

Abstract
Although p38 MAPK activation is essential for myogenesis, the upstream signaling mechanism that activates p38 during myogenesis remains undefined. We recently reported that p38 activation, myogenesis, and regeneration in cardiotoxin-injured soleus muscle are impaired in TNF-α receptor double-knockout (p55−/−p75−/−) mice. To fully evaluate the role of TNF-α in myogenic activation of p38, we tried to determine whether p38 activation in differentiating myoblasts requires autocrine TNF-α, and whether forced activation of p38 rescues impaired myogenesis and regeneration in the p55−/−p75−/− soleus. We observed an increase of TNF-α release from C2C12 or mouse primary myoblasts placed in low-serum differentiation medium. A TNF-α-neutralizing antibody added to differentiation medium blocked p38 activation and suppressed differentiation markers myocyte enhancer factor (MEF)-2C, myogenin, p21, and myosin heavy chain in C2C12 myoblasts. Conversely, recombinant TNF-α added to differentiation medium stimulated myogenesis at 0.05 ng/ml while inhibited it at 0.5 and 5 ng/ml. In addition, differentiation medium-induced p38 activation and myogenesis were compromised in primary myoblasts prepared from p55−/−p75−/− mice. Increased TNF-α release was also seen in cardiotoxin-injured soleus over the course of regeneration. Forced activation of p38 via the constitutive activator of p38, MKK6bE, rescued impaired myogenesis and regeneration in the cardiotoxin-injured p55−/−p75−/− soleus. These results indicate that TNF-α regulates myogenesis and muscle regeneration as a key activator of p38.
Keywords: myocyte enhancer factor-2C, myogenin, p21, myosin heavy chain, Akt, tumor necrosis factor-α, mitogen-activated protein kinase
Skeletal muscle has a remarkable ability to regenerate itself. However, muscle regeneration is a complex process comprising many highly coordinated events in a sequence of satellite cell activation, proliferation, and differentiation (myogenesis) that repairs damaged myofibers or forms new ones. The activation of satellite cells is characterized by the expression of myogenic regulatory factors (MRFs) Myf 5 and MyoD. After the proliferation phase, satellite cells express myogenin and MRF4 to initiate myogenic differentiation. This is followed by expression of the Cdk inhibitor p21 and a permanent exit from the cell cycle (reviewed in Ref. 11). The regenerative process requires a complicated array of intrinsic and extrinsic signals that regulate various satellite cell activities. Thus, despite many past efforts, our understanding of the intrinsic and extrinsic signals that regulate muscle regeneration remains limited.
Initiation of the myogenic program in adult muscle is a critical step in skeletal muscle regeneration, which requires chromatin remodeling in myogenic cells that allows transcriptional activation of myogenic target genes. The activation of p38 MAPK plays a critical role in chromatin remodeling and in the activation of key myogenic transcription factors and thus is considered a molecular switch for the activation of myogenesis. Activation of p38 is an early and essential event in myogenic differentiation in myoblasts and embryo (8, 16, 17, 46, 48, 68, 70). p38 activates myogenesis through multiple actions. p38-mediated phosphorylation activates the Brahma-related gene 1 (BRG1)-associated factor (BAF)60 subunit of the BRG1-based SWI/SNF chromatin remodeling complex, which permits the access of MyoD, E proteins, and myocyte enhancer factor (MEF)-2 to their binding sites in the myogenin promoter and, hence, the transcription of myogenin (19, 39, 53). In addition, p38 stimulates MyoD transactivation activity (48) by promoting the association of E47 with MyoD via the phosphorylation of E47 (39). Moreover, the MEF2 family of transcription factors, which bind to promoters of the majority of muscle-specific genes and interact with members of the MyoD family of proteins to activate myogenic differentiation (43), is activated by p38-mediated phosphorylation of their transactivation domain (17). Furthermore, p38 promotes cell cycle exit by inducing expression of the Cdk inhibitor p21 (8, 52, 68) so that terminal differentiation can proceed. However, the upstream signal that stipulates p38 activation for myogenesis has remained undefined, which represents a significant gap in our understanding of how myogenesis is initiated during muscle regeneration.
A number of autocrine or paracrine factors, mostly growth factors including fibroblast growth factor (FGF), hepatocyte growth factor (HGF), insulin-like growth factor (IGF)-I and -II, epidermal growth factor (EGF), and platelet-derived growth factor (PDGF), have been identified as important regulators of muscle regeneration. In addition, cytokines have been shown to participate in the regeneration process, including TGF-β, leukemia inhibitor factor (LIF), and IL-6. Although, remarkably, almost all of the above factors promote satellite cell activation/proliferation, most of them also inhibit myogenic differentiation; only IGF-I has been convincingly shown to promote both satellite cell differentiation and proliferation (11, 26). However, IGF-I is not able to activate p38 or to induce myogenesis when p38 is inhibited (68), which indicates the presence of a yet-to-be-identified mechanism that is critical for the initiation of myogenesis through the activation of p38.
Recently, the proinflammatory cytokine TNF-α has been shown to have a physiological role in muscle repair (13, 65) and myogenesis (38). As a mediator of inflammatory response, TNF-α is primarily synthesized by macrophages (63), and elevated circulating TNF-α is considered a pathological factor that mediates such disorders as cachectic muscle wasting, inflammatory myopathies, and insulin resistance (41, 49). On the other hand, it is now clear that myoblasts express TNF-α constitutively (51) and that this activity is transiently upregulated in differentiating myoblasts (38). In injured muscle, TNF-α levels increase dramatically because of enhanced TNF-α expression by injured muscle fibers as well as macrophage infiltration (14, 18, 31, 57, 65, 69). Interestingly, TNF-α expression by muscle fibers is correlated with regenerative activity (31). In addition, injured muscle fibers increase expression of the type I TNF-α receptor (18, 69), which mediates p38 activation (29). Although there are reports describing an inhibitory effect of exogenously added TNF-α on myoblast differentiation (24, 32, 33), it is difficult to reconcile such an inhibitory effect of TNF-α on myogenesis with the fact that muscle increases TNF-α production during regeneration and regenerates well in the high-TNF-α environment. In recent years, the significance of inflammation in mediating muscle regeneration became clear. Injured muscle releases factors that activate inflammatory cells residing within the muscle, and an inflammatory response is critical to muscle regeneration (58, 59). Independently of clearing cellular debris in injured muscle through phagocytosis, macrophages promote the activation, proliferation, and differentiation of myogenic cells through their release of soluble factors (9, 10, 12, 34). TNF-α has been shown to stimulate chemotactic response in mouse myogenic cells, which facilitates muscle regeneration (62). We have also observed that, during the early stages of differentiation, C2C12 myoblasts increase the expression of TNF-α, which is critical for the expression of the muscle-specific protein, the fast isoform of myosin heavy chain (MHCf) (38). TNF-α gene knockout in dystrophin-deficient mice (TNF−/mdx) resulted in a significantly lower muscle mass than control mdx mice (TNF+/mdx) at 8 wk of age, suggesting an attenuation of regenerative capacity (55). Warren et al. (65) reported an attenuation of muscle force recovery during regeneration in the hind-limb of TNF-α receptor double-knockout mice (p55−/−p75−/−) injured by freezing, suggesting a physiological role for TNF-α in muscle repair. Notwithstanding, the relevance and mechanism of TNF-α involvement in muscle regeneration remain undefined.
We recently found that, in the cardiotoxin-injured soleus muscle of wild-type (WT) mice, p38 is activated over the course of regeneration; however, in the cardiotoxin-injured soleus of p55−/−p75−/− mice, p38 activation is compromised and regeneration is impaired (13). These data indicate a significant role for TNF-α in the regulation of myogenesis and muscle regeneration. On the basis of these findings, we propose that muscle cell-produced TNF-α is a physiological regulator of muscle regeneration that is critical for the activation of p38 and myogenesis. Nevertheless, to fully establish this role of TNF-α, two critical questions must be answered. First, does autocrine TNF-α mediate p38 activation and myogenesis in myogenic cells? Second, does TNF-α-dependent p38 activation mediate myogenesis and regeneration in adult muscle? In the current study, we tested the following hypothesis: p38 activation and myogenesis in myogenic cells require autocrine TNF-α, and disruption of TNF-α-dependent p38 activation causes impaired myogenesis and regeneration in adult muscle. Here, we present evidence that 1) myoblast release of TNF-α and TNF-α receptor-mediated signaling are required for the activation of p38 and myogenic differentiation in myoblasts, and 2) compromised p38 activation is the cause of impaired myogenesis and regeneration in adult muscle, absent TNF-α receptor-mediated signaling.
