

Abstract
Recent data suggest a physiological role for the proinflammatory cytokine TNF-α in skeletal muscle regeneration. However, the underlying mechanism is not understood. In the present study, we analyzed TNF-α-activated signaling pathways involved in myogenesis in soleus muscle injured by cardiotoxin (CTX) in TNF-α receptor double-knockout mice (p55−/−p75−/−). We found that activation of p38MAPK, which is critical for myogenesis, was blocked in CTX-injured p55−/−p75−/− soleus on day 3 postinjury when myogenic differentiation was being initiated, while activation of ERK1/2 and JNK MAPK, as well as transcription factor NF-κB, was not reduced. Consequently, the phosphorylation of transcription factor myocyte enhancer factor-2C, which is catalyzed by p38 and crucial for the expression of muscle-specific genes, was blunted. Meanwhile, expression of p38-dependent differentiation marker myogenin and p21 were suppressed. In addition, expression of cyclin D1 was fivefold that in wild-type (WT) soleus. These results suggest that myogenic differentiation is blocked or delayed in the absence of TNF-α signaling. Histological studies revealed abnormalities in regenerating p55−/−p75−/− soleus. On day 5 postinjury, new myofiber formation was clearly observed in WT soleus but not in p55−/−p75−/− soleus. To the contrary, p55−/−p75−/− soleus displayed renewed inflammation and dystrophic calcification. On day 12 postinjury, the muscle architecture of WT soleus was largely restored. Yet, in p55−/−p75−/− soleus, multifocal areas of inflammation, myofiber death, and myofibers with smaller cross-sectional area were observed. Functional studies demonstrated an attenuated recovery of contractile force in injured p55−/−p75−/− soleus. These data suggest that TNF-α signaling plays a critical regulatory role in muscle regeneration.
Keywords: tumor necrosis factor-α receptor double-knockout mice, myogenesis, p38 mitogen-activated protein kinase, dystrophic calcification, contractility
In skeletal muscle, TNF-α has long been considered a pathological factor implicated in disorders such as cachectic muscle wasting, inflammatory myopathies, and insulin resistance (29, 37). On the other hand, recent evidence suggests that TNF-α also may have a role in skeletal muscle regeneration. Skeletal muscle regeneration is an adaptive response to muscle injury or disease that involves the degeneration of damaged myofibers, inflammation, and the formation of new myofibers through satellite cell proliferation and differentiation (myogenesis). Despite considerable efforts to understand the complex mechanism that controls muscle regeneration, the complete profile of intrinsic and extrinsic cues that regulate myogenesis during muscle regeneration remains to be understood. It has become increasingly clear that inflammation is a key response to muscle injury and critical for muscle regeneration (44). Inflammatory cells, particularly macrophages, facilitate muscle regeneration via phagocytosis of cellular debris and release of soluble factors that promote satellite cell proliferation and differentiation (3–5). Among those soluble factors, there are not only chemoattractants and growth factors that are traditionally recognized as factors promoting muscle regeneration but also cytokines that are known mainly as inflammatory mediators (16, 38). Some of the inflammatory cytokines, such as leukemia inhibitory factor and IL-6, have been shown to be part of the regulatory mechanism of muscle regeneration (16). Recent data suggest that TNF-α, a central proinflammatory cytokine, may also play a physiological role in the regulation of muscle regeneration.
Muscle regeneration takes place in an environment with unusually high TNF-α levels. Coincident with the onset of muscle regeneration, the TNF-α level in injured muscle rises dramatically because of a strong increase in TNF-α synthesis by injured myofibers as well as TNF-α released by infiltrating inflammatory cells (7, 10, 43, 46, 50), and myofiber synthesis of TNF-α is positively correlated to regenerating activity (21). At the same time, there is an increase in TNF-α receptor expression in injured muscle fibers (10, 50), suggesting an intrinsic need in injured muscle for increased TNF-α signaling. Recent evidence supports a physiological role for TNF-α in myogenesis. A rapid increase of TNF-α synthesis by C2C12 myoblasts during the early hours of myogenic differentiation is critical for muscle-specific gene expression, suggesting that TNF-α regulates myogenesis as an autocrine or paracrine function (28). TNF-α receptor double-knockout impairs strength recovery of mouse muscle injured by freezing, which suggests the participation of TNF-α in the regulation of muscle regeneration (46). However, the mechanism through which TNF-α participates in the regulation of muscle regeneration is not understood. In the present study, we demonstrate that 1) TNF-α signaling is critical for p38MAPK activation and p38-dependent signaling events required for myogenic differentiation and 2) deficiency in TNF-α signaling impairs muscle regeneration.
MATERIALS AND METHODS
Animal use
Experimental protocols were approved in advance by the Animal Protocol Review Committee of the Baylor Animal Program. Adult (8–11 wk old) TNF-α receptor double-knockout mice (p55−/−p75−/−) mice (B6; 129S-Tnfrsf1atm1Imx Tnfrsf1btm1Imx) and wild-type (WT) mice (B6; 129SF2/J) were purchased from Jackson Laboratory (Bar Harbor, ME). Cardiotoxin (CTX; Sigma Chemical, St. Louis, MO) dissolved in 100 μl of 10 μM PBS was injected into the hindlimb muscle while aiming at the soleus. Solei along with tendon were surgically removed for biochemical, histological, or contractile study at various times while the mice were under deep anesthesia induced by intraperitoneal (IP) injection of 85 mg/kg pentobarbital sodium. The animals were then killed by performing cervical dislocation.
Western blot analysis
Western blot analysis was performed as previously described (27). Antibodies for pan- and phosphorylated p38, ERK1/2, and JNK MAPK were obtained from Cell Signaling Technology (Beverly, MA). Antibodies for pan-myocyte enhancer factor (MEF)-2C (sc-13266), phosphorylated MEF-2C (sc-13920), and p21 (sc-397) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Antibody for myogenin was obtained from the Development Studies Hybridoma Bank at the University of Iowa (Iowa City, IA). Muscle protein extracts were used in all experiments, except for MEF-2C and phospho-MEF-2C, which were analyzed using muscle nuclear extracts prepared according to a protocol described previously (13). Detected protein bands were quantified using optical density (OD) measured with ImageQuant software (Molecular Dynamics). All Western blot analyses were performed two or three times to ensure that the results were repeatable, and representative blots are shown in Figs. 1, 2, and 3. Protein concentration in the extracts was determined using the Bio-Rad protein assay.
Fig. 1.
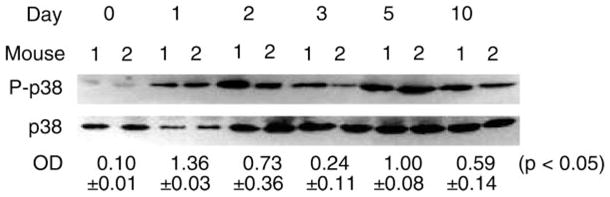
Activation of p38 during regeneration of cardiotoxin (CTX)-injured mouse soleus muscle. Total protein was extracted from soleus collected from wild-type (WT) mice on the indicated days after being injured by direct injection of 100 μl of 10 μM CTX. Western blot analysis was performed using antibodies that are specific for phosphorylated p38 or pan-p38. Average optical density (OD) data are expressed as the ratio of phospho-p38 to p38 and were assessed using ANOVA.
