

Abstract
Numerous studies support the notion that cumulative exposure to chronic stress is a risk factor for cardiovascular disease (CVD). Various stress-related hormones have been proposed as potential mediators of the relationship between psychological stress and CVD, including catecholamines and more indirectly, cortisol. Somewhat surprisingly, although aldosterone is also released in response to hypothalamic–pituitary–adrenal (HPA) axis activation, it has not been considered as relevant for this relationship. In the present review we will consider aldosterone as a potentially important mediator of the relationship between negative affective states and CVD. First, we will briefly review the known functions and roles of aldosterone, and then consider its actions in both the brain and the periphery. We will then review the available literature on the role of aldosterone in CVD, and also consider links between aldosterone and various forms of chronic psychological stress. Finally we will present an integrated model of how aldosterone may mediate effects of chronic stress on CVD, recommend new directions for research, and identify important methodological and design issues for this work.
Keywords: Aldosterone, Psychological distress, Stress, Cardiovascular disease, Stress-related hormones, Cortisol, Mineralocorticoid receptor
Cardiovascular disease (CVD) is one of the leading causes of death in the USA and other developed countries. In 2005, 80,700,000 people in the USA were estimated to have one or more forms of CVD, and the national burden of this disease is estimated to be $448.5 billion in 2008 (American Heart Association, 2008). Lifetime risk of CVD is 2 in 3 for men, and greater than 1 in 2 for women at age 40, and in 2004 32% of deaths from CVD occurred prematurely, prior to age 75 (American Heart Association, 2008). In a large-scale case control study conducted across 52 countries, 9 potentially modifiable risk factors were able to account for over 90% of the risk for an initial acute myocardial infarction (Yusuf et al., 2004). Among these risk factors, psychosocial distress was identified as conferring significant risk, even after accounting for the 8 other known behavioral and biological risk factors (Yusuf et al., 2004).
Numerous studies support the notion that cumulative exposure to chronic stress is a risk factor for CVD (Rozanski et al., 2005; Cohen et al., 2007; Dimsdale, 2008). Individuals who report higher levels of psychological stress are at greater risk of incident CVD than those who report less (Everson-Rose and Lewis, 2005; Rozanski et al., 2005; Kubzansky, 2007). Evidence exists for associations of incident coronary heart disease with three specific negative emotions related to psychological stress: anxiety, anger, and depression (Suls and Bunde, 2005; Kubzansky, 2007). While some work has considered how acute negative affective states may stimulate acute cardiac events, much of this research has been concerned with long-term etiology, and with demonstrating that exposure to chronic psychological stress promotes higher rates of CVD incidence (Steptoe and Brydon, 2009). Mechanisms explaining these relationships still have not been fully determined but among those proposed are recurring activation of the hypothalamic–pituitary–adrenal (HPA) axis and sympathetic-adrenomedullary system, as well as altered autonomic cardiac control, accelerated atherosclerosis, and health-related behaviors. Numerous stress-related hormones have been proposed as potential mediators of the relationship between psychological stress and CVD, including catecholamines and more indirectly, cortisol. Somewhat surprisingly, although aldosterone is also released in response to HPA axis activation, it has not been considered as relevant for this relationship.
In the present review we will consider aldosterone as a potentially important mediator of the relationship between psychological stress and CVD. First, we will briefly review the known functions and roles of aldosterone, and then consider its actions in both the brain and the periphery. We will then review the available literature on the role of aldosterone in CVD, and also consider links between aldosterone and various forms of chronic negative affect. Finally we will present an integrated model of how aldosterone may be involved in mediating effects of chronic psychological stress on CVD, recommend new directions for research, as well as identify both methodological and design issues that should be considered in this work.
1. Stress, hypothalamic–pituitary–adrenal axis, and CVD
One of the hallmarks of acute and chronic stress is activation of the HPA axis, and the corticotropin-releasing hormone (CRH)–adrenocorticotropic hormone (ACTH)–cortisol cascade represents the prototypic stress hormone system, often synonymous with the concept of stress for many investigators (Johnson and Grippo, 2006). Numerous studies have demonstrated how cortisol may influence hypertension via increases in the production of angiotensinogen by the liver (Vinson, 2007) and by effects on vascular smooth muscle, endothelial cells, kidney and the heart (Hadoke et al., 2006; Walker, 2007). Cortisol has also been linked with numerous measures of chronic stress and negative affect (including trait anxiety, depressive symptoms, caregiver stress, unemployment), and appears to have the greatest response to stressors that are uncontrollable and socially threatening (Dickerson and Kemeny, 2004). A great deal of work linking psychosocial stress and disease outcomes has focused on alterations in cortisol as one mechanism by which psychological stress might influence physical health outcomes. However, only a limited number of epidemiologic or psychological studies have successfully demonstrated direct links between psychological stress, cortisol and CVD outcomes (Girod and Brotman, 2004; Matthews et al., 2006). This may be because cortisol and dysregulated HPA axis activity are hypothesized to influence risk via indirect effects such as impairing endothelial and baroreflex function (Broadley et al., 2005), impairing insulin sensitivity (van Raalte et al., 2009), increasing risk of abdominal obesity (Bjorntorp, 1997; Epel et al., 2000), and altering inflammatory processes (Miller et al., 2002).
As consideration of effects of cortisol on cardiovascular outcomes has not yielded complete understanding of how psychological stress may influence CVD, it will be useful to consider other neurohumoral agents in relation to stress biology and CVD. In a scholarly review of effects of chronic stress, Selye identified the adrenal corticosteroid aldosterone, as another hormone intimately involved with HPA axis which also may have direct influence on CVD processes (Selye, 1955). Aldosterone is an adrenal corticosteroid that activates the type I glucocorticoid receptor (otherwise known as mineralocorticoid receptor [MR]). Levels of aldosterone are increased by three primary secretagogues: (1) ACTH; (2) angiotensin II; (3) potassium. Thus, activation of both HPA axis and the rennin–angiotensin system leads to increases in aldosterone. In addition, aldosterone is influenced by the sympathetic-adrenomedullary system (SNS), as activation of the SNS leads to activation of β1 adrenoreceptors on renal juxtaglomerular cells which release rennin and thus activate the rennin–angiotensin–aldosterone system (DiBona, 2003). The rennin–angiotensin–aldosterone system plays a key role in cardiovascular homeostasis by maintaining salt and water homeostasis and therefore blood volume, blood pressure, and electrolyte balance. Given that psychological stress activates both the HPA axis and the sympathetic-adrenomedullary system, aldosterone may be released in response to psychological stress and provide an additional pathway between negative affective states and cardiovascular health. Pathological consequences of excess mineralocorticoid activity in early studies of stress included high blood pressure and evidence of myocardial necrosis and fibrosis (Selye, 1946, 1960). Recent work has suggested that aldosterone is a hormone with widespread cardiovascular and metabolic effects, beyond its effects on fluid and electrolyte balance, and many of these processes promote cardiovascular injury (Garg and Adler, 2009).
