

Abstract
The plant plasma membrane H+-ATPase is regulated by an auto-inhibitory C-terminal domain that can be displaced by phosphorylation of the penultimate residue, a Thr, and the subsequent binding of 14-3-3 proteins. By mass spectrometric analysis of plasma membrane H+-ATPase isoform 2 (PMA2) isolated from Nicotiana tabacum plants and suspension cells, we identified a new phosphorylation site, Thr-889, in a region of the C-terminal domain upstream of the 14-3-3 protein binding site. This residue was mutated into aspartate or alanine, and the mutated H+-ATPases expressed in the yeast Saccharomyces cerevisiae. Unlike wild-type PMA2, which could replace the yeast H+-ATPases, the PMA2-Thr889Ala mutant did not allow yeast growth, whereas the PMA2-Thr889Asp mutant resulted in improved growth and increased H+-ATPase activity despite reduced phosphorylation of the PMA2 penultimate residue and reduced 14-3-3 protein binding. To determine whether the regulation taking place at Thr-889 was independent of phosphorylation of the penultimate residue and 14-3-3 protein binding, we examined the effect of combining the PMA2-Thr889Asp mutation with mutations of other residues that impair phosphorylation of the penultimate residue and/or binding of 14-3-3 proteins. The results showed that in yeast, PMA2 Thr-889 phosphorylation could activate H+-ATPase if PMA2 was also phosphorylated at its penultimate residue. However, binding of 14-3-3 proteins was not required, although 14-3-3 binding resulted in further activation. These results were confirmed in N. tabacum suspension cells. These data define a new H+-ATPase activation mechanism that can take place without 14-3-3 proteins.
Keywords: Membrane Proteins, Plasma Membrane, Plasma Membrane Proton Pumps, Protein Phosphorylation, Proton Pumps, Proton Transport
Introduction
The H+-ATPase4 is an electrogenic pump that couples ATP hydrolysis with the transport of protons across the plasma membrane of plant and fungi, thus establishing an electrochemical gradient that drives the secondary transport of ions and metabolites. The H+-ATPase is thus involved in many physiological roles, such as solute transport inside the cell and in the plant, cell pH homeostasis and elongation, stomatal opening, and salt stress tolerance. Because of the key role of this enzyme in plant cell function, multiple mechanisms have evolved to regulate H+-ATPase expression and activity (1, 2).
The H+-ATPase is encoded by a family of ∼10 genes (3, 4). At the post-translational level, enzyme activity is controlled by a C-terminal auto-inhibitory domain of about 100 residues. Site-directed mutagenesis and biochemical studies have shown that this domain can be functionally divided into two regions (I and II) (5) or three subdomains (A, B, and C) (6).
That the H+-ATPase is regulated by phosphorylation has been known for a long time. The best documented mechanism involves the phosphorylation of the penultimate residue, a Thr, and the subsequent binding of 14-3-3 regulatory proteins (7–10). This results in the formation of a dodecameric activated complex composed of six H+-ATPases and six 14-3-3 regulatory proteins (11, 12). Recently, the N-terminal region has also been proposed to be involved in enzyme auto-inhibition (13).
Other phosphorylated residues have been identified, and some have been shown to reduce or increase enzyme activity (1, 14–20). Mass spectrometric analysis of the purified His-tagged plasma membrane H+-ATPase isoform 2 (PMA2) from Nicotiana plumbaginifolia expressed in Nicotiana tabacum BY2 cells identified two phosphorylation sites, Thr-931 and Ser-938 (14). On mutation of these residues to aspartate, mimicking phosphorylation, the enzyme no longer binds 14-3-3 proteins and consequently cannot be activated, suggesting that both phosphorylation events negatively regulate the enzyme by interfering with 14-3-3 protein binding. Ser-931 of Arabidopsis H+-ATPase isoform 2 (AHA2), corresponding to PMA2 Ser-938, was shown to be phosphorylated by PKS5, a Ser/Thr protein kinase, both in vitro and in a yeast expression system, leading to lower enzyme activity due to prevention of 14-3-3 protein binding (15). Phospho-proteomic analysis of Arabidopsis plasma membrane proteins identified three additional AHA2 phosphorylation sites, Thr-881 (16, 21), Ser-889, and Ser-904 (17). Phosphorylation of Thr-881 was decreased in Arabidopsis cells treated with the bacterial elicitor flagellin (21) and increased in Arabidopsis seedlings supplied with sucrose (16). On mimicking the phosphorylation of this residue (Asp mutant) in a yeast expression system, enzyme activity was increased (6, 16). A proteomics study of rice identified two phosphorylation sites in Oryza sativa H+-ATPase isoform 3 (OSA3) at Thr-889 and Ser-870, OSA3 Thr-889 being homologous to AHA2 Thr-881 (20).