MATERIALS AND METHODS
Animal use
Experimental protocols were approved in advance by the Institutional Animal Care and Use Committee at Baylor College of Medicine. Adult (6 wk old) p55−/−p75−/− mice (B6;129S-Tnfrsf1atm1Imx Tnfrsf1btm1Imx) and WT mice (B6;129SF2/J) were purchased from The Jackson Laboratory (Bar Harbor, ME) for breeding. For primary myoblast cultures, hindlimb muscles were collected from 3- to 4-day-old p55−/−p75−/− or WT mice after euthanization. For muscle overexpression, infectious particles (1.5 × 109) of Ad5 cytomegalovirus encoding MKK6bE (27), a constitutively active mutant of mitogen-activated protein kinase kinase (MKK)6 (a gift from Dr. J. Han of Scripps Research Institute, La Jolla, CA), or green fluorescence protein (GFP) cDNA (prepared by The Vector Development Core of Baylor College of Medicine) in 10 μl of PBS were injected into the hindlimb of 3- to 4-day-old p55−/−p75−/− mice longitudinally in the region of the soleus. Subsequently, the mice were allowed to grow to 6 wk of age, and 100 μl of 10 μM cardiotoxin (Sigma-Aldrich) were dissolved in PBS and injected into the soleus to induce necrotic injury. At various time points, solei were collected from euthanized mice for biochemical and histology studies.
Cell cultures
Murine C2C12 cells (American Type Culture Collection) were cultured in growth medium (GM; DMEM supplemented with 10% fetal bovine serum and gentamicin) at 37°C under 5% CO2. At 85% confluence, cell differentiation was induced by replacing the preceding medium with differentiation medium (DM; DMEM supplemented with 4% heat-inactivated horse serum and gentamicin). A TNF-α-neutralizing antibody (R&D Systems) was included in DM at 5 μg/ml, when indicated, to block TNF-α signaling (38). This antibody neutralizes 0.025 μg/ml TNF-α at an ND50 of 0.02–0.08 μg/ml. Mouse recombinant TNF-α (Roche Applied Science) was added to differentiation medium as indicated and replenished at 12-h intervals. Primary myoblasts were isolated from 3- to 4-day-old p55−/−p75−/− or WT mice, as previously described (36), with minor modifications. Briefly, hindlimb skeletal muscles were excised, subsequently minced with razor blades in PBS, and enzymatically dissociated in dissociation buffer (0.1% trypsin, 0.1% collagenase type 2, and 0.025% DNase in PBS) at 37°C for 10 min. The dissociation buffer with released cells was collected and mixed with the same volume of DMEM-F-12 medium (Invitrogen) supplemented with 20% fetal bovine serum. Cells were pelleted by centrifugation at 300 g for 5 min at 4°C. The dissociation process was repeated two times. Collected cells were then resuspended in 1.082 g/ml Percoll (GE Healthcare) and subjected to a Percoll density gradient (1.050, 1.060, and 1.082 g/ml) purification procedure by centrifugation at 2,000 g for 25 min at room temperature. The Percoll gradient was adjusted with a buffer containing 6.8 g/l NaCl, 0.4 g/l KCl, 0.1 g/l MgSO4, 1.5 g/l NaH2PO4, 1.0 g/l dextrose, and 4.76 g/l HEPES (pH 7.3). After centrifugation, the band containing myocytes at the interface between the 1.060 and 1.082 g/ml Percoll layers was collected and resuspended in Ham’s F-10 nutrient mixture (Invitrogen) supplemented with 20% fetal bovine serum, 3% chicken embryo extract, and gentamicin. Cells were then plated in Matrigel (BD Biosciences/BD)-coated dishes and grown in the presence of 5% CO2. After 1 or 2 days, cells were released by mild trypsinization and preplated in noncoated dishes for 30 min to remove contaminating fibroblasts. The unattached cells were replated in Matrigel-coated dishes and grown at 37°C in growth medium (Ham’s F-10 nutrient mixture supplemented with 20% fetal bovine serum, 3% chicken embryo extract, and gentamicin) in the presence of 5% CO2. This replating procedure was repeated once. Primary myoblast differentiation was induced by shifting cells to differentiation medium [Ham’s F-10 nutrient mixture-DMEM, 1:1 vol/vol (supplemented with 5% heat-inactivated horse serum and gentamicin)] when cells reached ~60% confluence. The purity of the myoblast culture was verified as >90% through immunoperoxidase labeling (avidin-biotin complex and diaminobenzidine kits, Vector Laboratories) with the D3 desmin antibody (Developmental Studies Hybridoma Bank, University of Iowa, Iowa City, IA).
Determination of TNF-α concentration in culture medium
TNF-α concentration in DM and GM was determined by use of an ELISA kit (R&D Systems) according to the manufacturer’s protocol, after the medium was concentrated with a spin concentrator from Millipore (10K pore size).
Western blot analysis
Western blot analysis was performed as previously described (13), using either protein extracts or lysates prepared from cells or muscle. Antibodies for pan- and phosphorylated p38 (T181/Y182), ERK1/2 (T202/Y204), JNK (T183/Y185), and Akt (S473) were purchased from Cell Signaling Technology, and antibodies for pan-MEF2C, phosphorylated MEF2C (S387), and p21 were obtained from Santa Cruz Biotechnology. Myogenin (F5D), MHC (MF20), and embryonic MHC (F1.652) antibodies were obtained from the Development Studies Hybridoma Bank. The antibody against TNF-α was from Pierce Biotechnology, and the antibody against hemagglutinin (HA) was from Covance Research Products. Corresponding protein bands were quantified densitometrically and analyzed by ImageQuant software (GE Healthcare). Protein concentrations of the samples were determined using the Bio-Rad protein assay (Bio-Rad Laboratories).
Histology studies
Solei collected from mice were fixed in 4% formaldehyde, and paraffin sections were made and processed for hematoxylin and eosin staining by the Baylor Histology Service. Images of stained muscle sections were acquired using MetaVue computer software and a Zeiss Axioplan 2 microscope coupled to a Photometrics CoolSNAP charge-coupled device camera with a ×20 objective lens; images were edited using Adobe Photoshop software. Soleus myofiber cross-sectional area (XSA) was measured using ImageJ software (National Institutes of Health, Bethesda, MD) as described previously (13).
Statistics
Values were expressed as means ± SE. One-way ANOVA or Student’s t-test was used for comparisons, as indicated, using SigmaStat software. Differences were regarded as significant at a level of P < 0.05. When a significant difference was found by ANOVA, a multiple-comparison test was then performed, as indicated, to evaluate the difference between the groups.
RESULTS
Myoblasts release TNF-α to activate p38 and myogenesis
As an early signal of myogenesis, p38 is activated during the early stages of myoblast differentiation (68). On the basis of our previous observation that expression of MHCf during the early stages of myoblast differentiation is blocked by neutralizing TNF-α in the culture medium (38), we hypothesized that, on differentiation, myoblasts release TNF-α to activate myogenesis via the activation of p38. To test this hypothesis, we measured TNF-α concentration in the culture medium of C2C12 myoblasts induced to differentiate by a switch from serum-rich GM to low-serum DM. As shown in Fig. 1A, a very low level of TNF-α was detected in GM and DM at 0 h, which is at least partially due to the TNF-α present in the serum added to the medium. An increase in TNF-α concentration was observed in DM as early as at 3 h of differentiation, and the increase lasted to at least 48 h. During the same time period, TNF-α concentration in the GM incubated with control C2C12 myoblasts did not increase significantly. These data indicate that differentiating C2C12 myoblasts increase release of TNF-α. A more potent increase in TNF-α concentration was observed in the DM incubated with mouse primary myoblasts. At the same time, a smaller but statistically significant increase in TNF-α concentration in the GM incubated with control primary myoblasts was observed (Fig. 1B). These results verified that differentiating myoblasts strongly upregulate TNF-α release.
Fig. 1.

Differentiating myoblasts increase TNF-α release. Differentiation of C2C12 (A) or mouse primary myoblasts (B) was induced by switching from growth medium (GM) to differentiation medium (DM). At the same time, control cells were changed into fresh GM. At 0, 3, 10, 24, and 48 h, the media were collected and concentrated. TNF-α concentration was determined by ELISA. Two independent experiments were carried out for each type of myoblasts. Data within the same treatment were analyzed with ANOVA, and a difference is indicated next to the curve. The Fisher’s least-significant difference (LSD) multiple-comparison test was performed to compare TNF-α concentration at individual time points with 0 h within the same treatment (difference: *P < 0.01). Student’s t-test was performed to compare TNF-α concentration in DM and GM at each time point (difference: +P < 0.05).
To determine whether C2C12 myoblast-released TNF-α is critical to p38 activation, we evaluated the effect of a TNF-α-neutralizing antibody added to DM on p38 activation at 5 μg/ml, a concentration that was previously shown to block TNF-α stimulation of differentiation (38). As expected, Western blot analysis of the cell extracts with an antibody specific to phosphorylated p38 detected a dramatic activation of p38 at 24 and 48 h of differentiation, while total levels of p38 remained constant. On the other hand, the activation of p38 in DM was blocked by the TNF-α-neutralizing antibody present, whereas the preimmune IgG used as a control did not affect p38 activation (Fig. 2). Simultaneously, we observed a strong activation of Akt. However, Akt activity was not affected by the TNF-α-neutralizing antibody. In addition, ERK1/2 activity had a small but statistically significant increase, while JNK activity remained unchanged, and the TNF-α-neutralizing antibody did not alter the activity of these kinases (Fig. 2). These results indicate that p38 activation during serum restriction-induced differentiation is specifically mediated by TNF-α released from myoblasts and that the level of TNF-α required for p38 activation is in the lower picograms per milliliter range.
