

Abstract
Background
Predicting susceptibility to multiple sclerosis may have important clinical applications either as part of a diagnostic algorithm or as a tool with which to identify high-risk individuals for prospective studies. Here, we examine the utility of an aggregate measure of risk of multiple sclerosis (MS) based on genetic susceptibility loci. Secondarily, we assess the added effect of environmental risk factors that have been associated with susceptibility for MS.
Methods
We created a weighted genetic risk score (wGRS) that includes 16 MS susceptibility loci. We tested our model using data from (1) 2215 MS cases and 2189 controls (derivation samples), (2) a validation set of 1340 cases and 1109 controls taken from several MS therapeutic trials (TT samples), and (3) a second validation set of 143 cases and 281 controls from the U.S. Nurses’ Health Studies I and II (NHS) for whom we also have information regarding exposure to smoking and Epstein-Barr Virus (EBV).
Findings
. Patients with wGRS > 1.25 standard deviations from the mean had a significantly higher odds ratio for MS in all datasets. The area under the curve for a purely genetic model was 0.70 and for a gender + genetic model was 0.74 in the derivation samples (P <0.0001), 0.64 and 0.72 in the TT cohort (P <0.0001). Similarly, consideration of smoking and immune response to EBV enhanced the AUC of 0.64 for the genetic model to 0.68 in the NHS cohort (P =0.02). The wGRS does not appear to be correlated with conversion of a clinically isolated syndrome to MS.
Interpretation
The current combination of 16 susceptibility alleles into a wGRS modestly predicts MS risk and shows consistent discriminatory ability in independent subject samples and is enhanced by considering non-genetic risk factors.
Introduction
Multiple sclerosis (MS) is an inflammatory disease of the central nervous system for which several genetic and environmental susceptibility factors have been identified including both major histocompatibility complex (MHC) and non-MHC loci1. Given the results of genome-wide association studies in other inflammatory diseases such as Crohn’s disease and type 1 diabetes mellitus2,3 in which over 40 susceptibility alleles have already been described for each disease, it is likely that the association study strategy being implemented genome-wide in MS today will discover many more susceptibility loci. Aside from HLA DRB1*1501, most of these genetic risk factors have only a modest effect on susceptibility to MS (odds ratio 1.1 – 1.2); however, the risk alleles in these loci are relatively common in populations of European ancestry, being found at allele frequencies of 0.1– 0.9. As yet, there is little information on how this growing set of genetic susceptibility factors interacts with environmental risk factors that include infection with the Epstein-Barr Virus (EBV), smoking, and vitamin D levels4.
While the early results from whole genome association studies have not yet entered clinical use, the need for translation of genetic and epidemiologic risk factors to the clinic is compelling. An important goal of understanding the genetic basis of MS is to use these allelic variants to predict disease risk in subjects so that changes in the environment or therapeutic interventions can be initiated prior to the initiation of an inflammatory demyelinating process. Combining family history with a quantitative measure of genetic risk may eventually allow the implementation of a screening tool to discover clinically silent evidence of disease amongst first degree relatives of MS patients which are 20–50 times more likely to develop MS than the general population5. While such subjects are at higher risk of MS, the absolute risk remains low, between 2 and 5%5. Thus, both in these high-risk populations and in subjects experiencing an initial episode of neurologic deficit that is of unclear etiology, deployment of novel, screening clinical tools could have an impact by guiding the selection of subjects for which early imaging has a higher yield. Early detection of an inflammatory demyelinating illness is meaningful as early treatment of subjects with a single episode of inflammatory demyelination has been shown to be beneficial in reducing the accumulation of neurologic disability6.
Here, we report on the efficacy of a weighted genetic risk score (wGRS) that combines weighted odds ratios from each of 16 loci that have been associated with MS1 in predicting a diagnosis of MS in three independent cohorts of subjects. We also show that integrating clinical parameters and paraclinical measures of environmental exposure enhance the efficacy of our algorithm in predicting a diagnosis of demyelinating disease. This integrative approach based on only the fraction of genetic loci known today is therefore promising in exploring the question of susceptibility to MS.
Methods
Study Participants
All subjects were recruited in studies approved by the Institutional Review Boards or ethics committee of their respective institutions; all subjects gave an informed consent to participate in the collection of their DNA material for genetic analysis. All subjects with MS fulfill the revised McDonald diagnostic criteria7. The alleles and odds ratios included in our model were selected from the replication arm of a meta-analysis study1 that used UK and US cohorts outlined in Table 1a. Thus, we refer to this combined set of samples as the derivation samples for this wGRS study: 2215 MS or clinically isolated demyelinating syndrome (CIS) cases and 2189 healthy control subjects were included. The detailed provenance of these subjects has been described previously1. The characteristics of these subjects are outlined in Table 1a. The derivation samples are the only ones to have been previously published in terms of the MS susceptibility loci1; none of these subjects are found in any of the other three sample collections described below. Each of the four sample collections were collected for different purposes by different investigators and are clearly independent of one another.
Table 1a.
Clinical characteristics of the derivation samples
| Analysis stratum | US | UK | |||||
|---|---|---|---|---|---|---|---|
| Subject Source | BWH | WU | ACP | UCSF | RUSH | UC | 1958 BC |
| Number of controls | 4071 | 13 | 35 | 142 | 489 | - | 1030 |
| Number of Cases | 227 | 152 | 597 | 407 | - | 831 | - |
| Female: Male ratio | 2.8:1.0 | 3.1:1.0 | 3.1:1.0 | 5.6:1.0 | - | 2.28:1.0 | - |
| Mean Disease Duration, years (range) | 12 (<1–58) | 13 (<1–39) | 15 (1–56) | 12(<1–47) | - | 14 (<1–54) | - |
| Mean age at onset, years (range) | 33 (8–59) | 33 (13–71) | 33 (1–70) | 42 (3–60) | - | 32 (10–67) | - |
|
| |||||||
|
Disease course, n (%)
|
- | - | |||||
| “Bout onset” | 178 (78%) | 128 (84%) | 463 (78%) | 345 (85%) | - | 732 (88%) | - |
| Relapse Remitting | 136 (60%) | 106 (70%) | 358 (60%) | 237 (58%) | - | NA2 | - |
| Secondary Progressive | 42 (19%) | 22 (14%) | 105 (18%) | 111 (27%) | - | NA2 | - |
| Primary Progressive | 17 (8%) | 17 (11%) | 36 (6%) | 23 (6%) | - | 84 (10%) | - |
| Clinically Isolated Syndrome | 25 (11%) | 7 (4%) | 75 (13%) | 10 (2%) | - | 0 (0%) | - |
| Unknown | 7 (3%) | 0 | 23 (4%) | 26 (6%) | - | 15 (2%) | - |
BWH controls – these subjects of European ancestry recruited in the Boston area include (1) unaffected spouses from our MS Genetics collections (n=14), (2) The BWH PhenoGenetic Project subjects (n=292), and healthy subjects from the HPCGG collection (n=101) (see methods for details). These subjects do not overlap with BWH control subjects used in the meta-analysis.
