

Abstract
Antiviral innate immunity is triggered by sensing viral nucleic acids. RIG-I (retinoic acid-inducible gene-I) is an intracellular molecule that responds to viral nucleic acids and activates downstream signaling, resulting in the induction of members of the type I interferon (IFN) family, which are regarded among the most important effectors of the innate immune system. Although RIG-I is expressed ubiquitously in the cytoplasm, its levels are subject to transcriptional and post-transcriptional regulation. RIG-I belongs to the IFN-stimulated gene (ISG) family, but certain cells regulate its expression through IFN-independent mechanisms. Several lines of evidence indicate that deregulated RIG-I signaling is associated with autoimmune disorders. Further studies suggest that RIG-I has functions in addition to those directly related to its role in RNA sensing and host defense. We have much to learn and discover regarding this interesting cytoplasmic sensor so that we can capitalize on its properties for the treatment of viral infections, immune disorders, cancer, and perhaps other conditions.
Keywords: retinoic acid-inducible gene-I, innate immunity, RNA virus
I. Introduction
Vertebrate animals have developed innate and adaptive immune systems that defend them against microbial and other infections.1-3 The innate immune system is the first line of cellular defense against foreign invasion. When pathogens enter the cytoplasm, they are recognized by germline-encoded pattern-recognition receptors (PRRs) that initiate signaling events, resulting in the production of cytokines such as type I interferons (IFNs).2,3 IFNs have antiviral, antiproliferative, and immunomodulatory activities, and thus play crucial roles in host defense.4 The elucidation of mechanisms that link microbial invasion, engagement of PRRs, and activation of IFN responses has been an area of active investigation in recent years. The Toll-like receptor (TLR) family comprises a sub-group of highly conserved PRRs.5 Thus far, 10 isoforms (TLR1–TLR10) have been identified in humans, and mice are known to express an additional variant (TLR11). TLRs are roughly divided into two groups based on their subcellular localization. Cell surface-expressed TLRs (TLR1, TLR2, TLR4, TLR5, TLR6, and TLR11) recognize viral proteins and bacterial and fungal cell wall components. Other members of the family (TLR3, TLR7, TLR8, and TLR9) are expressed in intracellular compartments such as endosomes and sense mainly nucleic acids.5,6 Specifically, TLR3 senses double-stranded RNA (dsRNA) and TLR7 and TLR8 detect single-stranded RNA (ssRNA) of viral origin. However, TLR3-null cells respond to synthetic dsRNA, suggesting that additional mechanisms participate in the recognition of viral RNA.7
Two additional families have recently been characterized as members of the PRR superfamily: nucleotide-oligomerization domain (NOD)-like receptors (NLRs) and RIG-I (retinoic acid-inducible gene-I)-like receptors (RLRs). NLRs sense bacterial peptidoglycans in the cytoplasm, and RLRs detect viral nucleic acids in the cytosol.8 Three members of the RLR subfamily have been characterized thus far, including RIG-I, MDA5 (melanoma differentiation-associated gene 5), and LGP2 (laboratory of genetics and physiology 2). Each member senses specific viral RNA structures; the fact that RIG-I recognizes viruses that broadly impact human health, such as influenza and hepatitis C viruses, has sparked the interest of virologists and immunologists across the world.
This review is centered on structural and functional aspects of RIG-I. First, we discuss RNA features required for recognition by RLRs. Second, we provide current views regarding the role played by individual RIG-I domains in auto-repression, RNA binding, and signal transduction. Third, we present key studies that led to the elucidation of signaling events that follow RIG-I activation. Finally, we discuss viral- and host-mediated mechanisms that control the function and expression of RIG-I. Not surprisingly, deregulation of RIG-I-mediated events has been reported to contribute to the pathogenesis of human diseases, with an emphasis on autoimmune disorders. The final section of this review briefly discusses key work addressing this important issue.
II. Retinoic Acid-Inducible Gene-I-like Receptors
A. Family of RNA Sensors
RLRs (RIG-I, MDA5, and LGP2) are members of the DExH/D (Asp-Glu-X-His/Asp)-box RNA helicase family that function as cytoplasmic RNA sensors.9 These proteins participate in a wide range of biological processes, including transcription, translation, mRNA splicing, ribosome biogenesis, and RNA decay.7 DExH/D-box proteins harbor four highly conserved motifs and utilize hydrolysis of NTPs to NDPs as an energy source.10,11 RLRs are expressed in most cell types and are essential for the eradication of viruses through the innate antiviral activity of IFNs.9
B. Initial Studies
1. Retinoic Acid-Inducible Gene -I
In 1997, a group from the Shanghai Institute of Hematology cloned novel all-trans retinoic acid (ATRA)-inducible genes from leukemia cells and named them “RIG-A, RIG-B, RIG-C,” and so on. The nucleotide sequences were submitted to GenBank®, and RIG-I was identified as one of the genes whose expression was induced by ATRA treatment.12 Following the discovery of RIG-I, it was reported that stimulation of vascular cells with lipopolysaccharide (LPS), a well-recognized inducer of inflammation, led to up-regulation of RIG-I expression.13 Additional work showed that a variety of cells respond to IFN-γ stimulation by up-regulating RIG-I levels,14-17 thus supporting a role for the cytoplasmic sensor as a mediator of inflammation and innate immunity.
In 2002, Tabara et al. characterized features of the RNA-induced silencing complex, a protein complex that plays a key role in gene silencing in Caenorhabditis elegans. Mass-spectrometric studies revealed that one of the components of this complex was a helicase called Dicer-related helicase (DRH), which is required for dsRNA-mediated post-transcriptional gene silencing.18 The helicase domain of DRH is highly homologous to that of RLRs, suggesting evolutionary links between RNA interference and the RLR system with regard to the detection of foreign RNA.19
2. Melanoma Differentiation-Associated Gene 5
The discovery of MDA5, like that of RIG-I, was based on studies aimed at the identification of novel proteins involved in defined biological processes. Investigators focused on the characterization of proapoptotic signaling pathways and searched public databases to identify novel proteins harboring CARD (caspase recruitment domain) motifs.20 These studies led to the discovery of a CARD-containing mouse sequence that turned out to be Mda5.20 In addition, Kang et al. identified human MDA5 (also known as Helicard) as having dsRNA-dependent ATPase activity with melanoma growth-suppressive properties.21
3. Laboratory of Genetics and Physiology 2
The discovery of LGP2 was based on studies from a research team interested in mammary gland development and remodeling.22 These processes are regulated by two members of the STAT (signal transducer and activator of transcription) family, STAT3 and STAT5. The close proximity of Stat3, Stat5a, and Stat5b in a region of mouse chromosome 1123 led Cui et al. to hypothesize that this locus might harbor additional genes involved in mammary gland development.24 This approach led to the discovery of Lgp1 and Lgp2, two novel genes highly expressed in normal mammary tissue and mammary tumors. LGP2 was found to be a cytoplasmic protein harboring a DExH/D-box motif and a carboxy-terminal helicase domain.24
C. Comparative Analyses of Structural RLR Features
Structural examination of the three RLRs provided important clues related to the role played by individual domains in viral recognition, regulation of activity, and signal transduction. All RLRs harbor at least two functional domains: a central DExD/H-box helicase domain and a carboxy-terminal repressor domain (Fig. 1).25,26 Two amino-terminal CARDs are components of RIG-I and MDA5, but not LGP2 (Fig. 1). Overexpression of CARDs induces constitutive signaling independently of viral infection,9,27 demonstrating that these domains are required for the transduction of signals following activation of RIG-I and MDA5.26 The mechanism involves homotypic interaction with a CARD-containing adaptor molecule, MAVS (mitochondrial antiviral signaling),28 also known as IPS-1 (IFN-β promoter stimulator-1),29 VISA (virus-induced signaling adaptor),30 and Cardif (CARD adaptor-inducing interferon-β).31 Only the terminal CARD of RIG-I and MDA5 interacts physically with MAVS, but both CARDs are required for signaling.32 Constructs lacking these signatures have dominant negative effects.33 Conformational issues and interactions between CARDs and the carboxy-terminal domain of RLRs regulate homotypic interaction with MAVS, and thus modulate signal transduction (see below). CARDs negatively regulate the ATPase activity of RIG-I in the absence of dsRNA; this mechanism may constitute a safeguard to prevent signaling in the absence of viral RNA stimulation.27
FIGURE 1.
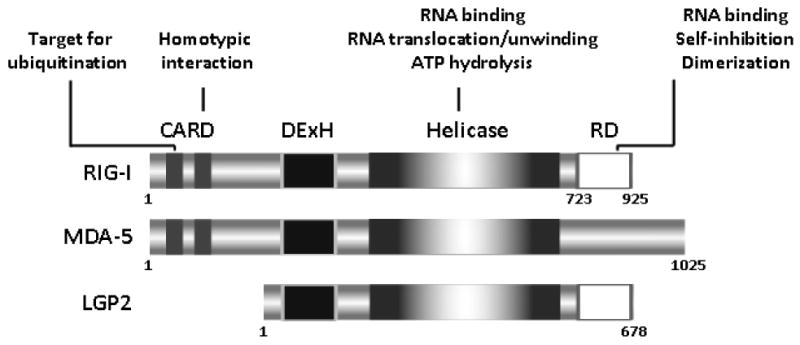
Domain structure of RIG-I-like receptors.
The helicase/translocase domain expressed by members of the RLR family was named owing to its characteristic structural signature (Fig. 1).34 Confirmation that RIG-I encodes a functional helicase was recently provided by Marques et al., who showed that full-length RIG-I exhibits complete unwinding activity toward dsRNA harboring a 3′ overhang longer than 15 nucleotides.35 ATP hydrolysis is necessary but not sufficient for unwinding in vitro.36-38 It is not clear, however, whether RIG-I unwinds dsRNA duplexes or simply translocates on the RNA molecule in vivo.32 In fact, it has been proposed that the helicase domain induces an ATP-dependent conformational change that facilitates signaling through CARDs.39 In addition, the helicase domain appears to bind distinct RNA motifs and cooperate with the carboxy-terminal domain to optimize the affinity of the interaction.38,40 The helicase domains of MDA5 and RIG-I are only about 35% identical, suggesting that these domains participate in the determination of substrate specificity.41 The primary RNA-binding site, however, is located in a different region of RIG-I (see below).38 In summary, the central helicase domain seems to utilize conformational changes to bridge RNA-binding events occurring at the carboxy-terminal end to the amino-terminal end, thus facilitating transduction of signals through the CARDs of RIG-I.
RIG-I and LGP2, but not MDA5, harbor a carboxy-terminal repressor domain that has been proposed to exert inhibitory functions by interacting with other protein regions such as the CARD and helicase domains (Fig. 1).33 In the absence of viral infection, the repressor domain represses RIG-I activity owing to its ability to fold in such a manner that CARDs are shielded from interaction with MAVS.32 A surface enriched with acidic patches has been associated with these repressor domain-regulatory functions.38 During viral infection, RIG-I activation and signaling occur through conformational changes that stimulate self-association and allow CARD/MAVS interaction.33,42,43
A stretch of amino acids comprised of carboxy-terminal domain residues 792 to 925 that overlap with the repressor domain has been demonstrated to be the primary RNA-binding domain.38 Structural analyses using X-ray crystallographic and nuclear magnetic resonance approaches revealed that this region harbors a positively charged groove that may be the RNA-binding site; in addition, a zinc-binding domain seems to be required for optimal interaction.38,44 Interestingly, the groove is different in LGP2 and MDA5, raising the possibility that this domain may account for the well-established ability of RIG-I and MDA5 to recognize different types of RNA viruses.44,45 Additional insights into the role of the carboxy-terminal domain were provided by recent studies in lgp2−/− mice.25 LGP2, a CARD-less RLR unable to transduce activation signals, has been proposed to negatively regulate RIG-I-mediated signaling through a variety of mechanisms, including sequestration of dsRNA, inhibition of RIG-I interaction with MAVS, and blockade of MAVS signaling downstream of RIG-I.33,46-49 However, Satoh et al. have shown that, with the exception of influenza virus, LGP2 acts as a positive regulator of viral recognition mediated by MDA5 and RIG-I.25 Their results suggest that LGP2 is involved in primary sensing of RNA viruses upstream of MDA5 and RIG-I. The authors speculate that LGP2 facilitates recognition of dsRNA by RIG-I and MDA5, either by removing proteins from viral ribonucleoprotein complexes or by unwinding complex RNA structures.25 These potential effects may require the carboxy-terminal domain of LGP2, as this region was shown to be responsible for binding to the termini of dsRNAs.38,44,50 It is possible that LGP2 has both positive and negative functions and that the type of response elicited is defined by the structure of the infecting virus.19,49 In summary, the carboxy-terminal end of RLRs is the primary site for RNA binding and it modulates RLR activity. These effects require conformations that either repress function under basal conditions or induce oligomerization following stimulation with RNA.
D. Ligand Recognition
Two recent reviews highlight important structural features required for viral sensing; readers are encouraged to consult these excellent articles for further details.32,51 Accumulating evidence indicates that RLRs can distinguish among different RNA ligands; this is evidenced when one compares structural features of MDA5 and RIG-I substrates. RIG-I has been reported to recognize RNA derived from 25 viruses (Table 1).9,46,52-65 It has specificity for RNA viruses and does not bind viral dsDNA (except Epstein-Barr virus, Table 1). Until recently, it was thought that RIG-I recognized only dsRNA, and it was assumed that ssRNA viruses were sensed when they replicate, a process during which dsRNA is produced. However, Habjan et al. recently showed that ss(−)RNA does not generate substantial amounts of dsRNA, suggesting that this mechanism may not account for the ability of RIG-I to recognize ssRNA viruses.55 This issue was resolved when two groups demonstrated that ssRNA harboring a 5′-triphosphate (5′ppp) group activates RIG-I-mediated type I IFN induction.62,66 Consistently, deficiency of RIG-I disrupted responses to ssRNA viruses that harbored this structural signature, such as Sendai virus, Newcastle disease virus, and hepatitis C virus.45 In contrast, ssRNA viruses harboring a single phosphate group at the 5′-end of their genomes (i.e., members of the Bornaviridae family) escape recognition by RIG-I.55,67 Most RNA viruses contain 5′ppp in their genomes and antigenomes, but cellular RNAs lack exposed 5′ppp as a result of mRNA capping or processing of 5′ppp into monophosphates.68 However, two groups recently showed that pure ssRNA with a 5′ppp group is unable to activate RIG-I, and demonstrated that the optimal agonist is blunt-ended 5′ppp dsRNA at least 20 base pairs in length (Fig. 2).36,37 The investigators proposed that a “panhandle” structure generated by base-pairing of 5′ and 3′ regions of the viral genome is recognized by RIG-I.36,37 This model provides a mechanism to explain how RIG-I detects negative-strand RNA viruses that lack long dsRNA but contain short double-stranded 5′ppp RNA in certain regions of their single-stranded genomes.36,37 Marq et al. contributed the observation that if the 5′ppp group is unpaired (as in members of the Arenavirus family), recognition by RIG-I is prevented.69
TABLE 1. Viruses Sensed by RIG-I.
| Systematics | Virus Genome | Host | Reference |
|---|---|---|---|
| Family Herpesviridae | dsDNA | ||
| Epstein-Barr virus | Human | 60 | |
| Family Reoviridae | dsRNA | ||
| Orthoreovirus | Human | 53 | |
| Rotavirus | Zoonosis | 58 | |
| Family Arenaviridae | ss(−)RNA | ||
| Lassa virus | Rodent, human | 55 | |
| Guanarito virus | Human | 52,55 | |
| Sabia virus | Human | 52 | |
| Machupo virus | Human | 52 | |
| Junín virus | Human | 52 | |
| Family Bunyaviridae | ss(−)RNA | ||
| Rift valley fever virus | Human | ||
| Family Filoviridae | ss(−)RNA | ||
| Ebola virus | Human | ||
| Family Orthomyxoviridae | ss(−)RNA | ||
| Influenza A virus | Human | ||
| Influenza B virus | Human | ||
| Family Paramyxoviridae | ss(−)RNA | ||
| Newcastle disease virus | Bird | ||
| Sendai virus | Mouse | ||
| Respiratory syncytial virus | Human | ||
| Measles virus | Human | ||
| Metapneumovirus | Human | ||
| Nipah virus | Zoonosis | ||
| Family Rhabdoviridae | ss(−)RNA | ||
| Vesicular stomatitis virus | Domestic animals | ||
| Rabies virus | Zoonosis | ||
| Family Flaviviridae | ss(+)RNA | ||
| Hepatitis C virus | Human | ||
| Japanese encephalitis virus | Human | ||
| Dengue virus | Human | ||
| West Nile virus | Human | ||
| GB virus/hepatitis G virus | Monkey |
FIGURE 2.