In this study, we purified a His-tagged PMA2 expressed in N. tabacum plants and in N. tabacum BY2 suspension cells, and mass spectrometry analysis showed that residue Thr-889 was phosphorylated in both cases. Analysis of PMA2 isoforms with a mutation only at this site or as double mutants with this and other mutations showed that the combined phosphorylation of this residue and the penultimate residue (PMA2 Thr-955) resulted in H+-ATPase activation in the absence of 14-3-3 protein binding, although 14-3-3 protein binding resulted in further activation.
EXPERIMENTAL PROCEDURES
Yeast Strains and Media
The YAK2 yeast strain has disrupted yeast PMA1 and PMA2 genes and contains a centromeric plasmid carrying the URA3 gene for selection and the yeast PMA1 gene under the control of the GAL1-10 promoter (22). Yeast strains were grown on rich medium containing 2% glucose and 2% yeast extract (YD medium) or on minimal medium containing 2% galactose, 0.7% yeast nitrogen base without amino acids (Difco) and 0.115% drop mix composed of all amino acids required for growth (dropout galactose medium) (23). The pH was adjusted to 6.5 (KOH), and 2% agar was added to obtain solid medium. 5′-Fluoroorotic acid medium was prepared as described in Ref. 23.
Plasmids and Genetic Constructions
The yeast plasmid 2μp(PMA1)6hispma2 contains the PMA2 gene with six His codons between the codons coding for residues 3 and 4 of PMA2 under the control of the Saccharomyces cerevisiae PMA1 promoter, the LEU2 gene for selection, and the 2μ plasmid-derived sequence for high copy number replication of the plasmid (22). The mutants PMA2-Glu14Asp (24), PMA2-Thr955Ala (9), PMA2-Ser938Asp (14), and PMA2-Δ52 (12) have been described previously. PMA2-ΔCter, coding for PMA2 lacking the last 102 amino acids, was obtained by introducing a stop codon at position 855 between the ApaI and BglII restriction sites of 2μp(PMA1)6hispma2 by triple PCR, and then the modified fragment was used to replace the corresponding ApaI/BglII fragment of 2μp(PMA1)6hispma2. Mutation of Thr-889 to Ala or Asp was achieved by amplifying a modified fragment between the BglII and SalI restriction sites of the 2μp(PMA1)6hispma2 plasmid by triple PCR and inserting it back into 2μp(PMA1)6hispma2 opened by BglII and SalI restriction. The PMA2 double mutants T889D/T955A, T889D/S938D, T889A-Δ52, and T889D-Δ52 were obtained using the same PCR strategy, except that the starting plasmids were 2μp(PMA1)6hispma2T955A, 2μp(PMA1)6hispma2S938D, or 2μp(PMA1)6hispma2Δ52. The plant binary vectors used for the N. tabacum BY2 cell transformation were obtained by first inserting an SmaI and XbaI fragment from the yeast 2μp(PMA1)6hispma2-T889A, 2μp(PMA1)6hispma2-T889D, and 2μp(PMA1)6hispma2-T889D-S938D plasmids corresponding, respectively, to the PMA2-Thr889Ala, PMA2-Thr889Asp, or PMA2-Thr889Asp-Ser938Asp coding sequence, into the pAUX3131 vector (25) between the N. plumbaginifolia PMA4 promoter reinforced with two copies of the cauliflower mosaic virus 35S enhancer (26) and the nos terminator and then transferring the PMA2 expression cassettes as an I-SceI fragment into the plant binary vector pPZP-RCS2 (25) containing the nptII marker gene.
