

Abstract
Objective
Inflammation is pivotal in atherosclerosis. Monocyte-macrophages are crucial in atherosclerosis. Monocytes can develop into subsets: classically (M1) or alternatively (M2) activated cells. Several studies point to a pro-inflammatory role for C-reactive protein (CRP). Since there is a paucity of data on the effects of CRP on macrophage phenotype, we tested effects of CRP on macrophage polarization.
Methods and Results
Monocytes were incubated with CRP (0–50ug/mL) and differentiated into macrophages for 7 days. Phenotypic characterization of M1 and M2 macrophages was performed using flow cytometry. CRP treatment resulted in increased population of M1 macrophages (TNF/IL12/CCR2 or TNF/IL-12/MCP-1 or TNF/IL-1/IL-12). These effects were not abrogated by polymixin B or siRNA to Toll-like receptor-4 (TLR-4) but by boiled CRP. Administration of human CRP to rats in-vivo increased polarization of macrophages to M1 phenotype when compared to HuSA. When macrophages were primed to M2 phenotype with IL-4, addition of CRP resulted in significantly increased secretion of TNF-α, MCP-1 and IL-1 and conversion of macrophages from M2 to M1 phenotype. CRP failed to prime macrophages to M1 phenotype in presence of CD32/CD64 siRNA or DN-NFKb.
Conclusions
Collectively, these results further support a role for CRP in promoting differentiation of human monocytes towards a pro-inflammatory M1 phenotype.
Keywords: CRP, Macrophage, M1 phenotype, M2 phenotype, inflammation
Introduction
Inflammation plays a pivotal role in atherosclerosis.1 Monocyte-macrophages are present in all stages of atherosclerosis from the nascent fatty streak lesion to the culmination in acute coronary syndromes. 1–3 A key early step in this inflammatory process is the infiltration of monocytes into the sub endothelial space of large arteries and their differentiation into tissue macrophages. Also, macrophage infiltration is observed in adipose tissue of obese subjects and appears to contribute to adipokine-induced inflammation. 4–5
Monocytes often function as control switches of the immune system and maintain the balance between pro- and anti-inflammatory activities. There is a novel concept emerging from numerous lines of evidence that these cells can develop into different subsets: classically (M1) or alternatively (M2) activated cells. 6–8 MI monocytes produce high levels of pro-inflammatory cytokines including interleukin (IL)-1, IL-6, IL-12/IL-23, tumor necrosis factor (TNF)and play a role in tissue destruction. Their M1 polarization can be mediated by lipopolysaccharide (LPS) and interferon (IFN) gamma. M2 monocytes are more prominent producers of anti-inflammatory cytokines, IL-1RA, IL-10 and transforming growth factor (TGF)-beta. They also express increased levels of scavenger and the mannose receptor (CD206) and appear to be involved in tissue remodeling. M2 polarization is mediated via IL-4 and IL-13. The number of M1 cells increases with obesity in rodents and analysis of macrophages that infiltrate adipose tissue of obese mice suggests that these cells are classically M1 phenotype and correlate inversely with insulin sensitivity of these mice. 6–12 Monocytes of the M1 phenotype infiltrate atherosclerotic lesions. In hypercholesterolemic apolipoprotein E (ApoE−/−) mice, monocytosis is mainly of the M1 subtype. 12–14
Among the numerous inflammatory biomarkers, the largest amount of published data supports a role for C-reactive protein (CRP) as a robust and independent risk marker in predicting adverse cardiovascular events in both primary and secondary prevention. 15 In addition to being a risk marker, there is much evidence indicating that CRP may indeed participate in atherogenesis. 16 CRP induces endothelial dysfunction. 17 CRP also induces pro-inflammatory cytokine release from human monocytes, upregulates CCR2 and NADPH oxidase activity.18–20 Ballou and Lozanski conducted a study in which they incubated human monocytes with CRP at different doses for 16 hours and were able to demonstrate significantly increased levels of IL-1, TNF, and IL-6. We have shown that incubation of human monocyte-derived macrophages with CRP significantly decreased LPS-induced second-wave IL-10 mRNA and intracellular and secreted IL-10 protein and destabilized IL-10 mRNA. 21–23 Also, CRP alone increased secretion of IL-8, IL-6, and TNF from human monocyte-derived macrophages and in-vivo from peritoneal macrophages of Wistar rats. 22–23 However, there is no data examining the direct effect of CRP on macrophage phenotypes. In the present study, we examined the hypothesis that CRP may exert its pro-inflammatory effects on human monocytes by priming to a pro-inflammatory M1 phenotype and prevent agonist-induced polarization to a M2 phenotype.
Materials and Methods
CRP purified from human ascitic or pleural fluids was passed through Detoxigel column to remove LPS and dialyzed extensively to remove azide as described previously.24 Our CRP preparations have been shown to have pro-inflammatory effects in TLR4siRNA knocked-down cells, while LPS loses its effect in these cells.24 Boiled CRP was also used as control.24
Cell culture
Following informed consent, human peripheral blood mononuclear cells were isolated from the blood of healthy volunteers by the Ficoll density gradient centrifugation and monocytes were isolated by negative magnetic separation as described previously and were seeded onto 6-well plates following suspension in RPMI 1640. Using this method, 92% of cells isolated are CD14+.22–23 After 2 h of incubation (37 °C, 5% CO2) for adherence, the medium was replaced with RPMI 1640 supplemented with 20% autologous human serum. Adhered monocytes were incubated at 37 °C in 5% CO2 for 7 days to induce differentiation into macrophages with change of medium every 3 days. CRP (0–50ug/mL) or human serum albumin (HuSA) was added during the 7 days of monocyte differentiation into macrophages. CRP specific effects: To ensure that the effects are in fact due to CRP per se and not some contaminant, we used boiled CRP as negative control. To examine if these effects were attributable to lipopolysaccharide/endotoxin contamination, we added polymixin B along with CRP as described previously.22–24
Phenotypic Characterization of Classical (M1) and Alternatively Activated Monocytes (M2)
Phenotypic characterization of M1 and M2 cells from subjects was performed using 4-color flow cytometry. For FACS analysis, cells (5× 106 cells) were washed twice and resuspended in 100ul of 1%BSA in PBS and incubated with respective fluorochrome labeled antibodies and isotype controls and then assessed by flow cytometry using the BD FACSArray. M1 cells were characterized by positivity for TNF, IL-12/23 and M2 cells were characterized by positivity for CD206 (mannose receptor) and IL-10. Percentage of classically versus alternatively monocytes was estimated. Furthermore, we also characterized M1 as TNF+/IL-1beta+; TNF+/IL-12+ cells and for M2, IL-10+/IL-1RA+ and IL-10+/CD163+ cells. Fluorescently labeled macrophages were analyzed under the same instrument settings (to eliminate measurement bias) and all data were corrected for background fluorescence intensity obtained with isotype control antibodies (all positive cells were over the first quadrant in the BDFACS array) and were usually less than 10%. While high positive expression of TNF, IL-12/23 and IL-1 beta was used for the M1 cells, these cells were also very low expressors of IL-10 and CD 206 (less than 10%) and hence classified as “M1 phenotype”. For “M2 phenotype”, characterization by flow cytometry used the same criteria.
