Google Scholar]
- 14.Maalej N, Osman HE, Shanmuganayagam D, Shebuski RJ, Folts JD. Antithrombotic properties of the thromboxane A2/prostaglandin H2 receptor antagonist S18886 on prevention of platelet-dependent cyclic flow reductions in dogs. J Cardiovasc Pharmacol. 2005;45:389–95. doi: 10.1097/01.fjc.0000157439.49612.83. [DOI] [PubMed] [Google Scholar]
- 15.Hong TT, Huang J, Driscoll E, Lucchesi BR. Preclinical evaluation of S18886 in an experimental model of coronary arterial thrombosis. J Cardiovasc Pharmacol. 2006;48:239–48. doi: 10.1097/01.fjc.0000248234.08277.7e. [DOI] [PubMed] [Google Scholar]
- 16.Osende JI, Shimbo D, Fuster V, Dubar M, Badimon JJ. Antithrombotic effects of S 18886, a novel orally active thromboxane A2 receptor antagonist. J Thromb Haemost. 2004;2:492–8. doi: 10.1111/j.1538-7933.2004.00639.x. [DOI] [PubMed] [Google Scholar]
- 17.Bal Dit Sollier C, Crassard I, Simoneau G, Bergmann JF, Bousser MG, Drouet L. Effect of the thromboxane prostaglandin receptor antagonist terutroban on arterial thrombogenesis after repeated administration in patients treated for the prevention of ischemic stroke. Cerebrovasc Dis. 2009;28:505–13. doi: 10.1159/000236915. [DOI] [PubMed] [Google Scholar]
- 18.D'Agostino RB, Sr, Vasan RS, Pencina MJ, Wolf PA, Cobain M, Massaro JM, Kannel WB. General cardiovascular risk profile for use in primary care: the Framingham Heart Study. Circulation. 2008;117:743–53. doi: 10.1161/CIRCULATIONAHA.107.699579. [DOI] [PubMed] [Google Scholar]
- 19.Yusuf S, Teo KK, Pogue J, Dyal L, Copland I, Schumacher H, Dagenais G, Sleight P, Anderson C. Telmisartan, ramipril, or both in patients at high risk for vascular events. N Engl J Med. 2008;358:1547–59. doi: 10.1056/NEJMoa0801317. [DOI] [PubMed] [Google Scholar]
- 20.Belhassen L, Carville C, Pelle G, Sediame S, Benacerraf S, Dubois-Rande JL, Adnot S. Molsidomine improves flow-dependent vasodilation in brachial arteries of patients with coronary artery disease. J Cardiovasc Pharmacol. 2000;35:560–3. doi: 10.1097/00005344-200004000-00008. [DOI] [PubMed] [Google Scholar]
- 21.Corretti MC, Anderson TJ, Benjamin EJ, Celermajer D, Charbonneau F, Creager MA, Deanfield J, Drexler H, Gerhard-Herman M, Herrington D, Vallance P, Vita J, Vogel R. Guidelines for the ultrasound assessment of endothelial-dependent flow-mediated vasodilation of the brachial artery: a report of the International Brachial Artery Reactivity Task Force. J Am Coll Cardiol. 2002;39:257–65. doi: 10.1016/s0735-1097(01)01746-6. [DOI] [PubMed] [Google Scholar]
- 22.Moens AL, Goovaerts I, Claeys MJ, Vrints CJ. Flow-mediated vasodilation: a diagnostic instrument, or an experimental tool? Chest. 2005;127:2254–63. doi: 10.1378/chest.127.6.2254. [DOI] [PubMed] [Google Scholar]
- 23.Halushka MK, Halushka PV. Why are some individuals resistant to the cardioprotective effects of aspirin? Could it be thromboxane A2? Circulation. 2002;105:1620–2. doi: 10.1161/01.cir.0000015422.86569.52. [DOI] [PubMed] [Google Scholar]
- 24.Gaussem P, Reny JL, Thalamas C, Chatelain N, Kroumova M, Jude B, Boneu B, Fiessinger JN. The specific thromboxane receptor antagonist S18886: pharmacokinetic and pharmacodynamic studies. J Thromb Haemost. 2005;3:1437–45. doi: 10.1111/j.1538-7836.2005.01468.x. [DOI] [PubMed] [Google Scholar]
- 25.McQuaid KR, Laine L. Systematic review and meta-analysis of adverse events of low-dose aspirin and clopidogrel in randomized controlled trials. Am J Med. 2006;119:624–38. doi: 10.1016/j.amjmed.2005.10.039. [DOI] [PubMed] [Google Scholar]
- 26.Patrono C, Bachmann F, Baigent C, Bode C, De Caterina R, Charbonnier B, Fitzgerald D, Hirsh J, Husted S, Kvasnicka J, Montalescot G, Garcia Rodriguez LA, Verheugt F, Vermylen J, Wallentin L, Priori SG, Alonso Garcia MA, Blanc JJ, Budaj A, Cowie M, Dean V, Deckers J, Fernandez Burgos E, Lekakis J, Lindahl B, Mazzotta G, Morais J, Oto A, Smiseth OA, Ferreira R, Steg PG, Teixeira F, Wilcox R. Expert consensus document on the use of antiplatelet agents. The task force on the use of antiplatelet agents in patients with atherosclerotic cardiovascular disease of the European society of cardiology. Eur Heart J. 2004;25:166–81. doi: 10.1016/j.ehj.2003.10.013. [DOI] [PubMed] [Google Scholar]