Fig. 2.

Myoblast-released TNF-α is critical to p38 activation in differentiating C2C12 myoblasts. Differentiation of C2C12 myoblasts was induced by replacing the GM with DM. TNF-α-neutralizing antibody (5 μg/ml) or preimmune IgG (pre-immun IgG; 5 μg/ml) was included in the DM, as indicated. At the indicated times, myoblasts were collected and processed for Western blot analysis with antibodies (Ab) against phosphorylated or pan-p38, Akt, ERK1/2, and JNK. Representative blots are shown for each of the kinases and were derived from 3 independent experiments. Kinase activation, normalized against the relevant total protein, was quantified by measuring the optical density of phosphorylated kinases on the X-ray film. Data are expressed as x-fold of the level at 0 h. Means ± SE (n = 3) were analyzed via ANOVA (P < 0.05), followed by the Fisher’s LSD multiple-comparison test. +Difference from 0 h (P < 0.05). *Difference from the control within the same time groups (24 or 48 h) (P < 0.05).
To evaluate whether TNF-α signaling is required for p38-mediated activation of differentiation markers, we analyzed the effect of neutralization of TNF-α on differentiation markers, including the activation of MEF2C, a prominent member of the MEF2 family that promotes myogenesis (43), and the expression of myogenin and p21. Similar to p38 activation, the TNF-α-neutralizing antibody blocked MEF2C activation at 24 and 48 h of differentiation without affecting the total levels of MEF2C (Fig. 3). Myogenin expression was severely attenuated by the antibody at 24 h and reached a level similar to the time-matched control at 48 h, indicating that myogenin expression was delayed (Fig. 3). Although the relatively low level of p21 expression was not affected by the antibody at 24 h of differentiation, the surge of p21 expression at 48 h was completely blocked by this antibody (Fig. 3). To determine whether the compromise of the differentiation markers resulted in impaired myogenic differentiation, we measured MHC expression by Western blot analysis. The presence of the TNF-α-neutralizing antibody attenuated MHC expression at 24 and 48 h of differentiation (Fig. 3). These results indicate that myoblast-released TNF-α is critical for the activation of myogenesis.
Fig. 3.

Myoblast-released TNF-α is critical to the activation of myogenesis in C2C12 myoblasts. Differentiation of C2C12 myoblasts was induced as described in Fig. 1. Myoblasts were collected at the indicated times and processed for Western blot analysis using antibodies against phosphorylated myocyte enhancer factor (MEF)-2C and myogenin, p21, and myosin heavy chain (MHC). Levels of these proteins were normalized against MEF2C or β-actin. Representative blots from 3 independent experiments are shown. Data were expressed and analyzed as described in Fig. 2.
Conversely, we tested the effect of exogenous TNF-α added to DM on p38 activation and myogenesis. Considering that TNF-α is known to have opposing bimodal effects in skeletal muscle depending on concentration (1), and that high levels of TNF-α (10–20 ng/ml) have been shown to inhibit myogenesis at 48–72 h of differentiation (24, 33), we tested the effect of mouse recombinant TNF-α ranging from 0.05 to 5 ng/ml at 48 h of C2C12 myoblast differentiation to simulate physiological as well as pathological conditions. While exogenously added TNF-α stimulated p38 activity (Fig. 4, left) over the entire concentration range, it stimulated the expression of myogenin (Fig. 4, middle) and MHC (Fig. 4, right) at 0.05 ng/ml but inhibited the expression of these differentiation markers at 0.5 and 5 ng/ml. It should be noted that, in the 48-h control, p38 activation and expression of myogenin and MHC are driven by endogenously released TNF-α, even though there was no exogenously added TNF-α, and that when exogenous TNF-α was added, the actual TNF-α concentrations are the sum of exogenous and endogenous TNF-α. These data indicate that TNF-α stimulates as well as inhibits myogenesis in a dose-dependent fashion. Therefore, TNF-α has bimodal effects on myogenesis depending on concentration: at low concentrations simulating physiological conditions, it stimulates myogenesis, and at high concentrations simulating pathological conditions, it inhibits myogenesis.
Fig. 4.

Exogenously added TNF-α exerts bimodal effects on myogenesis, depending on concentration. C2C12 myoblasts were induced to differentiation in DM with or without mouse recombinant TNF-α added at indicated concentrations. Myoblasts were collected at 48 h of differentiation and processed for Western blot analysis of phosphorylated p38, myogenin, and MHC. Levels of these proteins are normalized to pan-p38 or β-actin. Data from 3 independent experiments were analyzed by ANOVA (P < 0.05) combined with the Fisher’s LSD multiple-comparison test. Differences among the data points (P < 0.05) are indicated: a > b > c > d > e.
TNF-α has two distinct plasma membrane receptors known as p55 and p75 (22). To evaluate whether myogenic activation of p38 is mediated by TNF-α receptors, we prepared primary myoblasts from WT and p55−/−p75−/− mice. We chose to use myoblasts from 3- to 4-day-old neonatal mice rather than satellite cells from adult mice because adult satellite cells are quiescent and require about a week of time to reenter the cell cycle in culture medium richly supplied with growth factors. Once the cell cycle is initiated, the transition to myogenic differentiation, which is the focus of this study, in satellite cells is not different from that in neonatal myoblasts (11). A potent activation of p38 was observed in WT myoblasts at 48 h after switching to DM; however, p38 activation was blunted in p55−/−p75−/− myoblasts (Fig. 5). Meanwhile, the activity of ERK1/2 and JNK increased in both WT and p55−/−p75−/− myoblasts similarly. On the other hand, Akt activity appeared to be increased in WT as well as p55−/−p75−/− myoblasts, but the increase did not reach statistical significance (Fig. 5). Consequently, MHC expression was attenuated in p55−/−p75−/− myoblasts (Fig. 6). These data indicate that myogenic activation of p38 requires TNF-α receptor-mediated signaling.
Fig. 5.

TNF-α receptor-mediated signaling is essential to myogenic activation of p38. Primary myoblasts were isolated from 3- to 4-day-old wild-type (WT) or p55−/−p75−/− double-knockout (KO) mice. Differentiation was induced by replacing GM with DM. Myoblasts were collected at 48 h of differentiation. Activation of p38, Akt, ERK1/2, and JNK were analyzed as described in Fig. 2. Representative blots from 2 independent experiments are shown. Data are expressed as x-fold of the level at 0 h. Means ± SE were analyzed via ANOVA (P < 0.05) followed by the Fisher’s LSD multiple-comparison test. +Difference from WT at 0 h (P < 0.05). *Difference from WT within the same time groups (0 or 48 h) (P < 0.05).
Fig. 6.

TNF-α receptor-mediated signaling is essential to myogenic gene expression. Primary myoblasts from 3- to 4-day-old WT or p55−/−p75−/− (KO) mice were prepared, differentiated, and collected at 48 h of differentiation. MHC expression was determined by Western blot analysis and normalized to β-actin. A representative blot derived from 2 independent experiments is shown. Data were expressed and analyzed as described in Fig. 5.
Regenerating muscle increases TNF-α release
We previously showed that there is a TNF-α-dependent p38 activation over the course of regeneration in cardiotoxin-injured mouse soleus muscle (13). Although it is well established that injured muscle upregulates TNF-α expression (14, 18, 31, 57, 65, 69), whether injured muscle increases release of TNF-α so that it can activate p38 and myogenesis has not been determined. Utilizing Western blot analysis, we examined levels of both membrane-bound pro-TNF-α (26 kDa) and released TNF-α (17 kDa) in the cardiotoxin-injured mouse soleus. As shown in Fig. 7, we observed a rise in released TNF-α as well as pro-TNF-α over the course of regeneration. TNF-α release peaked on day 3 postinjury (~7.5-fold increase), a time at which myogenesis is being initiated (26). This result indicates that injured muscle indeed increases TNF-α release, and the time course of TNF-α release is consistent with our hypothesis concerning its role in myogenesis.
Fig. 7.