In vivo effects of CRP in Rat Macrophages
Wistar rats were injected with either Human CRP (20 mg/kg bodyweight) for 3 days or vehicle control (Human Serum Albumin) as described previously.19–22 Number of peritoneal macrophages that were of M1 phenotype (IL-23, TNF and IL-1 positive) vs. M2 phenotype (CD163/IL-10/CD206 positive) was assessed by flow cytometry and greater than 10% over isotype control was considered positive.
Flow Cytometric Analysis
The intracellular cytokine expression in human monocyte derived macrophages (HMDM) was determined by flow cytometry and cytokine levels in secreted supernatants were quantitated using multiplex high sensitivity cytokine array from Milliplex as described previously.22–24
Priming of monocytes to a M2 Phenotype
Monocytes were primed from Day 1, with 72 hour exchange of medium with RPMI containing IL-4 (10 ng/mL) or vehicle control for 7 days to allow for differentiation into macrophages. In some of the wells, CRP or HuSA was added.
Role of Fc gamma Receptors and NFKb
Cells were incubated with siRNA to CD32 or CD64 or CD16 or NFKb DN or scrambled ODN for 48 hours and M1/M2 phenotype in presence of CRP was examined.22–24
Statistical Analysis
Student’s paired t-test was used for parametric data and Wilcoxon signed rank test using Graph Pad Prizm software was used if data were non-parametric. ANOVA followed by appropriate multiple-comparisons post-test was carried out for experiments having more than two experimental groups. Data are represented as mean ± SD. P<0.05 was considered as statistically significant.
Results
To evaluate the effect of CRP on macrophage phenotype, we characterized the percentage of M1/M2 macrophages with increasing doses of CRP, boiled CRP or polymixin B (PMB, 25ug/mL) as control. CRP treatment resulted in increased population of M1 macrophages (Fig 1a–1c). We used different combinations of biomarkers to define the M1 phenotype. In Fig 1a, M1 phenotype was defined by positivity for TNF/IL-12 and CCR2. In Fig 1b, we defined M1 phenotype as positivity for TNF/IL-12 and MCP-1 and in Fig 1c, the M1 phenotype was defined by positivity for TNF/IL-1/IL-12. Using any of these combinations, CRP treatment resulted in a significant polarization of macrophages to the M1 phenotype. Addition of polymixin B did not abrogate CRP’s effects and boiled CRP had no effect (Fig 1). Furthermore, in order to prove that these effects of CRP were not due to LPS contamination, since LPS signals through TLR4, we transfected cells with siRNA to TLR4 or control siRNA before addition of LPS or CRP. We demonstrated that siRNA to TLR4 failed to abrogate CRP’s effects but abrogated the effects of LPS (Supplement I). Next, we evaluated levels of intracellular cytokines from these macrophages and also showed that CRP treatment resulted in increased pro-inflammatory cytokines/chemokines from human monocyte derived macrophages (HMDM), by flow cytometry, including TNF-alpha, IL-6, IL-1 beta and MCP-1 (Fig 2a–e). While IL-6 levels were significantly increased with CRP concentrations ≥ 12.5ug/mL, IL-1b, TNF, MCP-1 and CCR-2 levels were significantly increased with CRP concentrations ≥ 25ug/mL Also, levels of secreted cytokines in supernatants of macrophages treated with CRP was significantly increased with CRP treatment and these effects were not abrogated by addition of polymixin B (Table 1). All secreted biomediators were significantly increased at a CRP concentration of ≥ 12.5ug/mL. The observed changes in protein levels of M1 markers suggests that CRP primes macrophages into an M1 phenotype.
Figure 1.