Fig. 2.
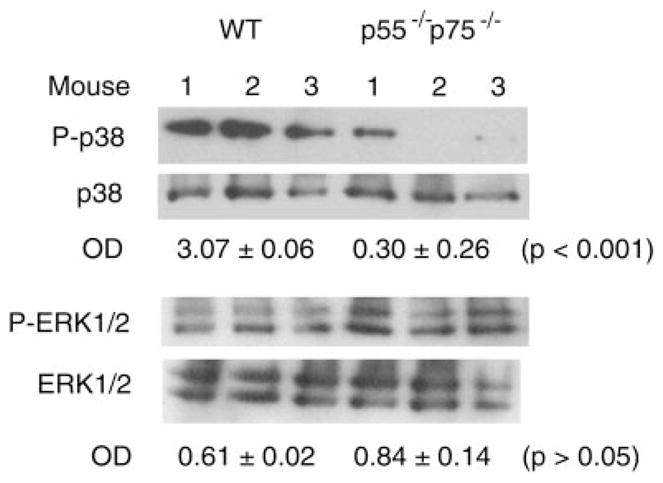
Blockade of p38 activation in TNF-α receptor double-knockout mice (p55−/−p75−/−) soleus muscle. Total protein was extracted from soleus collected from WT and p55−/−p75−/− mice 3 days after the mice were injured using CTX. Western blot analysis was performed using antibodies that are specific for phosphorylated p38 or ERK1/2 and pan-p38 or pan-ERK1/2. OD data are expressed as the ratio of phosphorylated protein to total protein and were analyzed using Student’s t-test.
Fig. 3.
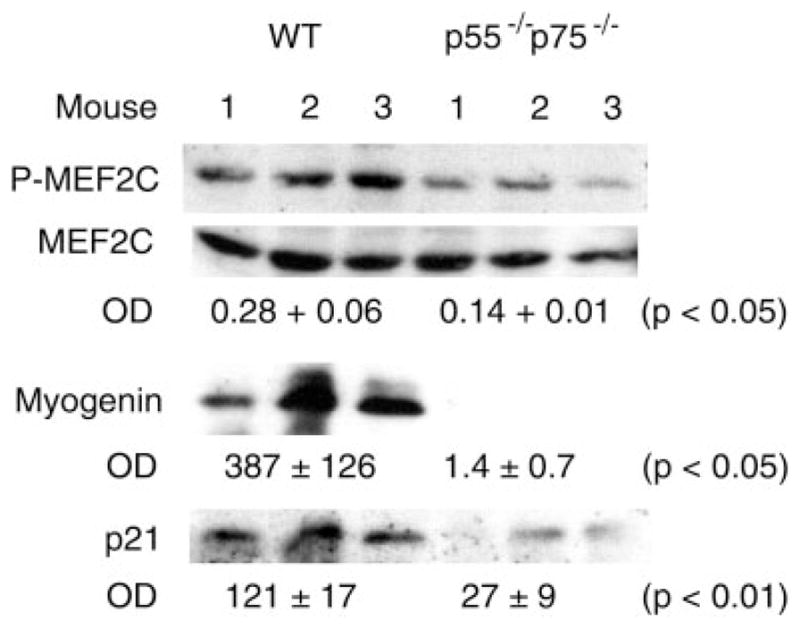
Blockade of p38-dependent myogenic signaling in regenerating p55−/−p75−/− soleus. Total protein or nuclear protein was extracted from soleus collected from WT and p55−/−p75−/− mice 3 days postinjury. Western blot analysis was performed using antibodies that are specific for phosphorylated myocyte enhancer factor (MEF)-2C and pan-MEF-2C to assess its activation in muscle nuclear extracts. Expression of myogenin and p21 was evaluated using Western blot analysis of muscle total protein extracts. OD data for MEF-2C activation are expressed as the ratio of phospho-MEF-2C to total MEF-2C, and OD data for myogenin and p21 expression are expressed as direct readings from the densitometer. Data were analyzed using Student’s t-test.
Real-time PCR
Total RNA was extracted from excised soleus using RNAzol reagent (TEL TEST, Friendswood, TX). Reverse transcription was performed using a kit from Applied Biosystems (Foster City, CA). Real-time PCR was performed using TaqMan Gene Expression primers and probes for mouse cyclin D1 (assay identification no. Mm00432357_m1; Applied Biosystems) using a 5700 sequence detection system (Applied Biosystems) according to the manufacturer’s protocol. The quantity of PCR product was normalized to 18S rRNA determined using TaqMan Gene Expression primers and probes for 18S (no. 4310893E; Applied Biosystems). The standard curve method was used to quantify the PCR products.
Electrophoretic mobility shift assay
Nuclear extracts were prepared from soleus muscle, and EMSA was performed for NF-κB as described previously (13).
Histological studies
Solei collected from mice were fixed in 4% formaldehyde, and paraffin-embedded sections were made and processed for hematoxylin and eosin (H&E) or Von Kassa staining performed by the Baylor Histology Service. Soleus myofiber cross-sectional area (XSA) was measured using ImageJ software (National Institutes of Health, Bethesda, MD). Mean XSA on day 12 were normalized to preinjury (day 0) XSA of soleus from the same type of mice. For immunohistochemical staining with anti-Mac-1, frozen sections of excised soleus (5 μM) were prepared and fixed in acetone for 10 min at −20°C. Sections were treated with 3% H2O2 for 10 min and blocked in 5% BSA for 1 h at room temperature. Incubation with anti-Mac-1 (M1/70.15.11.5.2; Developmental Studies Hybridoma Bank) was performed at 1:50 dilution in blocking buffer for 1 h at 37°C. After sections were washed in PBS, incubation with biotinylated anti-rat secondary antibody was conducted at room temperature for 30 min. Avidin-biotin complex and diaminobenzidine reagent kits (Vector Laboratories, Burlingame, CA) were used according to the manufacturer’s protocol to detect the secondary antibody. Counter-staining with hematoxylin was performed for 30 s. Images of stained muscle sections were acquired using MetaVue computer software and a Zeiss Axioplan 2 microscope coupled to a Photometric Cool Snap charge-coupled device camera and then edited using Adobe Photo-Shop software. The average number of Mac-1-positive cells observed under a microscope in 0.25-mm2 areas of soleus sections was obtained by counting multiple areas in each section. Means of the average derived from multiple mice were expressed as the inflammation index.