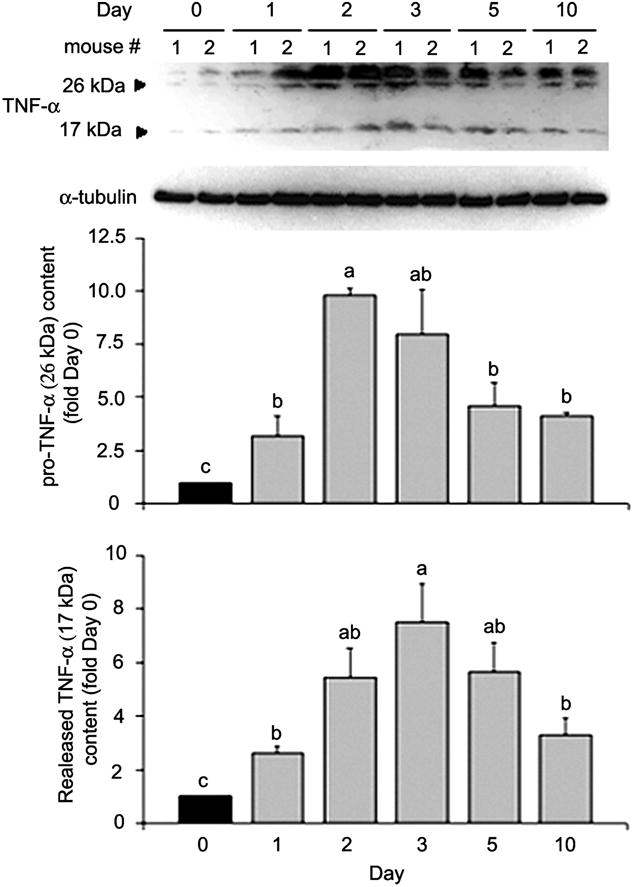
TNF-α release is increased in cardiotoxin-injured muscle. Cardiotoxin was injected into the soleus muscle of male adult mice (8 wk of age). Solei were collected before (day 0) or after injection at indicated times. Muscle lysates were analyzed by Western blot analysis with antibody against TNF-α, with antibody against α-tubulin as loading control. Optical density of TNF-α detected was normalized to α-tubulin and analyzed by ANOVA (P < 0.05 for both forms of TNF-α). Data were analyzed and expressed as described in Fig. 4; “ab” denotes that the value is not different from either a or b.
Forced activation of p38 rescues impaired myogenesis in p55−/−p75−/− soleus
Previously, we demonstrated that myogenesis in the cardiotoxin-injured soleus is impaired in p55−/−p75−/− mice (13). Notwithstanding, it remained unclear whether the impairment in myogenesis was due to the deficiency in p38 activation. Thus, in the present study, we evaluated whether forced activation of p38 can rescue impaired myogenesis in p55−/−p75−/− mice by overexpression of MKK6bE (27), a constitutively active mutant of MKK6 that mediates p38 activation by TNF-α (6). We chose the adenovirus vector (Adv) for the overexpression of MKK6bE because electric pulse-mediated delivery of plasmids causes muscle injury and regeneration (4), which could interfere with our experiment. To do so, we adopted a published protocol that allows sufficient adenovirus transduction in adult mouse skeletal muscle (20). An adenoviral construct encoding HA-tagged MKK6bE (a gift from Dr. Jiahuai Han of Scripps Research Institute) (27) was injected into the hindlimb of p55−/−p75−/− mouse pups; another adenoviral construct that encoded untagged GFP was injected to another group of pups as the control. At 6 wk of age, MKK6bE expression in soleus was confirmed by Western blot analysis of HA, as shown in Fig. 8A. Lower levels of phosphorylated p38 were observed in the p55−/−p75−/− and GFP-overexpressing p55−/−p75−/− soleus compared with the WT soleus. Overexpressed MKK6bE raised the level of phosphorylated p38 in the p55−/−p75−/− soleus, but not so high as to cause concern about unintended effects (Fig. 8B).
Fig. 8.

Adenovirus-mediated expression of mitogen-activated protein kinase kinase (MKK)6bE activates p38 in p55−/−p75−/− soleus. Adenovirus constructs encoding hemagglutin (HA)-tagged MKK6bE (Adv-MKK6bE) or untagged green fluorescence protein (Adv-GFP) cDNA were injected into the hindlimb muscle of 3- to 4-day-old p55−/−p75−/− mice (with 3 mice in each group). Soleus of the injected leg was collected when mice were 6 wk of age. A: adenovirus-mediated MKK6bE expression in the soleus muscle. MKK6bE expression was determined by analysis of HA levels in soleus extract via Western blot analysis using an antibody against HA. Level of β-actin was monitored as loading control. B: MKK6bE activates p38 in p55−/−p75−/− soleus. The activated form of p38 in virus-infected soleus was compared with that in uninfected soleus by Western blot analysis. Level of phosphorylated p38 was normalized to that of total p38 and analyzed by ANOVA (P < 0.05). The Fisher’s LSD multiple-comparison test was performed to evaluate the differences (P < 0.05) among groups: a > b > c.
To evaluate whether MKK6bE activation of p38 rescues myogenesis in regenerating muscle of p55−/−p75−/− mice, cardiotoxin was injected into the soleus of 6-wk-old WT, p55−/−p75−/−, and p55−/−p75−/− mice that overexpressed either MKK6bE or GFP to compare their effects on myogenesis. Consistent with our previous findings (13), we observed compromised p38 activation in the injured p55−/−p75−/− soleus at a time when myogenesis was underway (day 3 after cardiotoxin injection), accompanied by defective expression of myogenin and p21 (Fig. 9). Since regenerating muscle expresses embryonic/developmental forms of MHC (67), we further examined embryonic MHC expression in these soleus samples. A robust expression of embryonic MHC was seen in the WT but not in the p55−/−p75−/− soleus (Fig. 9). Conversely, the forced expression of MKK6bE activated p38 and restored the expression of myogenin, p21, and embryonic MHC in the p55−/−p75−/− soleus. However, GFP expression did not demonstrate these effects (Fig. 9). Although we examined activation of p38 and expression of the differentiation markers in total muscle extracts, the expression of the differentiation markers myogenin, p21, and embryonic MHC can only result from myogenic differentiation of satellite cells; therefore, we conclude that p38 in satellite cells was indeed activated by the overexpressed MKK6bE. Our findings indicate that TNF-α-mediated p38 activation is necessary and sufficient for the initiation of myogenesis in regenerating muscle.
Fig. 9.

MKK6bE overexpression in p55−/−p75−/− soleus restores myogenesis. Adeno-virus constructs encoding MKK6bE or GFP cDNA were injected into the hindlimb muscle of 3- to 4-day-old p55−/−p75−/− mice. At 6 wk of age, cardiotoxin was injected into soleus of these mice as well as WT and p55−/−p75−/− mice that were not infected with adenovirus. Soleus was collected 3 days after the cardiotoxin injection. Soleus extracts were analyzed for p38 activation and the expression of differentiation markers (i.e., myogenin, p21, and MHC) via Western blotting. The activation of p38 is expressed as the ratio of the optical density of phosphorylated p38 to that of pan-p38. Expression of the differentiation markers is conveyed as the optical density of the markers normalized to that of β-actin in arbitrary units. Data were analyzed via ANOVA followed by the Fisher’s LSD multiple-comparison test. Differences (P < 0.05) are indicated: a > b > c.
Forced activation of p38 rescues impaired regeneration in p55−/−p75−/− soleus
Previously, we demonstrated morphologically that muscle regeneration in cardiotoxin-injured soleus is impaired in TNF-α receptor double-knockout mice (p55−/−p75−/−), as evidenced by delayed myogenesis, sustained inflammation, severe calcification, and the death of muscle fibers (13). However, it remained unclear whether the impairment in regeneration is due to the deficiency in p38 activation. Thus, in the present study, one of our objectives was to evaluate whether forced activation of p38 can rescue impaired muscle regeneration in p55−/−p75−/− mice by overexpressing MKK6bE. To evaluate whether forced p38 activation rescues impaired regeneration, soleus sections before and 5 or 12 days after cardiotoxin injection were stained by hematoxylin and eosin to examine the muscle morphology (Fig. 10). Similar to our previous report (13), we observed signs of both injury and regeneration in WT soleus on day 5 after cardiotoxin injection, including infiltration of inflammatory cells and newly regenerated myofibers with centralized nuclei; on day 12, muscle architecture in WT soleus recovered nearly completely. On the other hand, p55−/−p75−/− soleus overexpressing GFP appeared similar to the p55−/−p75−/− soleus in the previous report, displaying more severe infiltration with calcium deposit on day 5 and sustained infiltration and calcified myofibers on day 12, indicating impaired regeneration. In dramatic contrast, p55−/−p75−/− soleus overexpressing MKK6bE displayed normal regeneration patterns that resembled those of WT on day 5 and day 12. To evaluate the efficiency of regeneration quantitatively, XSA of the myofibers on day 12 was compared with that on day 0 (in percentage) in each treatment. The mean XSA of WT myofibers was 90.0%, the mean XSA of myofibers in p55−/−p75−/− soleus that received Adv-GFP was 50.7% (P < 0.05 compared with WT), and the mean XSA of myofibers in p55−/−p75−/− soleus that received Adv-MKK6bE recovered to 79.7% (P < 0.05 compared with p55−/−p75−/− soleus that received Adv-GFP, as analyzed by ANOVA and Holm-Sidak multiple comparison test). These data indicate that forced activation of p38 rescues impaired regeneration in p55−/−p75−/− soleus. Therefore, deficiency in p38 activation is the cause of the impairment of muscle regeneration taking place in the absence of TNF-α signaling.