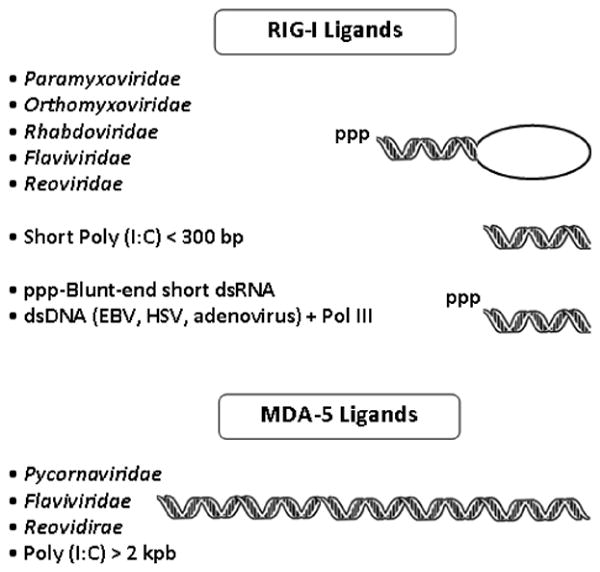
Structural representation of ligands recognized by RIG-I and MDA5.
A second factor that affects the suitability of RNAs as substrates for RLRs is the length of these molecules. For example, polyinosinic-polycytidylic acid, also called poly (I:C), a synthetic dsRNA, is thought to activate IFN responses following recognition by MDA5.41,45 However, when the length of poly (I:C) is decreased, it becomes a ligand for RIG-I (Fig. 2).70 This feature likely represents in vivo substrate preferences, as RIG-I and MDA5 are activated by viral dsRNAs in a length-dependent fashion.70
Structural features unique to certain families of RNA viruses may also affect recognition by RLRs. For example, encephalomyocarditis virus, a member of the Picornaviridae family, is a unique RNA virus that activates type I IFN induction exclusively through MDA5.41 It was suggested that structural characteristics unique to encephalomyocarditis virus, such as the presence of a polyC acid tract in the 5′-untranslated region (UTR), may define its suitability as an MDA5 substrate.41 Related studies have shown that, in addition to the 5′ppp, RIG-I recognizes the 3′-UTR of some viruses such as hepatitis C virus. Within this domain, a homopolymeric poly(U/UC) region unlikely to form stable secondary structures was mapped as that responsible for activation of IFN responses.71 Deletion of the 3′-UTR was shown to elicit decreased signaling, a result consistent with a critical role for this region in RIG-I-mediated innate immune responses.72 In summary, RIG-I recognizes the presence of 5′ppp groups in viral genomes and somewhat short, blunt-ended dsRNAs. Accordingly, dsRNA generated by infection of certain DNA viruses is also recognized by RIG-I. The elucidation of RNA sequences and structural features defining recognition by RLRs is likely to significantly affect our ability to capitalize on these properties to limit viral infection of humans, animals, and plants.
E. Signal Transduction Domains
Our understanding of the mechanisms through which RIG-I transduces signals following viral stimulation has been significantly advanced by two breakthrough discoveries. Pioneering work by Yoneyama et al. established that RIG-I has antiviral properties and plays a key role in dsRNA-mediated IFN-β expression.9 The IFN-β gene harbors one of the best-characterized higher eukaryotic enhancers.73 Gene expression requires coordinate activation and DNA binding of the activating transcription factor-2/c-Jun, interferon regulatory factor (IRF) 3, IRF7, and nuclear factor (NF)-κB (p50/p65).73 Upon viral infection, these transcription factors are recruited gradually on a specific enhancer sequence to form the “enhanceosome.”73 In the presence of cytoplasmic dsRNA, RIG-I activates NF-κB and IRF3. As noted above, CARDs are required for activation, and mutants in this region fail to transduce signals. Each CARD seems to have specificity for individual downstream targets.9
A second major breakthrough in this field was the discovery by four independent groups of MAVS,28,29,30,31 a CARD-containing adaptor molecule that, as previously mentioned (section IIC), links RIG-I and IFN-β signaling. MAVS was originally identified from large-scale screening of human genes that activate the NF-κB and MAPK signaling pathways.74 MAVS consists of an amino-terminal CARD, a proline-rich region near the amino-terminus, and a carboxy-terminal transmembrane domain. X-ray crystallographic studies revealed that the CARD in MAVS adopts the classic CARD fold and is homologous to the first CARD of RIG-I.75 The transmembrane domain allows MAVS to localize to the outer mitochondrial membrane, suggesting that the mitochondria are a platform for MAVS-mediated signaling.28 Indeed, the function of MAVS is impaired when the transmembrane domain is deleted or replaced; restoring mitochondrial localization by expression of the membrane targeting domain of Bcl-2 or Bcl-xL also restores function.28 Interaction between CARDs from RIG-I and MAVS results in the activation of type I IFNs,28-31 a process during which the signal originating in RIG-I is transferred to MAVS.
F. Modulation of RIG-I Activity Through Ubiquitin-Dependent and Related Mechanisms
Recent studies have led to the identification and characterization of host proteins that regulate the activity of RIG-I, and thus affect its ability to transduce signals following viral stimulation. Derivatization of proteins with lysine 63-linked ubiquitin chains is a post-translational modification known to regulate intracellular signaling.76 TRIM25, a member of the tripartite motif (TRIM) protein family,77 is an E3 ubiquitin and IFN-stimulated gene 15 (ISG15) ligase reported to bind to the amino-terminal CARD of RIG-I and conjugate lysine 172 located in the internal CARD to a lysine 63-linked ubiquitin chain (Fig. 3).32,78 Inhibition of this step by mutation or deletion of lysine 172 prevents interaction of RIG-I and MAVS, and thus inhibits IFN production.79 Cells lacking TRIM25 have impaired production of type I IFNs in response to viral infection. Interestingly, TRIM25 is inhibited by NS1, an influenza protein known for its ability to antagonize the immune system.80 NS1 directly interacts with TRIM25 and prevents activation of RIG-I,81 emphasizing the importance of TRIM25 in RIG-I function.
FIGURE 3.
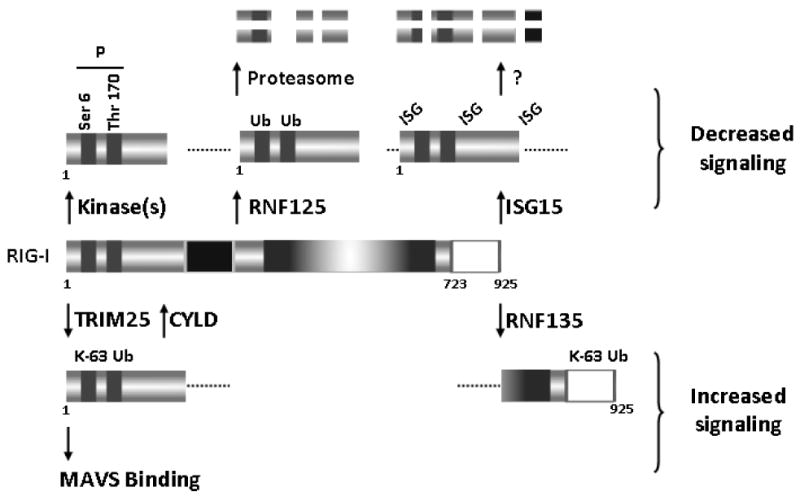
Modulation of RIG-I activity through ubiquitin-dependent and other mechanisms.
While it is clear that lysine 63 ubiquitin chains play an important role as RIG-I activators, a recent study by Zeng et al. challenged whether covalent derivatization of RIG-I is necessary for activation. This group developed a cell-free system consisting of RIG-I, mitochondria, cytosol, RNA, and ubiquitination enzymes (including TRIM25) that was used to mimic viral infection in intact cells.82 The studies showed that RNA promotes recruitment of lysine 63-linked ubiquitin chains to the CARDs of RIG-I and that unanchored ubiquitin chains potently activate RIG-I and function as endogenous RIG-I ligands in human cells.83 These results suggest that while the association between lysine 63-linked ubiquitin chains and CARDs of RIG-I is essential for activation, RIG-I ubiquitination per se may be dispensable.82 Recently, RNF135/Riplet/REUL was reported as another E3 ligase that can positively regulate RIG-I signaling (Fig. 3).84,85 The actions of RNF135 are centered on different domains of RIG-I, as this protein interacts with the repressor domain and with the helicase domain but not with CARD and catalyzes lysine 63-linked polyubiquitination of the carboxy-terminal region of RIG-I. It is not clear, however, how this modification regulates downstream signaling.
The cellular need to carefully monitor the activity of RIG-I and other RLRs likely resulted in the evolution of compensatory strategies to regulate ubiquitin-mediated activation of these sensors. The tumor suppressor CYLD, a deubiquitinating enzyme, associates with the CARD domain of RIG-I and removes lysine 63-linked ubiquitin conjugated by TRIM25 (Fig. 3).86 Thus, CYLD may provide a natural mechanism to limit RIG-I-mediated antiviral signaling.87 An additional strategy to limit activation through this pathway was recently proposed by Nistal-Villán et al.88 These investigators showed that phosphorylation of serine 8 located within the first CARD of RIG-I inhibits its ability to interact with TRIM25, and thus blunts IFN-β production (Fig. 3). This group also reported that phosphorylation of threonine 170 in the second CARD domain of RIG-I has a negative impact on RIG-I ubiquitination and activation.89 Derivatization of RIG-I with ubiquitin has also been shown to act as a negative regulator of RIG-I. A study using a yeast two-hybrid screen identified RNF125, a ubiquitin E3 ligase, as a protein that specifically interacts with the CARD and helicase domains of RIG-I (Fig. 3).86 RNF125 efficiently conjugates ubiquitin to the CARDs of RIG-I, and this modification leads to enhanced degradation of RIG-I through the proteasome.90 We found that this process can itself be inhibited under some circumstances. Our studies have shown that HSP90 (heat shock protein 90) interacts with RIG-I in a CARD-independent fashion and that disrupting this association prevents downstream signaling.91 Studies using HSP90 inhibitors suggested that the role of HSP90 in this setting is to protect RIG-I from ubiquitin-mediated proteasomal degradation. An additional mechanism to negatively regulate RIG-I involves modification with a ubiquitin-like protein known as ISG15 (Fig. 3).92,93 Overexpression of ISG15 combined with specific enzymes of the ubiquitination system decreases or delays basal and stimulated IFN-β promoter activity. Several ISG15-derivatized RIG-I species can be detected following ISG15 overexpression, but it is not clear whether these represent multiple sites of derivatization or if poly-ISG15 links to RIG-I at a single site. The mechanism whereby ISGylation dampens RIG-I signaling likely involves degradation of RIG-I through a proteasome-independent pathway.94 In summary, it is apparent that RIG-I is in a constant state of flux between free forms and species derivatized or associated with ubiquitin, ISG15, and possibly other proteins. The balance between these forms is likely dictated by multiple contributions that ensure optimal signaling when the need arises, and that prevent unnecessary responses under normal conditions.
III. Signaling Events Mediated by RIG-I
A. The MAVS Pathway
Following recognition of RNA by RIG-I, a conformational change allows interaction between the CARDs of RIG-I and MAVS.42,43 This process is independent of the helicase activity of RIG-I but requires its translocase activity and ATP.9,38,95 The assembly of RIG-I/MAVS has been thought to take place exclusively on mitochondria, but recent work by Dixit et al. has shown that MAVS is also found on peroxisomes.96 The data suggest that peroxisomal MAVS may establish an early and transient response to halt or delay viral replication, followed by robust and prolonged signaling driven by the mitochondrial MAVS pathway.97 A trans-membrane protein known as STING (stimulator of interferon genes) resides in the endoplasmic reticulum and the mitochondrial membrane, directly interacts with MAVS, and has been reported to be a critical scaffold for protein assembly.98,99 In addition, recent studies have shown that the DEAD (Asp-Glu-Ala-Asp)-box helicase DDX3 (DEAD box polypeptide 3) is a component of the MAVS complex owing to its ability to constitutively associate with MAVS through its carboxy-terminal end.100,101 DDX3 is proposed to serve as an initial sensor of RNA in unstimulated cells, and may intensify signaling from MAVS when the levels of RIG-I are low.100 In addition to the ubiquitin-mediated and related strategies (described in section IIF), naturally occurring mechanisms have evolved to inhibit interaction between MAVS and RIG-I, presumably to prevent unwanted or prolonged activation of antiviral signaling that may be harmful for host survival.99 An Atg5-Atg12 conjugate, a regulator of autophagy, has been shown to inhibit interactions between MAVS and RIG-I.102 NLRX1, a protein expressed on the outer mitochondrial membrane, is a negative regulator of RLR-mediated antiviral signal owing to its ability to directly interact with the CARD in MAVS.103 Finally, gC1qR, a C1q receptor implicated in the regulation of innate and adaptive immunity, has been shown to translocate to the mitochondria, interact with MAVS, inhibit the formation of RIG-I/MAVS complexes, and dampen antiviral signaling.104
The formation of the MAVS complex is followed by engagement of at least three signaling axes involved in the activation of IRFs, NF-κB, and apoptosis.19,105 We will discuss each of these pathways separately, with the understanding that significant overlap exists among them. This section is limited to a discussion of RIG-I-mediated events, but it is important to emphasize that the MAVS pathway can be engaged by a variety of receptors that detect viral nucleic acids.
B. TRAF-Mediated IRF Activation Through Non-Canonical IKKs
A number of tumor necrosis factor (TNF) receptor-associated factor (TRAF) family members have been shown to participate in signaling events downstream of RIG-I/MAVS complex formation (Fig. 4).99 TRAF3, an E3 ligase for lysine 63-linked polyubiquitination, directly interacts with the TIM (TRAF-interacting motif) in MAVS.106,107 TRAF3-dependent addition of K63-linked ubiquitin chains to components of the MAVS signaling complex is crucial for the activation of IFN production.108 In addition, auto-ubiquitination of TRAF3 enhances its binding to NF-κB essential modulator (NEMO, also known as IKKγ), a canonical I-κb kinase (IKK) that regulates the IKK complex, leading to activation of IRF3.109 Consistently, selective cleavage of lysine-63-linked polyubiquitin chains on TRAF3 through deubiquitinating enzyme A (DUBA) blunts IFN responses.110 During later phases of viral infection, the E3 ligase Triad3A adds lysine 48-linked polyubiquitin chains to TRAF3 and targets it for degradation through the proteasome.111 These observations suggest dual, temporal regulation of TRAF3 through derivatization with different types of ubiquitin polymers. It was recently reported that TRAF5 also plays an important role in IRF3/7 activation downstream of MAVS (Fig. 4).112
FIGURE 4.
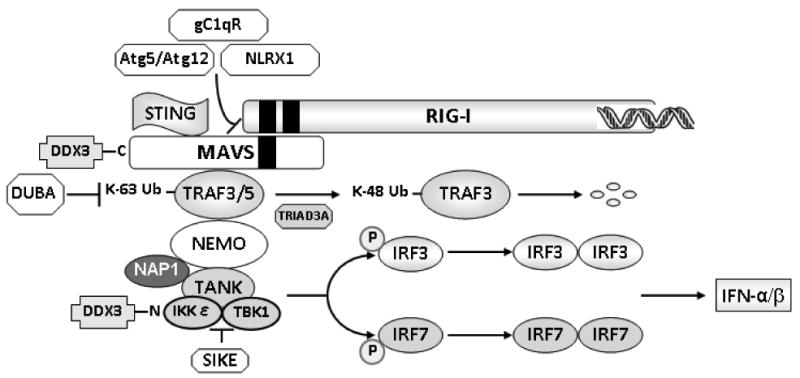
TRAF-mediated IRF activation through noncanonical IKKs.