Membrane Isolation
All buffers were supplemented with 1 mm phenylmethylsulfonyl fluoride and the protease inhibitors leupeptin, aprotinin, antipain, pepstatin, and chymostatin (2 μg/ml each). Yeast and plant microsomal and plasma membrane fractions were prepared as described previously (14). Microsomal membranes from N. tabacum BY2 cells at day 3 were prepared with a homogenization buffer supplemented with 1 μm fusicoccin to stabilize the H+-ATPase 14-3-3 complex during subcellular fractionation (11).
N. tabacum BY2 Cell Culture and Transformation
N. tabacum BY2 cells were maintained on MS medium (catalogue number 2610024; MP Biomedicals, LLC) supplemented with 3% sucrose, 0.2 mg/liter 2,4-dichlorophenoxyacetic acid, 0.2 g/liter KH2PO4, pH 5.8, 50 mg/liter myo-inositol, and 5 mg/liter thiamine-HCl. N. tabacum BY2 cells were diluted 20-fold each week with fresh medium and grown as described previously (27). Stable expression of the mutated PMA2 isoforms was achieved by a 48-h co-cultivation of 24 ml of a 4-day-old BY2 cell culture and 7 ml of Agrobacterium tumefaciens strain LBA4404virG (28) previously transformed with the different pPZP-PMA2 plasmids and grown to an A600 of 1.4. Washed A. tumefaciens and BY2 cells were co-cultured for 48 h in BY2 cell medium at pH 5.3 (KOH) supplemented with 10 mm glucose and 50 μm acetosyringone. Transformed lines were then obtained by plating 2 ml of the co-culture on solid BY2 cell medium supplemented with 0.8% agar, 500 μg/ml cefotaxime, 400 μg/ml carbenicillin, and 100 μg/ml kanamycin and growing the calli for 4–6 weeks at 24 °C in the dark. Transformed calli were kept on solid, or resuspended in liquid, BY2 cell medium supplemented with kanamycin (100 μg/ml).
PMA2 Purification
The His6-tagged PMA2 isoforms expressed in yeast or in N. tabacum BY2 cells were purified on Ni2+-NTA agarose (Qiagen) as described in Ref. 14.
Mass Spectrometric Analysis
Microsomal proteins were separated by SDS-PAGE, and the band of interest was excised, dried under vacuum, rehydrated in 100 μl of 200 mm ammonium bicarbonate with 250 ng of trypsin, and incubated for 16 h at 37 °C. The released peptides were analyzed by nano LC-MS/MS performed on a Dionex Ultimate capillary liquid chromatography system coupled to an Applied Biosystems 4000 QTRAP mass spectrometer. After collection on a C18 trap column, the peptides were separated on a PepMap C18 analytical column developed with a 30-min linear gradient from 0.1% formic acid, 6% acetonitrile and water to 0.1% formic acid, 40% acetonitrile, and water. In a first LC-MS run, putative phosphorylated peptides were detected by precursor 79 (−) ion scanning. In a second LC-MS run, multiple reaction monitoring of these peptides was used to induce product (+) ion scanning to determine the peptide sequence and localize the phosphorylated residue(s). Tandem MS spectra were interpreted manually.
ATPase Assays
ATPase assays were performed with 2 μg of protein in a 96-well microplate as described in Ref. 14.
Measurement of Acidification of the External Medium by Yeast or by N. tabacum BY2 Cells
Yeast cells were grown in YD medium, harvested at the late exponential phase, and washed three times in ice-cold water, and then 109 cells were added to 10 ml of 200 mm glucose, 20 mm KCl in either 0.2 mm MES, pH 6.5 (KOH) or 0.2 mm citrate, pH 4.0 (KOH) in a vial with a magnetic stirrer and a pH electrode at 30 °C. The pH was recorded for 20 min, and pumping activity was calculated as pH units × min−1 × (109 cells)−1. For N. tabacum BY2 cells, 8 ml of a 4-day-old 100-ml culture were centrifuged for 5 min at 1,000 g. The cells were resuspended in 8 ml of 200 mm glucose, 20 mm KCl (pH 5.8) and transferred into a vial fitted with a magnetic stirrer and a pH electrode. The pH was recorded at 25 °C.