Fig. 10.
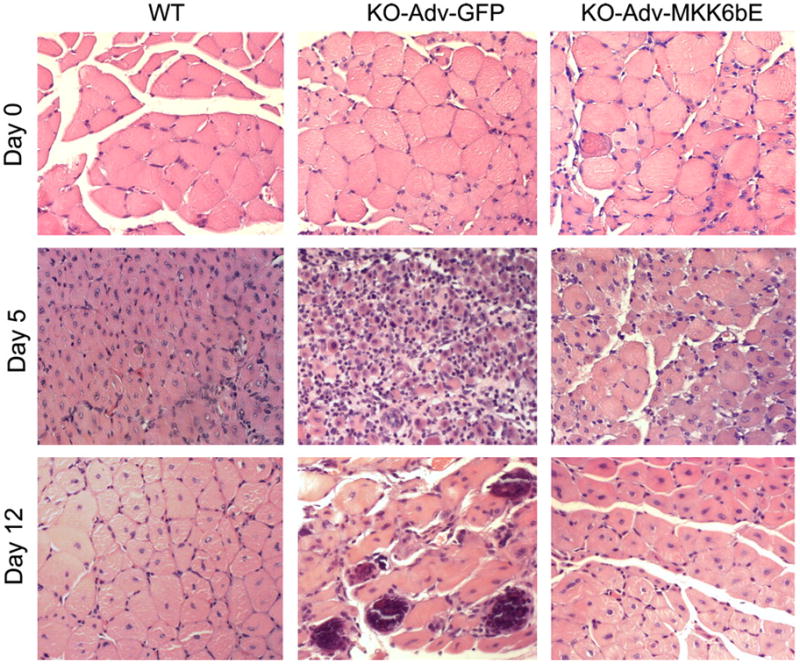
Forced activation of p38 restores muscle regeneration in p55−/−p75−/− soleus. Adenovirus constructs encoding MKK6bE or GFP cDNA were injected into the hindlimb muscle of 3- to 4-day-old p55−/−p75−/− (KO) mice. At 6 wk of age, cardiotoxin was injected into the soleus of these mice as well as WT mice. Soleus was collected before (day 0) and 5 or 12 days after cardiotoxin injection. Paraffin-embedded sections were prepared and stained with hematoxylin and eosin.
DISCUSSION
The present study demonstrates that differentiating myoblasts and injured muscle increase the release of TNF-α as a key mediator of p38 activation to induce myogenesis, and deficiency in TNF-α-mediated p38 activation causes impaired myogenesis and regeneration in adult muscle. These data depict TNF-α as a key regulator of myogenesis and muscle regeneration.
We have observed for the first time that differentiating myoblasts increase release of TNF-α, providing direct evidence that TNF-α is an autocrine factor associated with myogenic differentiation. The significantly more potent release of TNF-α by differentiating primary myoblasts compared with C2C12 is not surprising, considering that primary myoblasts replicate the properties of muscle cells more closely than C2C12 myoblasts and that myogenic responses including the expression of α-actin are much more potent in primary myoblasts than in C2C12 myoblasts (2, 5). The higher level of TNF-α release from primary myoblasts in GM (constitutive release), which reached >50 pg/ml, a level sufficient to activate p38 as shown in the present study, provides an explanation for the strong capacity of primary myoblasts to differentiate spontaneously without serum restriction.
By neutralizing TNF-α released into DM, we have demonstrated that, as an autocrine factor, TNF-α is critical for p38 activation during the early stages of myoblast differentiation, the ensuing activation of key mediators of differentiation including MEF2C, myogenin and p21, and the expression of such muscle-specific genes as MHC. The inability of p55−/−p75−/− myoblasts to activate p38 and myogenesis in DM indicates that these activities are indeed mediated by TNF-α receptor-mediated signaling. The complementary use of TNF-α-neutralizing antibody and p55−/−p75−/− myoblasts in blocking TNF-α signaling also rules out the possibility that the TNF-α-dependent activation of p38 observed is due to the potential nonspecificity of the antibody or the genetic background of the knockout mice. These results confirm the myogenic cell basis for the dependence of p38 activation and myogenesis on the TNF-α receptor-mediated signaling observed previously in intact muscle (13). In addition, these results indicate that endogenous TNF-α activates p38 and myogenesis at levels as low as the lower picograms per milliliter range.
The time course of TNF-α-dependent MEF2C activation is highly consistent with that of p38 activation, apparently because p38 directly phosphorylates MEF2C (43). Expression of myogenin was delayed by neutralization of TNF-α, indicating that TNF-α is a key upstream signal for the activation of myogenin expression via p38. Residual p38 activity or the presence of a redundant mechanism appears to activate myogenin expression at a later time. On the other hand, the bulk of p21 expression that takes place between 24 and 48 h of differentiation requires TNF-α signaling. Consequently, the blockade or delay in the activation of these differentiation markers attenuates MHC expression. These data indicate that myoblasts release TNF-α as a key upstream signal for myogenesis via the activation of p38.
We observed that Akt is activated in differentiating C2C12 myoblasts, which is consistent with a previous observation in differentiating C2C12 myoblasts (7), in a TNF-α-independent manner. Given that Akt also plays a critical role in myogenesis (35), its activation during myogenesis is expected, although, because of the larger standard error, the increase in Akt phosphorylation in primary myoblasts was not statistically significant (Fig. 5). Akt is known to be activated by such autocrine factors as IGF-I, which is released during myoblast differentiation (47). The observation that the TNF-α-neutralizing antibody blocks p38 activation and myogenesis while Akt phosphorylation is normally activated is consistent with the previous observations that Akt phosphorylation alone is unable to induce myogenesis (60, 68). Conversely, the observation that differentiation-induced Akt activation was not further increased when TNF-α signaling was blocked indicates that TNF-α may not inhibit IGF-I-mediated signaling under physiological conditions.
ERK1/2 mediates the growth factor stimulation of satellite cell proliferation (15, 25, 28), whereas the role of JNK in myogenesis has not been well defined, and both inhibitory and stimulatory effects have been reported (30, 44, 50). The observed activation of ERK1/2 and JNK during differentiation suggests that they may participate in the differentiation process in some way. The literature on the response of ERK1/2 and JNK activity to differentiation in C2C12 myoblasts is mixed, from decreased to unchanged to increased (3, 7, 23, 68), while we are not aware of any previous data on the response of these kinases in differentiating primary myoblasts. Although TNF-α is capable of activating ERK1/2 and JNK (37), the TNF-α-independent nature of ERK1/2 and JNK activation observed in the present study indicates that TNF-α is not a major influence on their activity during differentiation.
The observation that TNF-α has bimodal effects on myogenesis resolves a controversy in the literature. There have been reports that described an inhibitory effect of exogenously added TNF-α on myogenesis (24, 32, 33). The concentration of exogenously added TNF-α used in the above-referenced studies ranged from 10 to 20 ng/ml in culture medium, at least 1,000-fold higher than the physiological concentration of TNF-α in normal serum. As a pleiotropic cytokine, TNF-α is known to exert divergent actions, depending on concentration. Opposing effects of TNF-α at different concentrations have been observed in skeletal (1) and cardiac muscle (42). In normal muscle, the level of TNF-α is estimated in the range of several picograms per milliliter (21). Consistent with the observation that differentiating myoblasts increase the release of TNF-α, we have demonstrated in the present study that cardiotoxin-injured soleus increases the release of TNF-α over the course of regeneration, and that the release reaches its peak on day 3 postinjury, when myogenic activity is being initiated. The 7.5-fold peak level increase in TNF-α release after injury gives rise to an estimated level of released TNF-α of ~0.05 ng/ml, which is sufficient to activate p38, as shown in the present study. The 0.05 ng/ml recombinant TNF-α used to simulate the physiological TNF-α level in injured muscle further stimulated myogenesis in myoblasts on top of the endogenous TNF-α. On the other hand, at 0.5 or 5 ng/ml, levels seen in pathological conditions (45, 64), TNF-α inhibited myogenesis. These data indicate that whether TNF-α stimulates or inhibits myogenesis is dependent on concentration. In addition to the concentration-dependent divergence, the TNF-α effect on myogenesis is also known to be temporally divergent. We previously showed that the effect of exogenously added TNF-α on myogenesis is dependent on the differentiation stage. It simulates MHCf expression during the early stages of differentiation but inhibits MHCf expression at late stages of differentiation (38). Similarly, p38 has such temporal divergence in its effect on myogenesis. Although p38 activation during the early stages of myogenesis is essential to the initiation of myogenesis, p38-mediated phosphorylation inhibits myogenesis at late stages of myogenesis (56, 66). The studies that observed an inhibitory effect of TNF-α on myoblast differentiation only looked at the effect of pathological levels of TNF-α at late stages of myogenesis (24, 32, 33), which precludes the observation of the effect of TNF-α at physiological levels on the initiation of myogenesis. Therefore, we conclude that the transient increase of TNF-α release during muscle regeneration stimulates myogenesis. On the other hand, an unregulated and sustained increase of TNF-α release due to pathological conditions would impact myogenesis negatively, which may contribute to the muscle atrophy associated with inflammatory diseases.