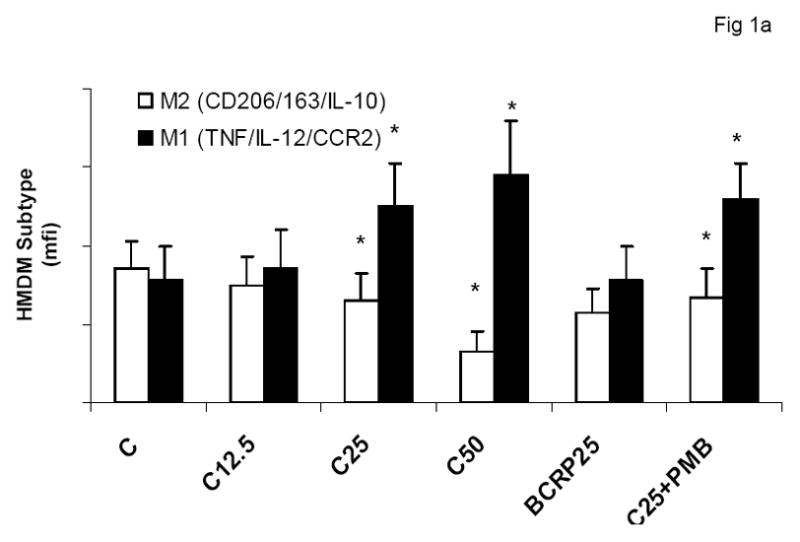
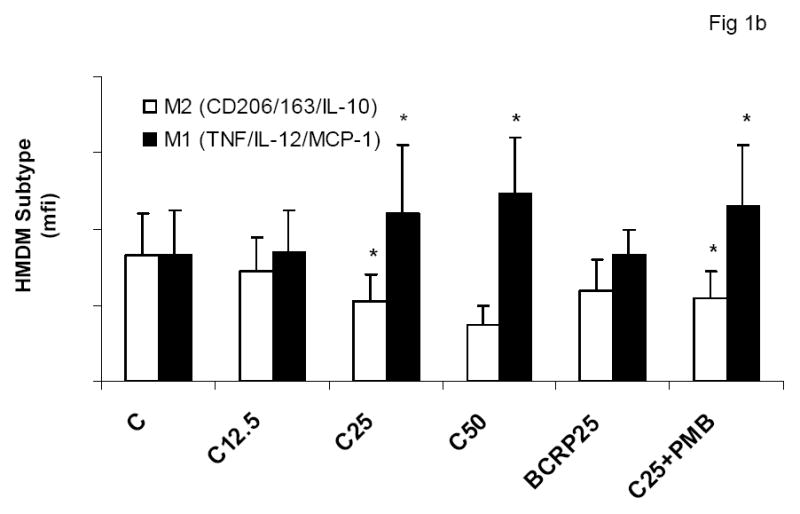
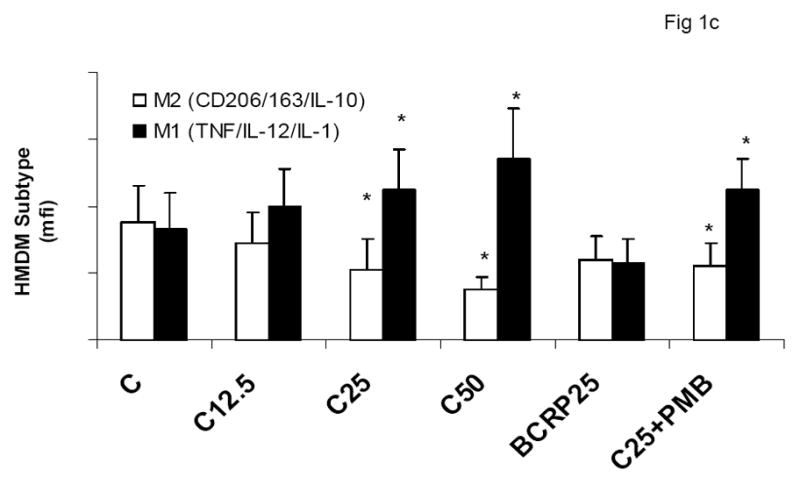
Effect of CRP treatment on HMDM Phenotype: Monocytes were isolated from healthy human volunteers and differentiated into macrophages for 7 days in absence and presence of different doses of CRP (0, 12.5, 25 and 50ug/mL) or boiled CRP (25ug/mL) or CRP and PMB (25ug/mL) and macrophage phenotype was assessed by flow cytometry as described in Methods, n=9, *p<0.05 compared to Control (C). In Fig 1a, M1 is expressed as positivity for TNF/IL-12 and CCR-2; In Fig 1b, M1 is expressed as positivity for TNF/IL-12 and MCP-1 and in Fig 1c, In Fig 1a, M1 is expressed as positivity for TNF/IL-12 and IL-1. Data are represented as mean ± SD.
Figure 2.
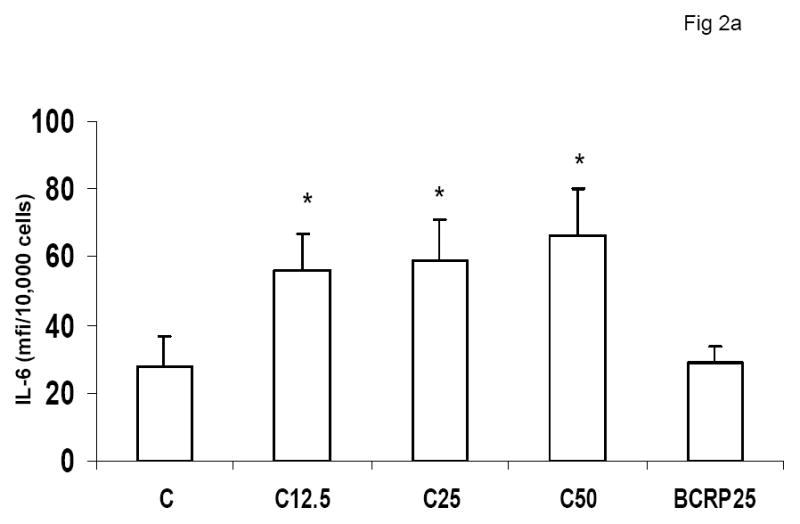