NEMO has been shown to physically interact with TANK (TRAF family member-associated NF-κB activator) to mediate recruitment of IKKε and TBK1 (TANK-binding kinase 1) to the complex (Fig. 4).113,114 A NEMO mutant lacking its TANK-binding domain failed to recruit IKKε to the RIG-I/MAVS complex, thus preventing IFN production induced by viral infection.113 In the absence of viral stimulation, SIKE (suppressor of IKKε) forms a complex with TBK1 and/or IKKε that prevents these kinases from interacting with RIG-I or IRF3.115
The transmission of activation signals to the non-canonical IKKs TBK1 and IKKε results in phosphorylation and dimerization of IRF3 and IRF7.107,116 This cascade leads to IFN-α production (Fig. 4) and contributes to IFN-β synthesis. IFN secretion from infected cells initiates autocrine-and paracrine-signaling cascades through type I IFN receptor that result in up-regulation of more than 100 different genes and the establishment of an antiviral state in both infected and neighboring uninfected cells.32
Additional regulatory components of this signaling axis include NAP1 (NAK-associated protein 1), initially characterized as an activator of IKK-related kinases, and DDX3 (see above).117 NAP1 interacts with IKKε and TBK1,118 and also physically interacts with MAVS. The coiled-coil motif of NAP1 is highly homologous to those of NEMO and TANK, suggesting that the three proteins utilize these domains for kinase binding and subsequent regulation of downstream signaling.118,119 The precise nature of the interaction(s) between NAP1 and other members of the complex remains to be established. The aminoterminal end of DDX3 associates with IKKε and exerts positive effects as a part of the TBK1/IKKε complex that activates IRF3.117
C. RIG-I/MAVS Signaling Through the Canonical IKK Complex
In addition to TRAF3/5, TRAF2 and TRAF6 have been reported to associate with MAVS and transmit signals to IKKs.30 Unlike TRAF3/5, which recruit canonical and non-canonical IKK complexes, TRAF2/6 are primarily involved in canonical signaling leading to NF-κB activation.30,107 The relay of signals through this axis involves participation of several proteins that harbor an intercellular death domain present in multiple proteins that trigger apoptosis (Fig. 5). These include FADD (Fas-associated death domain) and RIP1 (receptor interacting protein 1).29,120 Deficiency of these proteins prevents activation of IFN or NF-κB signaling in response to dsRNA treatment.120,121 FADD and RIP1 interact with the carboxy-terminal portion of MAVS, suggesting possible connections between MAVS signaling and death domain-expressing proteins during innate immune responses. In addition, TRADD (TNF receptor-associated death domain), a crucial adaptor of TNF receptor I, was also demonstrated to be essential for RIG-I-MAVS-dependent signaling.122 According to Michallet et al., a TRADD-containing complex that includes RIP1 and FADD forms a central molecular signaling unit that assembles on the intermediary region of MAVS following activation of RIG-I by viral nucleic acids.122 An additional step necessary for NF-κB activation is FADD-mediated engagement of caspase-8 and/or caspase-10. It is well established that these caspases are cleaved and become activated following dsRNA exposure, but the precise details that link caspase activation to the NF-κB pathway remain to be established.121 TRAFs recruited by MAVS induce RIP1 ubiquitination and association of NEMO, a key regulator of IKKs. The canonical IKK complex consisting of IKKα, IKKβ, and NEMO phosphorylates IκB, followed by its proteasomal degradation, translocation of NF-κB to the nucleus, and activation of proinflammatory cytokines and other target genes (Fig. 5).123
FIGURE 5.
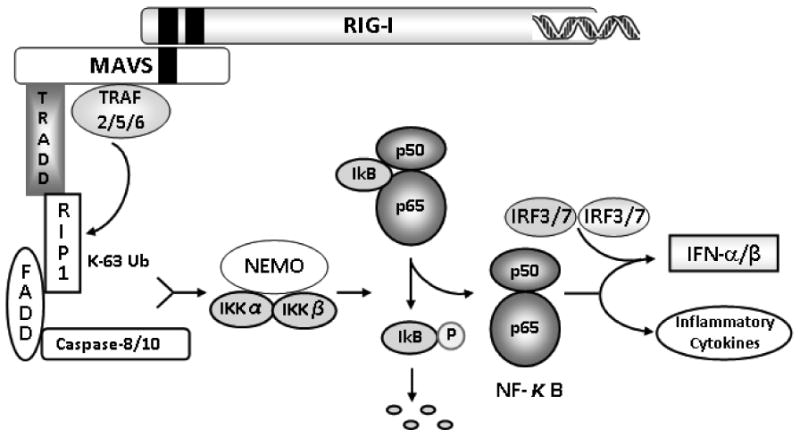
RIG-I/MAVS signaling through the canonical IKK complex.
D. RIG-I/MAVS Induces Two Effector Arms That Converge on NEMO
Sections IIIB and IIIC summarize evidence supporting RIG-I/MAVS-mediated activation of two distinct pathways: the IRF3/7 and the canonical NF-κB pathways (Figs. 4 and 5, respectively). A common feature between these axes is a requirement for NEMO as an adaptor protein that organizes the assembly of IKKs into activated high-molecular-weight complexes.124 In the NF-κB pathway, NEMO is required for recruitment of IKKα and IKKβ to RIG-I/MAVS, activation of NF-κB, and induction of inflammatory cytokine expression. In contrast, the IRF3 pathway requires interaction between NEMO and the TBK1 adapter TANK for efficient coupling of the RIG-I/MAVS complex to IRF3 activation.113 These considerations indicate that NEMO is the final component shared between the NF-κB and the IRF3 signaling pathways engaged following RIG-I/MAVS activation.125 Formal evidence supporting this paradigm was elegantly presented by Liu et al.125 These investigators used an alternatively spliced form of NEMO lacking an amino-terminal domain. The splice variant was found to efficiently mediate cytokine-induced, canonical NF-κB activation but failed to couple RIG-I/MAVS signaling to the TANK-IKKε complex, thus preventing IRF3 activation. This inability to uncouple IRF3- and NF-κB-dependent effects convincingly showed the existence of different mechanisms for NEMO-mediated regulation of canonical and non-canonical IKK signaling following RIG-I/MAVS activation. While both IRF3 and NF-κB signaling are important responses to viral infection, they serve different purposes: IRF3 plays a role in mucosal antiviral responses113 and NF-κB participates in the initiation of mucosal inflammation and in adaptive immunity.125 Appropriate regulation and temporal coordination of these two signaling arms downstream of NEMO likely plays an important role in the resolution of viral infection.
E. IRF3 Plays a Role as a Mediator of dsR-NA-Induced Apoptosis
In addition to transcription factor-driven up-regulation of IFN genes and ISGs, both of which are essential for innate and adaptive antiviral defenses, cells utilize apoptosis to limit viral spreading at the cost of cellular death. In recent elegant studies, Chattopadhyay et al. found that IRF3 has pro-apoptotic functions in addition to its role as a transcriptional activator (Fig. 6).126 Engagement of RIG-I was shown to activate the apoptotic activity of IRF3 through a distinct signaling pathway that required the participation of several proteins. The investigators clearly showed that RIG-I, MAVS, TRAF3, and TBK1, all of which are necessary for gene induction (Fig. 4), are also required for the apoptotic effects of IRF3 (Fig. 6). In addition, the adaptor proteins TRAF2 and TRAF6 were shown to be required for IRF3-mediated apoptosis but not for IRF3-mediated induction of gene expression (Fig. 6).126 Mechanistically, a BH3 domain located in the carboxy-terminal region of IRF3 was shown to enable interaction with pro-apoptotic Bax; the IRF3/Bax complex is subsequently translocated to the mitochondria, where activation of the intrinsic apoptotic pathway takes place (Fig. 6). Future studies should be centered on elucidating how TRAF2 and TRAF6 contribute to IRF3-dependent apoptotic responses.127
FIGURE 6.
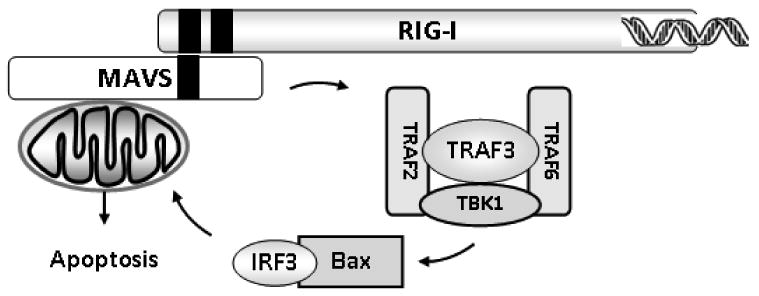
IRF3 as a mediator of dsRNA-induced apoptosis.
IV. Evasion of RIG-I-Mediated Antiviral Responses
Even though the innate immune system has developed sophisticated mechanisms for viral eradication, infections still occur.128 Viruses are skilled pathogens that have developed a number of survival mechanisms to effectively evade detection. First, structural features of viral RNAs and virally encoded proteins prevent recognition by RIG-I and allow certain viruses to escape detection. Second, some viruses produce proteins that inhibit key steps required for effective downstream signaling. Third, viral infection often leads to altered expression of key micro-RNAs (miRNAs). These mechanisms prevent host cell-mediated up-regulation of defense responses, thus inhibiting the ability of mammalian cells to successfully eradicate certain viruses.
A. Strategies Based on Interference of Viral RNA Recognition
We previously stated (section IID) that one of the features that allows recognition of viral RNA by RIG-I is the presence of a 5′ppp moiety.62,66 Viral RNAs that contain a 5′-monophosphate evade RIG-I sensing.55 All eukaryotic cellular mRNAs are blocked at their 5′-ends with a cap structure that precludes recognition by RIG-I (section IID).129 Many viruses use the host mRNA machinery to cap their newly synthesized mRNAs, encode their own capping enzymes, or hijack capped 5′ fragments from cellular mRNAs.130,131 While capped viral RNAs escape RIG-I recognition, viral RNA replication leads to transient cytosolic intermediates that harbor uncapped 5′ppp, thus allowing recognition by the host.132 Other viruses (e.g., members of the Picornaviridae and Calcioviridae families) covalently link a viral protein (VPg) to the 5′-end of viral RNAs to prevent recognition by RIG-I.133 Finally, it is apparent that viruses such as members of the Bunyaviridae and Bornaviridae families have developed mechanisms to remove their own 5′ppp group, thus evading detection by RIG-I.55
A number of viruses encode cytoplasmic proteins that have the ability to shield dsRNAs to prevent sensing by RIG-I (e.g., vaccinia virus E3L, Ebola virus VP35, and influenza A virus NS1).134-136 Interestingly, the dsRNA-binding domain of E3L inhibits 5′ppp ssRNA-, dsRNA-, and DNA-induced IFN-β synthesis.137,138 This motif binds to dsRNA but not to ssRNA or DNA, suggesting that E3L interferes with dsRNA binding and inhibits RIG-I/MAVS signaling through dual mechanisms. Some virally encoded proteins can both sequester RNA and bind to RIG-I directly, again suggesting the use of multiple strategies to inhibit host responses.131
B. Strategies Based on Inhibition of RIG-I-MAVS Signaling
In addition to strategies based on structure recognition, viruses have developed equally sophisticated mechanisms to inhibit functional aspects of RIG-I/MAVS signaling. First, RIG-I cleavage is used by members of the Picornaviridae family virus to evade recognition.128 This process involves a viral protease, 3Cpro, which has been shown to cleave RIG-I both in vitro and in vivo.128 Second, influenza A virus NS1 binds to the RIG-I/MAVS complex and blocks downstream signaling.136,139
An additional mechanism of viral defense against detection involves MAVS. NS3/4A is a protease encoded by hepatitis C virus that has been known to interfere with antiviral responses before the RIG-I/MAVS signaling axis was identified.140 It was later discovered that NS3/4A suppresses RIG-I-dependent IFN expression by cleaving MAVS from its mitochondrial tether.31,141-144 Picornaviruses and hepatitis A virus utilize similar, protease-based defense mechanisms, suggesting that specific cleavage of MAVS may be a common strategy to dampen antiviral activity.131,145 Other viruses (i.e., Poliovirus) trigger cleavage of MAVS through a caspase-dependent mechanism, suggesting that some viruses may activate caspases not only to facilitate shedding and replication, but also to impair antiviral responses.146 Additional viral countermeasures in the RIG-I-MAVS pathway, especially those related to strategies involving downstream signaling molecules, are described in more detail in excellent review articles.26,131
C. Strategies Based on Induction of MicroRNA Expression
Novel findings indicate that viruses utilize miRNA-based strategies to evade eradication initiated by the host. miRNAs are highly conserved, non-coding RNAs that affect biological processes such as development, infection, immunity, inflammation, and tumorigenesis by suppressing gene expression through their ability to bind to the 3′-UTR of target mRNAs.147,148 A recent interesting study by Hou et al. demonstrated that type I IFN production triggered by vesicular stomatitis virus infection is inhibited by up-regulation of miR-146a in a RIG-I-dependent manner.149 In addition, the authors showed that pro-inflammatory cytokine and chemokine expression and NF-κB activation were also attenuated by miR-146a. These events led to the promotion of viral replication. The targets of miR-146a included TRAF6 and also IRAK (IL-1R-associated kinase)-1 and IRAK-2, two kinases shown in this work to associate with FADD.149 Other viruses may utilize similar miRNA-based strategies to blunt type I IFN and perhaps other responses to negatively regulate host antiviral strategies.
V. Other Functions of RIG-I
RIG-I has been proposed to participate in a variety of intracellular events apparently unrelated to its role as a viral sensor. Some of these functions are summarized below.
A. Cellular Differentiation
The ability of ATRA to induce granulocytic differentiation during acute promyelocytic leukemia has been the basis for its use in the treatment of this malignancy.150 Interestingly, ATRA-induced differentiation and normal myelopoiesis are accompanied by induction of RIG-I.12 Conversely, disruption of the Rig-I gene (also known as dxd58) in mice leads to the development of a progressive myeloproliferative disorder.151 These observations suggest that RIG-I may play important roles in mammalian granulocyte production and differentiation.
B. Bacterial Phagocytosis
Studies by Kong et al. revealed that, in addition to its role in antiviral responses, RIG-I participates in TLR-stimulated phagocytosis.152 Stimulation with TLR4 ligands was shown to induce RIG-I expression in macrophages and depletion of RIG-I inhibited TLR4-induced bacterial phagocytosis. This effect seems to be mediated by activation of the small GT-Pase Cdc42/Rac1, actin polymerization, and recruitment of the actin regulator Arp2/3. Consistently, mice deficient in RIG-I expression are more susceptible to bacterial infection, suggesting that RIG-I plays an important role during antibacterial responses.152
C. Apoptosis
RIG-I is being actively investigated as a potential therapeutic target for cancer, particularly melanoma. Melanoma cells are known to have defects in programmed cell death in response to a wide range of external stimuli.153 IFN-α2β, a member of the type I IFN family, is widely used as an anticancer drug against melanoma; its efficacy is significant but limited.154 Poeck et al. designed a small RNA that silenced the anti-apoptotic gene Bcl2 and also activated RIG-I. To accomplish this, the investigators used an elegant strategy that consisted of replacing the 5′-monophosphate ends of the synthetic siRNA with a 5′ppp. The efficacy of this approach was tested in a mouse model of metastatic melanoma to the lung.155 Silencing Bcl2 promoted tumor cell apoptosis, and activating RIG-I stimulated the immune system to destroy the tumor through a mechanism that involved induction of type I IFN by RIG-I.155 These results suggest that single or combinatorial therapies that exploit the ability of RIG-I to stimulate beneficial immune responses may have potentially useful therapeutic applications for the treatment of melanoma and perhaps other types of cancer.