Limited Tryptic Digestion
Limited tryptic digestion was performed at 37 °C on 100 μg of purified plasma membrane fractions with a trypsin/protein ratio of 1/100 in 50 mm Tris-HCl (pH 8.5) in a final volume of 500 μl. After 0, 0.25, 0.5, 1, 2, 5, or 10 min, a 50-μl sample was taken and added to 10 μl of 10% SDS to stop the reaction, and then 20 μl of 4-fold concentrated Laemmli buffer (29) were added to each fraction.
RESULTS
Identification of a New PMA2 Phosphorylation Site
To identify phosphorylation sites in PMA2, one of the two most widely expressed H+-ATPases in N. tabacum, His6-tagged PMA2 was expressed in N. tabacum plants (30) and N. tabacum BY2 cell lines (31), solubilized from the microsomal fraction, purified by Ni2+-NTA chromatography, and analyzed by SDS-PAGE. The PMA2 band was then excised from the gel and digested with trypsin, and the resulting peptides were analyzed by nano LC-MS/MS as described under “Experimental Procedures.” Phosphorylated PMA2 Thr-889 was identified in both the BY2 cell (Fig. 1) and the plant (data not shown) samples. This threonine residue is very highly conserved within the H+-ATPase family and is located within the auto-inhibitory C-terminal region.
FIGURE 1.

Mass spectrometric analysis of PMA2 phosphopeptides. An analysis of a phosphopeptide from purified His-tagged PMA2 expressed in the N. tabacum BY2 cell line is shown. The product ion spectrum of m/z 803.4 (3+) identifies the peptide as mono-phosphorylated ELQWAHAQRTpLHGLQVPDTK and pinpoints threonine 889 as the site of phosphorylation. Intensity is in cps. amu, atomic mass units.
Characterization of PMA2 Thr-889 Mutants Expressed in Yeast
For further characterization, Thr-889 was mutated either to aspartate to mimic the negative charge of a phosphorylated residue or to alanine to prevent phosphorylation, and the resulting His6-tagged mutants were placed under the control of a constitutive promoter in a yeast plasmid and introduced into the S. cerevisiae YAK2 strain (22). This strain is deleted of its own H+-ATPase genes, but to allow growth, it contains a centromeric plasmid carrying the yeast PMA1 gene under the control of the GAL1-10 promoter and the URA3 gene for selection. To assess the ability of the PMA2 mutants to sustain yeast growth, yeast transformants were transferred to a glucose medium, which represses yeast PMA1 expression without affecting expression of PMA2. We assessed growth at pH 6.5, which was previously shown to be optimum for this yeast strain expressing PMA2 (32), and at pH 5.5 and 4.5, which are more challenging pHs. As shown in Fig. 2A, the PMA2-Thr889Asp (PMA2-T889D) mutant allowed better yeast growth than the wild type, whereas the PMA2-Thr889Ala (PMA2-T889A) mutant resulted in slightly slower growth than the wild type. To confirm these results, we selected the strains on 5′-fluoroorotic acid, which is converted into a toxic compound by the URA3 gene, and only strains that have lost the yeast PMA1 plasmid and carry a functional plant H+-ATPase gene on the other plasmid are able to grow (Fig. 2B). As a positive control, the constitutively activated mutant, PMA2-Glu14Asp, grew very well. Colonies were also obtained with the wild-type PMA2 and PMA2-Thr889Asp, the latter growing faster than the former. No colony was observed for PMA2-Thr889Ala (Fig. 2B), although slight growth was previously observed upon glucose shift (Fig. 2A), probably resulting from weak activity of this isoform combined with remaining endogenous PMA1. These data clearly suggest that phosphorylation of Thr-889 results in enzyme activation.
FIGURE 2.

