

Abstract
Resistin has been suggested to be involved in the development of diabetes and insulin resistance. We recently reported that resistin is expressed in diabetic hearts and promotes cardiac hypertrophy; however, the mechanisms underlying this process are currently unknown. Therefore, we wanted to elucidate the mechanisms associated with resistin-induced cardiac hypertrophy and myocardial insulin resistance. Overexpression of resistin using adenoviral vector in neonatal rat ventricular myocytes was associated with inhibition of AMP-activated protein kinase (AMPK) activity, activation of tuberous sclerosis complex 2/mammalian target of rapamycin (mTOR) pathway, and increased cell size, [3H]leucine incorporation (i.e. protein synthesis) and mRNA expression of the hypertrophic marker genes, atrial natriuretic factor, brain natriuretic peptide, and β-myosin heavy chain. Activation of AMPK with 5-aminoimidazole-4-carbozamide-1-β-d-ribifuranoside or inhibition of mTOR with rapamycin or mTOR siRNA attenuated these resistin-induced changes. Furthermore, resistin increased serine phosphorylation of insulin receptor substrate (IRS1) through the activation of the apoptosis signal-regulating kinase 1/c-Jun N-terminal Kinase (JNK) pathway, a module known to stimulate insulin resistance. Inhibition of JNK (with JNK inhibitor SP600125 or using dominant-negative JNK) reduced serine 307 phosphorylation of IRS1. Resistin also stimulated the activation of p70S6K, a downstream kinase target of mTOR, and increased phosphorylation of the IRS1 serine 636/639 residues, whereas treatment with rapamycin reduced the phosphorylation of these residues. Interestingly, these in vitro signaling pathways were also operative in vivo in ventricular tissues from adult rat hearts overexpressing resistin. These data demonstrate that resistin induces cardiac hypertrophy and myocardial insulin resistance, possibly via the AMPK/mTOR/p70S6K and apoptosis signal-regulating kinase 1/JNK/IRS1 pathways.
Keywords: AMP-activated Kinase (AMPK); Cardiac Hypertrophy; Diabetes; Gene Transfer; Insulin Resistance; Cardiomyocyte; Diabetic Cardiomyopathy; Resistin; mTOR, JNK Signaling
Introduction
Diabetic patients are at increased risk to develop cardiovascular diseases. Diabetes can affect cardiac structure and function independently of other risk factors, such as hypertension or coronary artery diseases (1, 2). Diabetes-induced cardiac dysfunction (known as diabetic cardiomyopathy) is characterized by cardiac hypertrophy and insulin resistance, among other elements (3).
Cardiac hypertrophy, defined as an increase in ventricular mass resulting from increased cardiomyocyte size, is an adaptive response of the heart to increased hemodynamic load arising from a variety of conditions, including exercise, hypertension, and valvular disease (4). Hallmark parameters of cardiac hypertrophy include increased myocardial cell size, a higher degree of sarcomeric organization, reactivation of the fetal genes (i.e. atrial natriuretic factor (ANF)2, brain natriuretic peptide (BNP), and β-myosin heavy chain (β-MHC) and enhanced protein synthesis (5). At the molecular level, mammalian target of rapamycin (mTOR), a kinase that responds to nutritional status and amino acid availability, is centrally involved in cell growth and proliferation (6, 7). Activated mTOR phosphorylates p70S6 kinase (p70S6K), which in turn phosphorylates the 40 S ribosomal protein S6 (8, 9), leading to stimulation of protein synthesis. Recently, it has been suggested that AMP-activated protein kinase (AMPK) can inhibit mTOR signaling through the phosphorylation of TSC2, an upstream regulator of mTOR (10). This observation raised the prospect that AMPK is also a key regulator of cardiac hypertrophy (11).
Myocardial insulin resistance has been recognized as a risk factor for the development of cardiovascular diseases. For instance, diabetes and insulin resistance aggravate myocardial ischemic injury in animal and human studies (2, 12). Although studies to date indicate multiple sites of impaired insulin signaling in various animal models, data support the efficacy of inhibition of insulin receptor substrate 1 (IRS1) serine-threonine kinases in the amelioration of insulin resistance. Stress-activated JNK is elevated in tissues of obese subjects, suggesting that it plays a crucial role in obesity and insulin resistance (13). JNK activation phosphorylates its downstream target, IRS1, at Ser-307, thus impairing insulin-induced IRS1 tyrosine phosphorylation and attenuating insulin sensitivity (14, 15). In addition, activation of the mTOR pathway along with enhanced phosphorylation of IRS1 at Ser-307 and Ser-636/Ser-639 has been observed. Ablation of p70S6K protects against high-fat diet-induced insulin resistance in mice (16), and rapamycin inhibition of mTOR/p70S6K action blunts insulin-induced Ser-632/Ser-635 phosphorylation of IRS1 (17).
Resistin, an adipocyte-secreted cytokine, has been suggested to be involved in the development of diabetes and insulin resistance (18–21). Deletion of the resistin gene or use of resistin antisense improves diet-induced insulin resistance (19, 20), whereas acute administration of resistin impairs glucose tolerance and insulin action in rodents (18–21). Resistin was shown to impair glucose transport in isolated mice cardiomyocytes (22) and to be up-regulated by cyclic stretch and aorta-caval shunt (23). We recently reported that overexpression of resistin induced hypertrophy in neonatal rat cardiomyocytes, increased serine phosphorylation of IRS1, and caused contractile dysfunction with impaired calcium handling in adult rat cardiomyocytes (24). However, the underlying mechanisms by which resistin induces cardiac hypertrophy and myocardial insulin resistance are currently unknown. Here we present evidence that resistin promotes cardiac hypertrophy and myocardial insulin resistance through activation of the AMPK/mTOR/p70S6K and ASK1/JNK/IRS1 signaling pathways.
EXPERIMENTAL PROCEDURES
Animal Care
Animals were handled as approved by the Mount Sinai Institutional Animal Care and Use Committee in accordance with the “Principles of Laboratory Animal Care by the National Society for Medical research and the Guide for the Care and Use of Laboratory Animals”(National Institutes of Health Publication No. 86–23, revised 1996).
Production of Recombinant Adenoviruses
Recombinant adenoviruses encoding full-length rat resistin (Ad.rRetn) and β-galactosidase (Ad.βGal) were prepared as described previously (24). A dominant-negative JNK adenovirus (Ad.dnJNK1) was purchased from Seven Hills Bioreagents. A multiplicity of infection of 100 has been used in all infection experiments.