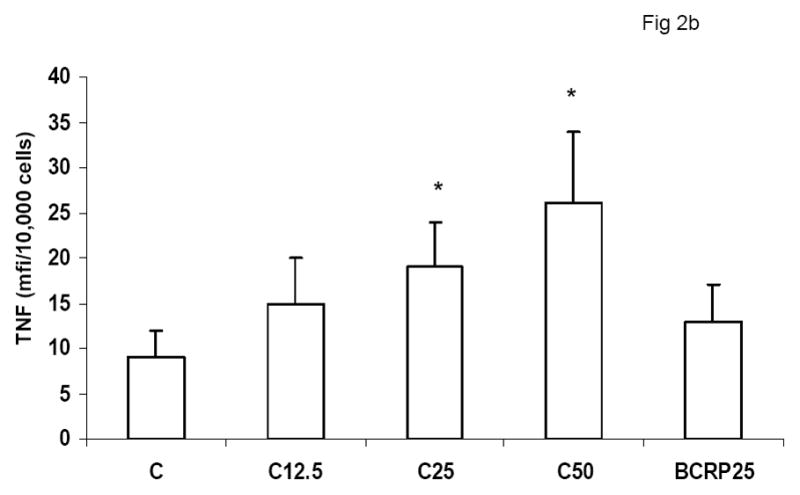
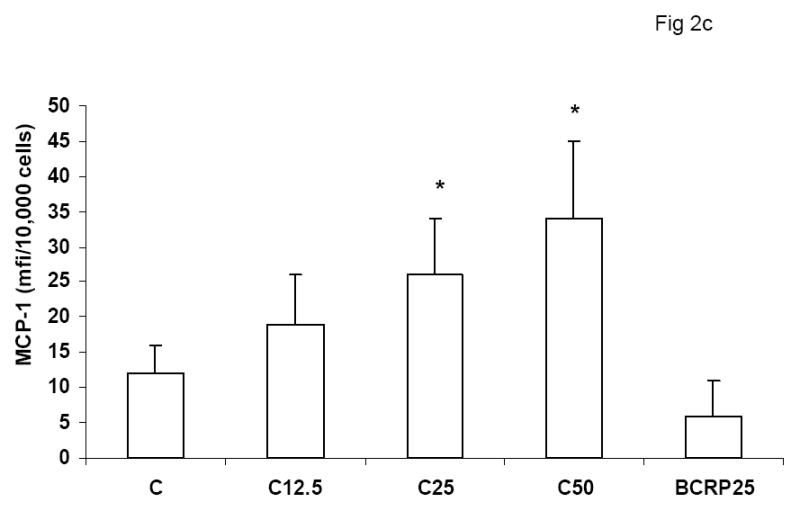
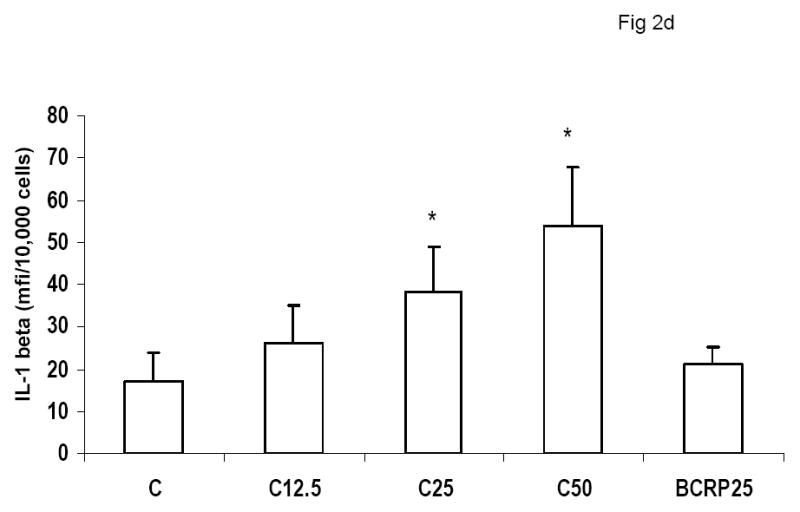
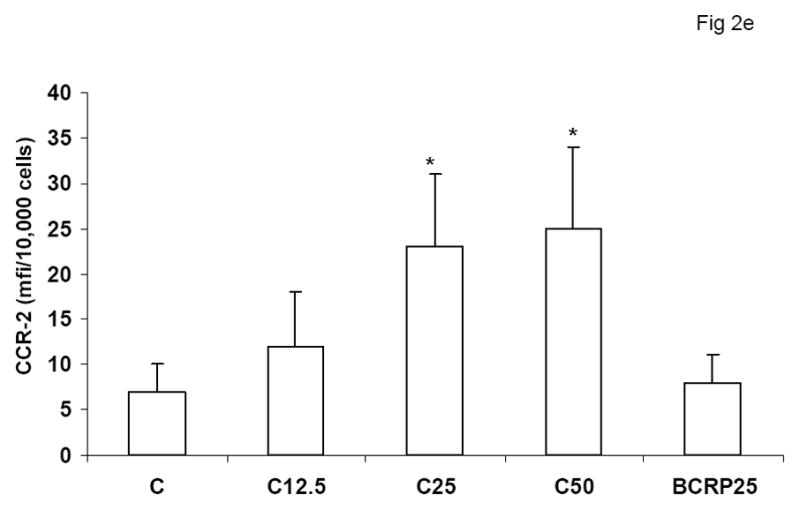
Effect of CRP treatment on Cytokine/chemokine release from HMDM: Monocytes were isolated from healthy human volunteers and differentiated into macrophages for 7 days in absence and presence of different doses of CRP (0, 12.5, 25 and 50ug/mL) or boiled CRP 25ug/mL) and macrophage cytokine release was assessed by multiplex analysis of supernatants and expressed per mg cell protein as described in Methods, n=9, *p<0.05 compared to Control (C). Fig 2a: IL-6 release; Fig 2b: TNF-alpha release; Fig 2c: MCP-1 release; Fig 2d: IL-1b release and Fig 2e: CCR2 expression. Data are represented as mean ± SD
Table 1.