Growth of yeast strains expressing PMA2 mutants. A, YAK2 yeast strains expressing yeast PMA1 and the indicated PMA2 isoform were grown overnight in dropout galactose medium and then were diluted to an A600 of 1, 0.1, or 0.01 and plated on solid YD medium at pH 6.5, 5.5, or 4.5 and allowed to grow for 3 days at 30 °C. B, same as in A, but strains were plated on solid 5′-fluoroorotic acid (5′-FOA) medium at pH 6.5 and allowed to grow for 10 days at 30 °C.
Usually, H+-ATPase activation is linked to higher levels of phosphorylation of the penultimate residue and increased binding of 14-3-3 proteins, as observed with PMA2-Glu14Asp (PMA2-E14D), a constitutively activated mutant (9, 24). We therefore wondered whether this was also the case for the T889D mutant. To assess the level of phosphorylation of the penultimate Thr-955, we used the monoclonal antibody mab-PMA2pT, which is specific for PMA2 phosphorylated at Thr-955 (30). Western blotting analysis of the yeast plasma membrane fraction showed a reduction in Thr-955 phosphorylation and 14-3-3 protein binding with the PMA2-Thr889Asp mutant when compared with the wild type (Fig. 3). However, because we could not exclude the possibility that 14-3-3 proteins were bound to other plasma membrane proteins, we performed the same analysis on Ni2+-NTA-purified isoforms and found an even larger decrease in Thr-955 phosphorylation and 14-3-3 protein binding when compared with the wild type than with the membranes, possibly reflecting that with lower 14-3-3 protein binding, the unprotected Thr-955 is partly dephosphorylated during purification. Remarkably, this decrease in Thr-955 phosphorylation and 14-3-3 binding was not correlated with a reduction in enzyme activity because the PMA2-Thr889Asp mutant in the plasma membrane fraction showed increased ATPase activity when compared with the wild-type PMA2 (Fig. 4A), in agreement with the observed better yeast growth. The activity of the PMA2-Thr889Asp mutant was not as high as that of the PMA2-Glu14Asp activated mutant, which showed high Thr-955 phosphorylation and 14-3-3 binding (Fig. 4A). The in vivo proton pumping activity of the PMA2 isoforms was indirectly monitored by measuring the rate of acidification of the external medium by yeast cells. Measurements performed at pH 6.5 or 4.0 (Fig. 4B) showed that, as with the ATPase activity, the PMA2-Thr889Asp mutant displayed pumping activity higher than that of wild-type PMA2, but lower than that of PMA2-Glu14Asp.
FIGURE 3.

Immunodetection of Thr-955 phosphorylation and bound 14-3-3 proteins for the wild-type PMA2 and the T889D and E14D mutants. A and C, the plasma membrane fraction (5 μg of protein) isolated from a 36-h culture (YD, pH 6.5) of yeast strains expressing wild-type or mutant PMA2 (A, left panel) or the corresponding His6-tagged PMA2 purified by Ni2+-NTA chromatography (A, right panel, and C) was analyzed by SDS-PAGE and Western blotting using antibodies against PMA2 (top), PMA2 phospho-Thr-955 (pPMA2; middle), or 14-3-3 proteins (bottom). Note that the double band identified by anti-14-3-3 protein antibodies corresponds to the two S. cerevisiae 14-3-3 proteins. B and D, the PMA2 phospho-Thr-955/PMA2 signal ratio (white bars) and the 14-3-3 protein/PMA2 signal ratio (black bars). The signals were quantified using the Image Station 4000R and Molecular Imaging software from Eastman Kodak Co. and were normalized to that for PMA2 (set at 100%). The data are the mean and S.E. for seven independent experiments in B and four independent experiments in D. The statistical significance of the difference between the indicated results is shown by the asterisks (*, p < 0.05; **, p < 0.01; ***, p < 0.001).
FIGURE 4.