Isolation, Culture, and Treatment of Neonatal Cardiomyocytes
Neonatal rat ventricular myocytes (NRVM) were prepared from 1- to 2-day-old Sprague-Dawley rat pups using the Worthington neonatal cardiomyocyte isolation system (Worthington). Cardiomyocytes were infected with Ad.rRetn or Ad.βGal for 24–36 h (MOI 100). After virus infection, cells were treated with various reagents for the indicated time as described in the figure legends.
Preparation of Protein Extracts and Western Blot Analysis
Protein samples were prepared from adult rat hearts or NRVM using a lysis buffer containing protease inhibitors (Roche) and phosphatase inhibitor mixtures (Sigma). Cell lysates were matched for protein concentration, separated by SDS-PAGE, and transferred onto nitrocellulose membranes. The membranes were blocked with 5% nonfat milk and incubated overnight at 4 °C with anti-resistin (Biovision), phospho-specific antibodies to AMPKα, acetyl-CoA carboxylase (ACC), Tuberin/TSC2, mTOR, p70S6K, ASK1, JNK, and IRS1 (Ser-636/Ser-639 or Ser-307), or their respective total antibodies (Cell Signaling Technology, Inc.). The membranes were incubated with appropriate secondary antibodies, and signal intensities were visualized with enhanced chemiluminescence (Pierce). Total protein contents of the corresponding proteins were analyzed after stripping the phospho-blots to verify protein loading. Films from at least three independent experiments were scanned, and densities of the immunoreactive bands were evaluated using National Institutes of Health ImageJ software. Densitometric values of phospho-protein bands were normalized against the corresponding total protein values (densitometric values are reported as arbitrary units (AU)). GAPDH was used as an internal control.
[3H]Leucine Incorporation Assay
Protein synthesis rates in NRVM were determined using [3H]leucine (n = 6 per group) as described previously (24). Radioactivity was normalized to total protein content measured by Micro BCA protein assay kit (Pierce).
Measurement of Cell Surface Area
Cell surface area was measured as we described previously (24) using ImageJ software (National Institutes of Health).
Real-time qRT-PCR
The expression of ANF, BNP, β-MHC, and 18s rRNA genes was quantified using real-time qRT-PCR (Applied Biosystems). The primers used are as follows: ANF, 5′-ACC TGC TAG ACC ACC TGG AGG AG-3′ and 5′-CCT TGG CTG TTA TCT TCG GTA CCG-3′; BNP, 5′-GCT GCT TTG GGC ACA AGA TAG-3′ and 5′-GGT CTT CCT ACA ACA ACT TCA-3′; β-MHC 5′-TTG GCA CGG ACT GCG TCA TC-3′ and 5′-GAG CCT CCA GAG TTT GCT GAA GGA-3′; and 18s rRNA, 5′-TCA AGA ACG AAA GTC GGA GG-3′ and 5′-GGA CAT CTA AGG GCA TCA C-3′. Fold changes in gene expression were determined using the relative comparison method with normalization to 18s rRNA.
Transfection of Myocytes with mTOR siRNA
Cardiomyocytes were transfected with 50 nm mTOR siRNA (I and II) (Cell Signaling Technology, Inc.) using Lipofectamine RNAiMAX according to the manufacturer's instructions (Invitrogen). Signal-SilenceControl siRNA was used as a control. After culturing for 48 h in serum-free medium, myocytes were then infected with Ad.rRetn or Ad.βGal and further cultured for 48 h in serum-free DMEM/F-12 Ham before harvesting.
Statistics
Where appropriate, the data were expressed as mean ± S.E., and comparisons of the group means were made with a Student's t test. p < 0.05 was considered statistically significant.
RESULTS
Resistin Is Increased in Ventricular Tissue from Pressure-overload Hypertrophic Rat Hearts
We first determined whether hypertrophic hearts express higher levels of resistin. We examined resistin expression in rat hearts subject to pressure overload because of thoracic aortic banding (25). Hypertrophy in this model was assessed by determining the ratio of left ventricular weight to body weight (LVW/BW). Aortic banded hearts showed significant increase in LVW/BW ratio (2.74 ± 0.20 mg/g, n = 5) compared with sham group (1.66 ± 0.11 mg/g, n = 3, p < 0.01) (Fig. 1A). Evaluation of resistin expression showed significant up-regulation of resistin protein levels in the hypertrophic hearts (Fig. 1B). For comparison, we also show resistin expression in diabetic hearts, which is also significantly elevated (Fig. 1B). Interestingly, in the rat model of hypertrophy, pressure overload stress seemed to affect the expression of resistin not only in the heart but also in other tissues, such as liver and lungs, which demonstrated increased resistin expression following aortic banding (supplemental figure). This finding, coupled with our earlier studies demonstrating resistin-induced cardiomyocyte hypertrophy and contractile abnormalities (24), prompted us to examine the molecular mechanism(s) underlying the promotion of cardiac hypertrophy of resistin.
FIGURE 1.

Resistin is increased in hypertrophic rat hearts. A, left ventricular hypertrophy measured by left ventricular weight (LVW, mg) to body weight (BW, g) ratio of ascending aortic banding (AAB, n = 5) hypertrophic heats or sham-operated (sham, n = 3) rats. B, representative Western blot analysis for resistin expression in sham-operated, aortic banded, normal non-diabetic (Norm) or type 2 diabetic (DM) rat hearts (DM hearts are from the Otsuka Long-Evans Tokushima Fatty rat model of type 2 diabetes, see Ref. 24). GAPDH is shown to verify protein loading. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
Resistin Modulates the AMPK/mTOR/p70S6K Pathway in NRVM
In initial preliminary experiments, we observed diminished AMPK activity by resistin, and because AMPK has been postulated to play a key role in cardiac hypertrophy, we sought to evaluate the role of resistin in modulating cardiac hypertrophy through the regulation of AMPK. NRVM were cultured and infected with recombinant adenovirus expressing the rat resistin gene (Ad.rRetn) or the β-galactosidase gene (Ad.βGal) as a control. Myocytes infected with Ad.rRetn displayed a sizable decrease in the level of AMPKα phosphorylated at Thr-172 compared with myocytes treated with Ad.βGal (Fig. 2). To confirm the inactivation of AMPKα, we also determined the phosphorylation state of ACC and TSC2, known downstream targets of AMPK. Phosphorylation of ACC at Ser-79 and TSC2 at Thr-1462 was markedly decreased in NRVM expressing resistin, paralleling the reduction of AMPK activity (Fig. 2). In addition, cardiac myocytes expressing resistin displayed significant increases in the level of mTOR phosphorylated at Ser-2448, indicating increased mTOR activity. p70S6K, a downstream effector of mTOR, was also significantly phosphorylated in cells expressing resistin (Fig. 2). Activated mTOR phosphorylates p70S6K at Thr-389 and promotes protein synthesis. AMPKα can modulate protein synthesis by inhibiting this pathway (10). These data demonstrate that resistin inhibits AMPK and activates mTOR/p70S6K signaling pathways in cardiomyocytes.