Cytokine Release from Human Macrophages with CRP
| IL-1b (pg/mg protein) | IL-6 (pg/mg protein) | TNF (pg/mg protein) | MCP-1 (pg/mg protein) | |
|---|---|---|---|---|
| Control | 48 ± 11 | 330 ± 101 | 451 ± 101 | 89 ± 33 |
| CRP 12.5 ug/mL | 98 ± 37# | 756 ± 126# | 1245 ± 345* | 124 ± 44† |
| CRP 25 ug/mL | 142 ± 71* | 890 ± 319* | 1879 ± 541* | 198 ± 83* |
| CRP 50 ug/mL | 156 ± 77* | 1124 ± 345* | 2141 ± 801* | 225 ± 45* |
| CRP 25 ug/mL +PMB | 138 ± 43* | 829 ± 411* | 1779 ± 468* | 186 ± 76* |
p<0.001 compared to Control and
p<0.05 compared to C; PMB-Polymixin B (25ug/mL)
We have previously shown in the rat model that administration of HCRP vs. HuSA results in pro-atherogenic effects in macrophages, including activation of NADPH oxidase, NFkb, MMP-9, tissue factor activity and pro-inflammatory cytokines, IL-1 and IL-6.19–22 Furthermore, the Pepys group has shown that hCRP in the rat increases infarct size, and this is prevented with a small molecule inhibitor to CRP, bisphosphocholine; Human CRP administration in the rat model results several pro-inflammatory, pro-atherogenic effects in vivo.25–28 Thus, we tested the effect of human CRP on macrophage phenotype in the rat model. As shown in Fig 3, intraperitoneal administration of human CRP to rats resulted in increased polarization of macrophages to M1 phenotype when compared to HuSA administration.
Figure 3.
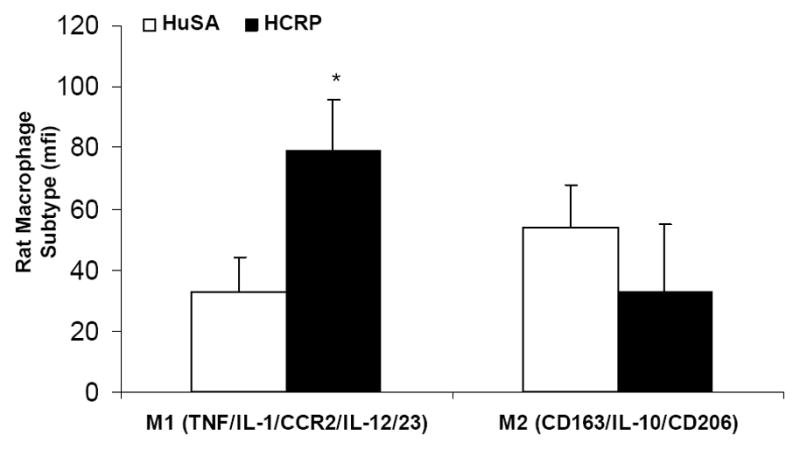
Effect of CRP administration in vivo on HMDM Phenotype: Peritoneal macrophages were obtained from Wistar rats (n=5/group) administered human CRP (20 mg/kg bw) or HuSA for 3 days and macrophage phenotype was assessed by flow cytometry as described in Methods, *p<0.01 compared to HuSA. Data are represented as mean ± SD
To determine if CRP could convert a M2 macrophage into a M1 phenotype, we primed macrophages to an M2 phenotype in presence of IL-4 and then tested the effect of CRP co-incubation on these cells. Incubation of M2 macrophages with CRP resulted in significantly increased secretion of the pro-inflammatory molecules TNF-α, MCP-1 and IL-1 and conversion of macrophages from M2 to M1 phenotype (Fig 4).
Figure 4.
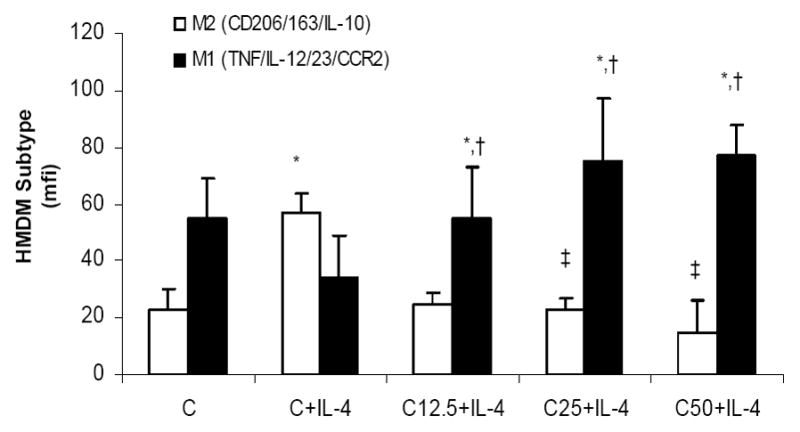
Effect of CRP on IL-4 Primed Macrophages: HMDM were primed with IL-4 to polarize to a M2 phenotype as decribed in Methods followed by incubation with CRP (0,12.5, 15 and 50ug/mL). *p<0.01 compared to Control; †: p<0.01 compared to C+IL-4 and ‡ p<0.05 compared to C+IL-4. n=7 experiments. Data are represented as mean ± SD
CRP has been shown to exert its effects via binding to the Fc gamma receptors, CD32 and CD64 on human monocyte-macrophages. Also, CRP has been shown to augment NFKb, the master switch of inflammation, which regulates expression of several pro-inflammatory cytokines in monocytes. Thus, we tested if downregulation of Fcgamma receptors or NFKb in HMDM results in abrogation of CRPs effects. As shown in Fig 5, CRP failed to prime monocytes to an M1 phenotype in presence of CD32 or CD64 siRNA but not CD16 siRNA. Furthermore, dominant negative NFKb also abrogated CRP’s effects on priming towards a M1 phenotype. Collectively, our results suggest that CRP-induced M1 phenotype polarization is mediated largely through CRP binding to Fc gamma receptors on HMDM and also is dependant on NFκB signaling. Taken together, these results provide further support for a role for CRP in promoting differentiation of monocytes towards a pro-inflammatory M1 phenotype.
Figure 5.