Force-frequency study
Immediately after excision of solei from mice, they were immersed in room temperature Krebs-Ringer solution containing (in mM) 137 NaCl, 5 KCl, 1 NaH2PO4, 24 NaHCO3, 2 CaCl2, and 1 MgSO4. The solution was aerated in a 95% O2-5% CO2 atmosphere. One soleus tendon was tied with 5-0 silk suture to a glass rod; the other tendon was tied to a force transducer (model BG 100; Kulite Instruments, Leonia, NJ) mounted on a micrometer. Soleus muscles sections were subjected to field stimulation using platinum electrodes. Muscle length was adjusted using the micrometer to produce optimum twitch force. The solution was then heated to 37°C, and the temperature was controlled using a digital water bath at 37°C throughout the remainder of the experiment. After a 30-min thermo-equilibration period, we determined the force-frequency relationship. Tetanic contractions were stimulated at 1-min intervals (500-ms train duration); between each intermediate frequency (15, 30, 50, 80, 120, 160, and 250 Hz), a maximum tetanic contraction (Po, 300 Hz) was elicited to serve as a reference for changes in force over time. Soleus muscle length that yielded optimum force was measured, and the muscle then was trimmed of tendons and weighed. Force measurements were later normalized for functional cross sections according to the method described by Close (6).
Statistics
Commercial software (SigmaStat; SPSS Science, Chicago, IL) was used to analyze data. Student’s t-test or ANOVA was used for analysis as indicated. Differences between groups were considered significant at the P < 0.05 level. Values are reported as means ± SE.
RESULTS
To assess the role of TNF-α in muscle regeneration, we chose to conduct the studies in p55−/−p75−/− mice instead of TNF-α-knockout mice to ensure the complete absence of TNF-α signaling, considering that lymphotoxin-α can activate TNF-α receptors (11) and that oligomerization of TNF-α receptors can occur in the absence of ligand binding, leading to receptor activation without actual ligand-receptor interaction (14). Muscle regeneration in soleus muscle was induced by direct injection of CTX derived from snake venom, which induces extensive and reproducible muscle necrotic injury. It is well documented that after CTX injection, satellite cell proliferation occurs within 2 days, myogenic differentiation is initiated within 3 days, new myotube formation is evident within 5 days, and muscle architecture is largely restored within 10 days (16).
Activation of p38MAPK during muscle regeneration is blocked in p55−/−p75−/− soleus
We previously showed that TNF-α promotes myogenic differentiation in an autocrine fashion in C2C12 myoblasts (28). Among TNF-α receptor-activated signaling events, p38MAPK (26) stands out as a necessary and sufficient mediator of myogenic differentiation (2, 9, 35, 36, 47, 51). The activity of p38 increases dramatically during myogenic differentiation in myoblasts (9, 47) and in injured human muscle (1) or injured myoblasts (49). However, the signal that is responsible for p38 activation during myogenesis in vivo has not been identified, while it is known that p38 activation is independent of the potent myogenic stimulus insulin-like growth factor I (47). To investigate the underlying mechanism for the potential regulatory role of TNF-α in muscle regeneration, we examined activation of p38MAPK in CTX-injured mouse soleus muscle during the course of regeneration as well as its relationship to TNF-α signaling. Using an antibody specific for phosphorylated p38, we found that Western blot analysis revealed that p38 was activated within 1 day after injury and lasted for at least 10 days in WT soleus (Fig. 1). Comparing p38 activation in WT and p55−/−p75−/− soleus on day 3 postinjury, when myogenic differentiation was being initiated, we found that activation of p38 was blunted in p55−/−p75−/− soleus, while total p38 levels were not different from those observed in WT soleus (Fig. 2), suggesting a dependence of p38 activation on TNF-α receptor activation during muscle regeneration. On the other hand, we observed no reduction in the activation of ERK1/2 (Fig. 2), a TNF-α-responsive MAPK that stimulates satellite cell proliferation but not differentiation (8). The activity of another TNF-α-responsive MAPK JNK whose role in myogenesis is not well defined, with both inhibitory and stimulatory effects having been reported (20, 32, 39), was not reduced in p55−/−p75−/− soleus either (data not shown). Because TNF-α activates transcription factor NF-κB, which also influences myogenic differentiation (19, 24, 28), we examined whether NF-κB is activated in CTX-injured muscle in a TNF-α signaling-dependent manner. Using EMSA, we observed a similar increase of NF-κB binding activity in WT and p55−/−p75−/− soleus muscle on day 3 postinjury compared with uninjured control (data not shown). Thus NF-κB is activated in injured muscle in a non-TNF-α-dependent manner. These data suggest that TNF-α receptor activation is a critical upstream signal for p38 activation during muscle regeneration.
p38-Dependent myogenic signaling during muscle regeneration is blocked in p55−/−p75−/− soleus
p38 regulates myogenic differentiation at multiple steps. It directly phosphorylates the transactivation domain of MEF-2 family of transcription factors that bind to the promoters of the majority of muscle-specific genes and interacts with members of the MyoD family of proteins to activate skeletal muscle differentiation (31), induces expression of the key differentiation signal myogenin (47, 48), and promotes cell cycle exit by inducing cdk inhibitor p21 expression (47) and suppressing cyclin D1 expression via downregulation of the Raf-ERK pathway (22, 23, 34) so that myogenic differentiation can take place. To determine whether blockade of p38 activation in p55−/−p75−/− soleus results in disruption of p38-dependent signaling events that are required for myogenic differentiation, we examined the phosphorylation of MEF-2C, a MEF-2 family member that regulates muscle gene expression (31), and expression of myogenin and p21. Western blot analysis using an antibody specific for MEF-2C that is phosphorylated at Ser387, a p38-phosphorylated site within the transactivation domain (15), revealed that phosphorylation of MEF-2C was attenuated in p55−/−p75−/− soleus on day 3 postinjury (Fig. 3). At the same time, both myogenin and p21 expression were suppressed in p55−/−p75−/− soleus as indicated by the protein levels detected by performing Western blot analysis (Fig. 3). We also determined cyclin D1 levels using real-time PCR. We observed a level of cyclin D1 mRNA in p55−/−p75−/− soleus that was fivefold that observed in WT soleus (Fig. 4), suggesting a deregulation of cyclin D1 expression and a blockade of cell cycle exit. These results consistently indicated that TNF-α signaling is critical for myogenesis because of its indispensable role in regulating p38-dependent signaling events.
Fig. 4.
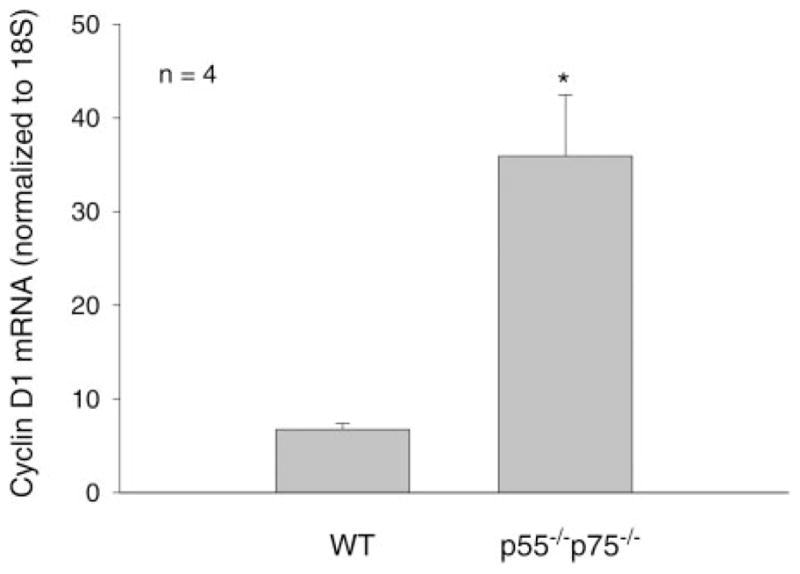
Deregulation of cyclin D1 expression in p55−/−p75−/− soleus. Total RNA was extracted from solei collected from WT and p55−/−p75−/− mice on day 3 after CTX injection. RT and real-time PCR were performed to evaluate cyclin D1 mRNA levels, which were normalized to 18S. The ratio of cyclin D1 expression to 18S is expressed in arbitrary units. Data are means ± SE derived from 5 mice in each group. *P < 0.01 (Student’s t-test).