NA – the breakdown of remitting relapsing and secondary progressive subjects is not available in this cohort.
Glossary: 1958 BC – 1958 birth cohort; ACP – Accelerated Cure Project; BWH – Brigham & Women’s Hospital; RUSH – RUSH University; UC – University of Cambridge, UK; UCSF – University California, San Francisco; UK – United Kingdom; US – United States; WU – Washington University, St. Louis
Similarly, details on the first validation set that includes subjects assembled by Bayer Schering Pharma from its therapeutic trials include a total of 1340 cases (Table 1b). This sample collection is therefore referred to as the Therapeutic Trial (TT) collection. They come from 3 studies evaluating IFNβ-1b8,9,10 and one study evaluating a CCR1-antagonist11. At the time of enrollment, 1132 of these subjects had a diagnosis of remitting-relapsing MS according to the McDonald criteria, and 208 had a diagnosis of CIS with MRI signs suggestive of demyelination. 95 of these CIS subjects developed MS according to the McDonald criteria over up to 5 years of observation. For the purpose of these analyses, they were matched to 1109 control subjects from a genome scan for myocardial infarction (the MIGEN study)12; since there is no known association between MS and early myocardial infarction (MI), these control subjects in the MS analysis include both healthy control subjects and subjects with early MI from the MIGEN study. These 1109 subjects are those subjects from the MIGEN study that were not included in our recent meta-analysis of MS genome-wide association studies1. These control subjects are therefore not specifically matched to the TT cases but represent a collection of subjects of European ancestry who, a priori, have the general prevalence rate of 0.001 for a diagnosis of MS or CIS.
Table 1b.
Clinical characteristics of the validation sample collections
| Subject Source | TT | MIGEN | NHS/NHS II | SET | |
|---|---|---|---|---|---|
| Analysis stratum | Case | Control | Case | Control | Cases |
| Number of Subjects | 1340 | 1109 | 143 | 281 | 182 |
| Female: Male ratio | 2.3:1.0 | 0.6:1.0 | All female | All female | 1.9:1.0 |
| Mean Disease Duration, years (range) | 5 (0–43) | - | 11 (0–40) | - | <1 |
| Mean age at onset, years (range) | 30 (8–54) | - | 40 (20–67) | - | 28 (14–54) |
|
| |||||
|
Disease course, n (%)
|
|||||
| “Bout onset” | 1340 | - | N/A | - | 182 (100)3 |
| Relapse Remitting | 1132 | - | N/A | - | - |
| Secondary Progressive | 0 | - | N/A | - | - |
| Primary Progressive | 0 | - | N/A | - | - |
| Clinically Isolated Syndrome | 2082 | - | N/A | - | 182 (100)3 |
Glossary: UK = United Kingdom; US = United States. “Bout onset MS” is used here to describe subjects whose disease course started with discrete attacks of inflammatory demyelination. Each subject with MS is used only once in our analyses.
95 of these subjects converted to MS during the course of the study.
41 of these CIS subjects converted to MS during the course of the study.
The second validation set consists of subjects from the Nurses Health Study (NHS) and Nurses’ Health Study II (NHS II) cohorts including 143 MS subjects and their 281 matched controls from these prospective studies. These subjects have been described in earlier publications13, and their characteristics are described in Table 1b.
To assess the role of our wGRS in the conversion of subjects with CIS to a diagnosis of MS, we analyzed a subset of 80 subjects from the TT collection that were in the placebo arm of the BENEFIT study9. All subjects were observed for 24 months and a diagnosis of clinical definite MS was recorded based on established criteria9. We similarly analyzed a cohort of 182 CIS subjects from the Observational Study of Early Interferon beta 1-a Treatment in High Risk Subjects after CIS (SET) that was recruited in Prague, Czech Republic. Inclusion criteria for this study included a diagnosis of CIS, at least 2 hyperintense lesions on MRI and positivity of oligoclonal bands in cerebrospinal fluid. The subjects had to be enrolled in the study within 4 months from the beginning of the first symptom. All subjects were treated with IFN-β 1a and were observed for 24 months, and all clinical events were recorded. 41 subjects from the SET study converted to a diagnosis of MS during the course of the study.
SNP selection and Genotypes
We have included 16 SNPs in our Genetic risk Score (GRS). These SNPs were picked in one of two ways at the conclusion of the replication effort of the meta-analysis: (1) 9 of the SNPs mark validated MS susceptibility loci that exceed a level of genome-wide significance (P< 5×10−8) either in the meta-analysis or in earlier publications or (2) 7 of the SNPs were strongly suggestive of association with MS in the meta-analysis (P<10−4 in the final joint analysis). Four of these SNPs had also previously been validated to be susceptibility markers for other inflammatory diseases.1 Since then, these 7 SNPs have presented evidence of validation in other sample collections (P. De Jager, unpublished data).
For the derivation samples, genotype data were produced using the Sequenom MASSarray platform and its iPLEX format as part of our earlier study1. The same platform was used to generate data on the subjects from the NHS/NHS II and SET studies. Genotypes for the TT and MIGEN subjects were available from genome-wide genotyping data collected using Affymetrix Genechip 6.0 arrays.
