

SUMMARY
Chloride intracellular channel (CLIC) 4 has diverse functions in membrane trafficking, apoptosis, angiogenesis and cell differentiation. CLIC4 is abundantly expressed in macrophages, but its role in innate immune functions is unclear. Here we show that primary murine macrophages expressed increased amounts of CLIC4 after exposure to bacterial lipopolysaccharide (LPS). The endogenous CLIC4 level is significantly elevated in the brain, heart, lung, kidney, liver and spleen after LPS injection of mice. Stable macrophage lines overexpressing CLIC4 produced more TNF, IL-6, IL-12 and CCL5 than mock transfectants when exposed to LPS. To explore the role of CLIC4 in vivo, we generated CLIC4-null mice. These mice were protected from LPS-induced death, had reduced serum levels of inflammatory cytokines. Upon infection with Listeria monocytogenes, CLIC4-deficient mice were impaired in their ability to clear infection, and their macrophages responded to Listeria by producing less inflammatory cytokines and chemokines than the wild type controls. When challenged with LPS in vitro, deletion of clic4 gene had little effect in MAPK and NF-κB activation, but led to a reduced accumulation of phosphorylated IRF3 within macrophages. Conversely, overexpression of CLIC4 enhanced LPS-mediated IRF3. Thus, CLIC4 is an LPS-induced product that can serve as a positive regulator of LPS signaling.
Keywords: inflammation, innate responses, macrophage, TLR-signaling
INTRODUCTION
Chloride intracellular channel (CLIC) is a unique family of seven intracellular proteins that are implicated in cytoskeletal function, mitosis, cell-cycle control and cell differentiations [1]. CLIC members share a conserved ~230 amino acid C-terminal module homologous to the glutathione S-transferase family [2, 3]. Several protein-protein interaction motifs, including SH2, SH3 and 14-3-3-binding domains, reside in this module [4]. Lacking a conventional signal sequence in their N-terminal, CLIC proteins are found in the cytosol as well as in the intracellular membranes within caveolae, trans-Golgi network [5], ER [6], mitochondria [7], and nuclear membranes [8]. This conserved structure can be traced to the invertebrates Drosophila melanogaster and Caenorhabditis elegans [9]. Mutations of EXC-4, a member of the CLIC family in C. elegans, lead to specific tubulogenesis defects in the worm [10]. The name of chloride channel came from the fact that the first discovered member in the family, p64, was identified on the basis of its association with chloride ion channel activity [11]. In reconstituted planar lipids however, CLIC1, 4 and 5 display weak channel activities with poor ion selectivity [12].
CLIC4 is the most studied member within the CLIC family. It is ubiquitously expressed in almost every cell type studied including endothelial cells, epithelial cells, neurons, messenchymal stem cells, keratinocytes, fibroblasts, skeletal muscle cells and leukocytes [13]. CLIC4 was reported to play a role in cellular processes as diverse as membrane trafficking [14], apoptosis [15], blood vessel formation [16] and cell differentiation [1, 8]. Little is known whether these functions of CLIC4 are related to its proposed chloride channel role. Instead, CLIC4 often serves as an adaptor/scaffold protein via association with microtubule, cytoskeleton proteins, or transcription factors during transduction of signals [4, 17, 18].
In a microarray study on the alteration of gene expression profiles in bone marrow-derived macrophages after exposure to LPS, we discovered that clic4 as a target gene whose expression was highly induced by LPS treatment (not shown). It is not known whether CLIC4 plays any role in macrophage innate immune function or whether induction of CLIC4 in macrophages has any physiological significance in host defenses. This study was undertaken to document the impact of over-expression or deletion of CLIC4 on macrophage innate immune responses. Our data suggest a positive regulatory role for CLIC4 in the host responses to LPS.
RESULTS
Induction of CLIC4 in macrophages by bacterial endotoxin and other microbial products
In keratinocytes, CLIC4 expression is induced by p53 expression, DNA damage or exposure to cytokines such as TNF and TGFβ [18–20]. The promoter of mouse Clic4 gene contains 4, 1, and 6 putative binding sites for NF-kB, IRF and AP-1, respectively. It is predicted that innate immune responses should augment CLIC4 expression. Indeed, studies from our lab and others [13] indicated that CLIC4 expression was also upregulated after exposure to bacterial endotoxin LPS in macrophages. To better characterize the regulation of CLIC4 expression in immune stimulated macrophages, we incubated BMDM from C57BL/6 mice with LPS and determined CLIC4 messenger RNA levels by Northern analysis. Primary macrophages expressed low resting levels of CLIC4. In response to LPS stimulation, BMDM increased expression of CLIC4 messenger RNA in the concentration- and time-dependent fashion (Fig. 1A,B). To determine whether CLIC4 expression was also regulated by other TLR ligands, we exposed wild type BMDM to the lipopeptide Pam3CysSerLys4 (Pam3, a ligand of TLR2), polyriboinosinic: polyribocytidylic acid (poly IC, a ligand of TLR3) and hypomethylated bacterial DNA (CpG, a ligand of TLR9) for 6 hours. CLIC4 expression increased in the response to each of the stimuli (Fig. 1C). CLIC4 appeared to be an LPS-induced early gene since its induction by LPS was insensitive to protein inhibitor cycloheximide (Fig. 1D). These results suggest that CLIC4 expression can be up-regulated during innate responses to various stimuli, indicating a possible role of CLIC4 in the early host defense again invading pathogens.
Figure 1. Induction of CLIC4 by LPS in vitro and in vivo.

(A–C) Bone marrow-derived macrophages (BMDMs) were exposed to the indicated concentrations of LPS (A), or to LPS (100ng/ml), synthetic analog of dsRNA (poly I:C, 1μg/ml), type C CpG oligonucleotide (CpG, 2μM), or synthetic bacterial lipoprotein Pam3CSK4 (Pam3, 200 ng/ml) (C) for 6 hours (, or to 100 ng/ml LPS for different times (B). RNA was extracted and subjected to Northern blot analyses for CLIC4 expression. (D) BMDMs pretreated with or without 10 μg/ml cycloheximide (CX) for 1 hour were exposed to 100 ng/ml LPS for 1 hour. CLIC4 mRNA levels were determined by real time RT-PCR. Results are mean ± SEM of triplicates from one of three experiments. (E) Northern blot analysis of CLIC4 in heart, liver, spleen, lung, kidney, and brain at 4 or 16 hours after intraperitoneal injection of LPS (40mg/kg). Equal loading was shown with ethidium bromide staining (EtBr). Northern blots are representative of two to three independent experiments.