Regenerating p55−/−p75−/− soleus displays morphological abnormalities
We investigated the morphological consequence of TNF-α receptor deficiency on muscle regeneration by comparing the histology of CTX-injured p55−/−p75−/− soleus with that of WT soleus. The CTX-induced regenerating process was highly consistent with the findings described previously in the literature. H&E-stained cross sections of WT soleus muscle showed typical signs of injury and regeneration on day 5 after CTX injection as indicated by chronic inflammation (infiltration) and newly formed myofibers with centralized nuclei, as well as near-completion of regeneration on day 12 as indicated by the largely restored muscle architecture (Fig. 5). However, injured p55−/−p75−/− soleus displayed a striking deficiency in regeneration. On day 5, damaged muscle architecture in p55−/−p75−/− soleus did not recover as well as it did in time-matched WT soleus, such that few newly formed myofibers that were well defined with centralized nuclei were observed. At the same time, more severe inflammatory infiltration, not only chronic but also acute, was observed (Fig. 5). To quantify the degree of inflammation, cross sections of soleus were stained with an antibody for Mac-1 (CD11b), a marker of macrophages and neutrophils. The density of Mac-1-positive cell (inflammation index) observed in p55−/−p75−/− soleus was 2.5-fold that observed in time-matched WT (Fig. 6). In addition to increased inflammatory infiltration, punctate staining patterns that appeared to be Ca2+ deposits were observed in sections from p55−/−p75−/− soleus, suggesting the presence of dystrophic calcification. To verify whether dystrophic calcification was indeed present, Von Kassa staining of soleus sections was performed to detect precipitated Ca2+ in black. Extensive Ca2+ deposits were observed in p55−/−p75−/− soleus on day 5 but not in WT (Fig. 7).
Fig. 5.
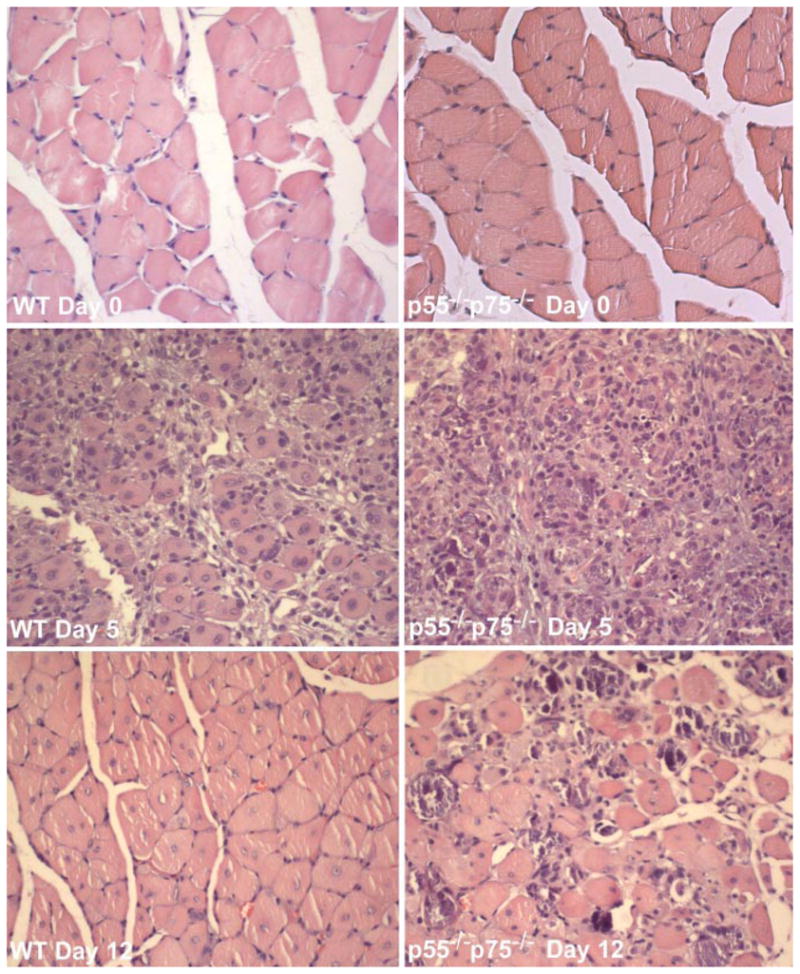
Abnormal morphology in regenerating p55−/−p75−/− soleus muscle. Solei of adult WT and p55−/−p75−/− mice were injured by CTX injection and collected surgically on day 5 or day 12 after injection. Uninjured soleus was collected as control (day 0). Cross sections of formalin-fixed and paraffin-embedded soleus muscles were processed for hematoxylin and eosin staining. Representative sections of regenerating WT and p55−/−p75−/− soleus are shown.
Fig. 6.
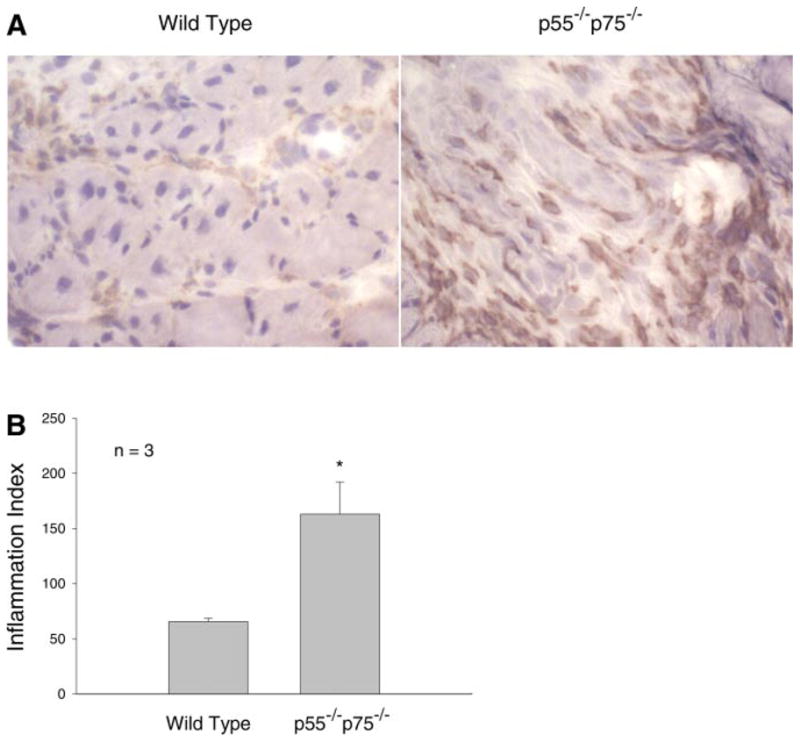
Increased inflammation in regenerating p55−/−p75−/− soleus muscle. Cross sections of frozen soleus that were prepared from WT and p55−/−p75−/− mice on day 5 after CTX injection were stained with anti-Mac-1 (A). The average number of Mac-1-positive cells observed in a 0.25-mm2 area under a microscope was expressed as the inflammation index (B). Means ± SE derived from solei of 3 mice in each group are shown and were analyzed using Student’s t-test. *P < 0.05.