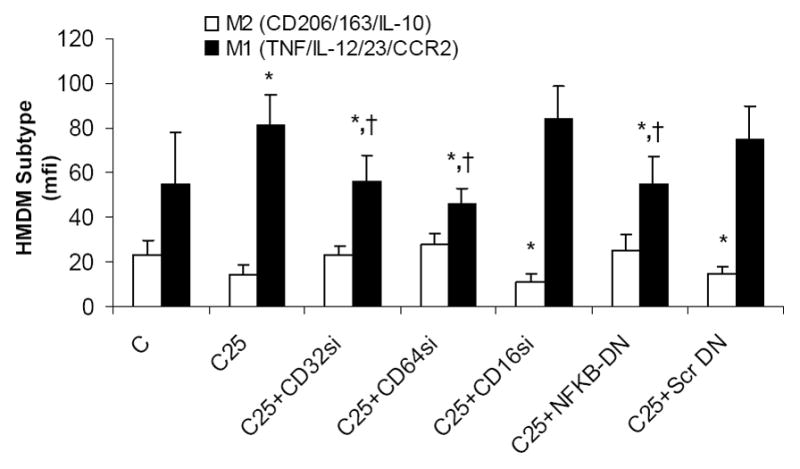
Mechanisms for CRP Polarization of HMDM to M1 Phenotype: HMDM were transfected with siRNA to CD32 or CD64 or CD16 or dominant negative to NFKb or scrambled DN as described in Methods followed by incubation with CRP 25ug/mL. *p<0.05 compared to C; †: p<0.05 compared to CRP 25ug/mL. Data are represented as mean ± SD
DISCUSSION
Monocytes function as control switches of the immune system, maintaining the balance between pro- and anti-inflammatory activities. Emerging evidence points to at least 2 different subsets of macrophages: classically (M1) or alternatively (M2) activated cells.6–12 The prototypic downstream marker of inflammation in man, CRP, is an accepted cardiovascular risk marker. Furthermore, both in vitro and in vivo data support a proatherogenic role for CRP via its effects on both endothelial and monocyte-macrophage biology.16 In this report, we provide novel in vitro and in vivo data further underscoring its proinflammatory effects by showing that it promotes the differentiation of monocytes into the pro-inflammatory M1 macrophage phenotype. In addition, we provide evidence that CRP treatment inhibits agonist-induced polarization to M2 macrophages and instead, promotes differentiation into the pro-inflammatory M1 phenotype. It should be noted that CRP levels of about 50–100ug/mL are attained in patients with acute coronary syndromes and high levels of CRP augur a poor prognosis in these patients.29–31 These effects in the vessel wall could result in an accumulation of pro-inflammatory M1 macrophages. Furthermore, monocytes of the M1 phenotype have been shown to be predominant in human atheroma and in ApoE−/− mice and thus could be implicated in atherogenesis.12–14
We have previously shown in the rat model that administration of HCRP vs. HuSA results in pro-atherogenic effects on macrophages, including activation of NADPH oxidase, NFkb, MMP-9, tissue factor activity and pro-inflammatory cytokines, IL-1 and IL-6.19–22 Furthermore, the Pepys group has shown that hCRP in the rat increases infarct size following coronary artery ligation, and this is prevented with a small molecule inhibitor to CRP, bisphosphocholine. Also, human CRP administration in the rat model increased stroke volume following middle cerebral artery occlusion and adenoviral vector induced human CRP synthesis in rats resulted in hypertension and was accompanied by decreased NO and impairment of endothelium dependent vasorelaxation.25–28 Thus, the rat is the preferred model to test the proatherogenic effects of CRP. We demonstrate in vivo, in this rat model that Human CRP administration results in polarization of macrophages to the M1 phenotype. This is in support of all the in vitro and in vivo data described previously. 19–22, 25–28
Previously, CRP’s proatherogenic activity in monocytes has been shown to be via the Fc gamma receptors CD32 and CD6432, thus we employed siRNA strategy to examine mechanisms by which CRP promoted a M1 phenotype.22–24 We show that both CD32 and CD64 blockade but not CD16 blockade prevents the CRP-induced transition of monocytes to the M1 phenotype.
At the molecular level, previous studies have shown that CRP promotes NFKb activation resulting in increased pro-inflammatory cytokines, thus we tested if inhibition of NFKb would abrogate CRP’s effects. Here, we show that NFKb downregulation using dominant negative NFKb abrogated CRP’s effects of promoting the polarization of HMDM to a M1 phenotype. Human atherosclerotic lesions appear to contain both M1 and M2 macrophages. Native monocytes, in the presence of cytokines such as IL-4 can be primed towards a M2 phenotype.12–14 Thus, in presence of IL-4, which resulted in an increased M2 phenotype; we tested if CRP could prevent polarization of such anti-inflammatory/reparative phenotype. We demonstrate that CRP indeed polarizes IL-4 M2 macrophages to a M1 phenotype, providing another molecular pathway by which it exerts pro-inflammatory effects and potentially promotes atherosclerosis by creating an abundant repository of M1 macrophages in the intima.
Previously, 2 groups have shown that adiponectin, an anti-inflammatory adipokine, promotes the M2 macrophage phenotype but fails to induce a M2 phenotype when differentiated macrophages have already differentiated into a M1 phenotype.33–36 In this report, we show that CRP, in addition to polarizing monocytes to a M1 phenotype, also prevents macrophages, in the presence of an agonist, IL-4, from differentiating to a M2 phenotype macrophage. Since CRP is increased and adiponectin is decreased in the metabolic syndrome, in diabetes and coronary artery disease this unfavorable ratio could promote atherosclerosis via accrual of M1 macrophages in the intima.35–36
In conclusion, the present study demonstrates that CRP polarizes human monocyte differentiation toward pro-inflammatory M1 macrophages via NFKb and by binding to the Fc gamma receptors CD32 and CD64. Furthermore, it reverses agonist-induced M2 macrophages by promoting their polarization to a pro-inflammatory M1 phenotype. These data, in addition to previous studies, further provide evidence for the role of CRP in atherothrombosis. Future studies will examine a complete chemokine array of the 2 subset of monocytes to determine which additional biomarkers best predict if monocytes are in pro-inflammatory M1 state or in an anti-inflammatory M2 state in our quest to identify cellular biomarkers of cardiovascular disease risk.