2. Mineralocorticoid and glucocorticoid receptors
Two receptors bind adrenal glucocorticoids (cortisol) and mineralocorticoids (aldosterone), the type I glucocorticoid receptor (MR) and the type II glucocorticoid receptor (generally referred to as the glucocorticoid receptor [GR]). MR has high affinity for both cortisol and aldosterone (Funder, 2009), whereas GR is selective for cortisol. In the majority of peripheral tissues including the kidney, the enzyme 11 beta-hydroxysteroid dehydrogenase 2 (11β-HSD2) is found in association with MR and prevents cortisol activation of MR by converting cortisol to inactive cortisone. Under conditions of excess cortisol (e.g., Cushing’s syndrome), the ability of 11β-HSD2 to inactivate cortisol is overwhelmed so that cortisol can actually activate MR. Further, under conditions of 11β-HSD2 inactivation, cortisol can activate MR. The intracellular redox state may also regulate cortisol activation of MR (Funder, 2009). Increases in oxidative stress (which preliminary work has also linked with psychological stress) are hypothesized to alter the intracellular redox state leading to activation of MR by glucocorticoids. While MR is expressed in multiple regions of the brain, 11β-HSD2 and MR are co-expressed only in the nucleus tractus solitarius (Funder, 2009). Thus, in most regions of the brain, which lack 11β-HSD2, MR are primarily occupied by cortisol. Activation of GR and MR in the hippocampus and the hypothalamus is thought to be important for glucocorticoid feedback inhibition of the HPA axis with corticosteroids mediating fast feedback inhibition via the MR (Atkinson et al., 2008).
Both GR and MR are present in brain structures that are involved in fear and anxiety (e.g., hippocampus and amygdala) (Korte, 2001). Central balance between MR and GR has been proposed as a critical ingredient for healthy adaptation and mitigating effects of stress (Korte, 2001). De Kloet (2004) has postulated that MR functions to limit disturbance of cellular homeostasis, control sensitivity of the stress–response system, and helps to select appropriate behavioral response, while GR facilitates recovery of cellular homeostasis, restrains stress-induced responses, promotes information storage and adaptation, and controls mobilization and storage of energy. Exposure to chronic psychosocial stress may lead to dysregulated central MR/GR balance by virtue of increases in cortisol and aldosterone (Korte, 2001). Moreover, whether MR is occupied by cortisol versus aldosterone may also have different effects in various tissues leading to potentially different outcomes. However, the role of MR in psychological stress or other behavioral functions has largely been investigated with regard to glucocorticoids and far less attention has been given to aldosterone (Korte, 2001).
3. Aldosterone and CVD
Numerous studies in animals have found that activation of the MR leads to cardiovascular injury, renovascular injury and stroke and that blockade of the MR reduces this injury (Joffe and Adler, 2005; Funder, 2006, 2009; Connell et al., 2008; Garg and Adler, 2009). First we consider how MR activation increases a range of processes involved with cardiovascular injury including oxidative stress, inflammation, insulin resistance, and central nervous system effects. Next we examine clinical situations suggesting that excess MR activation is associated with the pathophysiology of cardiovascular disease. Finally, consider whether these effects are likely to be depend on specific circumstances (e.g., high sodium intake or only evident within certain populations).
With regard to oxidative stress, in vitro studies have suggested that aldosterone increases superoxide generation (Leopold et al., 2007; Iwashima et al., 2008). This may involve decreases in glucose-6-phosphate dehydrogenase activity and increases in NAD(P)H oxidase. More evidence is available to suggest that aldosterone has pro-inflammatory actions. Animal models of cardiac and renal injury have demonstrated the pro-inflammatory effects of aldosterone as well as the anti-inflammatory effects of MR blockade in target organs, vessels, and adipose tissue (Joffe and Adler, 2005; Han et al., 2006a,b; Kang et al., 2006; Guo et al., 2008). Pre-clinical studies have demonstrated that aldosterone increases expression of the transcription factor, nuclear factor-kappa B (NF-κB) (Han et al., 2006b) and genes involved in inflammation, fibrosis, and atherosclerosis (Jaffe and Mendelsohn, 2005; Guo et al., 2008). MR blockade has also been demonstrated to reduce atherosclerosis in animal models (Rajagopalan et al., 2002; Takai et al., 2005). Studies in humans have shown that an aldosterone infusion increased circulating IL-6 levels while spironolactone blocked increases in IL-6 occurring with administration of angiotensin II (Luther et al., 2006).
With regard to insulin resistance, work in humans has demonstrated that obesity is associated with increased production of aldosterone, increased aldosterone correlates with increases in insulin resistance independent of body mass index (Goodfriend et al., 1995, 1998; Bentley-Lewis et al., 2007), and also that increased mineralocorticoid activity is one of the mechanisms by which obesity induces hypertension (Hiramatsu et al., 1981; Dustan, 1983). Weight loss has been demonstrated to reduce insulin resistance and also lower blood pressure and levels of aldosterone (Rocchini et al., 1986; Goodfriend et al., 1999; Engeli et al., 2005). Patients with primary hyperaldosteronism have been shown to be insulin resistant, but patients treated with surgery or MR antagonists, e.g., spironolactone or eplerenone, demonstrated improved sensitivity to insulin (Haluzik et al., 2002; Catena et al., 2006). Other work has shown that spironolactone also reduces insulin resistance in patients with polycystic ovarian syndrome (Moghetti et al., 1996; Ganie et al., 2004; Zulian et al., 2005). Aldosterone may lead to insulin resistance in a number of ways. Aldosterone increases expression of pro-inflammatory factors, decreases expression of the insulin-sensitizing factor adiponectin and has been shown to reduce insulin-stimulated glucose uptake in cultured adipocytes by degrading insulin receptor substrate (IRS) 1 and IRS2 (Kraus et al., 2005; Guo et al., 2008; Wada et al., 2009). MR also mediate corticosteroid-induced adipocyte differentiation (Caprio et al., 2007).