FIGURE 2.
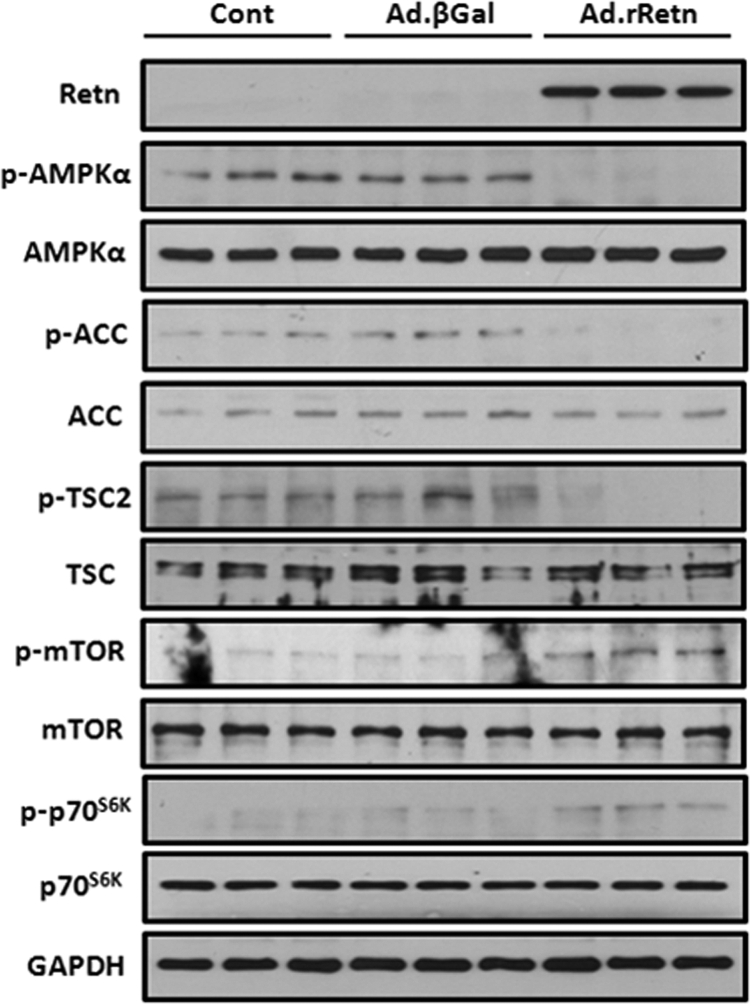
Resistin modulates AMPK/mTOR/p70S6K phosphorylation in NRVM. Representative Western blot analyses of cellular extracts from NRVM non-infected (Cont) or infected with 100 multiplicity of infection of Ad.βGal or Ad.rRetn for 36 h. Shown are phospho-proteins (p-) and their corresponding total levels. GAPDH is shown to verify protein loading.
AMPK Activation by AICAR Blocks Resistin-induced Cell Size, Protein Synthesis, and Expression of Hypertrophic Marker Genes by Inhibiting the mTOR/p70S6K Pathway in NRVM
To better understand the role of resistin in regulating the function of AMPK during hypertrophic growth, NRVM were treated with the pharmacological AMPK activator AICAR. Lysates from NRVM treated with Ad.rRetn or Ad.βGal in the presence or absence of 1 mm AICAR were analyzed by immunoblotting using an antibody against the α subunit of AMPK (AMPKα), phosphorylated at Thr-172. Treatment with Ad.rRetn showed decreased phosphorylation of AMPKα, ACC, and TSC2; however, in the presence of AICAR, resistin-suppressed AMPKα phosphorylation was clearly preserved (Retn, 0.109 ± 0.009 AU versus Retn+ AICAR, 0.244 ± 0.024 AU; p <0.001) (Fig. 3A).
FIGURE 3.

AMPK activation by AICAR reduces resistin-induced cell size, protein synthesis, and expression of hypertrophic marker genes by inhibiting mTOR/p70S6K phosphorylation in NRVM. A, representative Western blot analyses of cellular extracts from NRVM non-infected (Cont) or infected with Ad.βGal or Ad.rRetn in the absence (-) or presence (+) of 1 mm AICAR for 30 min. Shown are phospho-proteins (p-) and their corresponding total levels. B, cell surface area and C, leucine incorporation assay of cellular extracts from NRVM non-infected (PBS) or infected with Ad.βGal or Ad.rRetn in the absence (-) or presence (+) of 100 μm phenylephrine (PE) for 24 h (positive control) or 1 mm AICAR for 24 h. Measurement of protein synthesis using [3H]leucine incorporation with counts expressed in counts per minute as a percentage compared with the control. Values are mean ± S.E. with each experiment performed in duplicate (n = 6). D, quantitative RT-PCR analyses of ANF, BNP, and β-MHC expression in cellular extracts from control non-infected (PBS) or NRVM infected with Ad.βGal or Ad.rRetn in the absence (-) or presence (+) of 1 mm AICAR for 24 h. Expression of hypertrophic marker genes is presented as a ratio compared with the control. Values are mean ± S.E. with each experiment assessed in duplicate (n = 4). *, p < 0.05; **, p < 0.01; ***, p < 0.001.
Pharmacological and genetic activation of AMPK in myocytes can inhibit hypertrophy markers associated with the hypertrophic response (26–28). We therefore determined whether the decrease in AMPKα activity, by resistin, can influence cell size, protein synthesis, and expression of hypertrophic marker genes. Ad.rRetn increased cell size by about 53% compared with Ad.βGal as assessed by measuring the surface area of infected NRVM (Fig. 3B, p < 0.01); however, in the presence of AICAR, the resistin-induced increase in cell size was reduced significantly (Fig. 3B; p < 0.05). The effect of resistin on cell surface area was similar to that evoked by the potent hypertrophy agonist phenylephrine (PE) (Fig. 3B). Likewise, resistin induced a significant increase (1.75-fold, p < 0.001) in leucine incorporation over Ad.βGal treatment, but this effect was markedly reduced by AICAR addition (Fig. 3C, p < 0.01). In the presence of AICAR, AMPKα is activated 2- to 3-fold, which correlates with a 40–60% reduction in protein synthesis. Because resistin-induced protein synthesis was not completely inhibited by AICAR, this suggests that other regulatory pathways, independent of AMPK, may also contribute to resistin-induced protein synthesis. Furthermore, NRVM treated with Ad.rRetn showed a significant increase in mRNA expression of the hypertrophic marker genes ANF (3.48-fold), BNP (2.97-fold), and β-MHC (4.00-fold) (Fig. 3D). However, in the presence of AICAR, the resistin-induced gene expression was reduced significantly to basal levels (Fig. 3D).