D. Inflammation
As previously mentioned (section IIIC), activation of the NF-κB pathway through MAVS-dependent RIG-I signaling results in the production of cytokines, including the cytokine precursor pro-IL-1β.3 The generation of mature, biologically active IL-1β requires proteolytic processing through caspase-1, an enzyme activated by cytoplasmic multiprotein complexes known as inflammasomes.157 Pioneering work by Rintahaka et al. demonstrated that the RIG-I signaling pathway is involved in inflammasome-associated caspase-1 activation and apoptosis of virus infected cells.158 Poeck et al. later showed that RIG-I acts as a sensor for inflammasome activation, leading to caspase-1 activation and generation of mature IL-1β.159 These two studies represent the first examples of MA-VS-independent RIG-I signal transduction.
VI. Regulation of RIG-I Expression
A. The RIG-I Promoter
In 2007, Su et al. reported the cloning and characterization of essential elements regulating expression of the human RIG-I promoter.160 This work revealed the presence of two putative TATA boxes at positions −144 and −162 relative to the transcriptional initiation site. However, it is not clear which of these TATA boxes is functional in vivo. A canonical IRF1-binding site was identified between nucleotides −17 and −4; IRF2 was also shown to activate the RIG-I promoter.161 As expected, binding of IRF1 to the RIG-I promoter increased following stimulation, and mutational analyses identified a specific site essential for IFN-β-mediated RIG-I induction. An uncharacterized element between nucleotides −1141 and −362 was shown to interfere with IFN-β-stimulated RIG-I promoter activity.160 Additional experiments showed that under basal conditions, the levels of RIG-I and IRF1 are significantly higher in normal cells compared with their tumor counterparts.160 For example, abundant expression of RIG-I was detected in normal brain, but almost no RIG-I was present in tissues from an experimental mouse brain tumor. These results are consistent with the possibility that RIG-I may possess tumor-suppressor properties through its ability to activate IFN responses.
B. Regulation of RIG-I mRNA and Protein Expression
Under unstimulated conditions, RIG-I is expressed at relatively low levels and is thought to participate in cytoplasmic surveillance of viral nucleic acids. In addition, various agonists have been shown to upregulate RIG-I expression at the mRNA and protein level in cell culture. First, ATRA induced RIG-I expression in acute promyelocytic leukemia cells.12 Second, using subtraction hybridization techniques, RIG-I was identified as an LPS-inducible gene expressed by human endothelial cells (section IIB1).13 Recently, it was shown that LPS-mediated stimulation of RIG-I expression occurs through a TLR4-dependent mechanism whereby early production of IFN-β leads to the induction of RIG-I through autocrine stimulation.162
In addition to ATRA and LPS, type I IFNs (IFN-α and IFN-β) up-regulate RIG-I expression in a variety of cell types, including A549 human lung carcinoma cells,61 HEK293 human kidney embryonic cells,163 monocyte-derived dendritic cells,164 and HeLa human cervical cancer cells.165 IFN-γ, a member of the type II IFN family, also stimulates RIG-I expression in various cell types.14-17,164,166-169 Interestingly, cellular exposure to poly (I:C) increases RIG-I expression in human gingival fibroblasts170 and in human astrocytes.171 These findings, combined with fact that poly (I:C) triggers RIG-I-mediated IFN synthesis,70 suggest that RIG-I expression may be subject to positive feedback regulatory mechanisms. TNF-α also stimulates RIG-I expression in A549 cells,61 HeLa cells,165 and synoviocytes.172 Moreover, RIG-I is synergistically induced by IFN-α and TNF-α in A549 cells.61 Consistently, the expression of RIG-I is blocked by type I IFN receptor-specific neutralizing antibodies. In HeLa cells, RIG-I expression is mediated by IFN-ε,165 and in synoviocytes, IFN-β seems to play a key role in RIG-I induction.172 IL-1β up-regulates RIG-I expression in human gingival fibroblasts through an incompletely characterized mechanism.173
Listeria monocytogenes, an obligate intracellular parasite, infects a variety of cell types, including phagocytes.174,175 This parasite enters host cells by interacting with eukaryotic cell-surface adhesion molecules, including E-cadherin.174 Following internalization, parasites remain in a vacuole or escape to the cytosol, where they replicate and remain as intercellular pathogens. Imaizumi et al. found that infection with L. monocytogenes induces the expression of RIG-I in vitro and in vivo, and that NLRs likely mediate this effect.176 These results suggest that RIG-I may be involved in innate immune mechanisms to protect the host against parasitic infections.176
VII. RIG-I in Disease
In vivo studies and work in clinical samples suggest that RIG-I may play important roles in immune responses associated with both noninfectious and infectious diseases.
A. Atherosclerosis
Atherosclerosis is best described as a chronic inflammatory condition that can be converted into an acute clinical event by plaque rupture and thrombosis.177 The observation that IFN-γ production by proatherogenic/pro-inflammatory Th1 (T-helper 1) cells is a feature of atherosclerosis prompted Imaizumi et al. to characterize RIG-I expression in atherosclerotic plaques.164 These studies revealed robust immunohistochemical expression of RIG-I localized to intimal macrophages recruited to human aortic lesions. Interestingly, while the monocytic cell line THP-1 displayed modest responses to IFN-γ stimulation, differentiation into macrophage-like cells robustly increased IFN-γ-induced RIG-I expression. These studies suggest that RIG-I may play an active role in the pathogenesis of atherosclerosis through currently unknown mechanisms.
B. Psoriasis
Psoriasis vulgaris is an inflammatory disease of the skin and could arguably be considered the most prevalent cell-mediated inflammatory condition in humans.178 Clinically, psoriatic lesions present as erythematous, sharply demarcated, raised, scaly plaques. It is generally accepted that the presence of clonal populations of T cells in these lesions represents recruitment in response to unidentified cutaneous antigens.179,180 Interestingly, the levels of IFN-γ and RIG-I are significantly elevated in the epidermis of psoriasis patients181 compared with normal skin cells.182,183 It is tempting to speculate that expression of RIG-I in psoriatic lesions represents a regional immune response, and that IFN-γ may be important for this process. Future studies should characterize the functional consequences of RIG-I up-regulation in psoriatic patients. Work along these lines could generate important information to provide the basis for interventional approaches aimed at targeting this axis for the treatment of this autoimmune skin disease.
C. Rheumatoid Arthritis
Rheumatoid arthritis is a chronic autoimmune disease that affects multiple joints, and its pathogenesis is thought to involve Th1-type responses.184 In rheumatoid arthritis, joints express modest levels of IFN-γ,185 and mice deficient in IFN-γ receptors develop accelerated collagen-induced arthritis.186,187 These results do not support a clear role for IFN-γ in the pathogenesis of rheumatoid arthritis, and actually suggest possible beneficial effects of this cytokine in the pathogenesis of the disease.
van Holten et al. reported high levels of IFN-β in synovial tissues from rheumatoid arthritis patients and low levels of expression in tissues from patients with osteoarthritis.188 Interestingly, RIG-I expression mirrored that of IFN-β: that is, it was high in fibroblast-like synoviocytes from rheumatoid arthritis patients and low in tissues isolated from subjects diagnosed with osteoarthritis.189 TNF-α, one of the main cytokines produced during the development of rheumatoid arthritis and a prime target for immunotherapy, induces RIG-I expression in rheumatoid synoviocytes in an IFN-γ-dependent fashion.189 Further work will be required to rigorously test whether combinatorial approaches aimed at targeting IFNs or IFN-dependent signaling and RIG-I could be an effective therapeutic strategy for the treatment of rheumatoid arthritis.
D. Enterocolitis
The gastrointestinal mucosa is continuously being exposed to a variety of agents, including dietary antigens and microbiota. The ability of the gastrointestinal tract to distinguish pathogens from beneficial nutrients and normal microflora is a crucial homeostatic function.190 Membrane-bound TLRs are thought to be responsible for the immunotolerance characteristic of the healthy gut. Healthy intestinal epithelial cells acquire hyporesponsiveness to the immunostimulatory activity of bacterial components by down-regulating surface TLR expression.191,192 RIG-I is constitutively expressed on epithelial cells lining the gut mucosa,169 and has been proposed to be important for innate antiviral responses in this organ, including inhibition of both viral replication and virus-induced apoptosis.58 Evidence supporting a role for RIG-I in the intestine came from studies in Rig-I−/− mice. These animals are viable and fertile, but develop a colitis-like phenotype and display increased susceptibility to dextran sodium sulfate-induced colitis.193 The phenotype is similar to that observed in G protein αi2 subunit (Gαi2)-deficient mice, as it is characterized by a decrease in the number and size of Peyer's patches.193,194 In addition, Rig-I−/− mice display abnormal activation of peripheral T cells and decreased expression of the G protein αi2 subunit.193 Conversely, ATRA-induced RIG-I expression is accompanied by elevated Gαi2 levels in various tissues.193 These results suggest possible relationships in the control of RIG-I and Gαi2 expression, and reveal a potential novel role for RIG-I in the regulation of intestinal mucosal immunity.
E. Type 1 Diabetes
Type 1 diabetes is a chronic disease resulting from the autoimmune destruction of insulin-producing β cells in the pancreas.195 Accumulating evidence indicates that both genetic and environmental factors are important for the development of type 1 diabetes196,197; environmental agents involved in triggering this disease include toxins and food antigens.197 A variety of studies indicate that viral infections also play causal roles as modulators of type 1 diabetes development through multiple mechanisms.198 The presence of enterovirus or enteroviral RNA in the serum and in pancreatic islets of newly diagnosed type 1 diabetes in children and adults supports this possibility.199 Studies by Dogusan et al. demonstrated that stimulation of pancreatic β cells with poly (I:C) activates IRF-3 and NF-κB, inducing production of type I IFNs that contribute to cytosolic apoptosis in a TLR3-independent process.200 Additional work by the same group revealed that these responses occur, at least in part, through activation of RIG-I.199 Studies aimed at identifying variants associated with altered incidence of type 1 diabetes showed that four rare variants predicted to alter the expression levels and structure of another RLR (MDA5) are independently associated with decreased incidence of type 1 diabetes.201 These results firmly established that MDA5 is a disease-causing gene, and suggested that variants predicted to reduce MDA5 expression or activity would decrease the risk of type 1 diabetes.201 Recent molecular and functional analyses of seven naturally occurring RIG-I variants revealed that transversion of serine 183 into isoleucine inactivates the second CARD, and results in a dominant inhibitory phenotype.202 It remains to be determined whether this variant predisposes to type 1 diabetes, but these data, combined with the observation that attenuated function of MDA5 is associated with decreased incidence of the disease, strongly suggest that RLRs may play an important role in the pathogenesis of type 1 diabetes. Future studies should evaluate the potential utility of interventional approaches aimed at limiting expression and/or signaling through RLRs using animal models of type 1 diabetes.
VIII. Summary and Conclusions
Virologists and other scientists have dedicated much effort to the identification and characterization of mechanisms responsible for the recognition of viruses by mammalian hosts. The results of these studies led to the discovery of RIG-I and related proteins, and the elucidation of viral features required for recognition and stimulation of antiviral responses. These analyses also unveiled the existence of sophisticated downstream events necessary for signal activation and complex interactions between innate and adaptive immune responses. In addition, work from multiple laboratories provided novel information regarding the mechanisms that have evolved to optimally regulate signaling through RIG-I. Complementary analyses showed that viruses have themselves developed targeted strategies to evade detection by mammalian sensors. Continued investigations along these lines are likely to have a major impact on our ability to modulate the innate immune response so that we can effectively treat viral infections. For example, we may be able to capitalize on RIG-I- and other RLR-mediated signaling by developing specific agonists such as synthetic dsRNAs that could be used to treat viral infections and perhaps other conditions. These approaches could perhaps be complemented with inhibitors that target viral strategies known to limit host detection through RLRs. Finally, emerging functions of RIG-I that are independent of its role as a cytoplasmic viral sensor are opening new avenues for further discovery and development of interventional approaches. For example, inhibition of RIG-I-mediated signaling has potential utility for the treatment of exacerbated immune responses, such as those typical of autoimmune disorders. Much remains to be unveiled in this exciting, relatively new field of investigation. Continued molecular studies in this area will allow us to gain deeper understanding of how the immune system orchestrates its responses, and will likely result in the identification of new strategies for the treatment of immune-related disorders and viral infections.
Acknowledgments
We thank Dr. Tadaatsu Imaizumi for helpful discussions. This work was supported by National Institutes of Health grants HL35828 and CA073992, and by the Huntsman Cancer Foundation.