While many of these adverse effects of aldosterone are mediated through actions in peripheral tissues, activation of MR in the central nervous system also affects the cardiovascular system. For example, activation of MR in the anteroventral third ventricle region of the forebrain has been shown to lead to hypertension through increases in sympathetic drive to vascular smooth muscle, heart and kidneys and through stimulation of arginine vasopressin secretion (Brody et al., 1991; Francis et al., 2001; Gomez-Sanchez, 2004). Similarly, infusion into cerebral ventricles of an aldosterone synthase inhibitor or an MR antagonist reduced hypertension in animal models of salt-sensitive hypertension (Gomez-Sanchez, 2004).
Incidence of CVD is increased in clinical conditions that are associated with excess MR activation. For example, patients with primary aldosteronism are chronically exposed to high levels of aldosterone. Numerous studies have demonstrated that these individuals exhibit significantly higher rates of atrial fibrillation, stroke, and myocardial infarction compared with patients who had essential hypertension and who were matched according to their level of blood pressure elevation (Milliez et al., 2005). Further, evidence that aldosterone damages cardiac structure comes from work which found positive correlations between aldosterone levels and degree of left ventricular hypertrophy in patients with essential hypertension and in patients with primary aldosteronism (Duprez et al., 1993; Milliez et al., 2005).
Most importantly several large scale clinical studies have demonstrated the critical role for MR in the pathophysiology of CVD. These clinical studies using either the selective MR antagonist eplerenone or the nonselective antagonist spironolactone have provided some of the most convincing evidence for a relationship between aldosterone and cardiovascular health. For example, the Randomized Aldactone Evaluation Study (RALES) was designed to test the effect of a MR antagonist in addition to standard therapy on mortality in patients with severe heart failure secondary to systolic left ventricular dysfunction with ejection fraction < or =35% (Pitt et al., 1999). The trial enrolled 1663 patients who received either spironolactone or placebo. Although the follow-up was planned for 3 years, the trial was stopped after a mean follow-up of 2 years due to clear benefits with spironolactone. There was a 30% reduction in relative risk of death, hospitalization rate for worsening heart failure was 35% lower in the spironolactone group than in the placebo group, and spironolactone caused a significant improvement in symptoms of heart failure.
This benefit of MR blockade in heart disease was further demonstrated in the Eplerenone Post-Acute Myocardial Infarction Heart Failure Efficacy and Survival Study (EPHESUS). This double-blind placebo trial was designed to test the effect of the MR antagonist, eplerenone on mortality and hospitalization rates in patients with left ventricular dysfunction and heart failure after acute myocardial infarction (Pitt et al., 2003b). Treatment with eplerenone reduced overall mortality by 15% and reduced cardiovascular mortality by 17% over a mean follow-up of 16 months. Other studies have also demonstrated benefit from MR blockade in reducing left ventricular hypertrophy in patients with hypertensive heart disease and improving diastolic dysfunction (Grandi et al., 2002; Pitt et al., 2003a), suggesting a direct role of aldosterone in the pathophysiology of left ventricular hypertrophy as well as on diastolic function. Aldosterone infusion studies in humans have shown that aldosterone impairs baroreflex sensitivity, and impaired baroreflex sensitivity has been linked with increased mortality in post-MI populations (De Ferrari et al., 2007; Monahan et al., 2007). In patients with congestive heart failure, adverse vascular effects of aldosterone appeared to dominate as treatment with MR antagonist improved nitric oxide production and improved forearm blood flow response to acetylcholine (Farquharson and Struthers, 2000). MR blockade has been shown to improve coronary circulatory function in patients with diabetes (Joffe et al., 2007).
The normal response to dietary sodium involves activation of the rennin–angiotensin–aldosterone system and aldosterone-mediated increases in salt and water retention to maintain volume status. Many pre-clinical studies have suggested increased cardiovascular injury is related to aldosterone levels that are inappropriately elevated relative to dietary sodium intake (Weber, 2001; Martinez et al., 2002). Most individuals consume moderate to high sodium diets, and clinical trial evidence that demonstrated improved cardiac outcomes with MR blockade (i.e., alterations in effects of aldosterone) did not adjust for salt intake. This suggests that adverse effects of aldosterone have been observed in humans consuming habitual diets. Moreover, other evidence has suggested that effects of aldosterone may be evident within healthy populations. For example, healthy individuals with high levels of aldosterone (but within range of normal) exhibited a steeper rise in blood pressure over a 5-year period compared with individuals with low levels of aldosterone, with a dose–response relationship evident (Vasan et al., 2004). Those with the highest levels of aldosterone were the most likely to develop overt hypertension.
A number of factors beyond sodium intake have also been identified as influencing regulation of aldosterone secretion including genes, age, sex, ethnicity, and body mass index (Connell et al., 2008). It is also possible that other factors like psychosocial stress via changes in ACTH, sympathetic activation, or other mechanisms may increase aldosterone and therefore affect CVD risk. As a result and building on Selye’s early observations, the notion that aldosterone may be involved in the relation between psychological stress and CVD risk at a population level has gained new currency (Johnson and Grippo, 2006). As one considers the role of aldosterone in relation to CVD, it is important to remember that activation of MR by either aldosterone (mineralocorticoids) or cortisol (glucocorticoids) can lead to similar adverse cardiovascular effects. However, as much of the literature to date has focused on cortisol in the relation between psychological stress and poor health outcomes, our purpose here is to bring forward greater consideration of aldosterone and MR activation.
4. Aldosterone and psychological stress
Investigations of brain mechanisms underlying fear, anxiety, and psychopathology have frequently focused on interactions between corticosteroids and the brain (Korte, 2001). This work has suggested that rather than regulating behavior per se, corticosteroids induce chemical changes that alter the function of particular neural pathways which make certain behavioral outcomes more or less likely. Thus, the timing, duration, and context in which the hormonal action takes place are integrally related to its effects. As described above, MR and GR play an important role in maintaining homeostatic equilibrium (De Kloet et al., 1998; De Kloet, 2004), and extensive experimental work in animals has considered the different corticosteroid receptor mechanisms involved in various aspects of fear and anxiety and their sequelae (Korte, 2001). In fact, cortisol (or in the case of rodents, corticosterone) is the primary occupier of MR in many areas of the brain implicated in psychological response to stress as 11β-HSD2 is not expressed in these areas. However, in areas of the brain where 11β-HSD2 is expressed (e.g., nucleus tractus solitarius) aldosterone may be the primary corticosteroid activating MR and it may be these areas of the brain that mediate the central adverse cardiovascular effects of aldosterone on blood pressure and salt intake (Geerling et al., 2006). Taken together, these processes suggest an important role for aldosterone in addition to cortisol with regard to linking psychological stress and cardiovascular outcomes.