Supplementary Material
Acknowledgments
Financial support from NIH-K24AT00596 and NIH-HL74360
References
- 1.Libby P, Okamoto Y, Rocha VZ, Folco E. Inflammation in atherosclerosis: transition from theory to practice. Circ J. 2010;74:213–220. doi: 10.1253/circj.cj-09-0706. [DOI] [PubMed] [Google Scholar]
- 2.Ballantyne CM, Nambi V. Markers of inflammation and their clinical significance. Atheroscler Suppl. 2005;6:21–29. doi: 10.1016/j.atherosclerosissup.2005.02.005. [DOI] [PubMed] [Google Scholar]
- 3.Szalai AJ. The biological functions of C-reactive protein. Vascul Pharmacol. 2002;39:105–107. doi: 10.1016/s1537-1891(02)00294-x. [DOI] [PubMed] [Google Scholar]
- 4.Cancello R, Tordjman J, Poitou C, Guilhem G, Bouillot JL, Hugol D, Coussieu C, Basdevant A, Bar Hen A, Bedossa P, Guerre-Millo M, Clément K. Increased infiltration of macrophages in omental adipose tissue is associated with marked hepatic lesions in morbid human obesity. Diabetes. 2006;55:1554–1561. doi: 10.2337/db06-0133. [DOI] [PubMed] [Google Scholar]
- 5.Clément K, Viguerie N, Poitou C, Carette C, Pelloux V, Curat CA, Sicard A, Rome S, Benis A, Zucker JD, Vidal H, Laville M, Barsh GS, Basdevant A, Stich V, Cancello R, Langin D. Weight loss regulates inflammation-related genes in white adipose tissue of obese subjects. FASEB J. 2004;18:1657–1669. doi: 10.1096/fj.04-2204com. [DOI] [PubMed] [Google Scholar]
- 6.Mackman N. Lipopolysaccharide induction of gene expression in human monocytic cells. Immunol Res. 2000;21:247–251. doi: 10.1385/ir:21:2-3:247. [DOI] [PubMed] [Google Scholar]
- 7.Mantovani A. Molecular pathways linking inflammation and cancer. Curr Mol Med. 2010;10:369–373. doi: 10.2174/156652410791316968. [DOI] [PubMed] [Google Scholar]
- 8.Mantovani A, Locati M. Orchestration of macrophage polarization. Blood. 2009;114:3135–3136. doi: 10.1182/blood-2009-07-231795. [DOI] [PubMed] [Google Scholar]
- 9.Mantovani A, Garlanda C, Locati M. Macrophage diversity and polarization in atherosclerosis: a question of balance. Arterioscler Thromb Vasc Biol. 2009;29:1419–1423. doi: 10.1161/ATVBAHA.108.180497. [DOI] [PubMed] [Google Scholar]
- 10.Gordon S. Alternative activation of macrophages. Nat Rev Immunol. 2003;3:23–35. doi: 10.1038/nri978. [DOI] [PubMed] [Google Scholar]
- 11.Geissmann F, Gordon S, Hume DA, Mowat AM, Randolph GJ. Unravelling mononuclear phagocyte heterogeneity. Nat Rev Immunol. 2010;10:453–460. doi: 10.1038/nri2784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mantovani A, Garlanda C, Locati M. Macrophage diversity and polarization in atherosclerosis: a question of balance. Arterioscler Thromb Vasc Biol. 2009;29:1419–1423. doi: 10.1161/ATVBAHA.108.180497. [DOI] [PubMed] [Google Scholar]
- 13.Gautier EL, Jakubzick C, Randolph GJ. Regulation of the migration and survival of monocyte subsets by chemokine receptors and its relevance to atherosclerosis. Arterioscler Thromb Vasc Biol. 2009;29:1412–1418. doi: 10.1161/ATVBAHA.108.180505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Swirski FK, Weissleder R, Pittet MJ. Heterogeneous in vivo behavior of monocyte subsets in atherosclerosis. Arterioscler Thromb Vasc Biol. 2009;29:1424–1432. doi: 10.1161/ATVBAHA.108.180521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Verma S, Szmitko PE, Ridker PM. C-reactive protein comes of age. Nat Clin Pract Cardiovasc Med. 2005;2:29–36. doi: 10.1038/ncpcardio0074. [DOI] [PubMed] [Google Scholar]
- 16.Devaraj S, Singh U, Jialal I. The evolving role of C-reactive protein in atherothrombosis. Clin Chem. 2009;55:229–238. doi: 10.1373/clinchem.2008.108886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Valleggi S, Devaraj S, Dasu MR, Jialal I. C-reactive protein adversely alters the protein-protein interaction of the endothelial isoform of nitric oxide synthase. Clin Chem. 2010;56:1345–1348. doi: 10.1373/clinchem.2009.142364. [DOI] [PubMed] [Google Scholar]
- 18.Han KH, Hong KH, Park JH, Ko J, Kang DH, Choi KJ, Hong MK, Park SW, Park SJ. C-reactive protein promotes monocyte chemoattractant protein-1—mediated chemotaxis through upregulating CC chemokine receptor 2 expression in human monocytes. Circulation. 2004;109:2566–2571. doi: 10.1161/01.CIR.0000131160.94926.6E. [DOI] [PubMed] [Google Scholar]
- 19.Devaraj S, Dasu MR, Singh U, Rao LV, Jialal I. C-reactive protein stimulates superoxide anion release and tissue factor activity in vivo. Atherosclerosis. 2009;203:67–74. doi: 10.1016/j.atherosclerosis.2008.05.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Qamirani E, Ren Y, Kuo L, Hein TW. C-reactive protein inhibits endothelium-dependent NO-mediated dilation in coronary arterioles by activating p38 kinase and NAD (P) H oxidase. Arterioscler Thromb Vasc Biol. 2005;25:995–1001. doi: 10.1161/01.ATV.0000159890.10526.1e. [DOI] [PubMed] [Google Scholar]