Statistical Analyses
Weighted Genetic Risk Score
We developed a “weighted-GRS” (wGRS) that utilized the allelic odds ratios (OR) from a published study1 to account for the strength of the genetic association with each allele. We calculated a wGRS that included 2 MHC alleles and 14 non-MHC risk alleles. The weighted score is preferred over a simple count GRS equal to the sum the number of risk alleles carried, since the HLA DRB1*1501 allele has a substantially higher OR for MS susceptibility than do the more recently discovered SNPs (Table 2). The wGRS was calculated by multiplying the number of risk alleles for each SNP by the weight for that SNP and then taking the sum across the 16 SNPS. The formula and weights are presented in table 2; the weight is simply the natural log of the OR for each allele. By using the ORs from the replication study we have minimized the effect of over-inflation of the ORs. In addition the standard errors of those estimate were all very small given the large sample sizes from these analyses.1 Given comments from reviewers, we repeated our wGRS calculations taking into account the variance of the OR estimates but this did not affect our results.
Table 2.
SNPs that compose the wGRS and weights assigned to each marker
| Chr | SNP | Allele | base pair | OR | Gene | Weight | % of total weight |
|---|---|---|---|---|---|---|---|
| 1 | rs2300747 | A | 116905738 | 1.22 | CD58 | 0.2484614 | 7.5% |
| 1 | rs2760524 | G | 190797171 | 1.13 | RGS1 | 0.1392621 | 4.2% |
| 2 | rs882300 | C | 136692725 | 1.15 | CXCR4 | 0.1625189 | 4.9% |
| 3 | rs4680534 | C | 161181639 | 1.11 | IL12A | 0.10436 | 3.2% |
| 5 | rs6897932 | C | 35910332 | 1.11 | IL7R | 0.1165338 | 3.5% |
| 5 | rs6896969 | C | 40460183 | 1.09 | PTGER4 | 0.0943107 | 2.9% |
| 6 | rs2523393 | A | 29813638 | 1.21 | HLA B | 0.2357223 | 7.2% |
| 6 | rs3135388 | A | 32521029 | 2.77 | HLA DRB1 | 1.0188473 | 30.9% |
| 6 | rs9321619 | A | 137916101 | 1.11 | OLIG3/TNFAIP3 | 0.1165338 | 3.5% |
| 10 | rs2104286 | T | 6139051 | 1.14 | IL2RA | 0.1508229 | 4.6% |
| 10 | rs1250540 | G | 80706013 | 1.12 | ZMIZ1 | 0.1133287 | 3.4% |
| 11 | rs17824933 | G | 60517188 | 1.16 | CD6 | 0.14842 | 4.5% |
| 12 | rs1800693 | C | 6310270 | 1.22 | TNFRSF1A | 0.1988509 | 6.0% |
| 12 | rs1790100 | G | 122222678 | 1.10 | MPHOSPH9 | 0.0953102 | 2.9% |
| 16 | rs11865121 | C | 11074189 | 1.13 | CLEC16A | 0.1392621 | 4.2% |
| 16 | rs17445836 | G | 84575164 | 1.19 | IRF8 | 0.210721 | 6.4% |
Where i is the SNP, wi = weight for SNP i, and Xi = number of risk alleles (0,1 or 2).
To assess the effect of the HLA DRB1*1501 allele, we also calculated a wGRS15 (no DRB1*1501) that includes only the common variants with modest effects on MS susceptibility. The weights used in the wGRS were calculated as the natural log of the published OR with respect to the risk allele as presented in Table 2. The OR for the 16 risk alleles were derived from the replication arm of a recent meta-analysis of genome scans for MS susceptibility to minimize the inflation of effect size that is expected in a risk allele discovery study1. After removing individuals missing >10% genotype data, any individual with missing information for a particular SNP was assigned a value of twice the risk allele frequency for that SNP which is the expected value for that SNP in the population. Since there is currently no evidence of epistasis among these susceptibility loci, we have not added terms for interaction between the loci in our algorithm. In each set of samples, the distribution of the wGRS was plotted separately for cases and controls and compared using 2 independent sample t-tests. All analyses were done using SAS version 9.2 (SAS Institute, Cary, NC).
Partitioned wGRS
Since a continuous score is difficult to interpret on an individual level when a physician needs to explain the results of the wGRS to a patient, we partition subjects into different categories of risk. These categories are created using the means and standard deviations (SD) from the control samples specific to each population. The seven categories were defined as ± 0.25, ± 0.75 and ± 1.25 SDs from the mean, with the extreme categories being < 1.25 or >1.25 SDs from the mean. Dividing our score into 7 categories provided a robust distribution, allowing us to parse out the highest and lowest risk groups while assuring that there were statistically sufficient numbers of cases and controls in these extreme categories of interest. To avoid exaggerating the level of odds ratio associated with a given subject category, we use the largest subject subset, which contains the mean of the healthy control population (category 4), as the reference category. These category 4 subjects can be thought of representing the average risk of the assessed population. Within each dataset, we fit a single logistic regression model (controlling for gender in the derivation and TT samples and smoking status and anti-EBNA1 titers in the NHS/NHSII sample) using dummy variables for wGRS groups, to study the association of wGRS with MS, comparing each category of wGRS to a referent median category 4. (Table 3). An ordinal wGRS variable based on our categories was used to calculate a p-value for trend. Finally, we also calculated the odds of MS for the top category as compared to the bottom category.