Abbreviations
- 5′ppp
5′-triphosphate
- ATRA
all-trans retinoic acid
- CARD
caspase recruitment domain
- Cardif
CARD adaptor-inducing interferon-β
- DExH/D
Asp-Glu-X-His/Asp
- DEAD
Asp-Glu-Ala-Asp
- DDX3
DEAD box polypeptide 3
- DRH
Dicer-related helicase
- ds
double-stranded
- DUBA
de-ubiquitinating enzyme A
- FADD
Fas-associated via death-domain
- Gαi2
G protein αi2 subunit
- HSP90
heat shock protein 90
- IFN
interferon
- I-κB
inhibitor of NF-κB
- IKK
I-κb kinase
- IL
interleukin
- IPS-1
IFN-β promoter stimulator-1
- IRAK
IL-1R-associated kinase
- IRF
interferon regulatory factor
- ISG
interferon-stimulated gene
- LGP
Laboratory of Genetics and Physiology
- LPS
lipopolysaccharide
- MAVS
mitochondrial antiviral signaling
- MDA
melanoma differentiation-associated gene
- miRNA
micro-RNA
- NAK
NF-κB-activating kinase
- NAP
NAK-associated protein
- NEMO
NF-κB essential modulator
- NF-κB
nuclear factor κB
- NLR
NOD-like receptor
- NOD
nucleotide-binding oligomerization domain
- poly (I:C)
polyinosinic-polycytidylic acid
- PRR
pattern-recognition receptor
- RIG
retinoic acid-inducible gene
- RIP
receptor-interacting protein
- RISC
RNA-induced silencing complex
- RLR
retinoic acid-inducible gene-like receptor
- SIKE
suppressor of IKKε
- SNP
single nucleotide polymorphism
- ss
single-stranded
- STAT
signal transducer and activator of transcription
- STING
stimulator of interferon genes
- TANK
TRAF family member-associated NF-κB activator
- TBK
TANK-binding kinase
- Th1
T-helper 1
- TIM
TRAF-interacting motif
- TLR
Toll-like receptor
- TNF
tumor necrosis factor
- TRADD
TNF receptor-associated death domain
- TRAF
tumor necrosis factor receptor-associated factor
- TRIM
tripartite motif
- UTR
untranslated region
- VISA
virus-induced signaling adaptor
References
- 1.Abbas AK, Lichtman AH, Pober J, editors. Cellular and molecular immunology. Philadelphia: W.B. Saunders; 2000. [Google Scholar]
- 2.Iwasaki A, Medzhitov R. Toll-like receptor control of the adaptive immune responses. Nat Immunol. 2004;5:987–95. doi: 10.1038/ni1112. [DOI] [PubMed] [Google Scholar]
- 3.Kawai T, Akira S. Innate immune recognition of viral infection. Nat Immunol. 2006;7:131–7. doi: 10.1038/ni1303. [DOI] [PubMed] [Google Scholar]
- 4.Meurs EF, Breiman A. The interferon inducing pathways and the hepatitis C virus. World J Gastroenterol. 2007;13:2446–54. doi: 10.3748/wjg.v13.i17.2446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell. 2006;124:783–801. doi: 10.1016/j.cell.2006.02.015. [DOI] [PubMed] [Google Scholar]
- 6.Trinchieri G, Sher A. Cooperation of Toll-like receptor signals in innate immune defence. Nat Rev Immunol. 2007;7:179–90. doi: 10.1038/nri2038. [DOI] [PubMed] [Google Scholar]
- 7.Schwer B. A new twist on RNA helicases: DExH/D box proteins as RNPases. Nat Struct Biol. 2001;8:113–6. doi: 10.1038/84091. [DOI] [PubMed] [Google Scholar]
- 8.Kanneganti TD, Lamkanfi M, Nunez G. Intracellular NOD-like receptors in host defense and disease. Immunity. 2007;27:549–59. doi: 10.1016/j.immuni.2007.10.002. [DOI] [PubMed] [Google Scholar]
- 9.Yoneyama M, Kikuchi M, Natsukawa T, Shinobu N, Imaizumi T, Miyagishi M, Taira K, Akira S, Fujita T. The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat Immunol. 2004;5:730–7. doi: 10.1038/ni1087. [DOI] [PubMed] [Google Scholar]
- 10.Bork P, Koonin EV. An expanding family of helicases within the ‘DEAD/H’ superfamily. Nucleic Acids Res. 1993;21:751–2. doi: 10.1093/nar/21.3.751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jankowsky E, Jankowsky A. The DExH/D protein family database. Nucleic Acids Res. 2000;28:333–4. doi: 10.1093/nar/28.1.333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu TX, Zhang JW, Tao J, Zhang RB, Zhang QH, Zhao CJ, Tong JH, Lanotte M, Waxman S, Chen SJ, Mao M, Hu GX, Zhu L, Chen Z. Gene expression networks underlying retinoic acid-induced differentiation of acute promyelocytic leukemia cells. Blood. 2000;96:1496–504. [PubMed] [Google Scholar]
- 13.Imaizumi T, Aratani S, Nakajima T, Carlson M, Matsumiya T, Tanji K, Ookawa K, Yoshida H, Tsuchida S, McIntyre TM, Prescott SM, Zimmerman GA, Satoh K. Retinoic acid-inducible gene-I is induced in endothelial cells by LPS and regulates expression of COX-2. Biochem Biophys Res Commun. 2002;292:274–9. doi: 10.1006/bbrc.2002.6650. [DOI] [PubMed] [Google Scholar]
- 14.Cui XF, Imaizumi T, Yoshida H, Borden EC, Satoh K. Retinoic acid-inducible gene-I is induced by interferon-gamma and regulates the expression of interferon-gamma stimulated gene 15 in MCF-7 cells. Biochem Cell Biol. 2004;82:401–5. doi: 10.1139/o04-041. [DOI] [PubMed] [Google Scholar]
- 15.Imaizumi T, Hatakeyama M, Yamashita K, Yoshida H, Ishikawa A, Taima K, Satoh K, Mori F, Wakabayashi K. Interferon-gamma induces retinoic acid-inducible gene-I in endothelial cells. Endothelium. 2004;11:169–73. doi: 10.1080/10623320490512156. [DOI] [PubMed] [Google Scholar]
- 16.Imaizumi T, Yagihashi N, Hatakeyama M, Yamashita K, Ishikawa A, Taima K, Yoshida H, Inoue I, Fujita T, Yagihashi S, Satoh K. Expression of retinoic acid-inducible gene-I in vascular smooth muscle cells stimulated with interferon-gamma. Life Sci. 2004;75:1171–80. doi: 10.1016/j.lfs.2004.01.030. [DOI] [PubMed] [Google Scholar]
- 17.Imaizumi T, Yagihashi N, Hatakeyama M, Yamashita K, Ishikawa A, Taima K, Yoshida H, Yagihashi S, Satoh K. Upregulation of retinoic acid-inducible gene-I in T24 urinary bladder carcinoma cells stimulated with interferon-gamma. Tohoku J Exp Med. 2004;203:313–8. doi: 10.1620/tjem.203.313. [DOI] [PubMed] [Google Scholar]
- 18.Tabara H, Yigit E, Siomi H, Mello CC. The dsRNA binding protein RDE-4 interacts with RDE-1, DCR-1, and a DExH-box helicase to direct RNAi in C. elegans. Cell. 2002;109:861–71. doi: 10.1016/s0092-8674(02)00793-6. [DOI] [PubMed] [Google Scholar]
- 19.Yoneyama M, Fujita T. Recognition of viral nucleic acids in innate immunity. Rev Med Virol. 2010;20:4–22. doi: 10.1002/rmv.633. [DOI] [PubMed] [Google Scholar]
- 20.Kovacsovics M, Martinon F, Micheau O, Bodmer JL, Hofmann K, Tschopp J. Overexpression of Helicard, a CARD-containing helicase cleaved during apoptosis, accelerates DNA degradation. Curr Biol. 2002;12:838–43. doi: 10.1016/s0960-9822(02)00842-4. [DOI] [PubMed] [Google Scholar]
- 21.Kang DC, Gopalkrishnan RV, Wu Q, Jankowsky E, Pyle AM, Fisher PB. mda-5: An interferon-inducible putative RNA helicase with double-stranded RNA-dependent AT-Pase activity and melanoma growth-suppressive properties. Proc Natl Acad Sci U S A. 2002;99:637–42. doi: 10.1073/pnas.022637199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu X, Robinson GW, Wagner KU, Garrett L, Wynshaw-Boris A, Hennighausen L. Stat5a is mandatory for adult mammary gland development and lactogenesis. Genes Dev. 1997;11:179–86. doi: 10.1101/gad.11.2.179. [DOI] [PubMed] [Google Scholar]
- 23.Miyoshi K, Shillingford JM, Smith GH, Grimm SL, Wagner KU, Oka T, Rosen JM, Robinson GW, Hennighausen L. Signal transducer and activator of transcription (Stat) 5 controls the proliferation and differentiation of mammary alveolar epithelium. J Cell Biol. 2001;155:531–42. doi: 10.1083/jcb.200107065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cui Y, Li M, Walton KD, Sun K, Hanover JA, Furth PA, Hennighausen L. The Stat3/5 locus encodes novel endoplasmic reticulum and helicase-like proteins that are preferentially expressed in normal and neoplastic mammary tissue. Genomics. 2001;78:129–34. doi: 10.1006/geno.2001.6661. [DOI] [PubMed] [Google Scholar]
- 25.Satoh T, Kato H, Kumagai Y, Yoneyama M, Sato S, Matsushita K, Tsujimura T, Fujita T, Akira S, Takeuchi O. LGP2 is a positive regulator of RIG-I- and MDA5-mediated antiviral responses. Proc Natl Acad Sci U S A. 2010;107:1512–7. doi: 10.1073/pnas.0912986107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Barral PM, Sarkar D, Su ZZ, Barber GN, DeSalle R, Racaniello VR, Fisher PB. Functions of the cytoplasmic RNA sensors RIG-I and MDA-5: key regulators of innate immunity. Pharmacol Ther. 2009;124:219–34. doi: 10.1016/j.pharmthera.2009.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gee P, Chua PK, Gevorkyan J, Klumpp K, Najera I, Swinney DC, Deval J. Essential role of the N-terminal domain in the regulation of RIG-I ATPase activity. J Biol Chem. 2008;283:9488–96. doi: 10.1074/jbc.M706777200. [DOI] [PubMed] [Google Scholar]
- 28.Seth RB, Sun L, Ea CK, Chen ZJ. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. Cell. 2005;122:669–82. doi: 10.1016/j.cell.2005.08.012. [DOI] [PubMed] [Google Scholar]
- 29.Kawai T, Takahashi K, Sato S, Coban C, Kumar H, Kato H, Ishii KJ, Takeuchi O, Akira S. IPS-1, an adaptor triggering RIG-I- and Mda5-mediated type I interferon induction. Nat Immunol. 2005;6:981–8. doi: 10.1038/ni1243. [DOI] [PubMed] [Google Scholar]
- 30.Xu LG, Wang YY, Han KJ, Li LY, Zhai Z, Shu HB. VISA is an adapter protein required for virus-triggered IFN-beta signaling. Mol Cell. 2005;19:727–40. doi: 10.1016/j.molcel.2005.08.014. [DOI] [PubMed] [Google Scholar]
- 31.Meylan E, Curran J, Hofmann K, Moradpour D, Binder M, Bartenschlager R, Tschopp J. Cardif is an adaptor protein in the RIG-I antiviral pathway and is targeted by hepatitis C virus. Nature. 2005;437:1167–72. doi: 10.1038/nature04193. [DOI] [PubMed] [Google Scholar]
- 32.Baum A, Garcia-Sastre A. Induction of type I interferon by RNA viruses: cellular receptors and their substrates. Amino Acids. 2010;38:1283–99. doi: 10.1007/s00726-009-0374-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Saito T, Hirai R, Loo YM, Owen D, Johnson CL, Sinha SC, Akira S, Fujita T, Gale M., Jr Regulation of innate antiviral defenses through a shared repressor domain in RIG-I and LGP2. Proc Natl Acad Sci U S A. 2007;104:582–7. doi: 10.1073/pnas.0606699104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cordin O, Banroques J, Tanner NK, Linder P. The DEAD-box protein family of RNA helicases. Gene. 2006;367:17–37. doi: 10.1016/j.gene.2005.10.019. [DOI] [PubMed] [Google Scholar]
- 35.Marques JT, Devosse T, Wang D, Zamanian-Daryoush M, Serbinowski P, Hartmann R, Fujita T, Behlke MA, Williams BR. A structural basis for discriminating between self and nonself double-stranded RNAs in mammalian cells. Nat Biotechnol. 2006;24:559–65. doi: 10.1038/nbt1205. [DOI] [PubMed] [Google Scholar]
- 36.Schlee M, Roth A, Hornung V, Hagmann CA, Wimmenauer V, Barchet W, Coch C, Janke M, Mihailovic A, Wardle G, Juranek S, Kato H, Kawai T, Poeck H, Fitzgerald KA, Takeuchi O, Akira S, Tuschl T, Latz E, Ludwig J, Hartmann G. Recognition of 5′ triphosphate by RIG-I helicase requires short blunt double-stranded RNA as contained in panhandle of negative-strand virus. Immunity. 2009;31:25–34. doi: 10.1016/j.immuni.2009.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schmidt A, Schwerd T, Hamm W, Hellmuth JC, Cui S, Wenzel M, Hoffmann FS, Michallet MC, Besch R, Hopfner KP, Endres S, Rothenfusser S. 5′-triphosphate RNA requires base-paired structures to activate antiviral signaling via RIG-I. Proc Natl Acad Sci U S A. 2009;106:12067–72. doi: 10.1073/pnas.0900971106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Takahasi K, Yoneyama M, Nishihori T, Hirai R, Kumeta H, Narita R, Gale M, Jr, Inagaki F, Fujita T. Nonself RNA-sensing mechanism of RIG-I helicase and activation of antiviral immune responses. Mol Cell. 2008;29:428–40. doi: 10.1016/j.molcel.2007.11.028. [DOI] [PubMed] [Google Scholar]
- 39.Takeuchi O, Akira S. Innate immunity to virus infection. Immunol Rev. 2009;227:75–86. doi: 10.1111/j.1600-065X.2008.00737.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bamming D, Horvath CM. Regulation of signal transduction by enzymatically inactive antiviral RNA helicase proteins MDA5, RIG-I, and LGP2. J Biol Chem. 2009;284:9700–12. doi: 10.1074/jbc.M807365200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gitlin L, Barchet W, Gilfillan S, Cella M, Beutler B, Flavell RA, Diamond MS, Colonna M. Essential role of mda-5 in type I IFN responses to polyriboinosinic:polyribocy tidylic acid and encephalomyocarditis picornavirus. Proc Natl Acad Sci U S A. 2006;103:8459–64. doi: 10.1073/pnas.0603082103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kumar H, Kawai T, Kato H, Sato S, Takahashi K, Coban C, Yamamoto M, Uematsu S, Ishii KJ, Takeuchi O, Akira S. Essential role of IPS-1 in innate immune responses against RNA viruses. J Exp Med. 2006;203:1795–803. doi: 10.1084/jem.20060792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sun Q, Sun L, Liu HH, Chen X, Seth RB, Forman J, Chen ZJ. The specific and essential role of MAVS in antiviral innate immune responses. Immunity. 2006;24:633–42. doi: 10.1016/j.immuni.2006.04.004. [DOI] [PubMed] [Google Scholar]
- 44.Cui S, Eisenacher K, Kirchhofer A, Brzozka K, Lammens A, Lammens K, Fujita T, Conzelmann KK, Krug A, Hopfner KP. The C-terminal regulatory domain is the RNA 5′-triphosphate sensor of RIG-I. Mol Cell. 2008;29:169–79. doi: 10.1016/j.molcel.2007.10.032. [DOI] [PubMed] [Google Scholar]
- 45.Kato H, Takeuchi O, Sato S, Yoneyama M, Yamamoto M, Matsui K, Uematsu S, Jung A, Kawai T, Ishii KJ, Yamaguchi O, Otsu K, Tsujimura T, Koh CS, Reis e Sousa C, Matsuura Y, Fujita T, Akira S. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature. 2006;441:101–5. doi: 10.1038/nature04734. [DOI] [PubMed] [Google Scholar]
- 46.Yoneyama M, Kikuchi M, Matsumoto K, Imaizumi T, Miyagishi M, Taira K, Foy E, Loo YM, Gale M, Jr, Akira S, Yonehara S, Kato A, Fujita T. Shared and unique functions of the DExD/H-box helicases RIG-I, MDA5, and LGP2 in antiviral innate immunity. J Immunol. 2005;175:2851–8. doi: 10.4049/jimmunol.175.5.2851. [DOI] [PubMed] [Google Scholar]
- 47.Komuro A, Horvath CM. RNA- and virus-independent inhibition of antiviral signaling by RNA helicase LGP2. J Virol. 2006;80:12332–42. doi: 10.1128/JVI.01325-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rothenfusser S, Goutagny N, DiPerna G, Gong M, Monks BG, Schoenemeyer A, Yamamoto M, Akira S, Fitzgerald KA. The RNA helicase Lgp2 inhibits TLR-independent sensing of viral replication by retinoic acid-inducible gene-I. J Immunol. 2005;175:5260–8. doi: 10.4049/jimmunol.175.8.5260. [DOI] [PubMed] [Google Scholar]