Fig. 7.
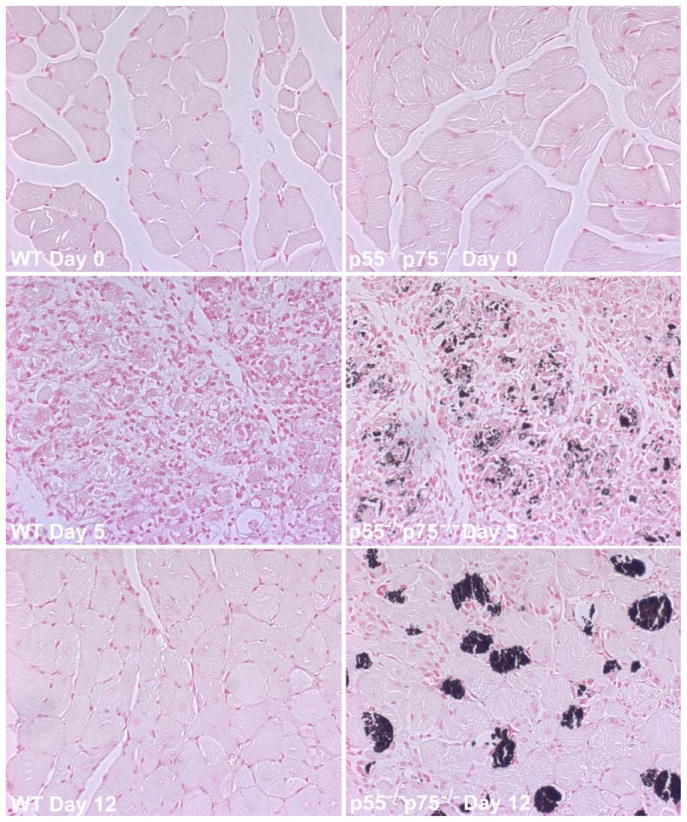
Calcification of regenerating p55−/−p75−/− soleus muscle. Solei of adult WT and p55−/−p75−/− mice were injured by direct injection of CTX and collected surgically on day 5 or day 12 after injection. Uninjured soleus was collected as control (day 0). Cross sections of formalin-fixed and paraffin-embedded soleus muscle were processed for Von Kassa staining, which stains solidified Ca2+ in black. Representative sections of regenerating WT and p55−/−p75−/− soleus are shown.
On day 12, multifocal areas of inflammation and severely calcified myofibers were observed among newly formed myofibers that appeared to be smaller than those in WT soleus (Fig. 5). Von Kassa staining again confirmed calcification of myofibers (Fig. 7). To determine whether the newly formed myofibers in p55−/−p75−/− soleus were indeed smaller than those in WT soleus, myofiber size was quantified by measuring XSA. The mean XSA of WT myofibers measured on day 12 was 86.4% of those measured on day 0. However, mean XSA of p55−/−p75−/− myofibers on day 12 was only 68.7% of that on day 0 (P < 0.05 compared with WT). These data indicate that muscle regeneration in CTX-injured p55−/−p75−/− soleus is impaired.
Restoration of contractile force is compromised in regenerating p55−/−p75−/− soleus
Next, we used a physiological approach to evaluate muscle regeneration by determining the restoration of contractile force generated by excised soleus by evaluating the force-frequency relationship. Uninjured soleus excised from WT and p55−/−p75−/− mice (day 0) produced comparable maximal force (Fig. 8), although the force-frequency curve was shifted slightly to the right in p55−/−p75−/− relative to WT. On day 5 postinjury, both types of soleus muscle lost at least 90% of maximal force. On day 12, the maximal force generated by WT soleus recovered to 75.8% of the preinjury level. However, force generated by p55−/−p75−/− soleus was only 53.9% of preinjury level (P < 0.05 compared with day 12 WT). This result corroborates the histological data and confirms that muscle regeneration is impaired in CTX-injured p55−/−p75−/− soleus muscle.
Fig. 8.

Compromised restoration of contractile force in injured p55−/−p75−/− soleus muscle. Soleus was excised from p55−/−p75−/− (B) and WT (A) mice before (day 0) or after CTX injection (days 5 and 12). Force developed by the muscle stimulated with 300-ms stimulus trains in Krebs-Ringer solution bath was recorded using an oscilloscope through a force transducer and normalized to muscle cross-sectional area (N/cm2). Data represent results from 3 or 4 mice (1 soleus per mouse) in each group. Data are means ± SE. The percentages of maximal force on day 12 compared with those on day 0 derived from WT and p55−/−p75−/− mice were compared using Student’s t-test.
DISCUSSION
The present study demonstrates for the first time that TNF-α signaling is required for p38 activation and p38-dependent signaling events during muscle regeneration, and it provides morphological and functional evidence that TNF-α signaling is required for normal muscle regeneration, thus suggesting a critical role for TNF-α in regulating muscle regeneration.
Given the negative image of TNF-α in skeletal muscle metabolism because of its involvement in muscle protein breakdown, inflammatory myopathies, and insulin resistance (29, 37), it appears counterintuitive that expression of TNF-α and its receptors by myofibers would increase dramatically during injury-induced regeneration (7, 10, 43, 46, 50), which, along with TNF-α released by infiltrating inflammatory cells, brings about an unusually high level of TNF-α receptor-mediated signaling. Several of the TNF-α-activated signaling molecules are involved in the regulation of myogenesis, including members of the MAPK family and NF-κB. Members of the MAPK family have different roles in myogenesis. p38MAPK is recognized as a necessary and sufficient “switch” that turns on the differentiation program (2, 36, 51), while ERK1/2 stimulates the proliferation of myocytes (8). On the other hand, the role of JNK in myogenesis is not well defined. Both inhibitory and stimulatory effects on myogenesis have been reported for JNK (20, 32, 39). NF-κB also influences myogenic differentiation (19, 24, 28). The dependence of p38 activation on TNF-α is remarkable, considering that the activation of ERK1/2, JNK, and NF-κB was not TNF-α dependent during muscle regeneration. These factors can be activated by multiple cytokines and growth factors in the absence of TNF-α signaling. Although p38 activation has been established as a key event in myogenic differentiation, the upstream signal for p38 activation during muscle regeneration was not identified. We have demonstrated herein for the first time that TNF-α receptors are required for p38 activation during muscle regeneration. Our observations suggest that the purpose of the surge of TNF-α synthesis in injured muscle fibers is to activate p38 and thus to turn on myogenic differentiation. Considering the importance of p38 activation in myogenic differentiation, the role of TNF-α in muscle regeneration appears to be more significant than previously thought.