Table 3.
wGRS scores and odd ratios of MS in derivation, TT and NHS/NHS II sample sets
| Derivation Sample | Validation Sample: TT | Validation Sample: NHS/NHS II | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Cases | Controls | OR (95%CI) | Cases | Controls | OR (95% CI) | Cases | Controls | OR (95% CI) | |
|
Unadjusted Model1
| |||||||||
| wGRS Groups | |||||||||
| 1 | 40(2) | 143(7) | 0.35 (0.24 – 0.51) | 41(3) | 76(7) | 0.53(0.35–0.81) | 23(8) | 4(3) | 0.43(0.13–1.41) |
| 2 | 127(6) | 332(15) | 0.48 (0.38 – 0.61) | 123(9) | 187(17) | 0.65(0.49–0.87) | 36(13) | 12(8) | 0.82(0.35–1.92) |
| 3 | 298(13) | 540(25) | 0.69 (0.57 – 0.84) | 205(15) | 240(22) | 0.84(0.65–1.09) | 80(29) | 28(20) | 0.87(0.44–1.72) |
| 4 | 378(17) | 475(22) | 1.0 (ref) | 239(18) | 236(21) | 1(ref) | 47(17) | 19(13) | 1(ref) |
| 5 | 315(14) | 255(12) | 1.55 (1.25 – 1.92) | 197(15) | 125(11) | 1.56(1.17–2.07) | 33(12) | 19(13) | 1.42(0.66–3.10) |
| 6 | 302(14) | 177(8) | 2.14 (1.70 – 2.70) | 205(15) | 110(10) | 1.84(1.37–2.47) | 26(9) | 29(20) | 2.76(1.30–5.85) |
| 7 | 755(34) | 267(12) | 3.55 (2.93 – 4.32) | 330(25) | 135(12) | 2.41(1.84–3.16) | 36(13) | 32(22) | 2.20(1.08–4.49) |
| 7 vs. 12 | 10.11 (6.93 – 14.74) | 4.53 (3.0 – 6.97) | 5.1 (1.60 – 16.36) | ||||||
Crude Odds ratio
Odds ratio using group 1 as referent group;
Predicting a Diagnosis of MS or CIS
To determine how well our wGRS predictors discriminate between cases of demyelinating disease and control subjects, we generated ROC curves by plotting sensitivity of the wGRS score (continuous) against 1-specificity, and calculated the area-under-the-curve (c-statistic) for each population. To assess the degree to which the HLA DRB1*1501 allele contributes to the wGRS, we plotted ROC curves for a wGRS that excluded HLA DRB1*1501 and ROC curves for the wGRS that includes HLA DRB1*1501. The c-statistics of each curve were compared using a non-parametric approach as described by De Long and colleagues14. Where available, additional susceptibility factors were considered: (1) in the TT subjects, gender was added to the model, and (2) in the NHS subjects (all women), smoking and EBV titers were added into the model.
Evaluating the role of the wGRS in the transition from CIS to MS
Eighty CIS subjects in the TT cohort are from the placebo arm of the BENEFIT study and were observed for 24 month for evidence of clinical disease activity. The last visit in the study was scheduled to take place between days 692 – 741. Therefore, the variable “time to a diagnosis of MS” is defined as a right-censored variable: a patient’s time to MS is censored if the patient was lost to follow up at any time up to day 692 or at day 692 if clinically definite MS has not been diagnosed up to this point in time. The association between the wGRS and the time to diagnosis of MS was assessed using the proportional hazards model. Non-linearity of the regressor wGRS was tested by incorporation of a quadratic term for wGRS and the proportional hazards assumption was tested by inclusion of a time-dependent covariate representing the interaction between wGRS and log(time). The model was also extended by the variables of age and gender. The analysis was repeated for the 182 subjects from the SET study which were observed up to 1271 days.
Role of the funding source
There is no study sponsor. The National MS Society and NIH have provided support for the analysis team which performed the study.
RESULTS
Distribution of the Genetic Risk Score
The weighted GRS (wGRS) that we have developed is based on odds ratios (ORs) among the MS susceptibility loci: these ORs are derived from the replication arm of our recent meta-analysis1. We refer to the US and UK subject collections included in this published replication effort as the “derivation” sample collections in the present study. We therefore expect our current wGRS model to be over fitted when applied to these cohorts. But empirically, the effect may be modest: The distribution of wGRS in the derivation sample is plotted in Figure 1, along with that of the validation sample collection assembled from several therapeutic trials (TT) that has not previously been used in genome-wide association studies. Both sets of curves show a clear separation between the distribution of the subjects with MS and that of the healthy control subjects (Figure 1). Within the derivation sample the mean ± standard deviation (SD) of the wGRS for MS subjects is 3.5 ± 0.8 and for controls is 2.9 ± 0.6 (P <0.0001). The TT validation sample is very consistent with this: the mean ± SD of the wGRS for MS subjects and controls are 3.3 ± 0.7 and 3.0 ± 0.7 (P <0.0001) respectively. Similarly, the smaller NHS/NHS II dataset has a mean ± SD of the wGRS for MS subjects and controls of 3.4 ± 0.8 and 3.0 ± 0.7 (P <0.0001) respectively
Figure 1. Distribution of wGRS in cases and controls.

A wGRS is calculated for each subject within each of the studied cohorts. Here, we illustrate the distribution of wGRS in the derivation samples, with cases in dark blue and controls in light blue. The TT sample collection is overlaid with cases in dark green and controls in light green. Finally, the Czech SET collection of CIS cases is plotted in red.
Another way to represent the difference in the distribution of wGRS between subjects with MS and controls is to partition the subjects: this has been performed by defining risk categories starting at the mean wGRS in the healthy control subjects of a sample collection and grouping subjects who are within 0.25 standard deviations of the mean as “category 4” subjects. Category 4 therefore approximates the group of subjects with an “average” population risk of MS (Figure 2a). Six subsequent categories (1–3 and 5–7) are defined by the subjects found in increasing intervals of wGRS. In the set of combined derivation samples, category 7 subjects, those with the largest wGRS had a 3.6 times increased odds developing MS as compared to category 4 (95% CI, 2.9 – 4.3), the category of “average” population risk (Table 3). When controlling for gender, given the excess in the proportion of women amongst MS cases, the odds for category 7 vs. category 4 is relatively unchanged at 3.7 (2.8 – 4.3). When comparing category 7 to category 1, the individuals with the lowest risk of MS, we see a 10.1 (6.9 – 17.4) fold increased odds of MS. In the validation TT sample collection, category 7 subjects, those with the largest wGRS, have, on average, a 2.4 (1.8 – 3.2) increase in odds of MS relative to category 4 subjects (Figure 2b). When controlling for gender, the odds again are relatively unchanged at 2.3 (1.7 – 3.0) for category 7 as compared to category 4. The risk is magnified to a 5-fold increase in odds when the comparison is made to category 1, the lowest risk category. Finally, results of the application of the wGRS method in the smaller NHS/NHS II collection provide very consistent results: a similar increase in odds of MS in category 7 subjects of 2.2 (1.1 – 4.5) relative to category 4 subjects is seen (Figure 2c). Since all of the subjects in the NHS/NHS II are female, we were unable to adjust for gender in this sample. Overall, this partitioning of subjects into seven categories may offer one avenue by which to facilitate the communication of the results of the wGRS to physicians and ultimately to patients.