To further dissect the role of AMPK during hypertrophic growth, cell lysates were subjected to immunoblot analysis using anti-phospho-mTOR and anti-phospho-p70S6K antibodies. As shown in Fig. 3A, elevated levels of phosphorylated mTOR and phosphorylated p70S6K were observed in NRVM treated with Ad.rRetn, indicating increased protein synthesis. However, treatment of NRVM with AICAR resulted in a considerable reduction in the phosphorylation states of mTOR and p70S6K (Fig. 3A), consistent with the observation that activation of AMPK inhibits the mTOR/p70S6K pathway. Because phosphorylation of the mTOR/p70S6K pathway results in an increase in mTOR and p70S6K activity and activation of protein synthesis, its inactivation seen in response to AICAR may, at least in part, explain the decrease in protein synthesis observed in Fig. 3C.
mTOR/p70S6K Pathway Inhibition Reduces Resistin-induced Hypertrophic Changes in NRVM
To further study mTOR signaling in resistin-induced cardiac hypertrophy, lysates from NRVM treated with Ad.rRetn in the presence or absence of rapamycin (100 nm) were subjected to immunoblot analysis using an antibody against mTOR phosphorylated at Ser-2448 or IRS1 phosphorylated at Ser-636/Ser-639 (a known p70S6K phosphorylation site). Ad.rRetn stimulated the phosphorylation of mTOR and IRS1; however, in the presence of rapamycin, resistin-induced mTOR and IRS1 phosphorylation was diminished (mTOR, 0.408 ± 0.033 AU versus 0.087 ± 0.005 AU; IRS1, 0.316 ± 0.015 AU versus 0.060 ± 0.011 AU; p < 0.001 for both) (Fig. 4A).
FIGURE 4.

Inhibition of mTOR/p70S6K pathway reduces resistin-induced hypertrophic markers in NRVM. A, representative Western blot analyses of phospho- and total mTOR and IRS1 (with the phospho-Ser-636/639 residues shown). B, cell surface area, C, leucine incorporation assay, and D, quantitative RT-PCR analyses of ANF, BNP, and β-MHC expression in cellular extracts from control non-infected (Cont) and NRVM infected with Ad.βGal or Ad.rRetn in the absence (-) or presence (+) of 100 nm rapamycin for 24 h. Measurement of protein synthesis using [3H]leucine incorporation with counts expressed in counts per minute as a percentage compared with the control (PBS) (C). E, a representative Western blot analysis demonstrating siRNA knockdown of mTOR expression (by the mTOR siRNA I and II constructs). F, quantitative RT-PCR analysis of β-MHC expression following siRNA knockdown of mTOR. Values are mean ± S.E. with each experiment performed in duplicate (n = 6). Expression of hypertrophic marker genes is presented as a ratio compared with the control (PBS) (C). Values are mean ± S.E. with each experiment assessed in duplicate (n = 4). *, p < 0.05; **, p < 0.01; ***, p < 0.001.
Treatment of NRVM with Ad.rRetn induced an increase in cell surface area by about 53% compared with Ad.βGal (Fig. 4B, p < 0.01); however, in the presence of rapamycin, resistin-induced increase in cell size was blunted significantly (Fig. 4B, p < 0.01). Similarly, resistin evoked a large increase in leucine incorporation (1.72-fold, p < 0.001) compared with control cells; however, in the presence of rapamycin, resistin-induced protein synthesis was reduced slightly but significantly (Fig. 4C). In addition, resistin induced a significant increase in mRNA expression of ANF (2.22-fold), BNP (2.75-fold), and β-MHC (1.98-fold); however, rapamycin significantly abrogated the observed effect of resistin (Fig. 4D). To further verify the involvement of the mTOR pathway in resistin-induced cardiomyocyte hypertrophy, we inhibited mTOR activity using siRNA targeted against mTOR. Like rapamycin, mTOR siRNA potently inhibited mTOR expression (0.585 ± 0.042 AU versus 0.019 ± 0.003 AU with siRNA II, p < 0.001) (Fig. 4E) and significantly decreased the expression of β-MHC, a marker of hypertrophy (-54%, p < 0.01, F). Although mTOR is a downstream target of AMPK signaling, as we showed in Fig. 3, it can also be regulated by other pathways (e.g. PI3K/Akt and MAPK) (29, 35). Hence, resistin-induced hypertrophic response is likely to be an AMPK-dependent (Fig. 3) and an AMPK-independent (Fig. 4) process.
Resistin Modulates the ASK1/JNK/IRS1 Pathway, and JNK Inhibition Diminishes Resistin-induced IRS1 Serine Phosphorylation in NRVM
To further elucidate the involvement of other mechanisms in the action of resistin on cardiac hypertrophy and insulin resistance, we investigated the activation of JNK in NRVM infected with Ad.rRetn or Ad.βGal. Resistin-transduced myocytes displayed elevated levels of JNK phosphorylation at Thr-183/Tyr-185 compared with myocytes treated with Ad.βGal (Fig. 5A). Furthermore, the dephosphorylation (i.e. activation) of ASK1, a known JNK kinase, was substantially induced by resistin (Fig. 5A), paralleling the observed increase in phosphorylation of JNK at Thr-183/Tyr-185.
FIGURE 5.

Resistin modulates the ASK1/JNK/IRS1 pathway in NRVM. Representative Western blot analyses of cellular extracts from control non-infected (Cont) and NRVM infected with Ad.βGal or Ad.rRetn for 36 h (A) and in the absence (-) or presence (+) of 10 μm JNK inhibitor SP600125 for 30 min (B) or a dominant-negative JNK adenovirus (Ad.dnJNK1) for 48 h (C). Shown are phospho-proteins (p-) and their corresponding total levels of ASK1, JNK, and IRS1 (Ser-307 and Ser-636/639). GAPDH is shown to verify protein loading.