The myogenic program is a highly complex signaling network, and p38 is involved in several key steps of the program. We show in the present study that multiple p38-mediated steps of the myogenic program are TNF-α signaling dependent, including MEF-2 phosphorylation, induction of myogenin and p21 expression, and suppression of cyclin D1 expression. MEF-2 phosphorylation is required for the expression of the majority of muscle-specific genes that are synergistically activated by MEF-2 and MyoD (31). Myogenin plays a key role in executing the myogenic differentiation program (33). On the other hand, p21 and cyclin D1 are cell cycle regulators for cell cycle exit that are critical so that differentiation can take place (41). Although we measured these signaling events in intact muscle, they actually take place in activated satellite cells, not in myofibers and infiltrating inflammatory cells, which are terminally differentiated cells. These data consistently demonstrate an impairment of myogenic differentiation in the absence of TNF-α signaling during muscle regeneration.
The morphological abnormalities observed in regenerating p55−/−p75−/− soleus muscle, including more severe and persistent inflammation, dystrophic calcification, and the formation of smaller myotubes, confirm that muscle regeneration is impaired in the absence of TNF-α signaling. The morphological abnormalities can result from impairment of various aspects of muscle regeneration, including impairment of myogenesis due to the lack of p38 activation.
The results of force-frequency studies provide functional evidence that supports the histological data. In Fig. 8, the force-frequency curve for undamaged soleus (day 0) was shifted slightly to the right in p55−/−p75−/− relative to WT. This response suggests that constitutive TNF-α signaling influences the contractile properties of soleus muscle. Most likely, TNF modulates the expression of one or more regulatory proteins that affect the force-frequency characteristic. A partial listing of candidate proteins includes the voltage-sensitive dihydropyridine receptor, ryanodine-sensitive sarcoplasmic reticulum (SR) Ca2+ release channel, SR Ca2+-ATPase, myosin heavy chains, myosin light chains, troponin C, and tropomyosin. This issue, while intriguing, was not the focus of our present study. No attempt has been made to identify the proteins responsible for differences between groups at day 0.
Inflammation is a key response to muscle injury (44) by virtue of its role in phagocytosis and satellite cell proliferation and differentiation (3–5). Because an important function of TNF-α signaling is to amplify inflammatory response, the presence of more severe inflammation in injured p55−/−p75−/− soleus appears counterintuitive. Yet, despite the presence of more severe inflammation, regeneration in p55−/−p75−/− soleus was still impaired. These observations suggest that there is a compensatory increase of inflammation to promote regeneration as a result of the deficiency in muscle regeneration in p55−/−p75−/− muscle; nevertheless, with the lack of TNF-α signaling, increased inflammation is still ineffective in promoting regeneration. Thus TNF-α signaling appears to be a key component of inflammation that promotes muscle regeneration. TNF-α is known to stimulate phagocytosis (30) and a chemotactic response (45), which facilitate muscle regeneration. The present study shows that in addition to the previously known effects, TNF-α signaling is required for p38-mediated myogenesis.
That skeletal muscle develops in TNF-α receptor-knockout mice almost normally, although with different mechanical properties (7), does not automatically preclude TNF-α from being a physiological regulator of myogenesis, because a compensatory mechanism may replace the role played by TNF-α. Previous studies showed that in TNF-α-null mice, any role played by TNF-α could be performed effectively by the upregulation of other cytokines. In the absence of TNF-α, a number of cytokines, including IL-12, INF-γ, and IL-1, are upregulated (12, 17, 42). The networking of these cytokines is capable of activating macrophages and modulating myoblast proliferation and fusion (12, 18). An analogy can be found in MyoD-knockout mice. MyoD-knockout mice still develop muscle because of a redundancy in the role of Myf-5 (40). A possible explanation for the difference observed between newborn animals and adult animals that undergo muscle regeneration is that in newborn animals, the compensatory mechanism plays out over time, whereas injury-induced regeneration is an acute response, especially with regard to regeneration induced by snake venom, in which degeneration and inflammation develop more quickly than other types of injury (25), so that a role of TNF-α signaling can more readily be appreciated.
In summary, the present study provides new evidence that TNF-α has an important physiological role in regulating skeletal muscle regeneration. These data are helpful in sorting out the details of the mechanism that initiates muscle regeneration, particularly myogenesis, in response to injury. The results described herein also have clinical implications. TNF-α has long been considered a therapeutic target for inflammatory diseases. Anti-TNF-α strategies have gained popularity clinically in treating a variety of inflammatory conditions. Our data suggest that long-term use of anti-TNF-α reagents may impair skeletal muscle adaptation. Thus caution should be exercised in using anti-TNF-α strategies.
Acknowledgments
We thank Dr. Roberto Barrios of Department of Pathology, Baylor College of Medicine, for providing the pathological diagnosis regarding the histology of the soleus sections.
GRANTS
This work was supported by National Institute of Arthritis and Musculoskeletal and Skin Diseases Grant AR-049022 (to Y.-P. Li) and National Heart, Lung, and Blood Institute Grant HL-59878 (to M. B. Reid).