Figure 2. Odds ratios for risk categories defined using the wGRS. (a) Derivation samples.
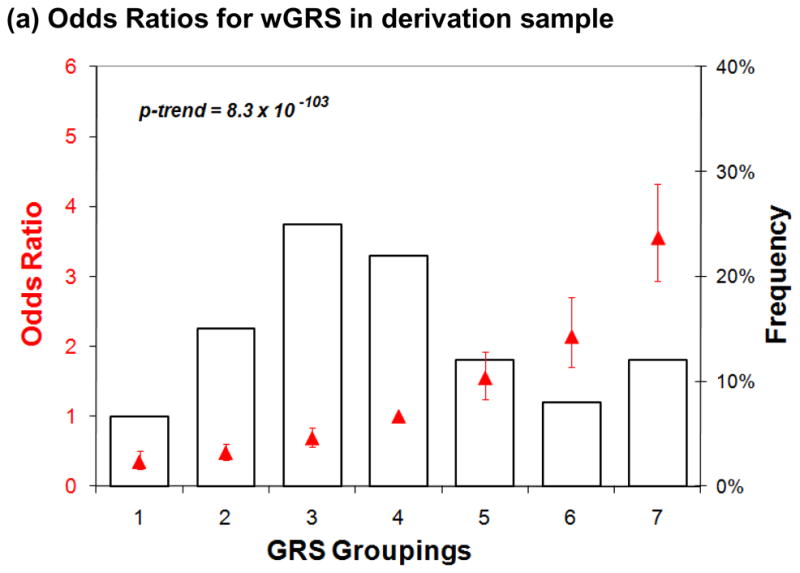

Data is presented only for subjects with MS. Seven categories of genetic risk are defined using the control subjects in the derivation samples, with “1” being the lowest risk category. The distribution of MS subjects amongst the seven risk categories is plotted in black as a histogram and is skewed since these subjects have a greater risk of MS than do healthy subjects. In red, we superimpose the log of the odds ratio (red triangle) for MS susceptibility of each risk category, along with a 95% confidence interval for that estimate. (b and c) TT and NHS/NHS II validation samples. Here, we present the distribution of wGRS in the seven categories of risk defined in each of the two sample collections used in validation exercises.
Discrimination ability of the MS wGRS
To estimate the discrimination ability of our algorithm, we calculate c-statistics in our different sample collections that are summarized in Table 4. As expected, the wGRS showed the best ability to differentiate between cases and controls when evaluated in our combined derivation sample, with a c-statistic of 0.697; this discrimination is significantly enhanced (p<0.0001) if we include gender in our model since there is a well-described excess of women among MS patients (c-statistic = 0.741). In both of our validation sample collections, our algorithm performed nearly as well (Figure 3b and c), with a C-statistic that is remarkably similar in the two sample collections: 0.636 for TT (0.721 when controlling for gender, a significant improvement, p<0.0001) and 0.637 for NHS/NHS II which consists of female subjects. This level of predictability remains modest, but our algorithm clearly and consistently performs better than chance even though it is based on just sixteen MS susceptibility loci.
Table 4.
C-statistics for ROC curves and comparisons between models predicting MS.
| Derivation Sample | TT Sample | NHS/NHS II Sample | ||||
|---|---|---|---|---|---|---|
| c-statistic | P value | c-statistic | P value | c-statistic | P value | |
| wGRS without HLA | 0.611 | 0.559 | 0.566 | |||
| wGRS with HLA | 0.697 | <0.00012 | 0.636 | <0.00012 | 0.637 | 0.01222 |
| wGRS with HLA & covariates1 | 0.741 | <0.00013 | 0.721 | <0.00013 | 0.683 | 0.01893 |
Glossary: HLA – HLA DRB1*1501 allele.
For derivation and TT samples adjusted for covariate sex, for the NHS/NHS II sample adjusted for smoking (never, ever, current) and EBNA titer.
p-value compared to wGRS without HLA model
p-value compared to wGRS with HLA model
Figure 3. ROC curves for models predicting a diagnosis of MS or CIS. (a) Derivation samples.


We plot the results for three separate models predicting a diagnosis of MS: “GRS w/o HLA DRB1” that includes 15 susceptibility loci and excludes the HLA DRB1*1501 allele (blue), “GRS w/HLA DRB1” that includes 15 susceptibility loci and the HLA DRB1*1501 allele (red), “GRS w/HLA DRB1+ female” that includes 15 susceptibility loci, the HLA DRB1*1501 allele, and gender (green). (b) TT samples. We repeat the analysis in (a) using the TT validation samples. The same presentation scheme is used. (c) NHS/NHS II samples. The analysis method is the same in the NHS/NHS II validation samples. These subjects are all women. The third model that is plotted (green line) includes all 15 susceptibility loci, the HLA DRB1*1501 allele, as well as terms for smoking and EBV titers.
As expected, much of the predictive power comes from the HLA DRB1*1501 allele which is both frequent in these sample collections of European ancestry and has a large effect on susceptibility to MS (odds ratio = 2.8)1. Nonetheless, as shown in Table 4, the other alleles do contribute to the wGRS, having a C-statistic of 0.611 in the combined derivation sample as well as 0.559 and 0.566 in the TT and NHS/NHS II samples respectively, once the HLA DRB1*1501 allele is excluded. A second MHC susceptibility allele, HLA B*44, of more modest effect exists; however, even after we remove HLA B*44 from our model, non-MHC alleles have a consistent contribution to the wGRS: derivation sample – 0.600, TT sample – 0.562, and NHS/NHSII – 0.546. The contribution of the non-MHC loci to the wGRS should increase substantially as more of the suspected susceptibility loci are identified.