Recent studies have linked components of the rennin–angiotensin–aldosterone system to psychological stress. For example, several studies have demonstrated that pharmacological blockade of the rennin–angiotensin system improves mood and reduces anxiety among humans and animals (Johnson and Grippo, 2006; Hlavacova and Jezova, 2008). While there is less research considering the direct relation between aldosterone and specific manifestations of psychological stress (e.g., anxiety, depression), some work has suggested the plausibility of a link. In a study of patients newly diagnosed with primary aldosteronism, investigators administered the Structured Clinical Interview for the Diagnostic and Statistical Manual of Mental Disorders (DSM-IV) and a structured interview for subclinical psychological syndromes. This study found 6 out of 10 individuals had generalized anxiety disorder, one had obsessive–compulsive disorder, with persistent somatization and among these individuals, irritable mood was also common (Sonino et al., 2006). In a comparison of sleep-related activity in the rennin–angiotensin–aldosterone system between depressed and non-depressed women, significantly higher levels of plasma aldosterone levels were evident among the depressed women (Murck et al., 2003). Other work has also found higher levels of plasma aldosterone among depressed patients compared with age- and sex-matched controls even after subjects in both groups followed a controlled sodium and potassium diet (Emanuele et al., 2005). Another study compared students practicing stress reduction techniques (transcendental meditation) with a similar group not engaging in systematic stress reduction (Walton et al., 1995). The stress reduction group had lower levels of aldosterone and mood disturbance and anxiety compared with the group not practicing stress reduction.
Most studies in humans to date have been cross-sectional. However findings from the animal literature are consistent with this work and have suggested that stress and distress may lead to higher levels of aldosterone. For example, animal studies have found that psychological stress stimulates the peripheral and central angiotensin II systems which in turn contribute to secretion of both ACTH and aldosterone (Saavedra et al., 2005; Johnson and Grippo, 2006). There may also be feedback effects whereby consistently high levels of aldosterone serve to enhance anxiety. In a recent animal study, male Wistar rats were subcutaneously implanted with osmotic minipumps and treated with aldosterone or vehicle for two weeks. Rats treated with aldosterone exhibited significant enhancement of anxious behaviors in the open field and elevated plus-maze tests (Hlavacova and Jezova, 2008).
We know of no studies in humans that have considered whether high levels of psychological stress might increase levels of aldosterone, either using a longitudinal study design or an experimental design considering acute effects. However, numerous experimental studies have demonstrated that acute psychosocial stress activates the HPA axis (Dickerson and Kemeny, 2004), and more specifically stimulates the release of ACTH (see for example Singh et al., 1999). In our own work, we have demonstrated that intravenous administration of ACTH in increasing doses (0, 0.05, 0.15, 0.5, 1.5, and 5.0 mIU/kg each for 30 min) leads to dose–response increases in serum cortisol (Adler et al., 1999) and aldosterone (see Fig. 1) in healthy women.
Fig. 1.
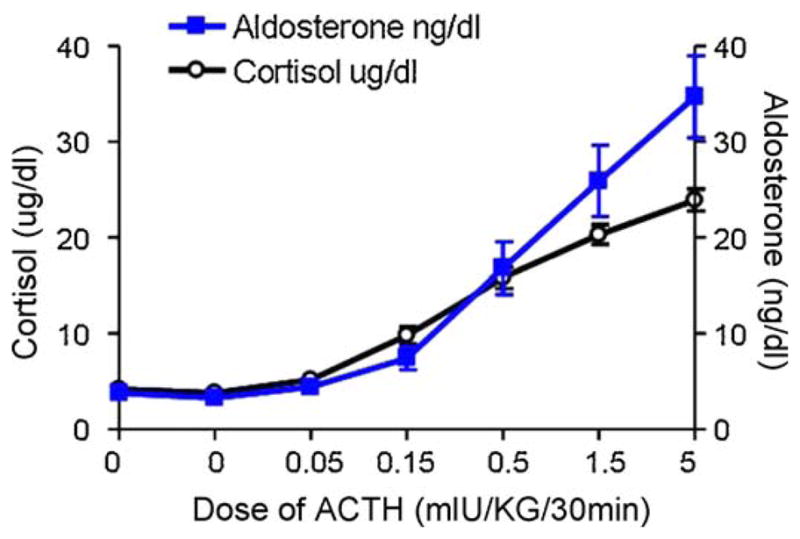
Effect of ACTH infusion on serum levels of cortisol (μg/dl) (open circles) and aldosterone (ng/dl) (squares). 13 healthy pre-menopausal women on ad lb diets received an intravenous infusion of ACTH at sequential doses of 0.00, 0.00, 0.05, 0.15, 0.50, 1.50 and 5 mIU/kg/30 min. 24 h urinary sodium levels were (156 ± 24 mEquib./24 h). Cortisol data was reported previously (Adler et al., 1999).
5. Aldosterone as a mediator of the relationship between psychological stress and CVD
Taken together, the emerging research linking aldosterone with CVD as well as with anxiety and depression, suggests it may be useful to consider aldosterone a key neurohormonal mechanism by which psychological stress and negative affective states may influence cardiovascular health. A model of how aldosterone may mediate effects of psychological stress on CVD is presented in Fig. 2. Psychological stress and negative affective states activate the HPA axis, leading to release of ACTH. Release of ACTH leads to release of both cortisol and aldosterone. Psychological stress also activates the sympathetic-adrenomedullary system which stimulates rennin release leading to increases in angiotensin II and aldosterone secretion. Aldosterone activates MR which in turn may lead to vascular injury and inflammation, and ultimately heart disease, renal disease, and stroke.
Fig. 2.

A model of aldosterone as a mediator of the relationship between psychological stress and CVD.
Empirical tests of the proposed model are needed. Given the similarities between cortisol and aldosterone in response to HPA axis activation, it is likely that many of the psychosocial factors involved in dysregulated cortisol are also involved in dysregulated aldosterone. Hundreds of studies have considered effects of psychological stress on cortisol (Dickerson and Kemeny, 2004), and many of these paradigms may be used for considering effects of psychological stress and negative affective states on aldosterone. Experimental designs may consider whether and how acute stress affects aldosterone levels, with additional consideration of the relation between aldosterone response and cardiovascular reactivity (e.g., changes in blood pressure, heart rate, heart rate variability, and other cardiac parameters) over the course of the experiment. Prospective cohort studies may be used to consider effects of chronic stress on aldosterone and changes in aldosterone levels over time. Other work may examine both chronic stress and levels of aldosterone in relation to pre-disease markers of CVD, including coronary calcification, atherosclerosis and progression, and development of hypertension. Additional research may focus on whether effects of psychological stress on CVD mediated by aldosterone can be reversed or mitigated if negative affective states are successfully treated.