Our data indicate that a physiological level of TNF-α activates both p38 and myogenesis, whereas a pathological level of TNF-α activates p38 but inhibits myogenesis; the latter effect may be attributable to TNF-α activation of certain signaling pathways that inhibit myogenesis at high concentrations. For example, at pathological concentrations, TNF-α induces loss of MyoD mRNA (24) and stimulates protein degradation by upregulating the ubiquitin ligase atrogin-1/MAFbx (37), which is expressed at around 48 h of differentiation and mediates the degradation of MyoD (61). These effects would inhibit myogenesis despite the activation of p38.
The identification of p38 as an essential signal for myogenic differentiation has been based on studies in cultured myoblasts (8, 16, 46, 48, 68, 70) and mouse embryos (17). To our knowledge, however, there have been no data regarding whether p38 is critical to myogenesis during adult muscle regeneration. We have demonstrated that forced activation of p38 by MKK6bE restores impaired myogenesis in the injured p55−/−p75−/− soleus. Although these analyses were done in muscle extracts, the activations of differentiation markers we measured are events taking place exclusively in satellite cells. These data show, for the first time, that TNF-α-mediated p38 activation is necessary and sufficient for myogenesis in adult muscle. Furthermore, we show here that forced p38 activation by MKK6bE rescues the abnormal morphology in the cardiotoxin-injured p55−/−p75−/− soleus, indicating that the lack of p38 activation is indeed the cause of impaired muscle regeneration in the p55−/−p75−/− soleus. Combining our in vitro and in vivo data, we conclude that a timely activation of p38 in satellite cells following muscle injury is crucial for muscle regeneration. Missing this window of time for p38 activation during the early phase of regeneration will irreversibly impair muscle regeneration.
The fact that the p55−/−p75−/− mice develop seemingly normal muscle suggests that there is a redundant mechanism that activates p38 during myogenesis, particularly in the developmental stage. For example, amphoterin (HMGB1) has been shown to stimulate myogenesis in rat L6 myoblasts by activating p38 through engaging the receptor for advanced glycation end products (RAGE), whose expression is developmentally regulated (54), although a dependence of myogenic activation of p38 on this pathway has not been demonstrated in vivo. Whereas myogenesis during embryonic growth is controlled internally by the developmental program, adult muscle regeneration is triggered by external stimuli, including injury, disease, or training. The two processes also transpire in different anatomical environments. A unique feature of muscle regeneration is that inflammation plays a critical role (58, 59). It is possible that, as a central inflammatory mediator, TNF-α may be more critical for muscle regeneration than for muscle development.
In summary, the present study identifies TNF-α as a key regulator of myogenesis and muscle regeneration through its activation of p38. Because of the implications of elevated circulating TNF-α generated by inflammatory diseases in cachectic muscle wasting, inflammatory myopathies, and insulin resistance (41, 49), TNF-α is traditionally viewed as a purely pathological factor in skeletal muscle. Now, the time has come to recognize the other side of TNF-α, as a physiological regulator critical to muscle regeneration. This role of TNF-α also calls for restraint in the long-term use of anti-TNF-α reagents in the treatment of inflammatory diseases to avoid their potential harmful effect on muscle maintenance.
Acknowledgments
We thank J. Han of the Scripps Research Institute for providing the adenovirus construct of MKK6bE and E. Rabinovsky of the Baylor College of Medicine for critical comments.
GRANTS
This work was supported by National Institute of Arthritis and Musculo-skeletal and Skin Diseases Grant AR-049022.
References
- 1.Alvarez B, Quinn LS, Busquets S, Lopez-Soriano FJ, Argiles JM. Direct effects of tumor necrosis factor alpha (TNF-alpha) on murine skeletal muscle cell lines. Bimodal effects on protein metabolism. Eur Cytokine Netw. 2001;12:399–410. [PubMed] [Google Scholar]
- 2.Bains W, Ponte P, Blau H, Kedes L. Cardiac actin is the major actin gene product in skeletal muscle cell differentiation in vitro. Mol Cell Biol. 1984;4:1449–1453. doi: 10.1128/mcb.4.8.1449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bennett AM, Tonks NK. Regulation of distinct stages of skeletal muscle differentiation by mitogen-activated protein kinases. Science. 1997;278:1288–1291. doi: 10.1126/science.278.5341.1288. [DOI] [PubMed] [Google Scholar]
- 4.Bertrand A, Ngo-Muller V, Hentzen D, Concordet JP, Daegelen D, Tuil D. Muscle electrotransfer as a tool for studying muscle fiber-specific and nerve-dependent activity of promoters. Am J Physiol Cell Physiol. 2003;285:C1071–C1081. doi: 10.1152/ajpcell.00104.2003. [DOI] [PubMed] [Google Scholar]
- 5.Blau HM, Pavlath GK, Hardeman EC, Chiu CP, Silberstein L, Webster SG, Miller SC, Webster C. Plasticity of the differentiated state. Science. 1985;230:758–766. doi: 10.1126/science.2414846. [DOI] [PubMed] [Google Scholar]
- 6.Brancho D, Tanaka N, Jaeschke A, Ventura JJ, Kelkar N, Tanaka Y, Kyuuma M, Takeshita T, Flavell RA, Davis RJ. Mechanism of p38 MAP kinase activation in vivo. Genes Dev. 2003;17:1969–1978. doi: 10.1101/gad.1107303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cabane C, Coldefy AS, Yeow K, Derijard B. The p38 pathway regulates Akt both at the protein and transcriptional activation levels during myogenesis. Cell Signal. 2004;16:1405–1415. doi: 10.1016/j.cellsig.2004.05.003. [DOI] [PubMed] [Google Scholar]
- 8.Cabane C, Englaro W, Yeow K, Ragno M, Derijard B. Regulation of C2C12 myogenic terminal differentiation by MKK3/p38α pathway. Am J Physiol Cell Physiol. 2003;284:C658–C666. doi: 10.1152/ajpcell.00078.2002. [DOI] [PubMed] [Google Scholar]
- 9.Cantini M, Carraro U. Macrophage-released factor stimulates selectively myogenic cells in primary muscle culture. J Neuropathol Exp Neurol. 1995;54:121–128. doi: 10.1097/00005072-199501000-00014. [DOI] [PubMed] [Google Scholar]
- 10.Cantini M, Massimino ML, Rapizzi E, Rossini K, Catani C, Dalla LL, Carraro U. Human satellite cell proliferation in vitro is regulated by autocrine secretion of IL-6 stimulated by a soluble factor(s) released by activated monocytes. Biochem Biophys Res Commun. 1995;216:49–53. doi: 10.1006/bbrc.1995.2590. [DOI] [PubMed] [Google Scholar]
- 11.Charge SB, Rudnicki MA. Cellular and molecular regulation of muscle regeneration. Physiol Rev. 2004;84:209–238. doi: 10.1152/physrev.00019.2003. [DOI] [PubMed] [Google Scholar]
- 12.Chazaud B, Sonnet C, Lafuste P, Bassez G, Rimaniol AC, Poron F, Authier FJ, Dreyfus PA, Gherardi RK. Satellite cells attract monocytes and use macrophages as a support to escape apoptosis and enhance muscle growth. J Cell Biol. 2003;163:1133–1143. doi: 10.1083/jcb.200212046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen SE, Gerken E, Zhang Y, Zhan M, Mohan RK, Li AS, Reid MB, Li YP. Role of TNF-α signaling in regeneration of cardiotoxin-injured muscle. Am J Physiol Cell Physiol. 2005;289:C1179–C1187. doi: 10.1152/ajpcell.00062.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Collins RA, Grounds MD. The role of tumor necrosis factor-alpha (TNF-alpha) in skeletal muscle regeneration. Studies in tnf-alpha(−/−) and tnf-alpha(−/−)/lt-alpha(−/−) mice. J Histochem Cytochem. 2001;49:989–1002. doi: 10.1177/002215540104900807. [DOI] [PubMed] [Google Scholar]