Integrating the wGRS with environmental risk factors
Previously, we and others have demonstrated that the effects on MS susceptibility of anti-EBV antibody titers (specifically anti-EBNA1 titers)9,15 is largely distinct from the effects contributed by the HLA DRB1*1501 allele. We have therefore expanded our wGRS to include this environmental susceptibility factor as well as smoking, which is independently associated with risk of MS4. These additional variables enhance our predictive power: the c-statistic for the model that incorporates both the wGRS and the environmental risk factors is 0.683 (vs. 0.637 for the model with just the wGRS) in the NHS/NHS II cohort of modest size for which these variables are available.
Conversion of a clinically isolated demyelinating syndrome to MS
We have also calculated the wGRS in a third collection of subjects: the SET study which consists of subjects with CIS recruited in Prague, Czech Republic (Table 1b). These subjects display the same distribution of wGRS (P =0.98 compared to NHS/NHS II samples that all have a diagnosis of MS), suggesting that there is little difference between the genetic architecture for susceptibility to CIS and MS. We also explored the possibility that the genetic factors associated with susceptibility – that is, with the onset of inflammatory demyelination – have a role in disease course by assessing whether the wGRS of subjects with CIS correlates with the time from the onset of symptoms to the time of a second demyelinating event and hence a diagnosis of MS in the Czech SET collection and the TT collection which contains subjects with CIS. In the subset of CIS subjects on placebo in the TT collection, the estimated hazard ratio from the univariate proportional hazards model for wGRS was 1.00 (95% confidence interval 0.71–1.41]), P =0.99. The estimate did not change substantially after the variables age and sex were taken into consideration. There is no evidence for a non-linear relation of wGRS to the log hazard ratio (P=0.74) or for a deviation from the proportional hazards assumption (p=0.087). The Czech SET study samples displayed a similar finding: the univariate proportional hazards model for wGRS was 0.82 (95% confidence interval 0.54–1.26]), P=0.36.
Discussion
The application of genome-wide association scan methods has been very successful in MS and other inflammatory diseases, and the rapidly evolving repertoire of susceptibility alleles will expand further at the conclusion of ongoing whole genome scans. For the most part, the susceptibility alleles that have been discovered to date fit the profile targeted by genome-wide association studies: we have discovered alleles with a frequency > 0.05 and modest odds ratios (1.1–1.3). It is then important to ask whether identification of these allelic variants are clinically meaningful. For an individual patient, a single polymorphism of modest effect that is common in the general population is not informative. However, because large numbers of MS susceptibility loci with common alleles exist, their aggregate risk is informative in estimating the probability that an individual will develop MS. Indeed, the first iteration of our algorithm, using just 16 loci, is robust, offering consistent predictive ability in two subject collections that had not previously been investigated as part of gene discovery efforts. It is flexible, as additional loci, whether containing common or rare alleles, can easily be added to enhance the predictive ability of our algorithm, and it includes both risk and protective loci using biallelic SNP markers. Currently, the algorithm does not include terms for interaction among the loci as there has been no validated evidence of such interaction in the genetic data examined to date. Such interactions may be robustly demonstrated over time, and the algorithm can be easily modified to accommodate such observations. As expected, the MHC currently plays a dominant role in the current algorithm (Table 4, Figure 3) given the large effect size and relatively high frequency of the HLA DRB1*1501 allele, but the other alleles clearly contribute to prediction of a diagnosis of MS. Of note, since the distribution of wGRS is not distinguishable between MS and CIS in subjects who are at high risk of MS based on MRI criteria (Figure 1), the model therefore appears to be applicable not only to MS but also to CIS.
Nonetheless, the algorithm’s predictive ability, while robust, remains modest and is not ready for clinical deployment. The c-statistics from our wGRS analyses demonstrate that much of the variance in MS susceptibility is not explained by the current models. Indeed, it is possible that, even with a larger complement of the fifty or more MS susceptibility loci that may exist, the algorithm may not be sufficiently effective. Our investigation of the NHS/NHS II sample collection that is of modest size suggests that the best strategy will probably involve the inclusion of non-genetic susceptibility factors to enhance the algorithm and make it useful in a clinical setting: the environmental risk factors assessed in this study appear to provide information that is non-redundant relative to the genetic data (Figure 3c). Thus, further prospective studies of the wGRS should include detailed environmental and immunologic characterizations. Overall, our best c-statistics for the model that includes gender is currently 0.721, which is near the performance of the Framingham Risk Score for coronary heart disease (c-statistic ~ 0.8) that is deemed clinically useful16. Thus, future iterations of the algorithm may reach a level of prediction that is useful in a clinical setting when assessing MS susceptibility.
On the other hand, our initial assessment of the wGRS in predicting relapse after an initial episode of inflammatory demyelination suggests that the current version of the algorithm is not useful for this critical question. Mechanisms involved recurrence of inflammatory demyelination may therefore be different from those involved in demyelinating disease onset. However, while we have assessed two different CIS populations (one treated with interferon beta 1a and one untreated) that show concordant results, both sample collections are small and have a limited duration of follow-up at this time and a low rate of clinical events. Our results should therefore be interpreted cautiously and further investigations of the wGRS with other parameters relating to disease course and disease activity are needed.
In the future, how could the wGRS be used in a clinical setting? In MS, the inciting event is not observed and remains unknown today. It is likely to occur months or years before the onset of clinical symptoms, which are often preceded by asymptomatic CNS lesions14. Identifying individuals with a high genetic risk for MS (category 7 in Figure 2) may therefore open new avenues of investigation and treatment seeking to prevent the onset of the disease rather than modifying its course once it has already become symptomatic, as is done today. Specifically, a version of the GRS may eventually be helpful in stratifying the risk of individuals who are already known to be at higher risk of developing MS, such as first degree relatives which have a rate of 2–5% risk of MS, which is 20–50 times greater than that of the general population of European ancestry5. Since vitamin D deficiency has been associated with MS susceptibility, relatively benign interventions such as vitamin D screening and vitamin D supplementation could be implemented in individuals in high-risk categories. Furthermore, given the benefit of early treatment on minimizing later disability in individuals having one episode of demyelination9 and the observation that many subjects with asymptomatic T2 hyperintense lesions go on to have radiographic or clinical relapses17, there may be an argument for MRI screening of first degree relatives of MS patients who are in the top stratum of the GRS distribution to capture patients at the clinically silent stage of the disease.