Similar to cortisol, aldosterone has a diurnal pattern with peak aldosterone occurring early in the morning, and declining throughout the day. Thus, studies of aldosterone in relation to psychological stress and CVD will face many of the same measurement challenges associated with studies of cortisol. For observational studies that are considering cardiovascular effects of chronic psychological stress, it will likely be more effective to obtain multiple aldosterone parameters (e.g., wake-up levels, bedtime levels, diurnal slope). An additional potential confounder for interpreting aldosterone levels relates to the strong effect of dietary sodium intake on aldosterone levels, and thus it is important to evaluate aldosterone levels relative to dietary sodium intake. Studies will also have to explore how different aldosterone parameters may relate to chronic psychological stress. For example, some work has suggested that cortisol response to awakening is a strong indicator of current strain, whereas average cortisol levels are not as strongly associated, while flattening of the diurnal rhythm is thought to be a good marker of exposure to chronic strain (Adam, 2005). Currently no salivary assay exists for aldosterone, although development of one is underway.
6. Conclusion
Few studies have directly addressed the mechanisms for the observed associations between psychological stress, HPA axis activation and CVD (Girod and Brotman, 2004). Building on emerging findings that excess activation of MR plays a key pathophysiologic role in the development of CVD, and the potential relationship between stress, negative affective states, and increased production of adrenal glucocorticoid and mineralocorticoid, we propose MR activation by adrenal steroids, in particular aldosterone, as a relevant mediator in the link between psychological stress and CVD. Current evidence for the link between aldosterone, MR activation, and increased risk of CVD is strong (Yoshimoto and Hirata, 2007; Garg and Adler, 2009), and work on the relation between aldosterone and psychological stress is promising. Important next steps will include experimental research to determine the link between high levels of psychological stress, aldosterone, and cardiovascular reactivity, epidemiologic studies considering aldosterone as a mediator of the observed relationship between chronic negative affect and development of CVD, and the relation of chronic negative affect and aldosterone with pre-disease markers of CVD, and the molecular mechanisms linking chronic negative affect to aldosterone. With this work, new insight may be gained into how psychological stress may impair cardiovascular health.
Acknowledgments
This work was supported by the Robert Wood Johnson Foundation Health and Society Scholars Program, and by an NIH grant R01-AR-43130.
References
- Adam E. Proper use vs. misuse of biomarker data in social science research: the case of cortisol. Chicago Workshop on Biomarkers in Population-Based Health and Aging Research; Chicago. Population Research Center at NORC and the University of Chicago; 2005. [Google Scholar]
- Adler GK, Kinsley BT, et al. Reduced hypothalamic–pituitary and sympathoadrenal responses to hypoglycemia in women with fibromyalgia syndrome. Am J Med. 1999;106:534–543. doi: 10.1016/s0002-9343(99)00074-1. [DOI] [PubMed] [Google Scholar]
- American Heart Association. Heart Disease and Stroke Statistics—2008 Update. American Heart Association; Dallas, TX: 2008. [DOI] [PubMed] [Google Scholar]
- Atkinson HC, Wood SA, et al. Corticosteroids mediate fast feedback of the rat hypothalamic–pituitary–adrenal axis via the mineralocorticoid receptor. Am J Physiol Endocrinol Metab. 2008;294 (6):E1011–E1022. doi: 10.1152/ajpendo.00721.2007. [DOI] [PubMed] [Google Scholar]
- Bentley-Lewis R, Adler GK, et al. Body mass index predicts aldosterone production in normotensive adults on a high-salt diet. J Clin Endocrinol Metab. 2007;92 (11):4472–4475. doi: 10.1210/jc.2007-1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjorntorp P. Body fat distribution, insulin resistance, and metabolic diseases. Nutrition. 1997;13 (9):795–803. doi: 10.1016/s0899-9007(97)00191-3. [DOI] [PubMed] [Google Scholar]
- Broadley AJ, Korszun A, et al. Inhibition of cortisol production with metyrapone prevents mental stress-induced endothelial dysfunction and baroreflex impairment. J Am Coll Cardiol. 2005;46 (2):344–350. doi: 10.1016/j.jacc.2005.03.068. [DOI] [PubMed] [Google Scholar]
- Brody MJ, Varner KJ, et al. Central nervous system and the pathogenesis of hypertension. Sites and mechanisms. Hypertension. 1991;18(5 Suppl):III7–III12. doi: 10.1161/01.hyp.18.5_suppl.iii7. [DOI] [PubMed] [Google Scholar]
- Caprio M, Feve B, et al. Pivotal role of the mineralocorticoid receptor in corticosteroid-induced adipogenesis. Faseb J. 2007;21 (9):2185–2194. doi: 10.1096/fj.06-7970com. [DOI] [PubMed] [Google Scholar]
- Catena C, Lapenna R, et al. Insulin sensitivity in patients with primary aldosteronism: a follow-up study. J Clin Endocrinol Metab. 2006;91 (9):3457–3463. doi: 10.1210/jc.2006-0736. [DOI] [PubMed] [Google Scholar]
- Cohen S, Janicki-Deverts D, et al. Psychological stress and disease. JAMA. 2007;298 (14):1685–1687. doi: 10.1001/jama.298.14.1685. [DOI] [PubMed] [Google Scholar]
- Connell JM, MacKenzie SM, et al. A lifetime of aldosterone excess: long-term consequences of altered regulation of aldosterone production for cardiovascular function. Endocr Rev. 2008;29 (2):133–154. doi: 10.1210/er.2007-0030. [DOI] [PubMed] [Google Scholar]