IRS1 serine phosphorylation is known to contribute to insulin resistance. Because JNK is known to phosphorylate serine residues of IRS1, thus promoting the insulin-mitogenic pathway and inducing insulin resistance, we also determined whether JNK activity, increased by resistin in cardiomyocytes, results in an increase in IRS1 serine phosphorylation at its primary activation site, Ser-307. Indeed, NRVM transduced with Ad.rRetn displayed increased phosphorylation of IRS1 at Ser-307 as well as increased JNK activity (Fig. 5A). Furthermore, immunoblot analysis of phosphorylated IRS1 at Ser-636/Ser-639 was also performed to investigate other serine phosphorylation of IRS1. Concomitant with increased IRS1 phosphorylation at Ser-307 in cells expressing resistin, phosphorylation of IRS1 at Ser-636/Ser-639 was also increased (Fig. 5A). These data confirm that increased activity of p70S6K is able to phosphorylate the serine residues of its downstream IRS1 target; thus, p70S6K can contribute to the induction of insulin resistance as well as increase protein synthesis.
To further assess the signaling mechanism by which the JNK/IRS1 pathway regulates insulin signaling during hypertrophic growth, NRVM were treated with Ad.rRetn in the presence or absence of SP600125 (10 μm), a known selective JNK inhibitor, or infected with a dominant-negative JNK1 adenoviral construct (Ad.dnJNK1). Immunoblot analysis using phospho-JNK and phospho-IRS1 (Ser-307) antibodies showed increased phosphorylation of JNK and of IRS1 at Ser-307 in cardiomyocytes expressing Ad.rRetn (Fig. 5, B and C). However, treatment with SP600125 (Fig. 5B) or Ad.dnJNK1 (C) almost completely reversed the resistin-induced phosphorylation of JNK and IRS1 back to basal level (phospho-JNK, 0.452 ± 0.027 AU versus 0.072 ± 0.006 AU, p < 0.001; phospho-IRS1, 1.302 ± 0.09 AU versus 0.598 ± 0.026 AU, p < 0.001 using dnJNK1; Fig. 5C). Altogether, these data demonstrate that resistin may contribute to the onset of myocardial insulin resistance and further support the idea that resistin is involved in the induction of insulin resistance in general.
Resistin Modulates the AMPK/p70S6K and JNK/IRS1 Pathways in adult Rat Hearts
We then wanted to know whether the above in vitro signaling pathways involved in resistin modulation of the hypertrophic response were also operative in vivo in ventricular tissues from adult rats overexpressing resistin. We have shown in preliminary findings that cardiac-targeted overexpression of resistin in vivo displayed features of diastolic dysfunction, hypertension, increased cardiac apoptosis, and hypertrophy (30). Proteins, isolated from left ventricular tissues from control animals or adult rats transduced with adenovirus-associated virus serotype 9-encoding rat resistin (AAV9.rRetn) for 10 weeks, were analyzed by immunoblotting using antibodies against AMPK phosphorylated at Thr-172, TSC2 phosphorylated at Thr-1462, p70S6K phosphorylated at Thr-389, JNK phosphorylated at Thr-183/Tyr-185, and IRS1 phosphorylated at Ser-307 and Ser-636/Ser-639. Interestingly, compared with control hearts, reduced phosphorylation of AMPK and increased phosphorylation of p70S6K, JNK, and IRS1 was observed in AAV9.rRetn hearts (Fig. 6). Therefore, resistin overexpression in ventricular tissues is associated with altered activities of the AMPK/p70S6K and JNK/IRS1 pathways, consistent with our findings in vitro.
FIGURE 6.
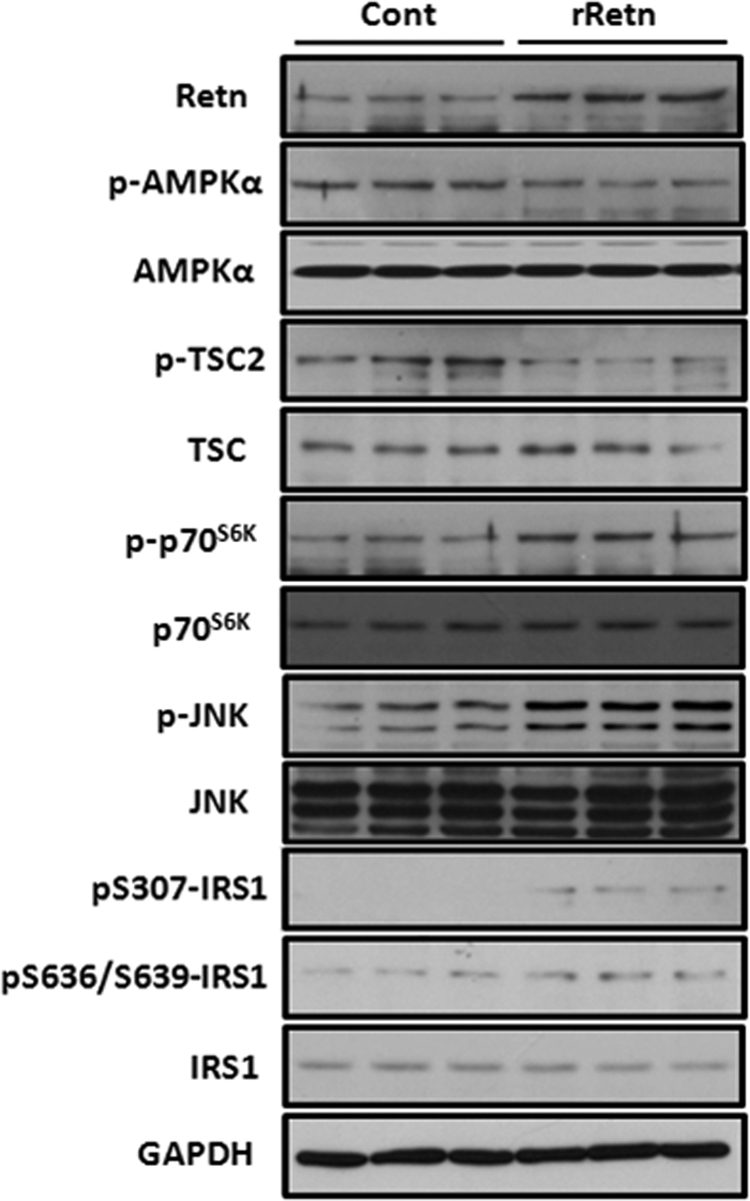
Resistin overexpression modulates the AMPK/p70S6K and JNK/IRS1 signaling pathways in adult rat hearts in vivo. Representative Western blot analyses showing phosphorylated and total levels of AMPK, TSC2, p70S6K, JNK, and IRS1 in ventricular tissues isolated from control hearts or AAV9.rRetn-transduced hearts. GAPDH is shown to verify protein loading.