References
- 1.Aronson D, Wojtaszewski JFP, Thorell A, Nygren J, Zangen D, Richter EA, Ljungqvist O, Fielding RA, Goodyear LJ. Extracellular-regulated protein kinase cascades are activated in response to injury in human skeletal muscle. Am J Physiol Cell Physiol. 1998;275:C555–C561. doi: 10.1152/ajpcell.1998.275.2.C555. [DOI] [PubMed] [Google Scholar]
- 2.Cabane C, Englaro W, Yeow K, Ragno M, Dérijard B. Regulation of C2C12 myogenic terminal differentiation by MKK3/p38α pathway. Am J Physiol Cell Physiol. 2003;284:C658–C666. doi: 10.1152/ajpcell.00078.2002. [DOI] [PubMed] [Google Scholar]
- 3.Cantini M, Carraro U. Macrophage-released factor stimulates selectively myogenic cells in primary muscle culture. J Neuropathol Exp Neurol. 1995;54:121–128. doi: 10.1097/00005072-199501000-00014. [DOI] [PubMed] [Google Scholar]
- 4.Cantini M, Massimino ML, Rapizzi E, Rossini K, Catani C, Dalla Libera L, Carraro U. Human satellite cell proliferation in vitro is regulated by autocrine secretion of IL-6 stimulated by a soluble factor(s) released by activated monocytes. Biochem Biophys Res Commun. 1995;216:49–53. doi: 10.1006/bbrc.1995.2590. [DOI] [PubMed] [Google Scholar]
- 5.Chazaud B, Sonnet C, Lafuste P, Bassez G, Rimaniol AC, Poron F, Authier FJ, Dreyfus PA, Gherardi RK. Satellite cells attract monocytes and use macrophages as a support to escape apoptosis and enhance muscle growth. J Cell Biol. 2003;163:1133–1143. doi: 10.1083/jcb.200212046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Close RI. Dynamic properties of mammalian skeletal muscles. Physiol Rev. 1972;52:129–197. doi: 10.1152/physrev.1972.52.1.129. [DOI] [PubMed] [Google Scholar]
- 7.Collins RA, Grounds MD. The role of tumor necrosis factor-α (TNF-α) in skeletal muscle regeneration: studies in TNF-α−/− and TNF-α−/−/LT-α−/− mice. J Histochem Cytochem. 2001;49:989–1001. doi: 10.1177/002215540104900807. [DOI] [PubMed] [Google Scholar]
- 8.Coolican SA, Samuel DS, Ewton DZ, McWade FJ, Florini JR. The mitogenic and myogenic actions of insulin-like growth factors utilize distinct signaling pathways. J Biol Chem. 1997;272:6653–6662. doi: 10.1074/jbc.272.10.6653. [DOI] [PubMed] [Google Scholar]
- 9.Cuenda A, Cohen P. Stress-activated protein kinase-2/p38 and a rapamycin-sensitive pathway are required for C2C12 myogenesis. J Biol Chem. 1999;274:4341–4346. doi: 10.1074/jbc.274.7.4341. [DOI] [PubMed] [Google Scholar]
- 10.De Bleecker JL, Meire VI, Declercq W, Van Aken EH. Immunolocalization of tumor necrosis factor-α and its receptors in inflammatory myopathies. Neuromuscul Disord. 1999;9:239–246. doi: 10.1016/s0960-8966(98)00126-6. [DOI] [PubMed] [Google Scholar]
- 11.Decoster E, Cornelis S, Vanhaesebroeck B, Fiers W. Autocrine tumor necrosis factor (TNF) and lymphotoxin (LT) α differentially modulate cellular sensitivity to TNF/LT-α cytotoxicity in L929 cells. J Cell Biol. 1998;143:2057–2065. doi: 10.1083/jcb.143.7.2057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.De Maeyer E, De Maeyer-Guignard J. Interferons. In: Thomson AW, editor. The Cytokine Handbook. 3. San Diego, CA: Academic; 1998. pp. 491–516. [Google Scholar]
- 13.Durham WJ, Li YP, Gerken E, Farid M, Arbogast S, Wolfe RR, Reid MB. Fatiguing exercise reduces DNA binding activity of NF-κB in skeletal muscle nuclei. J Appl Physiol. 2004;97:1740–1745. doi: 10.1152/japplphysiol.00088.2004. [DOI] [PubMed] [Google Scholar]
- 14.Engelmann H, Holtmann H, Brakebusch C, Avni YS, Sarov I, Nophar Y, Hadas E, Leitner O, Wallach D. Antibodies to a soluble form of a tumor necrosis factor (TNF) receptor have TNF-like activity. J Biol Chem. 1990;265:14497–14504. [PubMed] [Google Scholar]
- 15.Han J, Molkentin JD. Regulation of MEF2 by p38 MAPK and its implication in cardiomyocyte biology. Trends Cardiovasc Med. 2000;10:19–22. doi: 10.1016/s1050-1738(00)00039-6. [DOI] [PubMed] [Google Scholar]
- 16.Hawke TJ, Garry DJ. Myogenic satellite cells: physiology to molecular biology. J Appl Physiol. 2001;91:534–551. doi: 10.1152/jappl.2001.91.2.534. [DOI] [PubMed] [Google Scholar]
- 17.Hodge-Dufour J, Marino MW, Horton MR, Jungbluth A, Burdick MD, Strieter RM, Noble PW, Hunter CA, Puré E. Inhibition of interferon γ induced interleukin 12 production: a potential mechanism for the anti-inflammatory activities of tumor necrosis factor. Proc Natl Acad Sci USA. 1998;95:13806–13811. doi: 10.1073/pnas.95.23.13806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ji SQ, Neustrom S, Willis GM, Spurlock ME. Proinflammatory cytokines regulate myogenic cell proliferation and fusion but have no impact on myotube protein metabolism or stress protein expression. J Interferon Cytokine Res. 1998;18:879–888. doi: 10.1089/jir.1998.18.879. [DOI] [PubMed] [Google Scholar]
- 19.Kaliman P, Canicio J, Testar X, Palacín M, Zorzano A. Insulin-like growth factor-II, phosphatidylinositol 3-kinase, nuclear factor-κB and inducible nitric-oxide synthase define a common myogenic signaling pathway. J Biol Chem. 1999;274:17437–17444. doi: 10.1074/jbc.274.25.17437. [DOI] [PubMed] [Google Scholar]
- 20.Khurana A, Dey CS. Involvement of c-Jun N-terminal kinase activities in skeletal muscle differentiation. J Muscle Res Cell Motil. 2004;25:645–655. doi: 10.1007/s10974-004-7099-1. [DOI] [PubMed] [Google Scholar]
- 21.Kuru S, Inukai A, Kato T, Liang Y, Kimura S, Sobue G. Expression of tumor necrosis factor-α in regenerating muscle fibers in inflammatory and non-inflammatory myopathies. Acta Neuropathol (Berl) 2003;105:217–224. doi: 10.1007/s00401-002-0635-4. [DOI] [PubMed] [Google Scholar]
- 22.Lavoie JN, L’Allemain G, Brunet A, Müller R, Pouysségur J. Cyclin D1 expression is regulated positively by the p42/p44MAPK and negatively by the p38/HOGMAPK pathway. J Biol Chem. 1996;271:20608–20616. doi: 10.1074/jbc.271.34.20608. [DOI] [PubMed] [Google Scholar]
- 23.Lee J, Hong F, Kwon S, Kim SS, Kim DO, Kang HS, Lee SJ, Ha J, Kim SS. Activation of p38 MAPK induces cell cycle arrest via inhibition of Raf/ERK pathway during muscle differentiation. Biochem Biophys Res Commun. 2002;298:765–771. doi: 10.1016/s0006-291x(02)02562-7. [DOI] [PubMed] [Google Scholar]
- 24.Lee KH, Kim DG, Shin NY, Song WK, Kwon H, Chung CH, Kang MS. NF-κB-dependent expression of nitric oxide synthase is required for membrane fusion of chick embryonic myoblasts. Biochem J. 1997;324:237–242. doi: 10.1042/bj3240237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lefaucheur JP, Sébille A. The cellular events of injured muscle regeneration depend on the nature of the injury. Neuromuscul Disord. 1995;5:501–509. doi: 10.1016/0960-8966(95)00012-c. [DOI] [PubMed] [Google Scholar]