- De Ferrari GM, Sanzo A, et al. Baroreflex sensitivity predicts long-term cardiovascular mortality after myocardial infarction even in patients with preserved left ventricular function. J Am Coll Cardiol. 2007;50 (24):2285–2290. doi: 10.1016/j.jacc.2007.08.043. [DOI] [PubMed] [Google Scholar]
- De Kloet ER. Hormones and the stressed brain. Ann NY Acad Sci. 2004;1018:1–15. doi: 10.1196/annals.1296.001. [DOI] [PubMed] [Google Scholar]
- De Kloet ER, Vreugdenhil E, et al. Brain corticosteroid receptor balance in health and disease. Endocr Rev. 1998;19 (3):269–301. doi: 10.1210/edrv.19.3.0331. [DOI] [PubMed] [Google Scholar]
- DiBona GF. Neural control of the kidney: past, present, and future. Hypertension. 2003;41 (3 Pt 2):621–624. doi: 10.1161/01.HYP.0000047205.52509.8A. [DOI] [PubMed] [Google Scholar]
- Dickerson SS, Kemeny ME. Acute stressors and cortisol responses: a theoretical integration and synthesis of laboratory research. Psychol Bull. 2004;130 (3):355–391. doi: 10.1037/0033-2909.130.3.355. [DOI] [PubMed] [Google Scholar]
- Dimsdale JE. Psychological stress and cardiovascular disease. J Am Coll Cardiol. 2008;51 (13):1237–1246. doi: 10.1016/j.jacc.2007.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duprez DA, Bauwens FR, et al. Influence of arterial blood pressure and aldosterone on left ventricular hypertrophy in moderate essential hypertension. Am J Cardiol. 1993;71 (3):17A–20A. doi: 10.1016/0002-9149(93)90240-d. [DOI] [PubMed] [Google Scholar]
- Dustan HP. Mechanisms of hypertension associated with obesity. Ann Intern Med. 1983;98 (5 Pt 2):860–864. doi: 10.7326/0003-4819-98-5-860. [DOI] [PubMed] [Google Scholar]
- Emanuele E, Geroldi D, et al. Increased plasma aldosterone in patients with clinical depression. Arch Med Res. 2005;36 (5):544–548. doi: 10.1016/j.arcmed.2005.03.046. [DOI] [PubMed] [Google Scholar]
- Engeli S, Bohnke J, et al. Weight loss and the rennin–angiotensin–aldosterone system. Hypertension. 2005;45 (3):356–362. doi: 10.1161/01.HYP.0000154361.47683.d3. [DOI] [PubMed] [Google Scholar]
- Epel ES, McEwen B, et al. Stress and body shape: stress-induced cortisol secretion is consistently greater among women with central fat. Psychosom Med. 2000;62 (5):623–632. doi: 10.1097/00006842-200009000-00005. [DOI] [PubMed] [Google Scholar]
- Everson-Rose SA, Lewis TT. Psychosocial factors and cardiovascular diseases. Annu Rev Public Health. 2005;26:469–500. doi: 10.1146/annurev.publhealth.26.021304.144542. [DOI] [PubMed] [Google Scholar]
- Farquharson CA, Struthers AD. Spironolactone increases nitric oxide bioactivity, improves endothelial vasodilator dysfunction, and suppresses vascular angiotensin I/angiotensin II conversion in patients with chronic heart failure. Circulation. 2000;101 (6):594–597. doi: 10.1161/01.cir.101.6.594. [DOI] [PubMed] [Google Scholar]
- Francis J, Weiss RM, et al. Central mineralocorticoid receptor blockade improves volume regulation and reduces sympathetic drive in heart failure. Am J Physiol Heart Circ Physiol. 2001;281 (5):H2241–H2251. doi: 10.1152/ajpheart.2001.281.5.H2241. [DOI] [PubMed] [Google Scholar]
- Funder JW. Mineralocorticoid receptors and cardiovascular damage: it’s not just aldosterone. Hypertension. 2006;47 (4):634–635. doi: 10.1161/01.HYP.0000203732.03784.3b. [DOI] [PubMed] [Google Scholar]
- Funder JW. Reconsidering the roles of the mineralocorticoid receptor. Hypertension. 2009;53 (2):286–290. doi: 10.1161/HYPERTENSIONAHA.108.119966. [DOI] [PubMed] [Google Scholar]
- Ganie MA, Khurana ML, et al. Comparison of efficacy of spironolactone with metformin in the management of polycystic ovary syndrome: an open-labeled study. J Clin Endocrinol Metab. 2004;89 (6):2756–2762. doi: 10.1210/jc.2003-031780. [DOI] [PubMed] [Google Scholar]
- Garg R, Adler GK, editors. Textbook of Nephro-Endocrinology. Elsevier; Burlington, MA: 2009. Aldosterone and its cardiovascular effects. [Google Scholar]
- Geerling JC, Engeland WC, et al. Aldosterone target neurons in the nucleus tractus solitarius drive sodium appetite. J Neurosci. 2006;26 (2):411–417. doi: 10.1523/JNEUROSCI.3115-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girod JP, Brotman DJ. Does altered glucocorticoid homeostasis increase cardiovascular risk? Cardiovasc Res. 2004;1 (64):217–226. doi: 10.1016/j.cardiores.2004.07.006. [DOI] [PubMed] [Google Scholar]
- Gomez-Sanchez EP. Brain mineralocorticoid receptors: orchestrators of hypertension and end-organ disease. Curr Opin Nephrol Hypertens. 2004;13 (2):191–196. doi: 10.1097/00041552-200403000-00007. [DOI] [PubMed] [Google Scholar]
- Goodfriend TL, Egan B, et al. Relationships among plasma aldosterone, high-density lipoprotein cholesterol, and insulin in humans. Hypertension. 1995;25 (1):30–36. doi: 10.1161/01.hyp.25.1.30. [DOI] [PubMed] [Google Scholar]
- Goodfriend TL, Egan BM, et al. Aldosterone in obesity. Endocr Res. 1998;24 (3–4):789–796. doi: 10.3109/07435809809032689. [DOI] [PubMed] [Google Scholar]
- Goodfriend TL, Kelley DE, et al. Visceral obesity and insulin resistance are associated with plasma aldosterone levels in women. Obes Res. 1999;7 (4):355–362. doi: 10.1002/j.1550-8528.1999.tb00418.x. [DOI] [PubMed] [Google Scholar]
- Grandi AM, Imperiale D, et al. Aldosterone antagonist improves diastolic function in essential hypertension. Hypertension. 2002;40 (5):647–652. doi: 10.1161/01.hyp.0000036399.80194.d8. [DOI] [PubMed] [Google Scholar]