- 21.Ballou SP, Lozanski G. Induction of inflammatory cytokine release from cultured human monocytes by C-reactive protein. Cytokine. 1992;4:361–368. doi: 10.1016/1043-4666(92)90079-7. [DOI] [PubMed] [Google Scholar]
- 22.Singh U, Dasu MR, Yancey PG, Afify A, Devaraj S, Jialal I. Human C-reactive protein promotes oxidized low density lipoprotein uptake and matrix metalloproteinase-9 release in Wistar rats. J Lipid Res. 2008;49:1015–1023. doi: 10.1194/jlr.M700535-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Singh U, Devaraj S, Dasu MR, Ciobanu D, Reusch J, Jialal I. C-reactive protein decreases interleukin-10 secretion in activated human monocyte-derived macrophages via inhibition of cyclic AMP production. Arterioscler Thromb Vasc Biol. 2006;26:2469–2475. doi: 10.1161/01.ATV.0000241572.05292.fb. [DOI] [PubMed] [Google Scholar]
- 24.Dasu MR, Devaraj S, Du Clos TW, Jialal I. The biological effects of CRP are not attributable to endotoxin contamination: evidence from TLR4 knockdown human aortic endothelial cells. J Lipid Res. 2007;48:509–512. doi: 10.1194/jlr.C600020-JLR200. [DOI] [PubMed] [Google Scholar]
- 25.Pepys MB, Hirschfield GM, Tennent GA, Gallimore JR, Kahan MC, Bellotti V, Hawkins PN, Myers RM, Smith MD, Polara A, Cobb AJ, Ley SV, Aquilina JA, Robinson CV, Sharif I, Gray GA, Sabin CA, Jenvey MC, Kolstoe SE, Thompson D, Wood SP. Targeting C-reactive protein for the treatment of cardiovascular disease. Nature. 2006;440:1217–21. doi: 10.1038/nature04672. [DOI] [PubMed] [Google Scholar]
- 26.Verma S, Devaraj S, Jialal I. Is C-reactive protein an innocent bystander or proatherogenic culprit? C-reactive protein promotes atherothrombosis. Circulation. 2006;113:2135–50. [PubMed] [Google Scholar]
- 27.Gill R, Kemp JA, Sabin C, Pepys MB. Human C-reactive protein increases cerebral infarct size after middle cerebral artery occlusion in adult rats. J Cereb Blood Flow Metab. 2004;24:1214–8. doi: 10.1097/01.WCB.0000136517.61642.99. [DOI] [PubMed] [Google Scholar]
- 28.Guan H, Wang P, Hui R, Edin ML, Zeldin DC, Wang DW. Adeno-associated virus-mediated human C-reactive protein gene delivery causes endothelial dysfunction and hypertension in rats. Clin Chem. 2009;55:274–84. doi: 10.1373/clinchem.2008.115857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Caixeta A, Stone GW, Mehran R, Lee EA, McLaurin BT, Cox DA, Bertrand ME, Lincoff AM, Moses JW, White HD, Ohman EM, Palmerini T, Syros G, Kittas C, Fahy M, Hooper WC, Lansky AJ, Dangas GD. Predictive value of C-reactive protein on 30-day and 1-year mortality in acute coronary syndromes: an analysis from the ACUITY trial. J Thromb Thrombolysis. 2011;31:154–64. doi: 10.1007/s11239-010-0516-y. [DOI] [PubMed] [Google Scholar]
- 30.Ray KK, Nazer B, Cairns R, Michael Gibson C, Cannon CP. Association between percutaneous coronary intervention and long-term C-reactive protein levels in patients with acute coronary syndromes. J Thromb Thrombolysis. 2010;30:10–13. doi: 10.1007/s11239-010-0463-7. [DOI] [PubMed] [Google Scholar]
- 31.He LP, Tang XY, Ling WH, Chen WQ, Chen YM. Early C-reactive protein in the prediction of long-term outcomes after acute coronary syndromes: a meta-analysis of longitudinal studies. Heart. 2010;96:339–46. doi: 10.1136/hrt.2009.174912. [DOI] [PubMed] [Google Scholar]
- 32.Devaraj S, Du Clos TW, Jialal I. Binding and internalization of C-reactive protein by Fcgamma receptors on human aortic endothelial cells mediates biological effects. Arterioscler Thromb Vasc Biol. 2005;25:1359–1363. doi: 10.1161/01.ATV.0000168573.10844.ae. [DOI] [PubMed] [Google Scholar]
- 33.Ohashi K, Parker JL, Ouchi N, Higuchi A, Vita JA, Gokce N, Pedersen AA, Kalthoff C, Tullin S, Sams A, Summer R, Walsh K. Adiponectin promotes macrophage polarization toward an anti-inflammatory phenotype. J Biol Chem. 2010;285:6153–6160. doi: 10.1074/jbc.M109.088708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lovren F, Pan Y, Quan A, Szmitko PE, Singh KK, Shukla PC, Gupta M, Chan L, Al-Omran M, Teoh H, Verma S. Adiponectin primes human monocytes into alternative anti-inflammatory M2 macrophages. Am J Physiol Heart Circ Physiol. 2010;299:H656–H663. doi: 10.1152/ajpheart.00115.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ouchi N, Kihara S, Arita Y, Maeda K, Kuriyama H, Okamoto Y, Hotta K, Nishida M, Takahashi M, Nakamura T, Yamashita S, Funahashi T, Matsuzawa Y. Novel modulator for endothelial adhesion molecules: adipocyte-derived plasma protein adiponectin. Circulation. 1999;100:2473–2476. doi: 10.1161/01.cir.100.25.2473. [DOI] [PubMed] [Google Scholar]
- 36.Devaraj S, Swarbrick MM, Singh U, Adams-Huet B, Havel PJ, Jialal I. CRP and adiponectin and its oligomers in the metabolic syndrome: evaluation of new laboratory-based biomarkers. Am J Clin Pathol. 2008;129:815–822. doi: 10.1309/RN84K51B2JJY1Y0B. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.