- 26.Li YP, Chen Y, John J, Moylan J, Jin B, Mann DL, Reid MB. TNF-α acts via p38 MAPK to stimulate expression of the ubiquitin ligase atrogin1/MAFbx in skeletal muscle. FASEB J. 2005;19:362–370. doi: 10.1096/fj.04-2364com. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li YP, Lecker SH, Chen Y, Waddell ID, Goldberg AL, Reid MB. TNF-α increases ubiquitin-conjugating activity in skeletal muscle by up-regulating UbcH2/E220k. FASEB J. 2003;17:1048–1057. doi: 10.1096/fj.02-0759com. [DOI] [PubMed] [Google Scholar]
- 28.Li YP, Schwartz RJ. TNF-α regulates early differentiation of C2C12 myoblasts in an autocrine fashion. FASEB J. 2001;15:1413–1415. doi: 10.1096/fj.00-0632fje. [DOI] [PubMed] [Google Scholar]
- 29.Lundberg IE, Dastmalchi M. Possible pathogenic mechanisms in inflammatory myopathies. Rheum Dis Clin North Am. 2002;28:799–822. doi: 10.1016/s0889-857x(02)00025-x. [DOI] [PubMed] [Google Scholar]
- 30.Mandell GL. Cytokines, phagocytes, and pentoxifylline. J Cardiovasc Pharmacol. 1995;25(Suppl 2):S20–S22. doi: 10.1097/00005344-199500252-00005. [DOI] [PubMed] [Google Scholar]
- 31.McKinsey TA, Zhang CL, Olson EN. MEF2: a calcium-dependent regulator of cell division, differentiation and death. Trends Biochem Sci. 2002;27:40–47. doi: 10.1016/s0968-0004(01)02031-x. [DOI] [PubMed] [Google Scholar]
- 32.Meriane M, Roux P, Primig M, Fort P, Gauthier-Rouvière C. Critical activities of Rac1 and Cdc42Hs in skeletal myogenesis: antagonistic effects of JNK and p38 pathways. Mol Biol Cell. 2000;11:2513–2528. doi: 10.1091/mbc.11.8.2513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Molkentin JD, Olson EN. Defining the regulatory networks for muscle development. Curr Opin Genet Dev. 1996;6:445–453. doi: 10.1016/s0959-437x(96)80066-9. [DOI] [PubMed] [Google Scholar]
- 34.Page K, Li J, Hershenson MB. p38 MAP kinase negatively regulatescyclin D1 expression in airway smooth muscle cells. Am J Physiol Lung Cell Mol Physiol. 2001;280:L955–L964. doi: 10.1152/ajplung.2001.280.5.L955. [DOI] [PubMed] [Google Scholar]
- 35.Penn BH, Bergstrom DA, Dilworth FJ, Bengal E, Tapscott SJ. A MyoD-generated feed-forward circuit temporally patterns gene expression during skeletal muscle differentiation. Genes Dev. 2004;18:2348–2353. doi: 10.1101/gad.1234304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Puri PL, Wu Z, Zhang P, Wood LD, Bhakta KS, Han J, Feramisco JR, Karin M, Wang JYJ. Induction of terminal differentiation by constitutive activation of p38 MAP kinase in human rhabdomyosarcoma cells. Genes Dev. 2000;14:574–584. [PMC free article] [PubMed] [Google Scholar]
- 37.Reid MB, Li YP. Cytokines and oxidative signaling in skeletal muscle cells. Acta Physiol Scand. 2001;171:225–232. doi: 10.1046/j.1365-201x.2001.00824.x. [DOI] [PubMed] [Google Scholar]
- 38.Robertson TA, Maley MA, Grounds MD, Papadimitriou JM. The role of macrophages in skeletal muscle regeneration with particular reference to chemotaxis. Exp Cell Res. 1993;207:321–331. doi: 10.1006/excr.1993.1199. [DOI] [PubMed] [Google Scholar]
- 39.Rousse S, Lallemand F, Montarras D, Pinset C, Mazars A, Prunier C, Atfi A, Dubois C. Transforming growth factor-β inhibition of insulin-like growth factor-binding protein-5 synthesis in skeletal muscle cells involves a c-Jun N-terminal kinase-dependent pathway. J Biol Chem. 2001;276:46961–46967. doi: 10.1074/jbc.M104440200. [DOI] [PubMed] [Google Scholar]
- 40.Rudnicki MA, Braun T, Hinuma S, Jaenisch R. Inactivation of MyoD in mice leads to up-regulation of the myogenic HLH gene Myf-5 and results in apparently normal muscle development. Cell. 1992;71:383–390. doi: 10.1016/0092-8674(92)90508-a. [DOI] [PubMed] [Google Scholar]
- 41.Schafer KA. The cell cycle: a review. Vet Pathol. 1998;35:461–478. doi: 10.1177/030098589803500601. [DOI] [PubMed] [Google Scholar]
- 42.Storkus WJ, Tahara H, Lotze MT. Interleukin-12. In: Thomson AW, editor. The Cytokine Handbook. 3. San Diego, CA: Academic; 1998. pp. 391–426. [Google Scholar]
- 43.Tews DS, Goebel HH. Cytokine expression profile in idiopathic inflammatory myopathies. J Neuropathol Exp Neurol. 1996;55:342–347. doi: 10.1097/00005072-199603000-00009. [DOI] [PubMed] [Google Scholar]
- 44.Tidball JG. Inflammatory cell response to acute muscle injury. Med Sci Sports Exerc. 1995;27:1022–1032. doi: 10.1249/00005768-199507000-00011. [DOI] [PubMed] [Google Scholar]
- 45.Torrente Y, El Fahime E, Caron NJ, Del Bo R, Belicchi M, Pisati F, Tremblay JP, Bresolin N. Tumor necrosis factor-α (TNF-α) stimulates chemotactic response in mouse myogenic cells. Cell Transplant. 2003;12:91–100. doi: 10.3727/000000003783985115. [DOI] [PubMed] [Google Scholar]
- 46.Warren GL, Hulderman T, Jensen N, McKinstry M, Mishra M, Luster MI, Simeonova PP. Physiological role of tumor necrosis factor α in traumatic muscle injury. FASEB J. 2002;16:1630–1632. doi: 10.1096/fj.02-0187fje. [DOI] [PubMed] [Google Scholar]
- 47.Wu Z, Woodring PJ, Bhakta KS, Tamura K, Wen F, Feramisco JR, Karin M, Wang JY, Puri PL. p38 and extracellular signal-regulated kinases regulate the myogenic program at multiple steps. Mol Cell Biol. 2000;20:3951–3964. doi: 10.1128/mcb.20.11.3951-3964.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Xu Q, Yu L, Liu L, Cheung CF, Li X, Yee SP, Yang XJ, Wu Z. p38 Mitogen-activated protein kinase-, calcium-calmodulin-dependent protein kinase-, and calcineurin-mediated signaling pathways transcriptionally regulate myogenin expression. Mol Biol Cell. 2002;13:1940–1952. doi: 10.1091/mbc.02-02-0016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yeow K, Cabane C, Turchi L, Ponzio G, Dérijard B. Increased MAPK signaling during in vitro muscle wounding. Biochem Biophys Res Commun. 2002;293:112–119. doi: 10.1016/S0006-291X(02)00190-0. [DOI] [PubMed] [Google Scholar]
- 50.Zádor E, Mendler L, Takács V, de Bleecker J, Wuytack F. Regenerating soleus and extensor digitorum longus muscles of the rat show elevated levels of TNF-α and its receptors, TNFR-60 and TNFR-80. Muscle Nerve. 2001;24:1058–1067. doi: 10.1002/mus.1110. [DOI] [PubMed] [Google Scholar]
- 51.Zetser A, Gredinger E, Bengal E. p38 mitogen-activated protein kinase pathway promotes skeletal muscle differentiation: participation of the MEF2C transcription factor. J Biol Chem. 1999;274:5193–5200. doi: 10.1074/jbc.274.8.5193. [DOI] [PubMed] [Google Scholar]