Global increases of CLIC4 mRNA in endotoxic mice
To see whether CLIC4 expression could also be induced in vivo in response to systemic inflammation, we injected B57BL/6 mice intraperitoneally with E coli LPS (40 mg/kg body weight), and isolated the total RNAs from heart, liver, spleen, lung, kidney and brain 4 and 16 h following LPS challenge. CLIC4 expression in these organs was examined by Northern blot analyses. In naive mice, CLIC4 messenger RNA levels were detected in all the organs studied except liver (Fig. 1E, left panel). CLIC4 expression was dramatically increased in all the organs tested 4 hours after LPS injection (Fig. 1E middle panel). By 16 hours, the level of CLIC4 in the heart continued to increase, while CLIC4 levels in the liver, spleen, lung, kidney and brain were reduced from the peak, but remained higher than those in saline-injected mice (Fig. 1E, right panel). Saline injection did not lead to detectable changes in CLIC4 expression (data not shown). The results indicate that systemic inflammation can lead to global increases of CLIC4 expression in mice.
Increased expression of CLIC4 enhances macrophage responses to microbial products
Next we asked whether increased CLIC4 expression during inflammatory responses might affect the ability of macrophages to produce inflammatory mediators. RAW264.7 cells were transfected with the pEGFP-N1 vector expressing GFP alone or CLIC4-GFP fusion. Stably transfected clones were selected. Western blot analyses using anti-CLIC4 antibody confirmed that, whereas endogenous CLIC4 expression was comparable in all three samples including parental RAW cells, mock transfectants and stable CLIC4 transfectants, CLIC4-GFP was only detected in CLIC4-GFP expressing RAW cells (Fig. 2A). LPS responsiveness, judged by secretion of cytokines IL-6, IL-12 and chemokine CCL5 by LPS, was enhanced in CLIC4-GFP expressing cells as compared with GFP expressing controls (Fig. 2B). The augmented response to LPS in CLIC4-expressing cells was recapitulated with other microbial products such as poly IC, Pam3Cys and hypomethylated CpG using TNF release as a readout (Fig. 2C). Thus, increased CLIC4 expression, even at moderate levels, lead to enhanced macrophage inflammatory responses in vitro.
Figure 2. CLIC4 over-expression enhances macrophage responses to microbial products.

(A) CLIC4 protein expression in RAW264.7 cells stably expressing CLIC4-GFP was analysed by Western blot. CLIC4-EGFP, EGFP fused to CLIC4 protein. (B, C) ELISA assays of IL-6, IL-12p40 and CCL5 release after incubation of CLIC4-expressing RAW cells or mock transfectants with LPS (100ng/ml) for 24 hours. (C) ELISA assay of TNF release after incubation of CLIC4-expressing RAW cells or mock transfectants with LPS (100ng/ml), poly I:C (1μg/ml), Pam3 (200ng/ml), or CpG (2μM) for 6 hours. *P < 0.05 (Student’s t-test). Data are expressed as means ± SD. One representative of more than three experiments is shown.
Generation of CLIC4 knockout mice
To test whether deletion of CLIC4 leads to a decrease in inflammatory responses, we next generated CLIC4-null mice. To avoid the possibility of embryonic lethality, we chose to make conditional CLIC4 knockout mice. We flanked the clic4 locus exons 2–4 with loxP sites in a bacterial artificial chromosome (BAC) targeting vector (Fig. 3A). Heterozygous and homozygous floxed mice were confirmed by PCR (data not shown). CLIC4-null mice were generated by crossing male clic4 floxed mice with female mice transgenic for Cre recombinase driven by the promoter for the chicken actin gene (CAG). The breeding method was chosen because of Cre toxicity [21]. It is reported that mature oocytes of CAG-Cre transgenic females contain sufficient Cre activity to mediate the deletion of paternally derived LoxP-flanked DNA sequences upon fertilization, irrespective of the transmission of the Cre transgene [22]. We were able to select mice heterozygous for clic4 locus that contained no Cre transgene as the parental pair to generated universal CLIC4-null mice. The production of wild type, CLIC4 hemizygous-deficient, and CLIC4-null mice was confirmed by PCR of genomic DNA isolated from these animals (Fig. 3B). We detected no messenger for CLIC4 in BMDM isolated from the CLIC4 knockout mice even after LPS treatment (Fig. 3C). In contrast, LPS-induced CLIC4 expression in wild type macrophages was readily detectable (Fig. 3C). Western blot of BMDM after exposure to LPS confirmed that CLIC4 protein was absent in CLIC4-null macrophages (Fig. 3D).
Figure 3. Generation of CLIC4 conditional knockout mouse.

(A) Structure of the clic4 gene in wild type allele (top), the target vector (middle), and the mutant allele after cre-mediated recombination of the loxP sites (bottom). Numbered solid boxes, exons; open arrow, LoxP site; FNFLoxP, Frt-Neo-Frt-loxP cassette; DTA, diphtheria toxin fragment A (open circle); P1, P2 and P3, PCR primers employed for genotyping (small arrows) (see Materials and Methods). (B) PCR analysis of genomic DNA from wild type (WT), heterozygous (Het), and CLIC4 knockout (KO) mice using the indicated primers. (C) RT-PCR analysis of CLIC4 expression induced by LPS (100 ng/ml) for 6 hours in BMDMs from CLIC4 knockout mouse (KO) and wild type control (WT). (D) Immunoblot of LPS-induced CLIC4 protein in BMDMs from wild type (WT) and CLIC4 knockout (KO) mice. One of more than three experiments is shown.
CLIC4-null pups were born at the expected Mendelian frequency from hemizygous parents. Young adult CLIC4-null mice were grossly normal and fertile. Comparisons of the body weights between wild type, hemizygous-null and homozygous CLIC4-null mice at the young age revealed a slightly decrease of body weights in the absence of CLIC4 (not shown), consistent with a report from another independently generated CLIC4 knockout mice using a different strategy [23]. Evaluation of whole body pathology will be reported in a separate study (Sung et al., manuscript in preparation).
Deletion of the clic4 gene diminishes LPS responses in vitro and in vivo
To test the effect of deleting endogenous CLIC4 on macrophage function, we stimulated BMDM from CLIC4-null and wild type mice with LPS, and compared the induction of inflammatory mediators at both the messenger RNA and protein levels. Compared to their wild type counterparts, CLIC4-null BMDM responded to LPS with significantly lower transcript levels of a wide variety of cytokines and chemokines including TNF, IL-6, IL-1β, IFNβ, IP-10 and CCL5 (Fig. 4A), and releasing less TNF, IL-6, IL-12 and CCL5 into the medium (Fig. 4B). CLIC4-null macrophages also had a diminished TNF-induction when exposed to other microbial products (Fig. 4C). Some LPS-induced genes such as A20, an anti-inflammatory ubiquitin-editing enzyme, and inducible nitroxide synthase iNOS were not affected by the absence of CLIC4 (data not shown), indicating some selectivity for CLIC4’s action.