DISCUSSION
We have previously examined the role of resistin in modulating cardiac hypertrophy and cardiomyocyte physiology. We showed that adenovirus-mediated overexpression of rat resistin in cultured NRVM significantly increased sarcomere organization and cell size, enhanced protein synthesis, and increased the expression of ANF and β-MHC. Furthermore, overexpression of resistin in NRVM was associated with activation of MAPKs as well as increased serine phosphorylation of IRS1. Overexpression of resistin in adult cultured cardiomyocytes negatively altered myocyte mechanics and impaired cytoplasmic Ca2+ clearing (24). The data we obtained showed that resistin confers to primary cardiomyocytes all the features of the hypertrophic phenotype. In addition, we show in this study that resistin expression is elevated in hypertrophic hearts subjected to chronic pressure overload. Although these findings suggest, for the first time, that resistin may be involved in the induction of cardiac hypertrophy, the mechanisms underlying this effect are currently unknown. Therefore, we were interested in investigating the mechanisms underlying the actions of resistin in the setting of cardiac hypertrophy. Here we present evidence that resistin promotes cardiac hypertrophy through the modulation of the AMPK/mTOR/p70S6K and ASK1/JNK/IRS1 pathways in cardiomyocytes as we proposed in Fig. 7. Interestingly, these signaling pathways are also observed in vivo in adult hearts overexpressing resistin.
FIGURE 7.
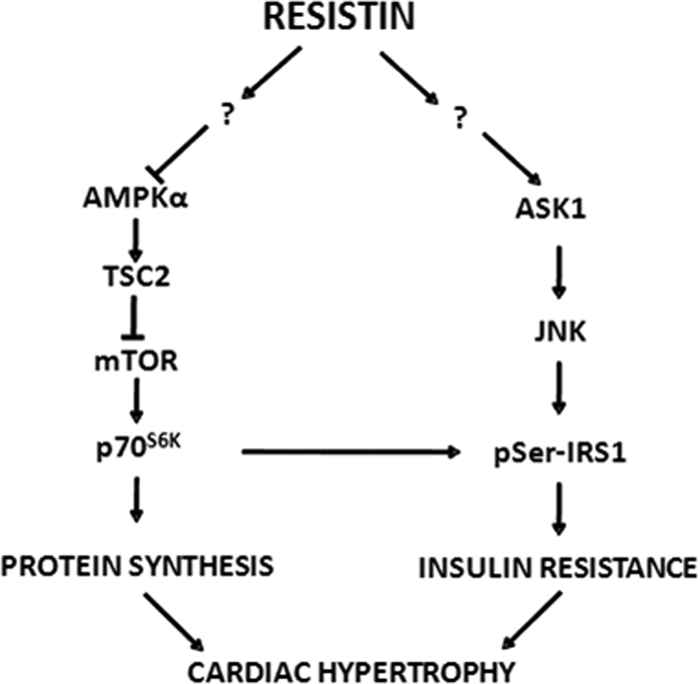
A diagram summarizing the proposed mechanisms underlying resistin modulation of cardiac hypertrophy and insulin resistance. Resistin, directly or through yet undetermined pathways (?), inhibits AMPK and TSC2 activity and stimulates mTOR and p70S6K, leading to increased protein synthesis, a hallmark feature of cardiomyocyte hypertrophy. On the other hand, resistin, either directly or through yet undetermined pathways (?), activates ASK1, which in turn induces JNK and the subsequent phosphorylation of IRS1 serine residues. p70S6K can also phosphorylate IRS1 on Ser-636/Ser-639, thus inducing insulin resistance. IRS1 serine phosphorylation promotes insulin resistance, which can contribute to cardiac hypertrophy.
The participation of AMPK pathway in the regulation of cardiac hypertrophy has emerged recently (31). Some observations have shown that the activity of AMPK is elevated in hypertrophic hearts subjected to chronic pressure overload, and this increase in AMPK activity was associated with enhanced glucose uptake, thus implicating AMPK activation as a cause of cardiac hypertrophy (32). However, substantial recent evidence indicates that AMPK, on the contrary, is involved in the attenuation of cardiac hypertrophy. The development of Akt1-induced hypertrophy in neonatal rat cardiac myocytes is associated with inactivation of AMPK, which may be permissive for cardiac growth (26). In agreement with this study, adiponectin has been shown to attenuate hypertrophic growth via activation of AMPK and an AMPK-dependent signaling mechanism (33). Further support of AMPK as a negative regulator of cardiac growth has been provided by other studies, showing that AMPK signaling is decreased or impaired in hearts from adiponectin or leptin deficient mice. This decrease in AMPK activity exacerbated hypertrophic growth and heart failure following transverse aortic constriction (34) or myocardial infarction (35). In addition, activation of AMPK inhibits protein synthesis associated with cardiac hypertrophy via two independent signaling pathways that are known to mediate protein synthesis in the heart, the mTOR-p70S6K-40S ribosomal protein S6 and the eEF2 kinase-eEF2 pathways (26). This implies that AMPK activation can inhibit protein synthesis associated with cardiac hypertrophy and may be able to prevent the development of pathological hypertrophy.
We showed that resistin decreases the activity of AMPK in cardiomyocytes (Figs. 2, 3, and 6). Whether resistin exacerbates the hypertrophic response through depressing the activity of AMPK remains to be determined. We hypothesize that restoring the activity of AMPK using pharmacological activators (such as AICAR or metformin) or through gene transfer might provide a beneficial mechanism to prevent or attenuate hypertrophic cardiomyopathy associated with diabetes and obesity in subjects with hyperresistinemia. The concept that AMPK may link adipokine (such as resistin) signaling and the regulation of the hypertrophic cardiomyopathies associated with diabetes/obesity is an exciting avenue of research that deserves further investigation. It is unclear how resistin alters the phosphorylation of AMPK; this is in part due to the fact that resistin receptors are yet to be identified. However, it is likely that the ability of resistin to decrease AMPK phosphorylation may be mediated by altering the activity of upstream kinases or phosphatases involved in deactivating AMPK, such as LKB1 and Ca2+/calmodulin-dependent kinase kinase. Whether resistin regulates the activities of LKB1 and/or calmodulin-dependent kinase kinase is undetermined at the moment, although we observed in preliminary experiments that resistin dephosphorylates LKB1 (not shown). In addition, we found that resistin increases the phosphorylation of the p70S6K (Figs. 2, 3, and 6). The phosphorylation and activation of p70S6K leads to an increase in protein synthesis and cell growth, which can induce hypertrophy, and in a negative feedback mechanism with mTOR, inhibits PI3K/Akt signaling by promoting reduction in IRS1 stability and transcription, leading to its disassociation from PI3K. In addition, activation of p70S6K can phosphorylate IRS1 on multiple inhibitory sites, promoting its degradation, thus inducing insulin resistance (36). It is therefore likely that some of the hypertrophic response observed in subjects with type 2 diabetes may be ascribed to shunting insulin signaling away from the PI3K/Akt metabolic pathway and toward the mitogenic pathway (i.e. insulin resistance).