In vitro ATPase activity and in vivo acidification rate of wild-type PMA2 and the T889D and E14D mutants. A, the vanadate-sensitive ATPase specific activity was measured using plasma membranes from YAK2 strains (after 5′-fluoroorotic acid selection) expressing the indicated wild-type or mutant PMA2. The specific activity (μmol of Pi·mg−1·min−1) was divided by the amount of PMA2 estimated by Western blotting and set at 1 for the PMA2 strain. The data are the mean and S.E. for three independent experiments, with several measurements per experiment. B, acidification of the external medium by yeast cells expressing PMA2 mutants was measured as indicated under “Experimental Procedures.” The data are the mean and S.E. for five independent experiments, with several replicates per experiment. The statistical significance of the difference between the indicated results is shown by the asterisks (*, p < 0.05; ***, p < 0.001).
Several single-point mutations in PMA2 that result in better yeast growth are characterized by a C-terminal region that is more accessible to trypsin digestion, indicating a loose auto-inhibitory domain (24). This high accessibility correlates with a high Thr-955 phosphorylation and high amounts of bound 14-3-3 proteins (9, 14). Because the activation of PMA2-Thr889Asp was not correlated with higher phosphorylation of Thr-955 and bound 14-3-3 proteins, we assessed the accessibility to trypsin of its C-terminal region. Limited tryptic digestion showed that degradation of both PMA2-Glu14Asp and PMA2-Thr889Asp was more rapid than that of wild-type PMA2 (Fig. 5), indicating increased accessibility of the C-terminal region in these two mutants. Because PMA2-Thr889Asp had much less bound 14-3-3 proteins, phosphorylation of Thr-889 might therefore activate the enzyme by displacing the C-terminal region without the need for 14-3-3 proteins.
FIGURE 5.
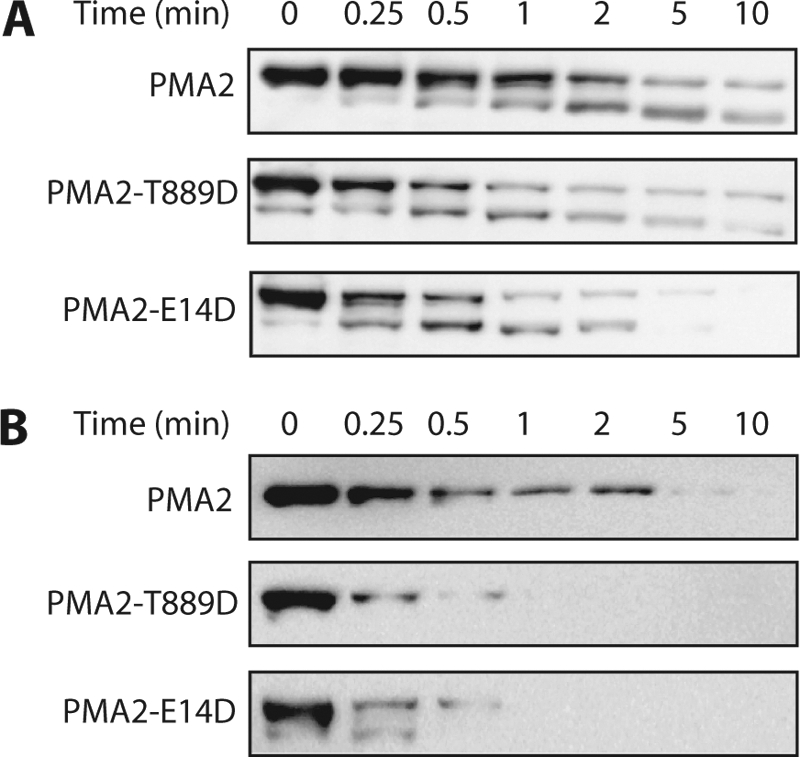
Limited tryptic digestion of wild-type and mutated PMA2. Purified plasma membranes from YAK2 strains (after 5′-fluoroorotic acid selection) expressing wild-type or mutated PMA2 were digested by trypsin as described under “Experimental Procedures” for the indicated times. A, immunodetection of PMA2 using an antibody against the whole protein, allowing the detection of both the full-length enzyme (100 kDa) and the 90-kDa degradation product. B, immunodetection of PMA2 with an antibody against the C-terminal part, detecting only the full-length enzyme (100 kDa).