Figure 4. Impaired induction of cytokines and chemokines by microbial products in CLIC4-null BMDMs.

(A) Expression of the indicated cytokines by CLIC4-null BMDMs after exposure to LPS (100ng/ml) for 1 h (for TNF, IL-1β and IFNβ) or 3 h (for IL-6, IP-10 and CCL5) was analysed by quantitative RT-PCR. (B and C) ELISAs on conditioned media collected 6 h (for TNF and CCL5) or 24 h (for IL-6 and IL-12) after adding LPS (100 ng/ml), poly I:C (1μg/ml), Pam3 (200 ng/ml), or CpG (2μM). Results are expressed as mean ± SD of triplicates from one of three similar experiments. * p < 0.05, student’s t-test.
To examine the role of CLIC4 in the systemic inflammation induced by LPS in vivo, we injected CLIC4-null and wild type control mice with LPS (30 mg/kg body weight) and monitored survival for 96 hours. Only ~15% of wild type mice survived, while ~55% of CLIC4-null mice recovered from the systemic cytokine storm induced by LPS administration (Fig. 5A). Analysis of circulating cytokine concentrations shortly after LPS injection showed that the levels of TNF, IL-6 and CCL5 were lower in CLIC4-null mice treated with LPS (Fig. 5B). IL-12 level at this early time was similar between wild type and CLIC4-null mice. Thus, CLIC4-null mice were less susceptible to systemic LPS toxicity, possibly due in part to their weakened macrophage responses to LPS.
Figure 5. CLIC4 knockout protects mice from LPS-induced endotoxemia.

(A) WT and CLIC4-null mice (n = 13 per group) were injected intraperitoneally with 30 mg/kg body weight of LPS. Kaplan-Meier survival curves represent the percentage of surviving individuals in each group over a 96 hours period. p = 0.0063 by the log-rank test. (B) ELISA analysis of TNF, IL-6, IL-12p40, and CCL5 in serum 60 or 120 min after LPS injection. Results are expressed as mean ± SE (n=6) from three independent experiments. *p < 0.05; **p < 0.001, student’s t-test.
Defective host defense of CLIC4 knockout mice against Listeria
To test the role of endogenous CLIC4 in host defense, we infected mice with Listeria monocytogenes by intravenous injection of 5000 bacteria/mouse as described [24, 25]. CLIC4-null mice had a high burden of Listeria in the spleen and liver 5 days after infection (Fig. 6A). The compromised host defense of CLIC4-null mice at early time points suggested that there might be a defect in their innate immune responses. Next, we exposed wild type or CLIC4-null BMDM to Listeria in vitro with an MOI of 1 for 6 or 24 hours, and measured the cytokine contents in the conditioned media using ELISA. CLIC4-deficient macrophages released less TNF, IL-6 and IL-12 in response to Listeria treatment (Fig. 6B), consistent with their defective responses to microbial products we described early (Fig. 4). Thus, compromises of host defense of CLIC4-null mice against Listeria infection may result from reduced innate responses of their macrophages to Listeria.
Figure 6. CLIC4 knockout mice are more susceptible to Listeria monocytogens (L. monocytogens) infection than wild type mice.

(A) WT and CLIC4 knockout mice were infected intravenously with 5 × 103 L. monocytogenes. Bacterial burdens in spleen and liver were determined as colony forming units 5 days after infection. Horizontal bars represent the means of each group mice. p- values were determined by student’s t-test. Data represent results pooled from three independent experiments. (B) ELISAs on conditioned media collected 6 h (for TNF) or 24 h (for IL-6 and IL-12) after adding Listeria. Results are expressed as means ± SE of triplicates from one of three similar experiments. * p < 0.05, student’s t-test.
LPS-induced IRF3 activation is repressed in the absence of CLIC4, but enhanced when CLIC4 is overexpressed
To investigate the mechanisms underlying defective responses of CLIC4-null macrophages to LPS, we first compared LPS-induced MAPK activation between wild type and CLIC4-null BMDM. Activation of ERK1/2, JNK and p38 was estimated via LPS-induced accumulation of phosphorylated forms of these kinases. Similar accumulations of phospho-ERK1/2, JNK and p38 in both groups with an identical kinetics were detected (Fig. 7A). Next, we compared LPS-induced activation of transcription factor NF-kB and interferon response factor 3 (IRF3) between wild type and CLIC4-null BMDM. In response to LPS treatment, CLIC4-null cells accumulated as much phosphorylated p65 with a similar kinetics as wild type cells (Fig. 7B). This was confirmed by an independent assay in which expression of CLIC4 did not alter the activity of a NF-kB-driven reporter in response to LPS (data not shown). By contrast, less phosphorylated IRF3 was detected in CLIC4-null throughout the entire period (Fig. 7B). To ascertain the specificity of CLIC4 on LPS signaling, we also compared interferon-gamma induced Stat1 and Stat3 activation between the wild type and CLIC4-null macrophages. Phosphorylation of stat1 and stat3 after IFNγ treatment followed a similar kinetics with an identical intensity regardless whether clic4 gene is deleted (Fig. 7C). Conversely, more phosphorylated IRF3 was accumulated in RAW 264.7 cells that express CLIC4-GFP fusion protein than mock controls in response to LPS (Fig. 7D, upper), confirming a positive correlation between CLIC4 expression and LPS-induced IRF3 phosphorylation. It is known that IRF3 translocates into nuclei upon activation [26]. To test whether expression of CLIC4 affects IRF3 nuclear translocation, we compared nuclear accumulations of IRF3 between CLIC4-GFP and GFP positive cells after LPS treatment. More prominent nuclear IRF3 signal was readily detectable in cells expressing CLIC4-GFP than cells expressing GFP in response to LPS (Fig. 7D, lower). These results collectively argued that CLIC4 modulates LPS-signaling pathway in macrophage, at least in part, through activation of IRF3.
Figure 7. CLIC4 participates in LPS-induced IRF3 activation in macrophages.