- 49.Venkataraman T, Valdes M, Elsby R, Kakuta S, Caceres G, Saijo S, Iwakura Y, Barber GN. Loss of DExD/H box RNA helicase LGP2 manifests disparate antiviral responses. J Immunol. 2007;178:6444–55. doi: 10.4049/jimmunol.178.10.6444. [DOI] [PubMed] [Google Scholar]
- 50.Pippig DA, Hellmuth JC, Cui S, Kirchhofer A, Lammens K, Lammens A, Schmidt A, Rothenfusser S, Hopfner KP. The regulatory domain of the RIG-I family ATPase LGP2 senses double-stranded RNA. Nucleic Acids Res. 2009;37:2014–25. doi: 10.1093/nar/gkp059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rehwinkel J, Reis e Sousa C. RIGorous detection: exposing virus through RNA sensing. Science. 2010;327:284–6. doi: 10.1126/science.1185068. [DOI] [PubMed] [Google Scholar]
- 52.Fan L, Briese T, Lipkin WI. Z proteins of New World arenaviruses bind RIG-I and interfere with type I interferon induction. J Virol. 2010;84:1785–91. doi: 10.1128/JVI.01362-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Loo YM, Fornek J, Crochet N, Bajwa G, Perwitasari O, Martinez-Sobrido L, Akira S, Gill MA, Garcia-Sastre A, Katze MG, Gale M., Jr Distinct RIG-I and MDA5 signaling by RNA viruses in innate immunity. J Virol. 2008;82:335–45. doi: 10.1128/JVI.01080-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Liao S, Bao X, Liu T, Lai S, Li K, Garofalo RP, Casola A. Role of retinoic acid inducible gene-I in human metapneumovirus-induced cellular signalling. J Gen Virol. 2008;89:1978–86. doi: 10.1099/vir.0.2008/000778-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Habjan M, Andersson I, Klingstrom J, Schumann M, Martin A, Zimmermann P, Wagner V, Pichlmair A, Schneider U, Muhlberger E, Mirazimi A, Weber F. Processing of genome 5′ termini as a strategy of negative-strand RNA viruses to avoid RIG-I-dependent interferon induction. PLoS One. 2008;3:e2032. doi: 10.1371/journal.pone.0002032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Plumet S, Herschke F, Bourhis JM, Valentin H, Longhi S, Gerlier D. Cytosolic 5′-triphosphate ended viral leader transcript of measles virus as activator of the RIG I-mediated interferon response. PLoS One. 2007;2:e279. doi: 10.1371/journal.pone.0000279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Liu P, Jamaluddin M, Li K, Garofalo RP, Casola A, Brasier AR. Retinoic acid-inducible gene I mediates early antiviral response and Toll-like receptor 3 expression in respiratory syncytial virus-infected airway epithelial cells. J Virol. 2007;81:1401–11. doi: 10.1128/JVI.01740-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hirata Y, Broquet AH, Menchen L, Kagnoff MF. Activation of innate immune defense mechanisms by signaling through RIG-I/IPS-1 in intestinal epithelial cells. J Immunol. 2007;179:5425–32. doi: 10.4049/jimmunol.179.8.5425. [DOI] [PubMed] [Google Scholar]
- 59.Chen Z, Benureau Y, Rijnbrand R, Yi J, Wang T, Warter L, Lanford RE, Weinman SA, Lemon SM, Martin A, Li K. GB virus B disrupts RIG-I signaling by NS3/4A-mediated cleavage of the adaptor protein MAVS. J Virol. 2007;81:964–76. doi: 10.1128/JVI.02076-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Samanta M, Iwakiri D, Kanda T, Imaizumi T, Takada K. EB virus-encoded RNAs are recognized by RIG-I and activate signaling to induce type I IFN. Embo J. 2006;25:4207–14. doi: 10.1038/sj.emboj.7601314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Matikainen S, Siren J, Tissari J, Veckman V, Pirhonen J, Severa M, Sun Q, Lin R, Meri S, Uze G, Hiscott J, Julkunen I. Tumor necrosis factor alpha enhances influenza A virus-induced expression of antiviral cytokines by activating RIG-I gene expression. J Virol. 2006;80:3515–22. doi: 10.1128/JVI.80.7.3515-3522.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hornung V, Ellegast J, Kim S, Brzozka K, Jung A, Kato H, Poeck H, Akira S, Conzelmann KK, Schlee M, Endres S, Hartmann G. 5′-Triphosphate RNA is the ligand for RIG-I. Science. 2006;314:994–7. doi: 10.1126/science.1132505. [DOI] [PubMed] [Google Scholar]
- 63.Fredericksen BL, Gale M., Jr West Nile virus evades activation of interferon regulatory factor 3 through RIG-I-dependent and -independent pathways without antagonizing host defense signaling. J Virol. 2006;80:2913–23. doi: 10.1128/JVI.80.6.2913-2923.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chang TH, Liao CL, Lin YL. Flavivirus induces interferon-beta gene expression through a pathway involving RIG-I-dependent IRF-3 and PI3K-dependent NF-kappaB activation. Microbes Infect. 2006;8:157–71. doi: 10.1016/j.micinf.2005.06.014. [DOI] [PubMed] [Google Scholar]
- 65.Sumpter R, Jr, Loo YM, Foy E, Li K, Yoneyama M, Fujita T, Lemon SM, Gale M., Jr Regulating intracellular antiviral defense and permissiveness to hepatitis C virus RNA replication through a cellular RNA helicase, RIG-I. J Virol. 2005;79:2689–99. doi: 10.1128/JVI.79.5.2689-2699.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Pichlmair A, Schulz O, Tan CP, Naslund TI, Liljestrom P, Weber F, Reis e Sousa C. RIG-I-mediated antiviral responses to single-stranded RNA bearing 5′-phosphates. Science. 2006;314:997–1001. doi: 10.1126/science.1132998. [DOI] [PubMed] [Google Scholar]
- 67.Garcin D, Lezzi M, Dobbs M, Elliott RM, Schmaljohn C, Kang CY, Kolakofsky D. The 5′ ends of Hantaan virus (Bunyaviridae) RNAs suggest a prime-and-realign mechanism for the initiation of RNA synthesis. J Virol. 1995;69:5754–62. doi: 10.1128/jvi.69.9.5754-5762.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Nallagatla SR, Toroney R, Bevilacqua PC. A brilliant disguise for self RNA: 5′-end and internal modifications of primary transcripts suppress elements of innate immunity. RNA Biol. 2008;5:140–4. doi: 10.4161/rna.5.3.6839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Marq JB, Kolakofsky D, Garcin D. Unpaired 5′ ppp-nucleotides, as found in arenavirus dsRNA panhandles, are not recognized by RIG-I. J Biol Chem. 2010;285:18208–16. doi: 10.1074/jbc.M109.089425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kato H, Takeuchi O, Mikamo-Satoh E, Hirai R, Kawai T, Matsushita K, Hiiragi A, Dermody TS, Fujita T, Akira S. Length-dependent recognition of double-stranded ribonucleic acids by retinoic acid-inducible gene-I and melanoma differentiation-associated gene 5. J Exp Med. 2008;205:1601–10. doi: 10.1084/jem.20080091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Uzri D, Gehrke L. Nucleotide sequences and modifications that determine RIG-I/RNA binding and signaling activities. J Virol. 2009;83:4174–84. doi: 10.1128/JVI.02449-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Saito T, Owen DM, Jiang F, Marcotrigiano J, Gale M., Jr Innate immunity induced by composition-dependent RIG-I recognition of hepatitis C virus RNA. Nature. 2008;454:523–7. doi: 10.1038/nature07106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Panne D. The enhanceosome. Curr Opin Struct Biol. 2008;18:236–42. doi: 10.1016/j.sbi.2007.12.002. [DOI] [PubMed] [Google Scholar]
- 74.Matsuda A, Suzuki Y, Honda G, Muramatsu S, Matsuzaki O, Nagano Y, Doi T, Shimotohno K, Harada T, Nishida E, Hayashi H, Sugano S. Large-scale identification and characterization of human genes that activate NF-kappaB and MAPK signaling pathways. Oncogene. 2003;22:3307–18. doi: 10.1038/sj.onc.1206406. [DOI] [PubMed] [Google Scholar]
- 75.Potter JA, Randall RE, Taylor GL. Crystal structure of human IPS-1/MAVS/VISA/Cardif caspase activation recruitment domain. BMC Struct Biol. 2008;8:11. doi: 10.1186/1472-6807-8-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Sun L, Chen ZJ. The novel functions of ubiquitination in signaling. Curr Opin Cell Biol. 2004;16:119–26. doi: 10.1016/j.ceb.2004.02.005. [DOI] [PubMed] [Google Scholar]
- 77.Reymond A, Meroni G, Fantozzi A, Merla G, Cairo S, Luzi L, Riganelli D, Zanaria E, Messali S, Cainarca S, Guffanti A, Minucci S, Pelicci PG, Ballabio A. The tripartite motif family identifies cell compartments. Embo J. 2001;20:2140–51. doi: 10.1093/emboj/20.9.2140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Gack MU, Kirchhofer A, Shin YC, Inn KS, Liang C, Cui S, Myong S, Ha T, Hopfner KP, Jung JU. Roles of RIG-I N-terminal tandem CARD and splice variant in TRIM25-mediated antiviral signal transduction. Proc Natl Acad Sci U S A. 2008;105:16743–8. doi: 10.1073/pnas.0804947105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Gack MU, Shin YC, Joo CH, Urano T, Liang C, Sun L, Takeuchi O, Akira S, Chen Z, Inoue S, Jung JU. TRIM25 RING-finger E3 ubiquitin ligase is essential for RIG-I-mediated antiviral activity. Nature. 2007;446:916–20. doi: 10.1038/nature05732. [DOI] [PubMed] [Google Scholar]
- 80.Garcia-Sastre A, Egorov A, Matassov D, Brandt S, Levy DE, Durbin JE, Palese P, Muster T. Influenza A virus lacking the NS1 gene replicates in interferon-deficient systems. Virology. 1998;252:324–30. doi: 10.1006/viro.1998.9508. [DOI] [PubMed] [Google Scholar]
- 81.Gack MU, Albrecht RA, Urano T, Inn KS, Huang IC, Carnero E, Farzan M, Inoue S, Jung JU, Garcia-Sastre A. Influenza A virus NS1 targets the ubiquitin ligase TRIM25 to evade recognition by the host viral RNA sensor RIG-I. Cell Host Microbe. 2009;5:439–49. doi: 10.1016/j.chom.2009.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Zeng W, Sun L, Jiang X, Chen X, Hou F, Adhikari A, Xu M, Chen ZJ. Reconstitution of the RIG-I pathway reveals a signaling role of unanchored polyubiquitin chains in innate immunity. Cell. 2010;141:315–30. doi: 10.1016/j.cell.2010.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Bennett EJ, Harper JW. Ubiquitin gets CARDed. Cell. 2010;141:220–2. doi: 10.1016/j.cell.2010.03.047. [DOI] [PubMed] [Google Scholar]
- 84.Gao D, Yang YK, Wang RP, Zhou X, Diao FC, Li MD, Zhai ZH, Jiang ZF, Chen DY. REUL is a novel E3 ubiquitin ligase and stimulator of retinoic-acid-inducible gene-I. PLoS One. 2009;4:e5760. doi: 10.1371/journal.pone.0005760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Oshiumi H, Matsumoto M, Hatakeyama S, Seya T. Riplet/RNF135, a RING finger protein, ubiquitinates RIG-I to promote interferon-beta induction during the early phase of viral infection. J Biol Chem. 2009;284:807–17. doi: 10.1074/jbc.M804259200. [DOI] [PubMed] [Google Scholar]
- 86.Komuro A, Bamming D, Horvath CM. Negative regulation of cytoplasmic RNA-mediated antiviral signaling. Cytokine. 2008;43:350–8. doi: 10.1016/j.cyto.2008.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Friedman CS, O'Donnell MA, Legarda-Addison D, Ng A, Cardenas WB, Yount JS, Moran TM, Basler CF, Komuro A, Horvath CM, Xavier R, Ting AT. The tumour suppressor CYLD is a negative regulator of RIG-I-mediated antiviral response. EMBO Rep. 2008;9:930–6. doi: 10.1038/embor.2008.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Nistal-Villan E, Gack MU, Martinez-Delgado G, Maharaj NP, Inn K, Yang H, Wang R, Aggarwal AK, Jung JU, Garcia-Sastre A. Negative role of RIG-I serine 8 phosphorylation in the regulation of IFN-{beta} production. J Biol Chem. 2010;285:20252–61. doi: 10.1074/jbc.M109.089912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Gack MU, Nistal-Villan E, Inn KS, Garcia-Sastre A, Jung JU. Phosphorylation-mediated negative regulation of RIG-I antiviral activity. J Virol. 2010;84:3220–9. doi: 10.1128/JVI.02241-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Arimoto K, Takahashi H, Hishiki T, Konishi H, Fujita T, Shimotohno K. Negative regulation of the RIG-I signaling by the ubiquitin ligase RNF125. Proc Natl Acad Sci U S A. 2007;104:7500–5. doi: 10.1073/pnas.0611551104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Matsumiya T, Imaizumi T, Yoshida H, Satoh K, Topham MK, Stafforini DM. The levels of retinoic acid-inducible gene I are regulated by heat shock protein 90-alpha. J Immunol. 2009;182:2717–25. doi: 10.4049/jimmunol.0802933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Zhao C, Denison C, Huibregtse JM, Gygi S, Krug RM. Human ISG15 conjugation targets both IFN-induced and constitutively expressed proteins functioning in diverse cellular pathways. Proc Natl Acad Sci U S A. 2005;102:10200–5. doi: 10.1073/pnas.0504754102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Kim MJ, Hwang SY, Imaizumi T, Yoo JY. Negative feedback regulation of RIG-I-mediated antiviral signaling by interferon-induced ISG15 conjugation. J Virol. 2008;82:1474–83. doi: 10.1128/JVI.01650-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Malakhov MP, Malakhova OA, Kim KI, Ritchie KJ, Zhang DE. UBP43 (USP18) specifically removes ISG15 from conjugated proteins. J Biol Chem. 2002;277:9976–81. doi: 10.1074/jbc.M109078200. [DOI] [PubMed] [Google Scholar]
- 95.Myong S, Cui S, Cornish PV, Kirchhofer A, Gack MU, Jung JU, Hopfner KP, Ha T. Cytosolic viral sensor RIG-I is a 5′-triphosphate-dependent translocase on double-stranded RNA. Science. 2009;323:1070–4. doi: 10.1126/science.1168352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Dixit E, Boulant S, Zhang Y, Lee AS, Odendall C, Shum B, Hacohen N, Chen ZJ, Whelan SP, Fransen M, Nibert ML, Superti-Furga G, Kagan JC. Peroxisomes are signaling platforms for antiviral innate immunity. Cell. 2010;141:668–81. doi: 10.1016/j.cell.2010.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Sharma S, Fitzgerald KA. Viral defense: it takes two MAVS to Tango. Cell. 2010;141:570–2. doi: 10.1016/j.cell.2010.04.043. [DOI] [PubMed] [Google Scholar]; Zhong B, Yang Y, Li S, Wang YY, Li Y, Diao F, Lei C, He X, Zhang L, Tien P, Shu HB. The adaptor protein MITA links virus-sensing receptors to IRF3 transcription factor activation. Immunity. 2008;29:538–50. doi: 10.1016/j.immuni.2008.09.003. [DOI] [PubMed] [Google Scholar]
- 98.Yoneyama M, Fujita T. RNA recognition and signal transduction by RIG-I-like receptors. Immunol Rev. 2009;227:54–65. doi: 10.1111/j.1600-065X.2008.00727.x. [DOI] [PubMed] [Google Scholar]