We then investigated in more depth the relationship between the phosphorylation of Thr-889 and Thr-955 and 14-3-3 protein binding by combining several mutants. The PMA2-Thr955Ala (PMA2-T955A) mutant, which cannot be phosphorylated at Thr-955 and thus cannot bind 14-3-3 proteins, is not able to sustain yeast growth (9). To test whether the activating effect of PMA2-Thr889Asp was independent of Thr-955 phosphorylation and 14-3-3 protein binding, the PMA2-Thr889Asp and PMA2-Thr955Ala mutations were combined as a double mutant PMA2-Thr889Asp-Thr955Ala (PMA2-T889D-T955A), which failed to allow growth of the YAK2 yeast strain in the glucose shift assay (Fig. 2A), showing that phosphorylation of Thr-889 alone does not result in sufficient activity to sustain yeast growth.
As neither PMA2-Thr889Ala nor PMA2-Thr889Asp-Thr955Ala could sustain yeast growth, phosphorylation of both Thr-889 and Thr-955 seems necessary for enzyme activation. However, whether 14-3-3 protein binding was required remains unanswered. Recently, it was shown that the PMA2-Ser938Asp (PMA2-S938D) mutation prevents 14-3-3 protein binding and fails to sustain yeast growth and that, in this mutant, Thr-955 phosphorylation is decreased, but not abolished (14). If we hypothesize that activation of H+-ATPase by phosphorylation of Thr-889 requires Thr-955 phosphorylation, but not the binding of 14-3-3 proteins, the double mutant PMA2-Thr889Asp-Ser938Asp (PMA2-T889D-S938D) should be devoid of bound 14-3-3 proteins but still sustain yeast growth. This was indeed the case as the double mutant sustained yeast growth on solid medium (Fig. 2A) or in liquid cultures (6.00 ± 0.15 h doubling time; data not shown) and even showed slightly better growth than wild-type PMA2 (6.90 ± 0.26 h doubling time). When compared with the single mutant PMA2-Thr889Asp (3.97 ± 0.04 h doubling time), the double mutant showed reduced growth. Western blot analysis of purified double mutant PMA2-Thr889Asp-Ser938Asp showed the absence of co-purifying 14-3-3 proteins (Fig. 3, C and D). Therefore, the ability of the double mutant to sustain yeast growth indicates that the mechanism of activation involving Thr-889 phosphorylation does not require 14-3-3 protein binding, but the reduced growth when compared with that of the PMA2-Thr889Asp mutant, which can still bind 14-3-3 proteins to some extent (Fig. 3, C and D), shows that 14-3-3 protein binding has an additional effect in proton pump activation.
The results with the double mutant strongly suggested that phosphorylation of both Thr-889 and Thr-955 was needed for activation. To study the effect of Thr-889 phosphorylation in isolation, we introduced the Thr-889 mutations into PMA2-Δ52, which codes for a PMA2 lacking the last C-terminal 52 amino acids (12). This isoform lacks part of the inhibitory domain and is therefore more active than unmodified PMA2. On this background, the PMA2-Thr889Ala mutation still allowed yeast growth, but retained its negative effect on enzyme activity, as the growth of PMA2-Thr889Ala-Δ52 was reduced when compared with that of PMA2-Δ52 (Fig. 2A). The other mutation, PMA2-Thr889Asp-Δ52, conferred better yeast growth at challenging pHs when compared with the PMA2-Δ52 isoform, suggesting that Thr-889 phosphorylation probably involves an activating mechanism in part distinct from that supported by Thr-955 phosphorylation and 14-3-3 protein binding and that phosphorylation of the Thr-889 and Thr-955 residues has a positive combined effect on enzyme activity.