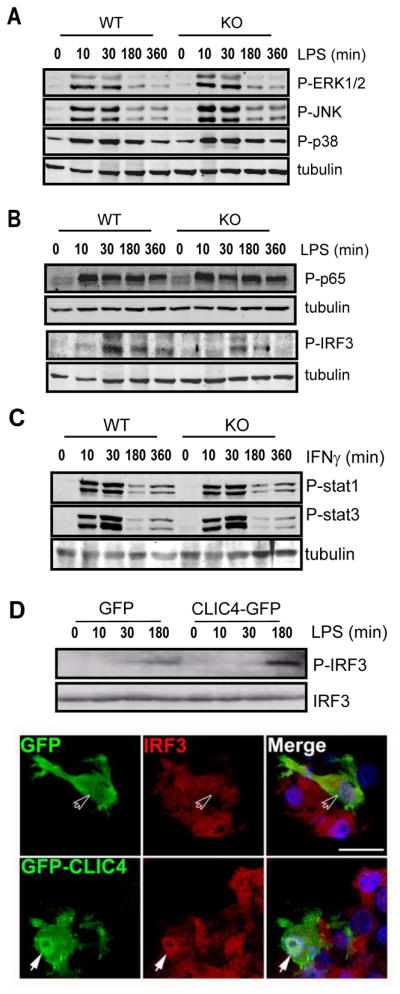
(A–C) Immunoblot analysis of phosphorylation of (A) Erk1/2, JNK1, p38, (B) NF-κB p65, IRF3 induced by LPS (100 ng/ml) or (C) Stat1 and Stat3 induced by IFNγ (100 U/ml) in BMDMs from WT and CLIC4-knockout mice. (D) Upper: Immunoblot analysis of phoosphorylation of IRF3 incuded by LPS (100 ng/ml). Lower: confocal images of IRF3 (red), GFP or CLIC4-GFP (green) and nuclei (blue) 2 hours after LPS treatment. Open arrows indicate nuclear staining of IRF3 in GFP-expressing cells. Closed arrows indicate nuclear enrichment of IRF3 in CLIC4-GFP expressing cells. Scale bar, 20 μm. Data are representative of at least three experiments.
DISCUSSION
Here, we report an unexpected role for CLIC4 in the immune responses of macrophage to microbial products and in the host defense against bacteria infection. Macrophages play an important role in initiating timely innate immune responses to protect the host from invading pathogens [27]. After sensing microbes or trace amounts of their shedding products within the host, macrophages produce an impressive array of inflammatory mediators that dictate the effectiveness and the appropriateness of host inflammatory responses [28]. The scale and the timing of such responses are tightly controlled by both positive and negative regulating molecules within macrophages [29]. Here, we provide three lines of evidence supporting a positive regulatory role of CLIC4 in macrophage innate responses. First, enforced expression of CLIC4 increased the response of RAW264.7 cells to microbial products LPS, Pam3Cys, poly IC and CpG by an enhanced production of proinflammatory mediators. Second, CLIC4-null macrophages, on the other hand, produced less proinflammatory mediators than wild type counterparts when exposed to microbial stimuli. Finally, the “cytokine-storm” induced lethality in experimental mice after LPS injection was reduced in CLIC4-null mice, suggesting a non-redundant role for CLIC4 in systemic innate immune responses in vivo.
CLIC4 was first cloned from rat endoplasmic reticulum of hippocampus and cerebellum but found to be ubiquitously expressed [6]. Expression of CLIC4 is tightly regulated and is often induced under conditions where CLIC4 has a role [4]. For example, CLIC4 could be induced by apoptosis inducing drugs etoposide and adriamycin [20]. Overexpression of CLIC4 in keratinocytes led to apoptosis that was inhibited by the pan-caspase inhibitor Z-VAD-FMK [20]. CLIC4 can also be induced by TGFβ [18]. Interestingly, TGFβ not only induces CLIC4, but also promotes its nuclear translocation. Nuclear CLIC4 then associates with phosphorylated Smad2 and Smad3, two transcription factors critical to TGFβ signaling, and protects them from dephosphorylation by nuclear phosphatase [18]. In accordance, we show here that CLIC4 was induced in macrophages when encountered innate immune stimuli such as LPS. Induction of CLIC4 was also reported in human macrophages [30] and dendritic cells [31–33] after exposure to LPS. Interestingly, an increased CLIC4 expression was only found in M1 but not M2 macrophages [34], suggesting a possible role of CLIC4 in human monocyte polarization. The enhanced CLIC4 expression in macrophages in turn led to augmented innate responses. However, deletion of CLIC4 reduced but not abolished LPS responses. Thus, CLIC4 is not indispensable for macrophage to mount an innate response, but is required for an optimal inflammatory reaction. The impairment of Listeria clearance in infected CLIC4-null mice underscores the importance of CLIC4 in host defense.
The molecular mechanism by which CLIC4 impacts macrophage innate responses remains unclear. Reprograming of the gene expression profile is a hallmark of inflammatory responses in macrophages [35]. Engagement of TLRs to their ligands activates several kinase cascades that are important in initiating transcription of many inflammatory mediators [36]. The MAPK cascade and the phosphorylation of intracellular signaling intermediates leading to activation of transcription factor NF-kB and IRFs are among the most studied signaling pathways in macrophages [37–39]. Here, we show that in CLIC4-null macrophages, LPS-mediated activation of MAP kinases and NF-kB was similar to that of wild type cells. However, LPS-mediated phosphorylation of IRF3 was reduced in CLIC4-null macrophages, but enhanced in CLIC4-expressing cells, suggesting a possible link between expression of CLIC4 and IRF3 activation.
IRF3 belongs to a master regulator family of signaling molecules for TLRs [39]. It is required for the transcriptional induction of the type I IFN genes and a subset of inflammatory cytokines and chemokines [40]. The finding that CLIC4 is required for optimal activation of IRF3 may help explain why CLIC4-null macrophages were compromised in their ability to producing IRF3-dependent cytokines and chemokines such as IFNβ, IP-10, and CCL5 when activated, and why CLIC4 knockout mice shared resistance to LPS-induced endotoxin shock with IRF3 knockout mice [41]. CLIC4 has been shown to associate with several nuclear proteins including transcription factor NTF2 [7], Schnurri-2, Smad2 and Smad-3 [18] within nuclei. A recent study reported that S-nitrosylation of CLIC4 regulates its nuclear translocation [42]. Although IRF-3 undergoes nuclear translocation following activation by TLRs [43–46], CLIC4 appears to be required in the events that precede nuclear translocation of IRF3 since we found that expression of CLIC4 influenced cytosolic phosphorylation of IRF3. Thus, the role of CLIC4 in IRF3 activation likely resides outside of the nucleus.