- 15.Coolican SA, Samuel DS, Ewton DZ, McWade FJ, Florini JR. The mitogenic and myogenic actions of insulin-like growth factors utilize distinct signaling pathways. J Biol Chem. 1997;272:6653–6662. doi: 10.1074/jbc.272.10.6653. [DOI] [PubMed] [Google Scholar]
- 16.Cuenda A, Cohen P. Stress-activated protein kinase-2/p38 and a rapamycin-sensitive pathway are required for C2C12 myogenesis. J Biol Chem. 1999;274:4341–4346. doi: 10.1074/jbc.274.7.4341. [DOI] [PubMed] [Google Scholar]
- 17.de Angelis L, Zhao J, Andreucci JJ, Olson EN, Cossu G, McDermott JC. Regulation of vertebrate myotome development by the p38 MAP kinase-MEF2 signaling pathway. Dev Biol. 2005;283:171–179. doi: 10.1016/j.ydbio.2005.04.009. [DOI] [PubMed] [Google Scholar]
- 18.De Bleecker JL, Meire VI, Declercq W, Van Aken EH. Immunolocalization of tumor necrosis factor-alpha and its receptors in inflammatory myopathies. Neuromuscul Disord. 1999;9:239–246. doi: 10.1016/s0960-8966(98)00126-6. [DOI] [PubMed] [Google Scholar]
- 19.de la Serna IL, Ohkawa Y, Berkes CA, Bergstrom DA, Dacwag CS, Tapscott SJ, Imbalzano AN. MyoD targets chromatin remodeling complexes to the myogenin locus prior to forming a stable DNA-bound complex. Mol Cell Biol. 2005;25:3997–4009. doi: 10.1128/MCB.25.10.3997-4009.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Durbeej M, Sawatzki SM, Barresi R, Schmainda KM, Allamand V, Michele DE, Campbell KP. Gene transfer establishes primacy of striated vs. smooth muscle sarcoglycan complex in limb-girdle muscular dystrophy. Proc Natl Acad Sci USA. 2003;100:8910–8915. doi: 10.1073/pnas.1537554100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Federici M, Hribal ML, Menghini R, Kanno H, Marchetti V, Porzio O, Sunnarborg SW, Rizza S, Serino M, Cunsolo V, Lauro D, Mauriello A, Smookler DS, Sbraccia P, Sesti G, Lee DC, Khokha R, Accili D, Lauro R. Timp3 deficiency in insulin receptor-haploinsufficient mice promotes diabetes and vascular inflammation via increased TNF-alpha. J Clin Invest. 2005;115:3494–3505. doi: 10.1172/JCI26052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fiers W, Beyaert R, Boone E, Cornelis S, Declercq W, Decoster E, Denecker G, Depuydt B, De Valck D, De Wilde G, Goossens V, Grooten J, Haegeman G, Heyninck K, Penning L, Plaisance S, Vancompernolle K, Van Criekinge W, Vandenabeele P, Vanden Berghe W, Van de CM, Vandevoorde V, Vercammen D. TNF-induced intra-cellular signaling leading to gene induction or to cytotoxicity by necrosis or by apoptosis. J Inflamm. 1995;47:67–75. [PubMed] [Google Scholar]
- 23.Gredinger E, Gerber AN, Tamir Y, Tapscott SJ, Bengal E. Mitogen-activated protein kinase pathway is involved in the differentiation of muscle cells. J Biol Chem. 1998;273:10436–10444. doi: 10.1074/jbc.273.17.10436. [DOI] [PubMed] [Google Scholar]
- 24.Guttridge DC, Mayo MW, Madrid LV, Wang CY, Baldwin AS., Jr NF-kappaB-induced loss of MyoD messenger RNA: possible role in muscle decay and cachexia. Science. 2000;289:2363–2366. doi: 10.1126/science.289.5488.2363. [DOI] [PubMed] [Google Scholar]
- 25.Halevy O, Cantley LC. Differential regulation of the phosphoinositide 3-kinase and MAP kinase pathways by hepatocyte growth factor vs. insulin-like growth factor-I in myogenic cells. Exp Cell Res. 2004;297:224–234. doi: 10.1016/j.yexcr.2004.03.024. [DOI] [PubMed] [Google Scholar]
- 26.Hawke TJ, Garry DJ. Myogenic satellite cells: physiology to molecular biology. J Appl Physiol. 2001;91:534–551. doi: 10.1152/jappl.2001.91.2.534. [DOI] [PubMed] [Google Scholar]
- 27.Huang S, Jiang Y, Li Z, Nishida E, Mathias P, Lin S, Ulevitch RJ, Nemerow GR, Han J. Apoptosis signaling pathway in T cells is composed of ICE/Ced-3 family proteases and MAP kinase kinase 6b. Immunity. 1997;6:739–749. doi: 10.1016/s1074-7613(00)80449-5. [DOI] [PubMed] [Google Scholar]
- 28.Jones NC, Fedorov YV, Rosenthal RS, Olwin BB. ERK1/2 is required for myoblast proliferation but is dispensable for muscle gene expression and cell fusion. J Cell Physiol. 2001;186:104–115. doi: 10.1002/1097-4652(200101)186:1<104::AID-JCP1015>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- 29.Jupp OJ, McFarlane SM, Anderson HM, Littlejohn AF, Mohamed AA, MacKay RH, Vandenabeele P, MacEwan DJ. Type II tumour necrosis factor-alpha receptor (TNFR2) activates c-Jun N-terminal kinase (JNK) but not mitogen-activated protein kinase (MAPK) or p38 MAPK pathways. Biochem J. 2001;359:525–535. doi: 10.1042/0264-6021:3590525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Khurana A, Dey CS. Involvement of c-Jun N-terminal kinase activities in skeletal muscle differentiation. J Muscle Res Cell Motil. 2004;25:645–655. doi: 10.1007/s10974-004-7099-1. [DOI] [PubMed] [Google Scholar]
- 31.Kuru S, Inukai A, Kato T, Liang Y, Kimura S, Sobue G. Expression of tumor necrosis factor-alpha in regenerating muscle fibers in inflammatory and non-inflammatory myopathies. Acta Neuropathol (Berl) 2003;105:217–224. doi: 10.1007/s00401-002-0635-4. [DOI] [PubMed] [Google Scholar]
- 32.Langen RC, Schols AM, Kelders MC, Van Der Velden JL, Wouters EF, Janssen-Heininger YM. Tumor necrosis factor-α inhibits myogenesis through redox-dependent and -independent pathways. Am J Physiol Cell Physiol. 2002;283:C714–C721. doi: 10.1152/ajpcell.00418.2001. [DOI] [PubMed] [Google Scholar]
- 33.Langen RC, Schols AM, Kelders MC, Wouters EF, Janssen-Heininger YM. Inflammatory cytokines inhibit myogenic differentiation through activation of nuclear factor-κB. FASEB J. 2001;15:1169–1180. doi: 10.1096/fj.00-0463. [DOI] [PubMed] [Google Scholar]
- 34.Lescaudron L, Peltekian E, Fontaine-Perus J, Paulin D, Zampieri M, Garcia L, Parrish E. Blood borne macrophages are essential for the triggering of muscle regeneration following muscle transplant. Neuromuscul Disord. 1999;9:72–80. doi: 10.1016/s0960-8966(98)00111-4. [DOI] [PubMed] [Google Scholar]
- 35.Li Y, Jiang B, Ensign WY, Vogt PK, Han J. Myogenic differentiation requires signalling through both phosphatidylinositol 3-kinase and p38 MAP kinase. Cell Signal. 2000;12:751–757. doi: 10.1016/s0898-6568(00)00120-0. [DOI] [PubMed] [Google Scholar]
- 36.Li YP. TNF-α is a mitogen in skeletal muscle. Am J Physiol Cell Physiol. 2003;285:C370–C376. doi: 10.1152/ajpcell.00453.2002. [DOI] [PubMed] [Google Scholar]
- 37.Li YP, Chen Y, John J, Moylan J, Jin B, Mann DL, Reid MB. TNF-α acts via p38 MAPK to stimulate expression of the ubiquitin ligase atrogin1/MAFbx in skeletal muscle. FASEB J. 2005;19:362–370. doi: 10.1096/fj.04-2364com. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li YP, Schwartz RJ. TNF-α regulates early differentiation of C2C12 myoblasts in an autocrine fashion. FASEB J. 2001;15:1413–1415. doi: 10.1096/fj.00-0632fje. [DOI] [PubMed] [Google Scholar]
- 39.Lluis F, Ballestar E, Suelves M, Esteller M, Munoz-Canoves P. E47 phosphorylation by p38 MAPK promotes MyoD/E47 association and muscle-specific gene transcription. EMBO J. 2005;24:974–984. doi: 10.1038/sj.emboj.7600528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lundberg IE, Dastmalchi M. Possible pathogenic mechanisms in inflammatory myopathies. Rheum Dis Clin North Am. 2002;28:799–822. doi: 10.1016/s0889-857x(02)00025-x. [DOI] [PubMed] [Google Scholar]