- 99.Mulhern O, Bowie AG. Unexpected roles for DEAD-box protein 3 in viral RNA sensing pathways. Eur J Immunol. 2010;40:933–5. doi: 10.1002/eji.201040447. [DOI] [PubMed] [Google Scholar]
- 100.Oshiumi H, Sakai K, Matsumoto M, Seya T. DEAD/H BOX 3 (DDX3) helicase binds the RIG-I adaptor IPS-1 to up-regulate IFN-beta-inducing potential. Eur J Immunol. 2010;40:940–8. doi: 10.1002/eji.200940203. [DOI] [PubMed] [Google Scholar]
- 101.Jounai N, Takeshita F, Kobiyama K, Sawano A, Miyawaki A, Xin KQ, Ishii KJ, Kawai T, Akira S, Suzuki K, Okuda K. The Atg5 Atg12 conjugate associates with innate antiviral immune responses. Proc Natl Acad Sci U S A. 2007;104:14050–5. doi: 10.1073/pnas.0704014104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Moore CB, Bergstralh DT, Duncan JA, Lei Y, Morrison TE, Zimmermann AG, Accavitti-Loper MA, Madden VJ, Sun L, Ye Z, Lich JD, Heise MT, Chen Z, Ting JP. NLRX1 is a regulator of mitochondrial antiviral immunity. Nature. 2008;451:573–7. doi: 10.1038/nature06501. [DOI] [PubMed] [Google Scholar]
- 103.Xu L, Xiao N, Liu F, Ren H, Gu J. Inhibition of RIG-I and MDA5-dependent antiviral response by gC1qR at mitochondria. Proc Natl Acad Sci U S A. 2009;106:1530–5. doi: 10.1073/pnas.0811029106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Irving A, Williams BR. Latest advances in innate antiviral defence. F1000 Biol Reports. 2009;1:nihpa103883. doi: 10.3410/B1-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Oganesyan G, Saha SK, Guo B, He JQ, Shahangian A, Zarnegar B, Perry A, Cheng G. Critical role of TRAF3 in the Toll-like receptor-dependent and -independent antiviral response. Nature. 2006;439:208–11. doi: 10.1038/nature04374. [DOI] [PubMed] [Google Scholar]
- 106.Saha SK, Pietras EM, He JQ, Kang JR, Liu SY, Oganesyan G, Shahangian A, Zarnegar B, Shiba TL, Wang Y, Cheng G. Regulation of antiviral responses by a direct and specific interaction between TRAF3 and Cardif. Embo J. 2006;25:3257–63. doi: 10.1038/sj.emboj.7601220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.O'Neill LA, Bowie AG. Sensing and signaling in antiviral innate immunity. Curr Biol. 2010;20:R328–33. doi: 10.1016/j.cub.2010.01.044. [DOI] [PubMed] [Google Scholar]
- 108.Zeng W, Xu M, Liu S, Sun L, Chen ZJ. Key role of Ubc5 and lysine-63 polyubiquitination in viral activation of IRF3. Mol Cell. 2009;36:315–25. doi: 10.1016/j.molcel.2009.09.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Kayagaki N, Phung Q, Chan S, Chaudhari R, Quan C, O'Rourke KM, Eby M, Pietras E, Cheng G, Bazan JF, Zhang Z, Arnott D, Dixit VM. DUBA: a deubiquitinase that regulates type I interferon production. Science. 2007;318:1628–32. doi: 10.1126/science.1145918. [DOI] [PubMed] [Google Scholar]
- 110.Nakhaei P, Mesplede T, Solis M, Sun Q, Zhao T, Yang L, Chuang TH, Ware CF, Lin R, Hiscott J. The E3 ubiquitin ligase Triad3A negatively regulates the RIG-I/MAVS signaling pathway by targeting TRAF3 for degradation. PLoS Pathog. 2009;5:e1000650. doi: 10.1371/journal.ppat.1000650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Tang ED, Wang CY. TRAF5 is a downstream target of MAVS in antiviral innate immune signaling. PLoS One. 2010;5:e9172. doi: 10.1371/journal.pone.0009172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Zhao T, Yang L, Sun Q, Arguello M, Ballard DW, Hiscott J, Lin R. The NEMO adaptor bridges the nuclear factor-kappaB and interferon regulatory factor signaling pathways. Nat Immunol. 2007;8:592–600. doi: 10.1038/ni1465. [DOI] [PubMed] [Google Scholar]
- 113.Chariot A, Leonardi A, Muller J, Bonif M, Brown K, Siebenlist U. Association of the adaptor TANK with the I kappa B kinase (IKK) regulator NEMO connects IKK complexes with IKK epsilon and TBK1 kinases. J Biol Chem. 2002;277:37029–36. doi: 10.1074/jbc.M205069200. [DOI] [PubMed] [Google Scholar]
- 114.Huang J, Liu T, Xu LG, Chen D, Zhai Z, Shu HB. SIKE is an IKK epsilon/TBK1-associated suppressor of TLR3-and virus-triggered IRF-3 activation pathways. Embo J. 2005;24:4018–28. doi: 10.1038/sj.emboj.7600863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Hemmi H, Takeuchi O, Sato S, Yamamoto M, Kaisho T, Sanjo H, Kawai T, Hoshino K, Takeda K, Akira S. The roles of two IkappaB kinase-related kinases in lipopolysaccharide and double stranded RNA signaling and viral infection. J Exp Med. 2004;199:1641–50. doi: 10.1084/jem.20040520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Schroder M, Baran M, Bowie AG. Viral targeting of DEAD box protein 3 reveals its role in TBK1/IKKepsilon-mediated IRF activation. Embo J. 2008;27:2147–57. doi: 10.1038/emboj.2008.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Fujita F, Taniguchi Y, Kato T, Narita Y, Furuya A, Ogawa T, Sakurai H, Joh T, Itoh M, Delhase M, Karin M, Nakanishi M. Identification of NAP1, a regulatory subunit of IkappaB kinase-related kinases that potentiates NF-kappaB signaling. Mol Cell Biol. 2003;23:7780–93. doi: 10.1128/MCB.23.21.7780-7793.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.May MJ, D'Acquisto F, Madge LA, Glockner J, Pober JS, Ghosh S. Selective inhibition of NF-kappaB activation by a peptide that blocks the interaction of NEMO with the IkappaB kinase complex. Science. 2000;289:1550–4. doi: 10.1126/science.289.5484.1550. [DOI] [PubMed] [Google Scholar]
- 119.Balachandran S, Thomas E, Barber GN. A FADD-dependent innate immune mechanism in mammalian cells. Nature. 2004;432:401–5. doi: 10.1038/nature03124. [DOI] [PubMed] [Google Scholar]
- 120.Takahashi K, Kawai T, Kumar H, Sato S, Yonehara S, Akira S. Roles of caspase-8 and caspase-10 in innate immune responses to double-stranded RNA. J Immunol. 2006;176:4520–4. doi: 10.4049/jimmunol.176.8.4520. [DOI] [PubMed] [Google Scholar]
- 121.Michallet MC, Meylan E, Ermolaeva MA, Vazquez J, Rebsamen M, Curran J, Poeck H, Bscheider M, Hartmann G, Konig M, Kalinke U, Pasparakis M, Tschopp J. TRADD protein is an essential component of the RIG-like helicase antiviral pathway. Immunity. 2008;28:651–61. doi: 10.1016/j.immuni.2008.03.013. [DOI] [PubMed] [Google Scholar]
- 122.Karin M, Ben-Neriah Y. Phosphorylation meets ubiquitination: the control of NF-[kappa]B activity. Annu Rev Immunol. 2000;18:621–63. doi: 10.1146/annurev.immunol.18.1.621. [DOI] [PubMed] [Google Scholar]
- 123.Yamamoto Y, Kim DW, Kwak YT, Prajapati S, Verma U, Gaynor RB. IKKgamma /NEMO facilitates the recruitment of the IkappaB proteins into the IkappaB kinase complex. J Biol Chem. 2001;276:36327–36. doi: 10.1074/jbc.M104090200. [DOI] [PubMed] [Google Scholar]
- 124.Liu P, Lu M, Tian B, Li K, Garofalo RP, Prusak D, Wood TG, Brasier AR. Expression of an IKKgamma splice variant determines IRF3 and canonical NF-kappaB pathway utilization in ssRNA virus infection. PLoS One. 2009;4:e8079. doi: 10.1371/journal.pone.0008079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Chattopadhyay S, Marques JT, Yamashita M, Peters KL, Smith K, Desai A, Williams BR, Sen GC. Viral apoptosis is induced by IRF-3-mediated activation of Bax. Embo J. 2010;29:1762–73. doi: 10.1038/emboj.2010.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Vince JE, Tschopp J. IRF-3 partners Bax in a viral-induced dance macabre. Embo J. 29:1627–8. doi: 10.1038/emboj.2010.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Barral PM, Sarkar D, Fisher PB, Racaniello VR. RIG-I is cleaved during picornavirus infection. Virology. 2009;391:171–6. doi: 10.1016/j.virol.2009.06.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Sonenberg N, Pelletier J. Poliovirus translation: a paradigm for a novel initiation mechanism. Bioessays. 1989;11:128–32. doi: 10.1002/bies.950110504. [DOI] [PubMed] [Google Scholar]
- 129.Plotch SJ, Bouloy M, Ulmanen I, Krug RM. A unique cap(m7GpppXm)-dependent influenza virion endonuclease cleaves capped RNAs to generate the primers that initiate viral RNA transcription. Cell. 1981;23:847–58. doi: 10.1016/0092-8674(81)90449-9. [DOI] [PubMed] [Google Scholar]
- 130.Bowie AG, Unterholzner L. Viral evasion and subversion of pattern-recognition receptor signalling. Nat Rev Immunol. 2008;8:911–2. doi: 10.1038/nri2436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Paul A. Possible unifying mechanism of Picornavirus genome replication. In: Semler BL, Wimmer E, editors. Molecular biology of picornaviruses. Washington, DC: ASM Press; 2002. pp. 227–46. [Google Scholar]
- 132.Lee YF, Nomoto A, Detjen BM, Wimmer E. A protein covalently linked to poliovirus genome RNA. Proc Natl Acad Sci U S A. 1977;74:59–63. doi: 10.1073/pnas.74.1.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Cardenas WB, Loo YM, Gale M, Jr, Hartman AL, Kimberlin CR, Martinez-Sobrido L, Saphire EO, Basler CF. Ebola virus VP35 protein binds double-stranded RNA and inhibits alpha/beta interferon production induced by RIG-I signaling. J Virol. 2006;80:5168–78. doi: 10.1128/JVI.02199-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Deng L, Dai P, Ding W, Granstein RD, Shuman S. Vaccinia virus infection attenuates innate immune responses and antigen presentation by epidermal dendritic cells. J Virol. 2006;80:9977–87. doi: 10.1128/JVI.00354-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Mibayashi M, Martinez-Sobrido L, Loo YM, Cardenas WB, Gale M, Jr, Garcia-Sastre A. Inhibition of retinoic acid-inducible gene I-mediated induction of beta interferon by the NS1 protein of influenza A virus. J Virol. 2007;81:514–24. doi: 10.1128/JVI.01265-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Chang HW, Watson JC, Jacobs BL. The E3L gene of vaccinia virus encodes an inhibitor of the interferon-induced, double-stranded RNA-dependent protein kinase. Proc Natl Acad Sci U S A. 1992;89:4825–9. doi: 10.1073/pnas.89.11.4825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Marq JB, Hausmann S, Luban J, Kolakofsky D, Garcin D. The double-stranded RNA binding domain of the vaccinia virus E3L protein inhibits both RNA- and DNA-induced activation of interferon beta. J Biol Chem. 2009;284:25471–8. doi: 10.1074/jbc.M109.018895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Guo Z, Chen LM, Zeng H, Gomez JA, Plowden J, Fujita T, Katz JM, Donis RO, Sambhara S. NS1 protein of influenza A virus inhibits the function of intracytoplasmic pathogen sensor, RIG-I. Am J Respir Cell Mol Biol. 2007;36:263–9. doi: 10.1165/rcmb.2006-0283RC. [DOI] [PubMed] [Google Scholar]
- 139.Foy E, Li K, Wang C, Sumpter R, Jr, Ikeda M, Lemon SM, Gale M., Jr Regulation of interferon regulatory factor-3 by the hepatitis C virus serine protease. Science. 2003;300:1145–8. doi: 10.1126/science.1082604. [DOI] [PubMed] [Google Scholar]
- 140.Loo YM, Owen DM, Li K, Erickson AK, Johnson CL, Fish PM, Carney DS, Wang T, Ishida H, Yoneyama M, Fujita T, Saito T, Lee WM, Hagedorn CH, Lau DT, Weinman SA, Lemon SM, Gale M., Jr Viral and therapeutic control of IFN-beta promoter stimulator 1 during hepatitis C virus infection. Proc Natl Acad Sci U S A. 2006;103:6001–6. doi: 10.1073/pnas.0601523103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Breiman A, Grandvaux N, Lin R, Ottone C, Akira S, Yoneyama M, Fujita T, Hiscott J, Meurs EF. Inhibition of RIG-I-dependent signaling to the interferon pathway during hepatitis C virus expression and restoration of signaling by IKKepsilon. J Virol. 2005;79:3969–78. doi: 10.1128/JVI.79.7.3969-3978.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Foy E, Li K, Sumpter R, Jr, Loo YM, Johnson CL, Wang C, Fish PM, Yoneyama M, Fujita T, Lemon SM, Gale M., Jr Control of antiviral defenses through hepatitis C virus disruption of retinoic acid-inducible gene-I signaling. Proc Natl Acad Sci U S A. 2005;102:2986–91. doi: 10.1073/pnas.0408707102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Lin R, Lacoste J, Nakhaei P, Sun Q, Yang L, Paz S, Wilkinson P, Julkunen I, Vitour D, Meurs E, Hiscott J. Dissociation of a MAVS/IPS-1/VISA/Cardif-IKKepsilon molecular complex from the mitochondrial outer membrane by hepatitis C virus NS3-4A proteolytic cleavage. J Virol. 2006;80:6072–83. doi: 10.1128/JVI.02495-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Yang Y, Liang Y, Qu L, Chen Z, Yi M, Li K, Lemon SM. Disruption of innate immunity due to mitochondrial targeting of a picornaviral protease precursor. Proc Natl Acad Sci U S A. 2007;104:7253–8. doi: 10.1073/pnas.0611506104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Rebsamen M, Meylan E, Curran J, Tschopp J. The antiviral adaptor proteins Cardif and Trif are processed and inactivated by caspases. Cell Death Differ. 2008;15:1804–11. doi: 10.1038/cdd.2008.119. [DOI] [PubMed] [Google Scholar]
- 146.Gottwein E, Cullen BR. Viral and cellular microRNAs as determinants of viral pathogenesis and immunity. Cell Host Microbe. 2008;3:375–87. doi: 10.1016/j.chom.2008.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–97. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 148.Hou J, Wang P, Lin L, Liu X, Ma F, An H, Wang Z, Cao X. MicroRNA-146a feedback inhibits RIG-I-dependent type I IFN production in macrophages by targeting TRAF6, IRAK1, and IRAK2. J Immunol. 2009;183:2150–8. doi: 10.4049/jimmunol.0900707. [DOI] [PubMed] [Google Scholar]
- 149.Leszczyniecka M, Roberts T, Dent P, Grant S, Fisher PB. Differentiation therapy of human cancer: basic science and clinical applications. Pharmacol Ther. 2001;90:105–56. doi: 10.1016/s0163-7258(01)00132-2. [DOI] [PubMed] [Google Scholar]
- 150.Zhang NN, Shen SH, Jiang LJ, Zhang W, Zhang HX, Sun YP, Li XY, Huang QH, Ge BX, Chen SJ, Wang ZG, Chen Z, Zhu J. RIG-I plays a critical role in negatively regulating granulocytic proliferation. Proc Natl Acad Sci U S A. 2008;105:10553–8. doi: 10.1073/pnas.0804895105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Kong L, Sun L, Zhang H, Liu Q, Liu Y, Qin L, Shi G, Hu JH, Xu A, Sun YP, Li D, Shi YF, Zang JW, Zhu J, Chen Z, Wang ZG, Ge BX. An essential role for RIG-I in tolllike receptor-stimulated phagocytosis. Cell Host Microbe. 2009;6:150–61. doi: 10.1016/j.chom.2009.06.008. [DOI] [PubMed] [Google Scholar]
- 152.Ivanov VN, Bhoumik A, Ronai Z. Death receptors and melanoma resistance to apoptosis. Oncogene. 2003;22:3152–61. doi: 10.1038/sj.onc.1206456. [DOI] [PubMed] [Google Scholar]