- Guo C, Ricchiuti V, et al. Mineralocorticoid receptor blockade reverses obesity-related changes in expression of adiponectin, peroxisome proliferator-activated receptor-gamma, and proinflammatory adipokines. Circulation. 2008;117 (17):2253–2261. doi: 10.1161/CIRCULATIONAHA.107.748640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hadoke PW, Macdonald L, et al. Intra-vascular glucocorticoid metabolism as a modulator of vascular structure and function. Cell Mol Life Sci. 2006;63 (5):565–578. doi: 10.1007/s00018-005-5427-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haluzik M, Sindelka G, et al. Serum leptin levels in patients with primary hyperaldosteronism before and after treatment: relationships to insulin sensitivity. J Hum Hypertens. 2002;16 (1):41–45. doi: 10.1038/sj.jhh.1001292. [DOI] [PubMed] [Google Scholar]
- Han KH, Kang YS, et al. Spironolactone ameliorates renal injury and connective tissue growth factor expression in type II diabetic rats. Kidney Int. 2006a;70 (1):111–120. doi: 10.1038/sj.ki.5000438. [DOI] [PubMed] [Google Scholar]
- Han SY, Kim CH, et al. Spironolactone prevents diabetic nephropathy through an anti-inflammatory mechanism in type 2 diabetic rats. J Am Soc Nephrol. 2006b;17 (5):1362–1372. doi: 10.1681/ASN.2005111196. [DOI] [PubMed] [Google Scholar]
- Hiramatsu K, Yamada T, et al. Changes in endocrine activities relative to obesity in patients with essential hypertension. J Am Geriatr Soc. 1981;29 (1):25–30. doi: 10.1111/j.1532-5415.1981.tb02389.x. [DOI] [PubMed] [Google Scholar]
- Hlavacova N, Jezova D. Chronic treatment with the mineralocorticoid hormone aldosterone results in increased anxiety-like behavior. Horm Behav. 2008;54 (1):90–97. doi: 10.1016/j.yhbeh.2008.02.004. [DOI] [PubMed] [Google Scholar]
- Iwashima F, Yoshimoto T, et al. Aldosterone induces superoxide generation via Rac1 activation in endothelial cells. Endocrinology. 2008;149 (3):1009–1014. doi: 10.1210/en.2007-0864. [DOI] [PubMed] [Google Scholar]
- Jaffe IZ, Mendelsohn ME. Angiotensin II and aldosterone regulate gene transcription via functional mineralocorticoid receptors in human coronary artery smooth muscle cells. Circ Res. 2005;96 (6):643–650. doi: 10.1161/01.RES.0000159937.05502.d1. [DOI] [PubMed] [Google Scholar]
- Joffe HV, Adler GK. Effect of aldosterone and mineralocorticoid receptor blockade on vascular inflammation. Heart Fail Rev. 2005;10 (1):31–37. doi: 10.1007/s10741-005-2346-0. [DOI] [PubMed] [Google Scholar]
- Joffe HV, Kwong RY, et al. Beneficial effects of eplerenone versus hydrochlorothiazide on coronary circulatory function in patients with diabetes mellitus. J Clin Endocrinol Metab. 2007;92 (7):2552–2558. doi: 10.1210/jc.2007-0393. [DOI] [PubMed] [Google Scholar]
- Johnson AK, Grippo AJ. Sadness and broken hearts: neurohumoral mechanisms and co-morbidity of ischemic heart disease and psychological depression. J Physiol Pharmacol. 2006;57 (Suppl 11):5–29. [PubMed] [Google Scholar]
- Kang YM, Zhang ZH, et al. Novel effect of mineralocorticoid receptor antagonism to reduce proinflammatory cytokines and hypothalamic activation in rats with ischemia-induced heart failure. Circ Res. 2006;99 (7):758–766. doi: 10.1161/01.RES.0000244092.95152.86. [DOI] [PubMed] [Google Scholar]
- Korte SM. Corticosteroids in relation to fear, anxiety and psychopathology. Neurosci Biobehav Rev. 2001;25 (2):117–142. doi: 10.1016/s0149-7634(01)00002-1. [DOI] [PubMed] [Google Scholar]
- Kraus D, Jager J, et al. Aldosterone inhibits uncoupling protein-1, induces insulin resistance, and stimulates proinflammatory adipokines in adipocytes. Horm Metab Res. 2005;37 (7):455–459. doi: 10.1055/s-2005-870240. [DOI] [PubMed] [Google Scholar]
- Kubzansky LD. Sick at heart: the pathophysiology of negative emotions. Cleve Clin J Med. 2007;74 (Suppl 1):S67–S72. doi: 10.3949/ccjm.74.suppl_1.s67. [DOI] [PubMed] [Google Scholar]
- Leopold JA, Dam A, et al. Aldosterone impairs vascular reactivity by decreasing glucose-6-phosphate dehydrogenase activity. Nat Med. 2007;13 (2):189–197. doi: 10.1038/nm1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luther JM, Gainer JV, et al. Angiotensin II induces interleukin-6 in humans through a mineralocorticoid receptor-dependent mechanism. Hypertension. 2006;48 (6):1050–1057. doi: 10.1161/01.HYP.0000248135.97380.76. [DOI] [PubMed] [Google Scholar]
- Martinez DV, Rocha R, et al. Cardiac damage prevention by eplerenone: comparison with low sodium diet or potassium loading. Hypertension. 2002;39 (2 Pt 2):614–618. [PubMed] [Google Scholar]
- Matthews K, Schwartz J, et al. Diurnal cortisol decline is related to coronary calcification: CARDIA study. Psychosom Med. 2006;68 (5):657–661. doi: 10.1097/01.psy.0000244071.42939.0e. [DOI] [PubMed] [Google Scholar]
- Miller GE, Cohen S, et al. Chronic psychological stress and the regulation of pro-inflammatory cytokines: a glucocorticoid-resistance model. Health Psychol. 2002;21 (6):531–541. doi: 10.1037//0278-6133.21.6.531. [DOI] [PubMed] [Google Scholar]
- Milliez P, Girerd X, et al. Evidence for an increased rate of cardiovascular events in patients with primary aldosteronism. J Am Coll Cardiol. 2005;45 (8):1243–1248. doi: 10.1016/j.jacc.2005.01.015. [DOI] [PubMed] [Google Scholar]
- Moghetti P, Tosi F, et al. The insulin resistance in women with hyperandrogenism is partially reversed by antiandrogen treatment: evidence that androgens impair insulin action in women. J Clin Endocrinol Metab. 1996;81 (3):952–960. doi: 10.1210/jcem.81.3.8772557. [DOI] [PubMed] [Google Scholar]
- Monahan KD, Leuenberger UA, et al. Aldosterone impairs baroreflex sensitivity in healthy adults. Am J Physiol Heart Circ Physiol. 2007;292 (1):H190–H197. doi: 10.1152/ajpheart.00622.2006. [DOI] [PubMed] [Google Scholar]