Negative regulation of insulin signaling occurs mainly at the level of the proximal tyrosine kinase substrates of the insulin receptor, namely IRS proteins, a family of adaptor proteins that are essential for insulin effects. When insulin binds to its receptor, IRS1 is activated via tyrosine phosphorylation to initiate downstream signaling pathways such as Akt (37). On the other hand, IRS1 contains more than 50 potential serine phosphorylation sites; serine phosphorylation of IRS1 or its degradation decreases its interaction with the insulin receptor, leading to a reduction in IRS1 tyrosine phosphorylation and a disturbance of insulin signaling (38). Among the various IRS1 serine phosphorylation sites, Ser-307 is phosphorylated by JNK (14, 15, 39), whereas Ser-636/Ser-639 is phosphorylated by the mTOR/p70S6K module (17, 40). In this study, we found that resistin induced higher phosphorylation of JNK and of the Ser 307 residue of IRS1 (Figs. 5 and 6). Anisomycin, a pharmacological activator of JNK, has been shown to inhibit insulin signal transduction by interrupting IRS1 binding to the insulin receptor tyrosine kinase following increased JNK phosphorylation (41). JNK has been shown to bind directly to IRS1 and to phosphorylate it on Ser-307, whereas phosphorylation of Ser-307 is reduced in cells lacking JNK1/2 (42). In addition, serine phosphorylation of IRS1 is demonstrated to be associated with decreased IRS1 protein stability. Mutation of Ser-312 to Ala in human IRS1 (equivalent to the Ser-307 site in rat IRS1) is partially resistant to insulin-induced degradation compared with wild-type IRS1, indicating that phosphorylation of this site might direct an increase in proteasomal degradation of IRS1 (43). Moreover, activation of mTOR/p70S6K by resistin and the subsequent induction of IRS1 Ser-636/Ser-639 phosphorylation also suggest that these pathways are involved in controlling IRS1 signaling mechanisms (Fig. 4–6). As stated earlier, mTOR, through the activation of p70S6K, inhibits insulin signal transduction by promoting reduction of IRS1 stability and transcription and enhancing its degradation. Therefore, nonspecific or regulated degradation of elements in the insulin signaling pathway might cause insulin resistance. Our observation that resistin stimulates serine phosphorylation of IRS1 through activation of JNK and or mTOR/p70S6K may provide a rational mechanism to explain, at least in part, the myocardial insulin resistance that occurs in diabetic cardiomyopathy. However, a direct effect of resistin on IRS1 protein stability and degradation remains to be determined.
Our study further supports a role for resistin in the regulation of cardiac hypertrophy and myocardial insulin resistance. The actions of resistin in the setting of cardiac hypertrophy and insulin resistance are attributed to the modulation of intracellular growth signals in cardiac cells involving the AMPK/mTOR/p70S6K and JNK/IRS1 signaling pathways.
Supplementary Material
This study was supported, in whole or in part, by an NHLBI National Institutes of Health grant (to D. L. and R. J. H.) and a National Institutes of Health training grant (to E. R. C.).

The on-line version of this article (available at http://www.jbc.org) contains a supplemental figure.
- ANF
- atrial natriuretic factor
- BNP
- brain natriuretic peptide
- β-MHC
- β-myosin heavy chain
- mTOR
- mammalian target of rapamycin
- AMPK
- AMP-activated protein kinase
- TSC2
- tuberous sclerosis complex 2
- IRS1
- insulin receptor substrate 1
- ASK1
- apoptosis signal-regulating kinase 1
- NRVM
- neonatal rat ventricular myocytes
- ACC
- acetyl-CoA carboxylase
- AU
- arbitrary unit(s)
- qRT-PCR
- quantitative RT-PCR
- LVW
- left ventricular weight
- BW
- body weight
- AICAR
- 5-aminoimidazole-4-carbozamide-1-β-D-ribifuranoside
- dn
- dominant-negative
- JNK
- c-Jun N-terminal Kinase.