Several lines of findings here, however, also suggest that CLIC4 can regulate macrophage innate responses via mechanisms other than promoting IRF3 activation. First, inflammatory mediators, whose induction by LPS was affected by deletion or overexpression of CLIC4, also include some IRF3-independent gene products such as TNF, IL-1β and IL-6. In addition, CLIC4 expression seemed to impact macrophage responses not only to LPS, but also to ligands of other TLRs including TLR2 and TLR9, which do not utilize IRF3 pathway. Finally, IRF3-deficient mice were protected from Listeria infection [47] while CLIC4-null mice were more susceptible to this infection, suggesting a non-overlapping role of CLIC4 and IRF3 in this model. It is likely that ubiquitously expressed CLIC4 may contribute to the innate immune responses via multiple routes in macrophages and other cell types.
In summary, our findings demonstrate an unexpected role of CLIC4 in innate immune responses to microbial products. CLIC4 is required for an optimal macrophage response to diverse pathogens. Whether CLIC4 has any channel activity in macrophages and if yes, whether this activity relates to its function in macrophage activation remains to be determined.
MATERIALS AND METHODS
Mice and primary macrophages
C57BL/6 mice were purchased from the Jackson Labolatories. Bone marrow-derived macrophages (BMDM) were prepared as described [48]. Animal studies were approved by the Institutional Animal Care and Use Committee of Weill Cornell Medical College.
Northern blot analysis
Total RNA was extracted from cell culture or from mouse tissues with TRIzolR Reagent (Invitrogen), and analysed with northern blotting using a 300 bp probe corresponding to 460–759 nt of CLIC4 cDNA. The primer sequences for RT-PCR of CLIC4 probe are as follows: forward primer 5′-GCGCTGTCGATGCCCCTGAACGGGC-3′ and reverse primer 5′-CTTGGTAAGTCTCTTGGCGACA-3′.
Generation of CLIC4 expressing RAW264.7 cell line
cDNA for the CLIC4 open reading frame was generated by RT-PCR from total RNA from LPS-treated RAW 264.7 cells, and inserted into Eco RI and Bam HI sites of pEGFP-N1 (Clontech) to obtain a sequence coding for the CLIC4-GFP fusion protein that is driven by CMV promoter. Plasmids with or without CLIC4 insert (10 μg) was added into 107 RAW 264.7 cells followed by electroporation at the 975 μF, 300 V (Gene Pulser, Bio-Rad). Two days later, 600 μg/ml of G418 was added to the transfected cells. Stable transfectants were selected for 7~10 days. GFP (controls) or CLIC4-GFP expressing cells were sorted with FACSCalibur (BD Biosciences) and GFP positve cells were collected for experiments.
ELISA
The levels of cytokines and chemokines in the conditioned media of cell cultures or in the sera collected from LPS injected mice were measured with ELISA kits from R&D systems according to the manufacturer’s protocol.
Construction of CLIC4 targeting vector
A Bacterial Artificial Chromosome (BAC, clone RP23–230K10) containing the mouse WT clic4 locus, was from the Children’s Hospital of Oakland Research Institute. The BAC modifications were made in SW102 strain in a three-step strategy as described previously [48]. First, a PCR product containing a loxP site fused with an FRT-flanked neomycin/kanamycin (neo/kan) cassette and two 50 bps of sequences homologous to the targeting site at both ends was introduced into the fourth intron of the clic4 gene on the BAC clone. Then a single loxP site was inserted into the first intron using galactose positive/negative selection. Finally, a negative selection cassette for diphtheria toxin/ampicillin (DTA) was inserted into the sequence after the last exon of clic4 gene with the selection of both kanamycin-and ampicillin- resistantrecombinants.
Generation of CLIC4-null mice
Fourty micrograms of the BAC targeting vector was linearized and electroporated into CY2.4 embryonic stem (ES) cells with homozygous C57BL/6J/TyrC-21 genetic background (http://jaxmice.jax.org/strain/000058.html). 200 ES cell clones selected for G418 resistance were screened by Southern analysis for both the 5′- and 3′-insertion of loxP sites. 3 out of 6 targeted ES clones were injected into C57BL/6J blastocysts to produce chimeras using standard procedure. Two male chimeras from independently targeted ES clones were backcrossed with C57BL/6J/TyrC-21 female mice and resulted in the germline-transmitted heterozygous floxed mice (CLIC4flox). CAG-Cre mice were from the Mouse Genetics Core Facility at WMC/Sloan-Kettering Institute. Mating of CLIC4flox/+ mice with CAG-Cre mice resulted in both Cre-negative and Cre-positive CLIC4 heterozygous knockout mice (CLIC4+/−). Cre-negative heterozygous clic4 deleted male and female mice (cre−, CLIC4+/−) were mated to generate homozygous clic4 gene-deleted mice (CLIC4−/−) without cre. All the experiments were performed using age- and sex-matched cre-negative homozygous CLIC4 knockout mice (cre−, clic4−/−) and their WT littermates.
Genotyping of CLIC4 knockout mice
The primer sequences used for genotyping of CLIC4 knockout mice are as follows: P1, 5′-CATGTGCCACCACCACCAGA-3′, P2, 5′-GACCAAGCTGGCCTCCAATTA-3′ and P3, 5′-TCAGTTATAGTTTGTGTTAA -3′.
Quantitative real-time RT-PCR
Total RNA was extracted with RNeasy mini kit (Qiagen) and real time PCR was carried out on the ABI PRISM 7900HT sequence detection system (PerkinElmer). The primer and probe sequences for TNF, IL-6, IL-12p40, CCL5, and GAPDH were described previously [48]. The amount of mRNA was expressed as relative mRNA, normalized to GAPDH mRNA.
Western blot
Cell proteins were separated by 10% or 15% SDS-PAGE, transferred onto nitrocellulose membranes for immunoblotting with respective antibodies. The sources of the antibodies are as follows. Rabbit anti-phospho-IRF3 (Ser396) (cat# 4947), rabbit anti-phospho-NF-κB p65 (Ser536) (cat# 3031), rabbit anti-phospho-JNK (Thr183/Tyr185) (cat# 9251), rabbit anti-phospho-Stat1 (Tyr701) (cat# 9171), mouse anti-phospho-Stat3 (Tyr705) (cat# 9138), and rabbit anti-phospho-Erk1/2 (Tyr202/Tyr204) (cat# 4370) were purchased from Cell Signaling Technology. Rabbit anti-phospho-p38 (sc-7975-R) was from Santa Cruz, Inc. Rabbit anti-CLIC4 antibody was raised against CLIC4-GST fusion protein and affinity purified as described [49].