Overall, this study suggests that information gleaned from MS susceptibility loci may offer useful information if used in the context of clinical algorithms with other dimensions of information such as environmental risk factors. We note that our estimate of genetic risk is still crude, as it contains only a minority of susceptibility loci that are suspected to exist. Further assessments of this methodology are therefore warranted, particularly as loci correlated with disease course in MS begin to emerge and a separate algorithm targeting progression can be designed.
Acknowledgments
P.L.D. is a Harry Weaver Neuroscience Scholar Award Recipient of the National MS Society (NMSS). DAH is a Jacob Javits Scholar of the NIH. DH, EH, MT and PL are partially supported by the Czech Ministry of Education (Research Program MSM 0021620849) and by the Czech Ministry of Health (grant IGA MZCR 1A/8713 – 5). KCS is supported by training grant T32 ES16645-01. The SET study was supported by Biogen IDEC, Inc. We thank the International MS Genetics Consortium for the use of genotype data. We thank the Myocardial Infarction Genetics Consortium (MIGen) study for the use of their genotype data as control data in our study. The MIGen study was funded by the U.S. National Institutes of Health and National Heart, Lung, and Blood Institute’s STAMPEED genomics research program and a grant from the National Center for Research Resources. We acknowledge use of genotype data from the British 1958 Birth Cohort DNA collection, funded by the Medical Research Council grant G0000934 and the Wellcome Trust grant 068545/Z/02.
Additional Authors
Principle investigators as members of the steering committees of studies evaluating IFNβ-1b and a CCR1-antagonist (studies sponsored by Bayer Schering Pharma) include
The BEtaferon®/BEtaser•on ® in Newly Emerging Multiple Sclerosis For Initial Treatment (BENEFIT) study
Prof. Gilles Edan
Service Neurologie, C.H.U. Rennes Hôpital Pontchaillou/France
Prof. Ludwig Kappos
Neurologische Universitätsklinik, Kantonsspital Basel/Switzerland
Prof. David Miller
Institute of Neurology, The National Hospital, Queen Square, London/GB
Prof. Javier Montalbán
Unitat de Neuroimmunología Clínica Hospitals Vall dHebrón, Madrid/Spain
Prof. Chris H. Polman
Dept. Neurology Free University Hospital, Amsterdam/The Netherlands
Prof. Mark S. Freedman
General Campus - Division of Neurology, The Ottawa Hospital, Ottawa/Canada
Prof. Dr. Hans-Peter Hartung
Neurologische Klinik der Heinrich-Heine-Universität, Düsseldorf/Germany
The Betaferon® Efficacy Yielding Outcomes of a New Dose (BEYOND) study
Prof. Dr. Barry G.W. Arnason
Department of Neurology, Surgery Brain Research Institutes, Chicago/USA
Prof. Dr. Giancarlo Comi
Divisione Neurologia e Neurofisiologia; Ospedale San Raffaele, Milano/Italy
Prof. Dr. Stuart Cook
Department of Neurology and Neuroscience, UMDNJ-New Jersey Medical School, Newark, New Jersey/USA
Prof. Dr. Massimo Filippi
Neuroimaging Research Unit, Ospedale San Raffaele, Milano/Italy
Prof. Dr. Douglas S. Goodin
Department of Neurology, University of California San Francisco, San Francisco/USA
Prof. Dr. Hans-Peter Hartung
Neurologische Klinik der Heinrich-Heine-Universität, Düsseldorf/Germany
Prof. Dr. Douglas Jeffery
Wake Forest University, Department of Neurology, Winston-Salem/USA
Prof. Dr. Ludwig Kappos
Neurologische Universitätsklinik, Kantonsspital Basel/Switzerland
Prof. Dr. Paul O’Connor
Division of Neurology, St. Michael’s Hospital, Toronto/Canada Central MRI (also SC)
Prof. Dr. Massimo Filippi
Neuroimaging Research Unit, University Hospital San Raffaele, Milano/Italy
The 16 year long term follow-up of the pivotal study on IFNB-1b in RRMS:
Prof. George C. Ebers
University Dept of Clinical Neurology, Oxford/UK
Dr. Douglas S. Goodin
Department of Neurology, University of California San Francisco, San Francisco/USA
Dr. Dawn Langdon
Psychology Department, Royal Holloway, University of London/UK
Dr. Anthony T. Reder
The University of Chicago, Department of Neurology, Chicago/USA
Dr. Anthony Traboulsee
UBC Hospital, Vancouver/Canada
The CCR1-antagonist study:
Prof. Frauke Zipp
Cecilie-Vogt-Klinik, Charite – University Hospital Berlin, Germany
Dr. Jan Schimrigk,
University Hospital Bochum/Germany
Prof. Hans-Peter Hartung
Neurologische Klinik der Heinrich-Heine-Universität, Düsseldorf/Germany
Prof. Jan Hillert
Karolinska Institutet at Huddinge University Hospital, Huddinge/Sweden
Prof. Dr. Massimo Filippi
Neuroimaging Research Unit, University Hospital San Raffaele, Milano/Italy
Footnotes
Author Contributions
Study design and manuscript preparation: PLD, LBC and EK. Data Analysis: LBC, JC, SL, and PLD. Data Generation: JR, CA, and PLD. Subject recruitment and clinical characterization: KCS, JH, RS, CP, MT, PL, EH, DH, AA, and the steering committees of studies evaluating IFNβ-1b and a CCR1-antagonist. Critical review of the manuscript: DAH, JR, CP, EWK, DH, EH, and the steering committees of studies evaluating IFNβ-1b and a CCR1-antagonist. All authors have read and reviewed the manuscript.
The corresponding author has had full access to all the data in the study and had final responsibility for the decision to submit for publication.
Conflict of Interest
Dr. Cook has received honoraria and consulting fees from Merck-Serono and Bayer Healthcare.