- 42.Mann DL. Stress-activated cytokines and the heart: from adaptation to maladaptation. Annu Rev Physiol. 2003;65:81–101. doi: 10.1146/annurev.physiol.65.092101.142249. [DOI] [PubMed] [Google Scholar]
- 43.McKinsey TA, Zhang CL, Olson EN. MEF2: a calcium-dependent regulator of cell division, differentiation and death. Trends Biochem Sci. 2002;27:40–47. doi: 10.1016/s0968-0004(01)02031-x. [DOI] [PubMed] [Google Scholar]
- 44.Meriane M, Roux P, Primig M, Fort P, Gauthier-Rouviere C. Critical activities of Rac1 and Cdc42Hs in skeletal myogenesis: antagonistic effects of JNK and p38 pathways. Mol Biol Cell. 2000;11:2513–2528. doi: 10.1091/mbc.11.8.2513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nakashima J, Tachibana M, Ueno M, Baba S, Tazaki H. Tumor necrosis factor and coagulopathy in patients with prostate cancer. Cancer Res. 1995;55:4881–4885. [PubMed] [Google Scholar]
- 46.Penn BH, Bergstrom DA, Dilworth FJ, Bengal E, Tapscott SJ. A MyoD-generated feed-forward circuit temporally patterns gene expression during skeletal muscle differentiation. Genes Dev. 2004;18:2348–2353. doi: 10.1101/gad.1234304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Perrone CE, Fenwick-Smith D, Vandenburgh HH. Collagen and stretch modulate autocrine secretion of insulin-like growth factor-1 and insulin-like growth factor binding proteins from differentiated skeletal muscle cells. J Biol Chem. 1995;270:2099–2106. doi: 10.1074/jbc.270.5.2099. [DOI] [PubMed] [Google Scholar]
- 48.Puri PL, Wu Z, Zhang P, Wood LD, Bhakta KS, Han J, Feramisco JR, Karin M, Wang JY. Induction of terminal differentiation by constitutive activation of p38 MAP kinase in human rhabdomyosarcoma cells. Genes Dev. 2000;14:574–584. [PMC free article] [PubMed] [Google Scholar]
- 49.Reid MB, Li YP. Cytokines and oxidative signaling in skeletal muscle cells. Acta Physiol Scand. 2001;171:225–232. doi: 10.1046/j.1365-201x.2001.00824.x. [DOI] [PubMed] [Google Scholar]
- 50.Rousse S, Lallemand F, Montarras D, Pinset C, Mazars A, Prunier C, Atfi A, Dubois C. Transforming growth factor-beta inhibition of insulin-like growth factor-binding protein-5 synthesis in skeletal muscle cells involves a c-Jun N-terminal kinase-dependent pathway. J Biol Chem. 2001;276:46961–46967. doi: 10.1074/jbc.M104440200. [DOI] [PubMed] [Google Scholar]
- 51.Saghizadeh M, Ong JM, Garvey WT, Henry RR, Kern PA. The expression of TNF alpha by human muscle. Relationship to insulin resistance. J Clin Invest. 1996;97:1111–1116. doi: 10.1172/JCI118504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Schafer KA. The cell cycle: a review. Vet Pathol. 1998;35:461–478. doi: 10.1177/030098589803500601. [DOI] [PubMed] [Google Scholar]
- 53.Simone C, Forcales SV, Hill DA, Imbalzano AN, Latella L, Puri PL. p38 pathway targets SWI-SNF chromatin-remodeling complex to muscle-specific loci. Nat Genet. 2004;36:738–743. doi: 10.1038/ng1378. [DOI] [PubMed] [Google Scholar]
- 54.Sorci G, Riuzzi F, Arcuri C, Giambanco I, Donato R. Amphoterin stimulates myogenesis and counteracts the antimyogenic factors basic fibroblast growth factor and S100B via RAGE binding. Mol Cell Biol. 2004;24:4880–4894. doi: 10.1128/MCB.24.11.4880-4894.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Spencer MJ, Marino MW, Winckler WM. Altered pathological progression of diaphragm and quadriceps muscle in TNF-deficient, dystrophin-deficient mice. Neuromuscul Disord. 2000;10:612–619. doi: 10.1016/s0960-8966(00)00160-7. [DOI] [PubMed] [Google Scholar]
- 56.Suelves M, Lluis F, Ruiz V, Nebreda AR, Munoz-Canoves P. Phosphorylation of MRF4 transactivation domain by p38 mediates repression of specific myogenic genes. EMBO J. 2004;23:365–375. doi: 10.1038/sj.emboj.7600056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tews DS, Goebel HH. Cytokine expression profile in idiopathic inflammatory myopathies. J Neuropathol Exp Neurol. 1996;55:342–347. doi: 10.1097/00005072-199603000-00009. [DOI] [PubMed] [Google Scholar]
- 58.Tidball JG. Inflammatory cell response to acute muscle injury. Med Sci Sports Exerc. 1995;27:1022–1032. doi: 10.1249/00005768-199507000-00011. [DOI] [PubMed] [Google Scholar]
- 59.Tidball JG. Inflammatory processes in muscle injury and repair. Am J Physiol Regul Integr Comp Physiol. 2005;288:R345–R353. doi: 10.1152/ajpregu.00454.2004. [DOI] [PubMed] [Google Scholar]
- 60.Tiffin N, Adi S, Stokoe D, Wu NY, Rosenthal SM. Akt phosphorylation is not sufficient for insulin-like growth factor-stimulated myogenin expression but must be accompanied by down-regulation of mitogen-activated protein kinase/extracellular signal-regulated kinase phosphorylation. Endocrinology. 2004;145:4991–4996. doi: 10.1210/en.2004-0101. [DOI] [PubMed] [Google Scholar]
- 61.Tintignac LA, Lagirand J, Batonnet S, Sirri V, Leibovitch MP, Leibovitch SA. Degradation of MyoD mediated by the SCF (MAFbx) ubiquitin ligase. J Biol Chem. 2005;280:2847–2856. doi: 10.1074/jbc.M411346200. [DOI] [PubMed] [Google Scholar]
- 62.Torrente Y, El Fahime E, Caron NJ, Del Bo R, Belicchi M, Pisati F, Tremblay JP, Bresolin N. Tumor necrosis factor-alpha (TNF-alpha) stimulates chemotactic response in mouse myogenic cells. Cell Transplant. 2003;12:91–100. doi: 10.3727/000000003783985115. [DOI] [PubMed] [Google Scholar]
- 63.Tracey KJ, Cerami A. Pleiotropic effects of TNF in infection and neoplasia: beneficial, inflammatory, catabolic, or injurious. In: Aggarwal BBVJ, editor. Tumor Necrosis Factors Structure, Function, and Mechanism of Action. New York: Marcel Dekker; 1992. pp. 431–452. [PubMed] [Google Scholar]
- 64.Vreugdenhil G, Lowenberg B, Van Eijk HG, Swaak AJ. Tumor necrosis factor alpha is associated with disease activity and the degree of anemia in patients with rheumatoid arthritis. Eur J Clin Invest. 1992;22:488–493. doi: 10.1111/j.1365-2362.1992.tb01495.x. [DOI] [PubMed] [Google Scholar]
- 65.Warren GL, Hulderman T, Jensen N, McKinstry M, Mishra M, Luster MI, Simeonova PP. Physiological role of tumor necrosis factor α in traumatic muscle injury. FASEB J. 2002;16:1630–1632. doi: 10.1096/fj.02-0187fje. [DOI] [PubMed] [Google Scholar]
- 66.Weston AD, Sampaio AV, Ridgeway AG, Underhill TM. Inhibition of p38 MAPK signaling promotes late stages of myogenesis. J Cell Sci. 2003;116:2885–2893. doi: 10.1242/jcs.00525. [DOI] [PubMed] [Google Scholar]
- 67.Whalen RG, Harris JB, Butler-Browne GS, Sesodia S. Expression of myosin isoforms during notexin-induced regeneration of rat soleus muscles. Dev Biol. 1990;141:24–40. doi: 10.1016/0012-1606(90)90099-5. [DOI] [PubMed] [Google Scholar]
- 68.Wu Z, Woodring PJ, Bhakta KS, Tamura K, Wen F, Feramisco JR, Karin M, Wang JY, Puri PL. p38 and extracellular signal-regulated kinases regulate the myogenic program at multiple steps. Mol Cell Biol. 2000;20:3951–3964. doi: 10.1128/mcb.20.11.3951-3964.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zador E, Mendler L, Takacs V, de Bleecker J, Wuytack F. Regenerating soleus and extensor digitorum longus muscles of the rat show elevated levels of TNF-alpha and its receptors, TNFR-60 and TNFR-80. Muscle Nerve. 2001;24:1058–1067. doi: 10.1002/mus.1110. [DOI] [PubMed] [Google Scholar]
- 70.Zetser A, Gredinger E, Bengal E. p38 mitogen-activated protein kinase pathway promotes skeletal muscle differentiation. Participation of the Mef2c transcription factor. J Biol Chem. 1999;274:5193–5200. doi: 10.1074/jbc.274.8.5193. [DOI] [PubMed] [Google Scholar]