Immunostaining and confocal analysis
RAW 264.7 cells (4 × 104) seeded on a converslip in 24-well place were transfected with 0.2 μg of CLIC4-GFP or GFP plasmids using Fugene 6 (Roche) according to the manufacture’s instruction. Twenty-four hours after transfection, converslips were washed twice with PHEM buffer (60 mM PIPES, 25 mM HEPES, 10 mM EGTA, and 2 mM MgCl2) then incubated with PHEM containing 0.1% TritonX-100 for 15 second to remove excess GFP. Cells were then fixed with 4% paraformaldehyde in PHEM for 10 min. After quenched with 5 mM NH4Cl for 10 min, converslips were blocked for 30 min with a buffer containing 0.5% bovine serum albumin, 0.25% TritonX-100 in PBS, then with rabbit anti-IRF3 Antibody (1:200, Cell Signaling) and chicken anti-GFP Ab (1:2000, Abcam) at 4°C overnight. After extensive washings, cells were incubated with the Alexa 488 conjugated Donkey anti-chicken IgG and Alexa 568 conjugated Donkey anti-rabbit IgG (1:400, Invitrogen) in PBS for 2 hr. Final images were taken with a Leica Confocal Microscope.
LPS-induced endotoxic shock
Six to ten week-old CLIC4 knockout and wild type mice were intraperitoneally injected with 30 mg/kg of LPS from Escherichia coli serotype 0111:B4 (Sigma Cat # L-2630). The survival of mice was monitored every 12 hours for a period of 96 hours.
Listeria monocytogens infection
Sex and age-matched mice were infected intravenously with 5,000 L. monocytogenes, prepared as described (28). At various times after infection, stainless steel wire mesh screens are placed into sterile 100-mm Petri dishes containing 1 ml of 0.1% Triton X-100 in PBS each. Mice were sacrificed, and their spleen and liver homogenized on the sterile wire mesh screen with the rubber plungers of 5-ml syringes. Four serial 1/10 dilutions of the organ homogenates were made and plated on BHI agar plates. The number of viable bacteria was determined by counting overnight (37°C) outgrowth of bacteria.
Statistical analyses were done using two-tailed Student’s t test for independent samples except for the survival study in the endotoxic shock experiment, where the log-rank test was used. P values < 0.05 were considered statistically significant. All the data are presented as means ± S.E. or S.D.
Acknowledgments
This work was supported by National Institutes of Health grants AI030165 (to AD), EY016805 and a research grant from “Research To Prevent Blindness” (to CHS). The Department of Microbiology and Immunology is supported by the William Randolph Hearst Foundation. We thank C Nathan for helpful discussion and K Rhee for critical reading of the manuscript.
Abbreviations used
- BAC
bacterial artificial chromosome
- BMDM
bone marrow-derived macrophage
- CAG
chicken actin gene
- CLIC
chloride intracellular channel
- ES cell
embryonic stem cell
- IRF
interferon response factor
- MOI
multiplicity of infection, Pam3, lipopeptide Pam3CysSerLys4
- poly IC
polyriboinosinic: polyribocytidylic acid
Footnotes
Conflict of interest: The authors declare no financial or commercial conflict of interest.
References
- 1.Suh KS, Mutoh M, Gerdes M, Yuspa SH. CLIC4, an intracellular chloride channel protein, is a novel molecular target for cancer therapy. J Investig Dermatol Symp Proc. 2005;10:105–109. doi: 10.1111/j.1087-0024.2005.200402.x. [DOI] [PubMed] [Google Scholar]
- 2.Dulhunty A, Gage P, Curtis S, Chelvanayagam G, Board P. The glutathione transferase structural family includes a nuclear chloride channel and a ryanodine receptor calcium release channel modulator. J Biol Chem. 2001;276:3319–3323. doi: 10.1074/jbc.M007874200. [DOI] [PubMed] [Google Scholar]
- 3.Harrop SJ, DeMaere MZ, Fairlie WD, Reztsova T, Valenzuela SM, Mazzanti M, Tonini R, Qiu MR, Jankova L, Warton K, Bauskin AR, Wu WM, Pankhurst S, Campbell TJ, Breit SN, Curmi PM. Crystal structure of a soluble form of the intracellular chloride ion channel CLIC1 (NCC27) at 1.4-A resolution. J Biol Chem. 2001;276:44993–45000. doi: 10.1074/jbc.M107804200. [DOI] [PubMed] [Google Scholar]
- 4.Suh KS, Malik M, Shukla A, Yuspa SH. CLIC4, skin homeostasis and cutaneous cancer: surprising connections. Mol Carcinog. 2007;46:599–604. doi: 10.1002/mc.20324. [DOI] [PubMed] [Google Scholar]
- 5.Edwards JC. A novel p64-related Cl- channel: subcellular distribution and nephron segment-specific expression. Am J Physiol. 1999;276:F398–408. doi: 10.1152/ajprenal.1999.276.3.F398. [DOI] [PubMed] [Google Scholar]
- 6.Duncan RR, Westwood PK, Boyd A, Ashley RH. Rat brain p64H1, expression of a new member of the p64 chloride channel protein family in endoplasmic reticulum. J Biol Chem. 1997;272:23880–23886. doi: 10.1074/jbc.272.38.23880. [DOI] [PubMed] [Google Scholar]
- 7.Suh KS, Mutoh M, Nagashima K, Fernandez-Salas E, Edwards LE, Hayes DD, Crutchley JM, Marin KG, Dumont RA, Levy JM, Cheng C, Garfield S, Yuspa SH. The organellular chloride channel protein CLIC4/mtCLIC translocates to the nucleus in response to cellular stress and accelerates apoptosis. J Biol Chem. 2004;279:4632–4641. doi: 10.1074/jbc.M311632200. [DOI] [PubMed] [Google Scholar]
- 8.Suh KS, Mutoh M, Mutoh T, Li L, Ryscavage A, Crutchley JM, Dumont RA, Cheng C, Yuspa SH. CLIC4 mediates and is required for Ca2+-induced keratinocyte differentiation. J Cell Sci. 2007;120:2631–2640. doi: 10.1242/jcs.002741. [DOI] [PubMed] [Google Scholar]