REFERENCES
- 1. Fox C. S., Coady S., Sorlie P. D., D'Agostino R. B., Sr., Pencina M. J., Vasan R. S., Meigs J. B., Levy D., Savage P. J. (2007) Circulation 115, 1544–1550 [DOI] [PubMed] [Google Scholar]
- 2. Boudina S., Abel E. D. (2007) Circulation 115, 3213–3223 [DOI] [PubMed] [Google Scholar]
- 3. Lebeche D., Davidoff A. J., Hajjar R. J. (2008) Nat. Clin. Pract. Cardiovasc. Med. 5, 715–724 [DOI] [PubMed] [Google Scholar]
- 4. Frey N., Olson E. N. (2003) Annu. Rev. Physiol. 65, 45–79 [DOI] [PubMed] [Google Scholar]
- 5. Sugden P. H., Clerk A. (1998) J. Mol. Med. 76, 725–746 [DOI] [PubMed] [Google Scholar]
- 6. Proud C. G. (2004) Biochem. Biophys. Res. Commun. 313, 429–436 [DOI] [PubMed] [Google Scholar]
- 7. Fingar D. C., Salama S., Tsou C., Harlow E., Blenis J. (2002) Genes Dev. 16, 1472–1487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Isotani S., Hara K., Tokunaga C., Inoue H., Avruch J., Yonezawa K. (1999) J. Biol. Chem. 274, 34493–34498 [DOI] [PubMed] [Google Scholar]
- 9. Proud C. G. (1996) Trends Biochem. Sci. 21, 181–185 [PubMed] [Google Scholar]
- 10. Inoki K., Zhu T., Guan K. L. (2003) Cell 115, 577–590 [DOI] [PubMed] [Google Scholar]
- 11. Wong A. K., Howie J., Petrie J. R., Lang C. C. (2009) Clin. Sci. 116, 607–620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ferdinandy P., Schulz R., Baxter G. F. (2007) Pharmacol. Rev. 59, 418–458 [DOI] [PubMed] [Google Scholar]
- 13. Bennett B. L., Satoh Y., Lewis A. J. (2003) Curr. Opin. Pharmacol. 3, 420–425 [DOI] [PubMed] [Google Scholar]
- 14. Hirosumi J., Tuncman G., Chang L., Görgün C. Z., Uysal K. T., Maeda K., Karin M., Hotamisligil G. S. (2002) Nature 420, 333–336 [DOI] [PubMed] [Google Scholar]
- 15. Aguirre V., Uchida T., Yenush L., Davis R., White M. F. (2000) J. Biol. Chem. 275, 9047–9054 [DOI] [PubMed] [Google Scholar]
- 16. Um S. H., Frigerio F., Watanabe M., Picard F., Joaquin M., Sticker M., Fumagalli S., Allegrini P. R., Kozma S. C., Auwerx J., Thomas G. (2004) Nature 431, 200–205 [DOI] [PubMed] [Google Scholar]
- 17. Khamzina L., Veilleux A., Bergeron S., Marette A. (2005) Endocrinology 146, 1473–1481 [DOI] [PubMed] [Google Scholar]
- 18. Steppan C. M., Bailey S. T., Bhat S., Brown E. J., Banerjee R. R., Wright C. M., Patel H. R., Ahima R. S., Lazar M. A. (2001) Nature 409, 307–312 [DOI] [PubMed] [Google Scholar]
- 19. Banerjee R. R., Rangwala S. M., Shapiro J. S., Rich A. S., Rhoades B., Qi Y., Wang J., Rajala M. W., Pocai A., Scherer P. E., Steppan C. M., Ahima R. S., Obici S., Rossetti L., Lazar M. A. (2004) Science 303, 1195–1198 [DOI] [PubMed] [Google Scholar]
- 20. Muse E. D., Obici S., Bhanot S., Monia B. P., McKay R. A., Rajala M. W., Scherer P. E., Rossetti L. (2004) J. Clin. Invest. 114, 232–239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Rajala M. W., Obici S., Scherer P. E., Rossetti L. (2003) J. Clin. Invest. 111, 225–230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Graveleau C., Zaha V. G., Mohajer A., Banerjee R. R., Dudley-Rucker N., Steppan C. M., Rajala M. W., Scherer P. E., Ahima R. S., Lazar M. A., Abel E. D. (2005) J. Biol. Chem. 280, 31679–31685 [DOI] [PubMed] [Google Scholar]
- 23. Wang B. W., Hung H. F., Chang H., Kuan P., Shyu K. G. (2007) Am. J. Physiol. Heart Circ. Physiol. 293, H2305–2312 [DOI] [PubMed] [Google Scholar]
- 24. Kim M., Oh J. K., Sakata S., Liang I., Park W., Hajjar R. J., Lebeche D. (2008) J. Mol. Cell. Cardiol. 45, 270–280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Del Monte F., Butler K., Boecker W., Gwathmey J. K., Hajjar R. J. (2002) Physiol. Genomics 9, 49–56 [DOI] [PubMed] [Google Scholar]
- 26. Chan A. Y., Soltys C. L., Young M. E., Proud C. G., Dyck J. R. (2004) J. Biol. Chem. 279, 32771–32779 [DOI] [PubMed] [Google Scholar]
- 27. Noga A. A., Soltys C. L., Barr A. J., Kovacic S., Lopaschuk G. D., Dyck J. R. (2007) Am. J. Physiol. Heart Circ. Physiol. 292, H1460–H1469 [DOI] [PubMed] [Google Scholar]
- 28. Li H. L., Yin R., Chen D., Liu D., Wang D., Yang Q., Dong Y. G. (2007) J. Cell. Biochem. 100, 1086–1099 [DOI] [PubMed] [Google Scholar]
- 29. Tzatsos A., Kandror K. V. (2006) Mol. Cell. Biol. 26, 63–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Chemaly E. R., Kim M., Hadri L., Hajjar R. J., Lebeche D. (2008) Circulation 118, S487–S488 [Google Scholar]
- 31. Dyck J. R., Lopaschuk G. D. (2006) J. Physiol. 574 (Pt 1), 95–112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Tian R., Musi N., D'Agostino J., Hirshman M. F., Goodyear L. J. (2001) Circulation 104, 1664–1669 [DOI] [PubMed] [Google Scholar]
- 33. Shibata R., Ouchi N., Ito M., Kihara S., Shiojima I., Pimentel D. R., Kumada M., Sato K., Schiekofer S., Ohashi K., Funahashi T., Colucci W. S., Walsh K. (2004) Nat. Med. 10, 1384–1389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Liao Y., Takashima S., Maeda N., Ouchi N., Komamura K., Shimomura I., Hori M., Matsuzawa Y., Funahashi T., Kitakaze M. (2005) Cardiovasc. Res. 67, 705–713 [DOI] [PubMed] [Google Scholar]
- 35. McGaffin K. R., Witham W. G., Yester K. A., Romano L. C., O'Doherty R. M., McTiernan C. F., O'Donnell C. P. (2011) Cardiovasc. Res. 89, 60–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Harrington L. S., Findlay G. M., Lamb R. F. (2005) Trends Biochem. Sci. 30, 35–42 [DOI] [PubMed] [Google Scholar]
- 37. Virkamäki A., Ueki K., Kahn C. R. (1999) J. Clin. Invest. 103, 931–943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Greene M. W., Garofalo R. S. (2002) Biochemistry 41, 7082–7091 [DOI] [PubMed] [Google Scholar]
- 39. Rui L., Aguirre V., Kim J. K., Shulman G. I., Lee A., Corbould A., Dunaif A., White M. F. (2001) J. Clin. Invest. 107, 181–189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ozes O. N., Akca H., Mayo L. D., Gustin J. A., Maehama T., Dixon J. E., Donner D. B. (2001) Proc. Natl. Acad. Sci. U.S.A. 98, 4640–4645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Aguirre V., Werner E. D., Giraud J., Lee Y. H., Shoelson S. E., White M. F. (2002) J. Biol. Chem. 277, 1531–1537 [DOI] [PubMed] [Google Scholar]
- 42. Lee Y. H., Giraud J., Davis R. J., White M. F. (2003) J. Biol. Chem. 278, 2896–2902 [DOI] [PubMed] [Google Scholar]
- 43. Greene M. W., Sakaue H., Wang L., Alessi D. R., Roth R. A. (2003) J. Biol. Chem. 278, 8199–8211 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.