- Murck H, Held K, et al. The rennin–angiotensin–aldosterone system in patients with depression compared to controls—a sleep endocrine study. BMC Psychiatry. 2003;3:15. doi: 10.1186/1471-244X-3-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitt B, Reichek N, et al. Effects of eplerenone, enalapril, and eplerenone/enalapril in patients with essential hypertension and left ventricular hypertrophy: the 4E-left ventricular hypertrophy study. Circulation. 2003a;108 (15):1831–1838. doi: 10.1161/01.CIR.0000091405.00772.6E. [DOI] [PubMed] [Google Scholar]
- Pitt B, Remme W, et al. Eplerenone, a selective aldosterone blocker, in patients with left ventricular dysfunction after myocardial infarction. N Engl J Med. 2003b;348 (14):1309–1321. doi: 10.1056/NEJMoa030207. [DOI] [PubMed] [Google Scholar]
- Pitt B, Zannad F, et al. The effect of spironolactone on morbidity and mortality in patients with severe heart failure. Randomized Aldactone Evaluation Study Investigators. N Engl J Med. 1999;341 (10):709–717. doi: 10.1056/NEJM199909023411001. [DOI] [PubMed] [Google Scholar]
- Rajagopalan S, Duquaine D, et al. Mineralocorticoid receptor antagonism in experimental atherosclerosis. Circulation. 2002;105 (18):2212–2216. doi: 10.1161/01.cir.0000015854.60710.10. [DOI] [PubMed] [Google Scholar]
- Rocchini AP, Katch VL, et al. Role for aldosterone in blood pressure regulation of obese adolescents. Am J Cardiol. 1986;57 (8):613–618. doi: 10.1016/0002-9149(86)90845-3. [DOI] [PubMed] [Google Scholar]
- Rozanski A, Blumenthal JA, et al. The epidemiology, pathophysiology, and management of psychosocial risk factors in cardiac practice: the emerging field of behavioral cardiology. J Am Coll Cardiol. 2005;45 (5):637–651. doi: 10.1016/j.jacc.2004.12.005. [DOI] [PubMed] [Google Scholar]
- Saavedra JM, Ando H, et al. Anti-stress and anti-anxiety effects of centrally acting angiotensin II AT1 receptor antagonists. Regul Pept. 2005;128 (3):227–238. doi: 10.1016/j.regpep.2004.12.015. [DOI] [PubMed] [Google Scholar]
- Selye H. The general adaptation syndrome and the diseases of adaptation. J Clin Endocr. 1946;6:117–230. doi: 10.1210/jcem-6-2-117. [DOI] [PubMed] [Google Scholar]
- Selye H. Stress and disease. Science. 1955;122 (3171):625–631. doi: 10.1126/science.122.3171.625. [DOI] [PubMed] [Google Scholar]
- Selye H. Protection by a steroid-spirolactone against certain types of cardiac necrosis. Proc Soc Exp Biol Med. 1960;104:212–213. doi: 10.3181/00379727-104-25782. [DOI] [PubMed] [Google Scholar]
- Singh A, Petrides JS, et al. Differential hypothalamic–pituitary–adrenal axis reactivity to psychological and physical stress. J Clin Endocrinol Metab. 1999;84 (6):1944–1948. doi: 10.1210/jcem.84.6.5746. [DOI] [PubMed] [Google Scholar]
- Sonino N, Fallo F, et al. Psychological aspects of primary aldosteronism. Psychother Psychosom. 2006;75 (5):327–330. doi: 10.1159/000093956. [DOI] [PubMed] [Google Scholar]
- Steptoe A, Brydon L. Emotional triggering of cardiac events. Neurosci Biobehav Rev. 2009;33:63–70. doi: 10.1016/j.neubiorev.2008.04.010. [DOI] [PubMed] [Google Scholar]
- Suls J, Bunde J. Anger, anxiety, and depression as risk factors for cardiovascular disease: the problems and implications of overlapping affective dispositions. Psycholog Bull. 2005;131 (2):260–300. doi: 10.1037/0033-2909.131.2.260. [DOI] [PubMed] [Google Scholar]
- Takai S, Jin D, et al. Eplerenone inhibits atherosclerosis in nonhuman primates. Hypertension. 2005;46 (5):1135–1139. doi: 10.1161/01.HYP.0000184640.81730.22. [DOI] [PubMed] [Google Scholar]
- van Raalte DH, Ouwens DM, et al. Novel insights into glucocorticoid-mediated diabetogenic effects: towards expansion of therapeutic options? Eur J Clin Invest. 2009;39 (2):81–93. doi: 10.1111/j.1365-2362.2008.02067.x. [DOI] [PubMed] [Google Scholar]
- Vasan RS, Evans JC, et al. Serum aldosterone and the incidence of hypertension in nonhypertensive persons. N Engl J Med. 2004;351 (1):33–41. doi: 10.1056/NEJMoa033263. [DOI] [PubMed] [Google Scholar]
- Vinson GP. Angiotensin II, corticosteroids, type II diabetes and the metabolic syndrome. Med Hypotheses. 2007;68 (6):1200–1207. doi: 10.1016/j.mehy.2006.09.065. [DOI] [PubMed] [Google Scholar]
- Wada T, Ohshima S, et al. Aldosterone inhibits insulin-induced glucose uptake by degradation of insulin receptor substrate (IRS) 1 and IRS2 via a reactive oxygen species-mediated pathway in 3T3-L1 adipocytes. Endocrinology. 2009;150 (4):1662–1669. doi: 10.1210/en.2008-1018. [DOI] [PubMed] [Google Scholar]
- Walker BR. Glucocorticoids and cardiovascular disease. Eur J Endocrinol. 2007;157 (5):545–559. doi: 10.1530/EJE-07-0455. [DOI] [PubMed] [Google Scholar]
- Walton KG, Pugh ND, et al. Stress reduction and preventing hypertension: preliminary support for a psychoneuroendocrine mechanism. J Altern Complement Med. 1995;1 (3):263–283. doi: 10.1089/acm.1995.1.263. [DOI] [PubMed] [Google Scholar]
- Weber KT. Aldosterone in congestive heart failure. N Engl J Med. 2001;345 (23):1689–1697. doi: 10.1056/NEJMra000050. [DOI] [PubMed] [Google Scholar]
- Yoshimoto T, Hirata Y. Aldosterone as a cardiovascular risk hormone. Endocr J. 2007;54 (3):359–370. doi: 10.1507/endocrj.kr-80. [DOI] [PubMed] [Google Scholar]
- Yusuf S, Hawken S, et al. Effect of potentially modifiable risk factors associated with myocardial infarction in 52 countries (the INTERHEART study): case–control study. Lancet. 2004;364 (9438):937–952. doi: 10.1016/S0140-6736(04)17018-9. [DOI] [PubMed] [Google Scholar]
- Zulian E, Sartorato P, et al. Spironolactone in the treatment of polycystic ovary syndrome: effects on clinical features, insulin sensitivity and lipid profile. J Endocrinol Invest. 2005;28 (1):49–53. doi: 10.1007/BF03345529. [DOI] [PubMed] [Google Scholar]