- 9.Littler DR, Harrop SJ, Brown LJ, Pankhurst GJ, Mynott AV, Luciani P, Mandyam RA, Mazzanti M, Tanda S, Berryman MA, Breit SN, Curmi PM. Comparison of vertebrate and invertebrate CLIC proteins: the crystal structures of Caenorhabditis elegans EXC-4 and Drosophila melanogaster DmCLIC. Proteins. 2008;71:364–378. doi: 10.1002/prot.21704. [DOI] [PubMed] [Google Scholar]
- 10.Berry KL, Bulow HE, Hall DH, Hobert O. A C. elegans CLIC-like protein required for intracellular tube formation and maintenance. Science. 2003;302:2134–2137. doi: 10.1126/science.1087667. [DOI] [PubMed] [Google Scholar]
- 11.Landry D, Sullivan S, Nicolaides M, Redhead C, Edelman A, Field M, al-Awqati Q, Edwards J. Molecular cloning and characterization of p64, a chloride channel protein from kidney microsomes. J Biol Chem. 1993;268:14948–14955. [PubMed] [Google Scholar]
- 12.Singh H, Ashley RH. CLIC4 (p64H1) and its putative transmembrane domain form poorly selective, redox-regulated ion channels. Mol Membr Biol. 2007;24:41–52. doi: 10.1080/09687860600927907. [DOI] [PubMed] [Google Scholar]
- 13.Ogawa S, Lozach J, Benner C, Pascual G, Tangirala RK, Westin S, Hoffmann A, Subramaniam S, David M, Rosenfeld MG, Glass CK. Molecular determinants of crosstalk between nuclear receptors and toll-like receptors. Cell. 2005;122:707–721. doi: 10.1016/j.cell.2005.06.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jentsch TJ, Stein V, Weinreich F, Zdebik AA. Molecular structure and physiological function of chloride channels. Physiol Rev. 2002;82:503–568. doi: 10.1152/physrev.00029.2001. [DOI] [PubMed] [Google Scholar]
- 15.Suh KS, Mutoh M, Gerdes M, Crutchley JM, Mutoh T, Edwards LE, Dumont RA, Sodha P, Cheng C, Glick A, Yuspa SH. Antisense suppression of the chloride intracellular channel family induces apoptosis, enhances tumor necrosis factor {alpha}-induced apoptosis, and inhibits tumor growth. Cancer Res. 2005;65:562–571. [PubMed] [Google Scholar]
- 16.Bohman S, Matsumoto T, Suh K, Dimberg A, Jakobsson L, Yuspa S, Claesson-Welsh L. Proteomic analysis of vascular endothelial growth factor-induced endothelial cell differentiation reveals a role for chloride intracellular channel 4 (CLIC4) in tubular morphogenesis. J Biol Chem. 2005;280:42397–42404. doi: 10.1074/jbc.M506724200. [DOI] [PubMed] [Google Scholar]
- 17.Suhara W, Yoneyama M, Kitabayashi I, Fujita T. Direct involvement of CREB-binding protein/p300 in sequence-specific DNA binding of virus-activated interferon regulatory factor-3 holocomplex. J Biol Chem. 2002;277:22304–22313. doi: 10.1074/jbc.M200192200. [DOI] [PubMed] [Google Scholar]
- 18.Shukla A, Malik M, Cataisson C, Ho Y, Friesen T, Suh KS, Yuspa SH. TGF-beta signalling is regulated by Schnurri-2-dependent nuclear translocation of CLIC4 and consequent stabilization of phospho-Smad2 and 3. Nat Cell Biol. 2009;11:777–784. doi: 10.1038/ncb1885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fernandez-Salas E, Sagar M, Cheng C, Yuspa SH, Weinberg WC. p53 and tumor necrosis factor alpha regulate the expression of a mitochondrial chloride channel protein. J Biol Chem. 1999;274:36488–36497. doi: 10.1074/jbc.274.51.36488. [DOI] [PubMed] [Google Scholar]
- 20.Fernandez-Salas E, Suh KS, Speransky VV, Bowers WL, Levy JM, Adams T, Pathak KR, Edwards LE, Hayes DD, Cheng C, Steven AC, Weinberg WC, Yuspa SH. mtCLIC/CLIC4, an organellular chloride channel protein, is increased by DNA damage and participates in the apoptotic response to p53. Mol Cell Biol. 2002;22:3610–3620. doi: 10.1128/MCB.22.11.3610-3620.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schmidt-Supprian M, Wunderlich FT, Rajewsky K. Excision of the Frt-flanked neo (R) cassette from the CD19cre knock-in transgene reduces Cre-mediated recombination. Transgenic Res. 2007;16:657–660. doi: 10.1007/s11248-007-9100-4. [DOI] [PubMed] [Google Scholar]
- 22.Sakai K, Miyazaki J. A transgenic mouse line that retains Cre recombinase activity in mature oocytes irrespective of the cre transgene transmission. Biochem Biophys Res Commun. 1997;237:318–324. doi: 10.1006/bbrc.1997.7111. [DOI] [PubMed] [Google Scholar]
- 23.Ulmasov B, Bruno J, Gordon N, Hartnett ME, Edwards JC. Chloride intracellular channel protein-4 functions in angiogenesis by supporting acidification of vacuoles along the intracellular tubulogenic pathway. Am J Pathol. 2009;174:1084–1096. doi: 10.2353/ajpath.2009.080625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Serbina NV, Jia T, Hohl TM, Pamer EG. Monocyte-mediated defense against microbial pathogens. Annu Rev Immunol. 2008;26:421–452. doi: 10.1146/annurev.immunol.26.021607.090326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pamer EG. Immune responses to Listeria monocytogenes. Nat Rev Immunol. 2004;4:812–823. doi: 10.1038/nri1461. [DOI] [PubMed] [Google Scholar]
- 26.Taniguchi T, Ogasawara K, Takaoka A, Tanaka N. IRF family of transcription factors as regulators of host defense. Annu Rev Immunol. 2001;19:623–655. doi: 10.1146/annurev.immunol.19.1.623. [DOI] [PubMed] [Google Scholar]
- 27.Gordon S. The macrophage: past, present and future. Eur J Immunol. 2007;37 (Suppl 1):S9–17. doi: 10.1002/eji.200737638. [DOI] [PubMed] [Google Scholar]
- 28.Nathan C. Points of control in inflammation. Nature. 2002;420:846–852. doi: 10.1038/nature01320. [DOI] [PubMed] [Google Scholar]
- 29.Nathan C, Ding A. Nonresolving inflammation. Cell. 2010;140:871–882. doi: 10.1016/j.cell.2010.02.029. [DOI] [PubMed] [Google Scholar]