- 153.Kirkwood JM, Ibrahim JG, Sondak VK, Richards J, Flaherty LE, Ernstoff MS, Smith TJ, Rao U, Steele M, Blum RH. High- and low-dose interferon alfa-2b in high-risk melanoma: first analysis of intergroup trial E1690/S9111/C9190. J Clin Oncol. 2000;18:2444–58. doi: 10.1200/JCO.2000.18.12.2444. [DOI] [PubMed] [Google Scholar]
- 154.Poeck H, Besch R, Maihoefer C, Renn M, Tormo D, Morskaya SS, Kirschnek S, Gaffal E, Landsberg J, Hellmuth J, Schmidt A, Anz D, Bscheider M, Schwerd T, Berking C, Bourquin C, Kalinke U, Kremmer E, Kato H, Akira S, Meyers R, Hacker G, Neuenhahn M, Busch D, Ruland J, Rothenfusser S, Prinz M, Hornung V, Endres S, Tuting T, Hartmann G. 5′-Triphosphate-siRNA: turning gene silencing and Rig-I activation against melanoma. Nat Med. 2008;14:1256–63. doi: 10.1038/nm.1887. [DOI] [PubMed] [Google Scholar]
- 155.Petrocca F, Lieberman J. RIG-ing an antitumor response. Nat Med. 2008;14:1152–3. doi: 10.1038/nm1108-1152. [DOI] [PubMed] [Google Scholar]
- 156.Yu HB, Finlay BB. The caspase-1 inflammasome: a pilot of innate immune responses. Cell Host Microbe. 2008;4:198–208. doi: 10.1016/j.chom.2008.08.007. [DOI] [PubMed] [Google Scholar]
- 157.Rintahaka J, Wiik D, Kovanen PR, Alenius H, Matikainen S. Cytosolic antiviral RNA recognition pathway activates caspases 1 and 3. J Immunol. 2008;180:1749–57. doi: 10.4049/jimmunol.180.3.1749. [DOI] [PubMed] [Google Scholar]
- 158.Poeck H, Bscheider M, Gross O, Finger K, Roth S, Rebsamen M, Hannesschlager N, Schlee M, Rothenfusser S, Barchet W, Kato H, Akira S, Inoue S, Endres S, Peschel C, Hartmann G, Hornung V, Ruland J. Recognition of RNA virus by RIG-I results in activation of CARD9 and inflammasome signaling for interleukin 1 beta production. Nat Immunol. 2010;11:63–9. doi: 10.1038/ni.1824. [DOI] [PubMed] [Google Scholar]
- 159.Su ZZ, Sarkar D, Emdad L, Barral PM, Fisher PB. Central role of interferon regulatory factor-1 (IRF-1) in controlling retinoic acid inducible gene-I (RIG-I) expression. J Cell Physiol. 2007;213:502–10. doi: 10.1002/jcp.21128. [DOI] [PubMed] [Google Scholar]
- 160.Masumi A, Ito M, Mochida K, Hamaguchi I, Mizukami T, Momose H, Kuramitsu M, Tsuruhara M, Takizawa K, Kato A, Yamaguchi K. Enhanced RIG-I expression is mediated by interferon regulatory factor-2 in peripheral blood B cells from hepatitis C virus-infected patients. Biochem Biophys Res Commun. 2010;391:1623–8. doi: 10.1016/j.bbrc.2009.12.092. [DOI] [PubMed] [Google Scholar]
- 161.Wang J, Wu S, Jin X, Li M, Chen S, Teeling JL, Perry VH, Gu J. Retinoic acid-inducible gene-I mediates late phase induction of TNF-alpha by lipopolysaccharide. J Immunol. 2008;180:8011–9. doi: 10.4049/jimmunol.180.12.8011. [DOI] [PubMed] [Google Scholar]
- 162.Imaizumi T, Tanaka H, Tajima A, Tsuruga K, Oki E, Sashinami H, Matsumiya T, Yoshida H, Inoue I, Ito E. Retinoic acid-inducible gene-I (RIG-I) is induced by IFN-{gamma} in human mesangial cells in culture: possible involvement of RIG-I in the inflammation in lupus nephritis. Lupus. 2010;19:830–6. doi: 10.1177/0961203309360540. [DOI] [PubMed] [Google Scholar]
- 163.Imaizumi T, Yagihashi N, Kubota K, Yoshida H, Sakaki H, Yagihashi S, Kimura H, Satoh K. Expression of retinoic acid-inducible gene-I (RIG-I) in macrophages: possible involvement of RIG-I in atherosclerosis. J Atheroscler Thromb. 2007;14:51–5. doi: 10.5551/jat.14.51. [DOI] [PubMed] [Google Scholar]
- 164.Matsumiya T, Prescott SM, Stafforini DM. IFN-epsilon mediates TNF-alpha-induced STAT1 phosphorylation and induction of retinoic acid-inducible gene-I in human cervical cancer cells. J Immunol. 2007;179:4542–9. doi: 10.4049/jimmunol.179.7.4542. [DOI] [PubMed] [Google Scholar]
- 165.Imaizumi T, Kumagai M, Taima K, Fujita T, Yoshida H, Satoh K. Involvement of retinoic acid-inducible gene-I in the IFN-{gamma}/STAT1 signalling pathway in BEAS-2B cells. Eur Respir J. 2005;25:1077–83. doi: 10.1183/09031936.05.00102104. [DOI] [PubMed] [Google Scholar]
- 166.Hatakeyama M, Imaizumi T, Terasaki F, Mori F, Tanji K, Sato F, Kijima H, Suma H, Wakabayashi K, Yoshida H, Fukuda I, Satoh K. Interferon-gamma upregulates retinoic acid-inducible gene-I in human pericardial mesothelial cells. Acta Cardiol. 2007;62:553–7. doi: 10.2143/AC.62.6.2024013. [DOI] [PubMed] [Google Scholar]
- 167.Yuzawa E, Imaizumi T, Matsumiya T, Yoshida H, Fukuhara R, Kimura H, Fukui A, Tanji K, Mori F, Wakabayashi K, Fujii S, Mizunuma H, Satoh K. Retinoic acid-inducible gene-I is induced by interferon-gamma and regulates CXCL11 expression in HeLa cells. Life Sci. 2008;82:670–5. doi: 10.1016/j.lfs.2007.12.025. [DOI] [PubMed] [Google Scholar]
- 168.Kawaguchi S, Ishiguro Y, Imaizumi T, Mori F, Matsumiya T, Yoshida H, Ota K, Sakuraba H, Yamagata K, Sato Y, Tanji K, Haga T, Wakabayashi K, Fukuda S, Satoh K. Retinoic acid-inducible gene-I is constitutively expressed and involved in IFN-gamma-stimulated CXCL9-11 production in intestinal epithelial cells. Immunol Lett. 2009;123:9–13. doi: 10.1016/j.imlet.2009.01.008. [DOI] [PubMed] [Google Scholar]
- 169.Kubota K, Sakaki H, Imaizumi T, Nakagawa H, Kusumi A, Kobayashi W, Satoh K, Kimura H. Retinoic acid-inducible gene-I is induced in gingival fibroblasts by lipopolysaccharide or poly IC: possible roles in interleukin-1beta, -6 and -8 expression. Oral Microbiol Immunol. 2006;21:399–406. doi: 10.1111/j.1399-302X.2006.00326.x. [DOI] [PubMed] [Google Scholar]
- 170.Yoshida H, Imaizumi T, Lee SJ, Tanji K, Sakaki H, Matsumiya T, Ishikawa A, Taima K, Yuzawa E, Mori F, Wakabayashi K, Kimura H, Satoh K. Retinoic acid-inducible gene-I mediates RANTES/CCL5 expression in U373MG human astrocytoma cells stimulated with double-stranded RNA. Neurosci Res. 2007;58:199–206. doi: 10.1016/j.neures.2007.02.017. [DOI] [PubMed] [Google Scholar]
- 171.Imaizumi T, Matsumiya T, Yoshida H, Naraoka T, Uesato R, Ishibashi Y, Ota K, Toh S, Fukuda S, Satoh K. Tumor-necrosis factor-alpha induces retinoic acid-inducible gene-I in rheumatoid fibroblast-like synoviocytes. Immunol Lett. 2009;122:89–93. doi: 10.1016/j.imlet.2008.12.005. [DOI] [PubMed] [Google Scholar]
- 172.Sakaki H, Imaizumi T, Matsumiya T, Kusumi A, Nakagawa H, Kubota K, Nishi N, Nakamura T, Hirashima M, Satoh K, Kimura H. Retinoic acid-inducible gene-I is induced by interleukin-1beta in cultured human gingival fibroblasts. Oral Microbiol Immunol. 2005;20:47–50. doi: 10.1111/j.1399-302X.2005.00181.x. [DOI] [PubMed] [Google Scholar]
- 173.Alexander J, Satoskar AR, Russell DG. Leishmania species: models of intracellular parasitism. J Cell Sci. 1999;112(Pt 18):2993–3002. doi: 10.1242/jcs.112.18.2993. [DOI] [PubMed] [Google Scholar]
- 174.Cossart P, Sansonetti PJ. Bacterial invasion: the paradigms of enteroinvasive pathogens. Science. 2004;304:242–8. doi: 10.1126/science.1090124. [DOI] [PubMed] [Google Scholar]
- 175.Imaizumi T, Sashinami H, Mori F, Matsumiya T, Yoshida H, Nakane A, Wakabayashi K, Oyama C, Satoh K. Listeria monocytogenes induces the expression of retinoic acid-inducible gene-I. Microbiol Immunol. 2006;50:811–5. doi: 10.1111/j.1348-0421.2006.tb03857.x. [DOI] [PubMed] [Google Scholar]
- 176.Lusis AJ. Atherosclerosis. Nature. 2000;407:233–41. doi: 10.1038/35025203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Lebwohl M. Psoriasis. Lancet. 2003;361:1197–204. doi: 10.1016/S0140-6736(03)12954-6. [DOI] [PubMed] [Google Scholar]
- 178.Lew W, Bowcock AM, Krueger JG. Psoriasis vulgaris: cutaneous lymphoid tissue supports T-cell activation and “Type 1” inflammatory gene expression. Trends Immunol. 2004;25:295–305. doi: 10.1016/j.it.2004.03.006. [DOI] [PubMed] [Google Scholar]
- 179.Vollmer S, Menssen A, Prinz JC. Dominant lesional T cell receptor rearrangements persist in relapsing psoriasis but are absent from nonlesional skin: evidence for a stable antigen-specific pathogenic T cell response in psoriasis vulgaris. J Invest Dermatol. 2001;117:1296–301. doi: 10.1046/j.0022-202x.2001.01494.x. [DOI] [PubMed] [Google Scholar]
- 180.Livden JK, Nilsen R, Bjerke JR, Matre R. In situ localization of interferons in psoriatic lesions. Arch Dermatol Res. 1989;281:392–7. doi: 10.1007/BF00455323. [DOI] [PubMed] [Google Scholar]
- 181.Kitamura H, Matsuzaki Y, Kimura K, Nakano H, Imaizumi T, Satoh K, Hanada K. Cytokine modulation of retinoic acid-inducible gene-I (RIG-I) expression in human epidermal keratinocytes. J Dermatol Sci. 2007;45:127–34. doi: 10.1016/j.jdermsci.2006.11.003. [DOI] [PubMed] [Google Scholar]
- 182.Prens EP, Kant M, van Dijk G, van der Wel LI, Mourits S, van der Fits L. IFN-alpha enhances poly-IC responses in human keratinocytes by inducing expression of cytosolic innate RNA receptors: relevance for psoriasis. J Invest Dermatol. 2008;128:932–8. doi: 10.1038/sj.jid.5701087. [DOI] [PubMed] [Google Scholar]
- 183.Sato K, Suematsu A, Okamoto K, Yamaguchi A, Morishita Y, Kadono Y, Tanaka S, Kodama T, Akira S, Iwakura Y, Cua DJ, Takayanagi H. Th17 functions as an osteoclastogenic helper T cell subset that links T cell activation and bone destruction. J Exp Med. 2006;203:2673–82. doi: 10.1084/jem.20061775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Husby G, Williams RC., Jr Immunohistochemical studies of interleukin-2 and gamma-interferon in rheumatoid arthritis. Arthritis Rheum. 1985;28:174–81. doi: 10.1002/art.1780280212. [DOI] [PubMed] [Google Scholar]
- 185.Manoury-Schwartz B, Chiocchia G, Bessis N, Abehsira-Amar O, Batteux F, Muller S, Huang S, Boissier MC, Fournier C. High susceptibility to collagen-induced arthritis in mice lacking IFN-gamma receptors. J Immunol. 1997;158:5501–6. [PubMed] [Google Scholar]
- 186.Vermeire K, Heremans H, Vandeputte M, Huang S, Billiau A, Matthys P. Accelerated collagen-induced arthritis in IFN-gamma receptor-deficient mice. J Immunol. 1997;158:5507–13. [PubMed] [Google Scholar]
- 187.van Holten J, Smeets TJ, Blankert P, Tak PP. Expression of interferon beta in synovial tissue from patients with rheumatoid arthritis: comparison with patients with osteoarthritis and reactive arthritis. Ann Rheum Dis. 2005;64:1780–2. doi: 10.1136/ard.2005.040477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Imaizumi T, Arikawa T, Sato T, Uesato R, Matsumiya T, Yoshida H, Ueno M, Yamasaki S, Nakajima T, Hirashima M, Sakata K, Ishibashi Y, Toh S, Ohyama C, Satoh K. Involvement of retinoic acid-inducible gene-I in inflammation of rheumatoid fibroblast-like synoviocytes. Clin Exp Immunol. 2008;153:240–4. doi: 10.1111/j.1365-2249.2008.03685.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Fukata M, Abreu MT. Pathogen recognition receptors, cancer and inflammation in the gut. Curr Opin Pharmacol. 2009;9:680–7. doi: 10.1016/j.coph.2009.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Melmed G, Thomas LS, Lee N, Tesfay SY, Lukasek K, Michelsen KS, Zhou Y, Hu B, Arditi M, Abreu MT. Human intestinal epithelial cells are broadly unresponsive to Toll-like receptor 2-dependent bacterial ligands: implications for host-microbial interactions in the gut. J Immunol. 2003;170:1406–15. doi: 10.4049/jimmunol.170.3.1406. [DOI] [PubMed] [Google Scholar]
- 191.Otte JM, Cario E, Podolsky DK. Mechanisms of cross hyporesponsiveness to Toll-like receptor bacterial ligands in intestinal epithelial cells. Gastroenterology. 2004;126:1054–70. doi: 10.1053/j.gastro.2004.01.007. [DOI] [PubMed] [Google Scholar]
- 192.Wang Y, Zhang HX, Sun YP, Liu ZX, Liu XS, Wang L, Lu SY, Kong H, Liu QL, Li XH, Lu ZY, Chen SJ, Chen Z, Bao SS, Dai W, Wang ZG. Rig-I-/- mice develop colitis associated with downregulation of G alpha i2. Cell Res. 2007;17:858–68. doi: 10.1038/cr.2007.81. [DOI] [PubMed] [Google Scholar]
- 193.Ohman L, Franzen L, Rudolph U, Birnbaumer L, Hornquist EH. Regression of Peyer's patches in G alpha i2 deficient mice prior to colitis is associated with reduced expression of Bcl-2 and increased apoptosis. Gut. 2002;51:392–7. doi: 10.1136/gut.51.3.392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Gillespie KM. Type 1 diabetes: pathogenesis and prevention. Cmaj. 2006;175:165–70. doi: 10.1503/cmaj.060244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Hyttinen V, Kaprio J, Kinnunen L, Koskenvuo M, Tuomilehto J. Genetic liability of type 1 diabetes and the onset age among 22,650 young Finnish twin pairs: A nationwide follow-up study. Diabetes. 2003;52:1052–5. doi: 10.2337/diabetes.52.4.1052. [DOI] [PubMed] [Google Scholar]
- 196.Akerblom HK, Vaarala O, Hyoty H, Ilonen J, Knip M. Environmental factors in the etiology of type 1 diabetes. Am J Med Genet. 2002;115:18–29. doi: 10.1002/ajmg.10340. [DOI] [PubMed] [Google Scholar]
- 197.van der Werf N, Kroese FG, Rozing J, Hillebrands JL. Viral infections as potential triggers of type 1 diabetes. Diabetes Metab Res Rev. 2007;23:169–83. doi: 10.1002/dmrr.695. [DOI] [PubMed] [Google Scholar]
- 198.Garcia M, Dogusan Z, Moore F, Sato S, Hartmann G, Eizirik DL, Rasschaert J. Regulation and function of the cytosolic viral RNA sensor RIG-I in pancreatic beta cells. Biochim Biophys Acta. 2009;1793:1768–75. doi: 10.1016/j.bbamcr.2009.09.005. [DOI] [PubMed] [Google Scholar]
- 199.Dogusan Z, Garcia M, Flamez D, Alexopoulou L, Goldman M, Gysemans C, Mathieu C, Libert C, Eizirik DL, Rasschaert J. Double-stranded RNA induces pancreatic beta-cell apoptosis by activation of the toll-like receptor 3 and interferon regulatory factor 3 pathways. Diabetes. 2008;57:1236–45. doi: 10.2337/db07-0844. [DOI] [PubMed] [Google Scholar]
- 200.Nejentsev S, Walker N, Riches D, Egholm M, Todd JA. Rare variants of IFIH1, a gene implicated in antiviral responses, protect against type 1 diabetes. Science. 2009;324:387–9. doi: 10.1126/science.1167728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Shigemoto T, Kageyama M, Hirai R, Zheng J, Yoneyama M, Fujita T. Identification of loss of function mutations in human genes encoding RIG-I and MDA5: Implications for resistance to type I diabetes. J Biol Chem. 2009;284:13348–54. doi: 10.1074/jbc.M809449200. [DOI] [PMC free article] [PubMed] [Google Scholar]