Dr. Filippi has received grant/research support from TEVA, Merck-Serono, Bayer Schering Pharma, Biogen IDEC, and GENMAB. He has been a consultant or received speaker fees from: TEVA, Merck-Serono, Bayer Schering Pharma, Biogen IDEC, and GENMAB.
Dr. Freedman has received honoraria or consulting fees from Merck-Serono, Novartis, Bayer Healthcare, TEVA, Biogen IDEC, and Sanofi Aventis.
Dr. Havrdova received speaker’s honoraria from Biogen IDEC, TEVA, Novartis, Merck and Bayer Schering Pharma.
Dr. Langdon has received honoraria from Merck-Serono, Novartis, Bayer Healthcare, Sanofi-Aventis, Hoffman LaRoche. She performs contract work for Merck-Serono, and Bayer Healthcare
Dr. O’Connor has received consulting fees from TEVA, Sanofi-Aventis, Novartis, Bayer. He receives grant support from Bayer, Biogen IDEC, Novartis, and Sanofi Aventis.
Dr. Polman has received consulting and/or lecturing fees from Biogen IDEC, Bayer Schering Pharma AG, TEVA, Serono, Novartis, GlaxoSmithKline, UCB, AstraZeneca, Roche, and antisense therapeutics. He receives grant support from Biogen IDEC, Bayer Schering Pharma, TEVA, Serono, Novartis, GlaxoSmithKline.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Bibliography
- 1.De Jager PL, Jia X, Wang J, et al. Meta-analysis of genome scans and replication identify CD6, IRF8, and TNRSF1A as new multiple sclerosis susceptibility loci. Nat Genet. 2009;41:776–782. doi: 10.1038/ng.401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Barrett JC, Hansoul S, Nicolae DL, et al. Genome-wide association defines more than 30 distinct susceptibility loci for Crohn’s disease. Nat Genet. 2008;40:955–962. doi: 10.1038/NG.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Barrett JC, Clayton DG, Goncannon P, et al. Genome-wide association study and meta-analysis find that over 40 loci affect risk of type I diabetes. Nat Genet. 2009 doi: 10.1038/ng.381. online. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ascherio A, Munger KL. Environmental risk factors for multiple sclerosis: from risk factors to prevention. Semin Neurol. 2008;28:17–28. doi: 10.1055/s-2007-1019126. [DOI] [PubMed] [Google Scholar]
- 5.Compston A, Coles A. Multiple Sclerosis. Lancet. 2008;359:1221–1231. doi: 10.1016/S0140-6736(02)08220-X. [DOI] [PubMed] [Google Scholar]
- 6.Kieseier BC, Wiendl H, Leussink VI, Stuve O. Immunomodulatory treatment strategies in multiple sclerosis. J Neurol. 2008;255 (suppl 6):15–21. doi: 10.1007/s00415-008-6004-z. [DOI] [PubMed] [Google Scholar]
- 7.Polman CH, Reingold SC, Edan G, et al. Diagnostic criteria for multiple sclerosis: 2005 revisions to the “McDonald” criteria. Ann Neurol. 2005;58:840–846. doi: 10.1002/ana.20703. [DOI] [PubMed] [Google Scholar]
- 8.The Interferon beta-1b study group. Interferon beta-1b is effective in relapsing-remitting multiple sclerosis. Neurology. 2005;67:1242–1249. [Google Scholar]
- 9.Kappos L, Polman CH, Freedman MS, et al. Long-term effect of early treatment with interferon beta-1b after a first clinical event suggestive of multiple sclerosis: 5 year active treatment extension of the phase 3 BENEFIT trial. Lancet Neurol. 2009 doi: 10.1016/S1474-4422(09)70237-6. online. [DOI] [PubMed] [Google Scholar]
- 10.O’Connor P, Filippi M, Arnason B, et al. 250 mug or 500 mug interferon beta-1b versus 20 mg glatiramer acetate in relapsing-remitting multiple sclerosis: a prospective, multicentre study. Lancet Neurol. 2009 doi: 10.1016/S1474-4422(09)70226-1. online. [DOI] [PubMed] [Google Scholar]
- 11.Zipp F, Hartung HP, Hillert J, et al. Blockade of chemokine signaling in patients with multiple sclerosis. Neurology. 2006;67:1880–1883. doi: 10.1212/01.wnl.0000244420.68037.86. [DOI] [PubMed] [Google Scholar]
- 12.Myocardial Infarction Genetics Consortium. Genome-wide association of early-onset myocardial infarction with single nucleotide polymorphisms and copy number variants. Nat Genet. 2009;41:334–341. doi: 10.1038/ng.327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.De Jager, Simon KC, Munger KL, et al. Integrating risk factors: HLA DRB1*1501 and Epstein-Barr virus in multiple sclerosis. Neurol. 2008;70:1113–1118. doi: 10.1212/01.wnl.0000294325.63006.f8. [DOI] [PubMed] [Google Scholar]
- 14.DeLong ER, DeLong DM, Clarke-Pearson DL. Comparing the areas under two or more correlated receiver operating characteristic curves: a nonparametric approach. Biometrics. 1988;44:837–845. [PubMed] [Google Scholar]
- 15.Sundstrom P, Nystrom L, Jidell E, Hallmans G. EBNA-1 reactivity and HLA DRB1*1501 as statistically independent risk factors for multiple sclerosis: a case-control study. Mult Scler. 2008;14:1120–1122. doi: 10.1177/1352458508092353. [DOI] [PubMed] [Google Scholar]
- 16.Wilson PWF, D’Agostino RB, Levy D, et al. Prediction of coronary heart disease using risk factor categories. Circulation. 1998;97:1837–1847. doi: 10.1161/01.cir.97.18.1837. [DOI] [PubMed] [Google Scholar]
- 17.Okuda DT, Mowry EM, Behestian A, et al. Incidental MRI abnormalities suggestive of multiple sclerosis: the radiologically isolated syndrome. Neurol. 2009;72:800–805. doi: 10.1212/01.wnl.0000335764.14513.1a. [DOI] [PubMed] [Google Scholar]