- 30.Hofer TP, Frankenberger M, Mages J, Lang R, Meyer P, Hoffmann R, Colige A, Ziegler-Heitbrock L. Tissue-specific induction of ADAMTS2 in monocytes and macrophages by glucocorticoids. J Mol Med. 2008;86:323–332. doi: 10.1007/s00109-007-0284-0. [DOI] [PubMed] [Google Scholar]
- 31.Napolitani G, Rinaldi A, Bertoni F, Sallusto F, Lanzavecchia A. Selected Toll-like receptor agonist combinations synergistically trigger a T helper type 1-polarizing program in dendritic cells. Nat Immunol. 2005;6:769–776. doi: 10.1038/ni1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fulcher JA, Hashimi ST, Levroney EL, Pang M, Gurney KB, Baum LG, Lee B. Galectin-1-matured human monocyte-derived dendritic cells have enhanced migration through extracellular matrix. J Immunol. 2006;177:216–226. doi: 10.4049/jimmunol.177.1.216. [DOI] [PubMed] [Google Scholar]
- 33.Macagno A, Molteni M, Rinaldi A, Bertoni F, Lanzavecchia A, Rossetti C, Sallusto F. A cyanobacterial LPS antagonist prevents endotoxin shock and blocks sustained TLR4 stimulation required for cytokine expression. J Exp Med. 2006;203:1481–1492. doi: 10.1084/jem.20060136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Martinez FO, Gordon S, Locati M, Mantovani A. Transcriptional profiling of the human monocyte-to-macrophage differentiation and polarization: new molecules and patterns of gene expression. J Immunol. 2006;177:7303–7311. doi: 10.4049/jimmunol.177.10.7303. [DOI] [PubMed] [Google Scholar]
- 35.Ehrt S, Schnappinger D, Bekiranov S, Drenkow J, Shi S, Gingeras TR, Gaasterland T, Schoolnik G, Nathan C. Reprogramming of the macrophage transcriptome in response to interferon-gamma and Mycobacterium tuberculosis: signaling roles of nitric oxide synthase-2 and phagocyte oxidase. J Exp Med. 2001;194:1123–1140. doi: 10.1084/jem.194.8.1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dunne A, O’Neill LA. The interleukin-1 receptor/Toll-like receptor superfamily: signal transduction during inflammation and host defense. Sci STKE. 2003;2003:re3. doi: 10.1126/stke.2003.171.re3. [DOI] [PubMed] [Google Scholar]
- 37.Jefferies CA, O’Neill LA. Bruton’s tyrosine kinase (Btk)-the critical tyrosine kinase in LPS signalling? Immunol Lett. 2004;92:15–22. doi: 10.1016/j.imlet.2003.11.017. [DOI] [PubMed] [Google Scholar]
- 38.Kawai T, Akira S. Signaling to NF-kappaB by Toll-like receptors. Trends Mol Med. 2007;13:460–469. doi: 10.1016/j.molmed.2007.09.002. [DOI] [PubMed] [Google Scholar]
- 39.Honda K, Taniguchi T. IRFs: master regulators of signalling by Toll-like receptors and cytosolic pattern-recognition receptors. Nat Rev Immunol. 2006;6:644–658. doi: 10.1038/nri1900. [DOI] [PubMed] [Google Scholar]
- 40.Sato M, Suemori H, Hata N, Asagiri M, Ogasawara K, Nakao K, Nakaya T, Katsuki M, Noguchi S, Tanaka N, Taniguchi T. Distinct and essential roles of transcription factors IRF-3 and IRF-7 in response to viruses for IFN-alpha/beta gene induction. Immunity. 2000;13:539–548. doi: 10.1016/s1074-7613(00)00053-4. [DOI] [PubMed] [Google Scholar]
- 41.Sakaguchi S, Negishi H, Asagiri M, Nakajima C, Mizutani T, Takaoka A, Honda K, Taniguchi T. Essential role of IRF-3 in lipopolysaccharide-induced interferon-beta gene expression and endotoxin shock. Biochem Biophys Res Commun. 2003;306:860–866. doi: 10.1016/s0006-291x(03)01049-0. [DOI] [PubMed] [Google Scholar]
- 42.Mori M, Yoneyama M, Ito T, Takahashi K, Inagaki F, Fujita T. Identification of Ser-386 of interferon regulatory factor 3 as critical target for inducible phosphorylation that determines activation. J Biol Chem. 2004;279:9698–9702. doi: 10.1074/jbc.M310616200. [DOI] [PubMed] [Google Scholar]
- 43.Doyle S, Vaidya S, O’Connell R, Dadgostar H, Dempsey P, Wu T, Rao G, Sun R, Haberland M, Modlin R, Cheng G. IRF3 mediates a TLR3/TLR4-specific antiviral gene program. Immunity. 2002;17:251–263. doi: 10.1016/s1074-7613(02)00390-4. [DOI] [PubMed] [Google Scholar]
- 44.Alexopoulou L, Holt AC, Medzhitov R, Flavell RA. Recognition of double-stranded RNA and activation of NF-kappaB by Toll-like receptor 3. Nature. 2001;413:732–738. doi: 10.1038/35099560. [DOI] [PubMed] [Google Scholar]
- 45.Andersen J, VanScoy S, Cheng TF, Gomez D, Reich NC. IRF-3-dependent and augmented target genes during viral infection. Genes Immun. 2008;9:168–175. doi: 10.1038/sj.gene.6364449. [DOI] [PubMed] [Google Scholar]
- 46.Reich NC. Nuclear/cytoplasmic localization of IRFs in response to viral infection or interferon stimulation. J Interferon Cytokine Res. 2002;22:103–109. doi: 10.1089/107999002753452719. [DOI] [PubMed] [Google Scholar]
- 47.O’Connell RM, Saha SK, Vaidya SA, Bruhn KW, Miranda GA, Zarnegar B, Perry AK, Nguyen BO, Lane TF, Taniguchi T, Miller JF, Cheng G. Type I interferon production enhances susceptibility to Listeria monocytogenes infection. J Exp Med. 2004;200:437–445. doi: 10.1084/jem.20040712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yin F, Banerjee R, Thomas B, Zhou P, Qian L, Jia T, Ma X, Ma Y, Iadecola C, Beal MF, Nathan C, Ding A. Exaggerated inflammation, impaired host defense, and neuropathology in progranulin-deficient mice. J Exp Med. 207:117–128. doi: 10.1084/jem.20091568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chuang JZ, Milner TA, Zhu M, Sung CH. A 29 kDa intracellular chloride channel p64H1 is associated with large dense-core vesicles in rat hippocampal neurons. J Neurosci. 1999;19:2919–2928. doi: 10.1523/JNEUROSCI.19-08-02919.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]