Characterization of PMA2 Thr-889 Mutants Expressed in Tobacco Cells
To verify whether the dramatic decrease in 14-3-3 protein binding seen with PMA2-Thr889Asp also occurred in plants, we stably expressed wild-type PMA2 and the mutants PMA2-Thr889Ala, PMA2-Thr889Asp, PMA2-Ser938Asp, and PMA2-Thr889Asp-Ser938Asp in N. tabacum BY2 cells by co-cultivation with transformed A. tumefaciens. Under standard culture conditions for N. tabacum BY2 cells, both Thr-955 phosphorylation and 14-3-3 protein binding are moderate and decrease during culture growth (30). We therefore characterized cells collected at a young stage (i.e. 3-day-old cultures) to assess the amount of Thr-955 phosphorylation of, and 14-3-3 proteins co-eluting with, purified His6-tagged PMA2. In addition, to prevent dephosphorylation of the penultimate residue and the loss of 14-3-3 proteins during the purification, fusicoccin, a fungal toxin stabilizing the H+-ATPase 14-3-3 protein complex, was added to the homogenization buffer during membrane preparation (11, 33). Western blotting of the purified PMA2 isoforms (Fig. 6) showed that PMA2-Ser938Asp had a lower level of phosphorylated Thr-955 and less bound 14-3-3 proteins than the wild type, as described previously (14). When compared with unmodified PMA2, the mutation of Thr-889 into alanine or aspartate did not modify significantly the level of Thr-955 phosphorylation, but significantly decreased the amount of co-purified 14-3-3 proteins. The PMA2-Thr889Asp-Ser938Asp double mutant showed a significant decrease in the amount of 14-3-3 proteins when compared with either the wild-type PMA2 or the PMA2-Thr889Asp single mutant, whereas Thr-955 phosphorylation was higher than that of the PMA2-Ser938Asp single mutant, but not different from that of the wild-type PMA2 or the PMA2-Thr889Asp mutant. Thus, although the phenotypes were less severe in terms of 14-3-3 protein binding and Thr-955 phosphorylation, the results obtained in BY2 cells supported the data obtained in the yeast.
FIGURE 6.

Immunodetection of Thr-955 phosphorylation and bound 14-3-3 proteins for purified PMA2 mutants expressed in N. tabacum BY2 culture cells. A, 3-day-old cultures of N. tabacum BY2 cells expressing the indicated wild-type or mutant His-tagged PMA2 were harvested and homogenized in the presence of 1 μm fusicoccin, and a microsomal fraction was prepared, and then the proteins were solubilized, and the tagged PMA2 was purified by Ni2+-NTA chromatography and analyzed by SDS-PAGE and Western blotting using antibodies against PMA2 (top), PMA2 phospho-Thr-955 (pPMA2; middle), or 14-3-3 proteins (bottom). B, the PMA2 phospho-Thr-955/PMA2 signal ratio (white bars) and the 14-3-3 protein/PMA2 signal ratio (black bars). The signals were quantified using the Image Station 4000R and Molecular Imaging software from Kodak and were each normalized to that for PMA2 (set at 100%). The data are the mean and S.E. for four independent experiments. The statistical significance of differences between results is indicated by the letters, results with different letters being significantly different by analysis of variance (p < 0.05).
To determine whether H+-ATPase activation of PMA2-Ser938Asp resulted in increased in vivo proton pumping activity, we measured the acidification rate of the external medium by BY2 cells. Comparison of the four lines, WT, PMA2, PMA2-Thr889Asp (PMA2-T889D), and PMA2-Thr889Ala (PMA2-T889A), showed that the expression of the mutated or the unmodified PMA2 did not result in a significant modification of the external acidification rate (Fig. 7A). This is probably due to the low contribution of the ectopic PMA2 when compared with that of the endogenous PMA2 and other PMA isoforms. Indeed, Western blotting of microsomal fractions showed that although the His-tagged PMA2 could be clearly identified using His tag antibodies, the total PMA2 amount was not significantly increased when compared with the untransformed cells (Fig. 7B). We cannot rule out that the ectopic PMA2 expression resulted in a lower expression of the endogenous PMA2. However, this is unlikely because the amount of PMA4, which is the other most expressed isoform in N. tabacum, remained unchanged (Fig. 7B).
FIGURE 7.

In vivo acidification rate and immunodetection of ectopic PMA2 in N. tabacum BY2 cells expressing PMA2, PMA2 T889D, or PMA2 T889A. A, acidification of the external medium by N. tabacum BY2 cells was measured (pH unit·min−1·g−1 (UpH.min−1g−1, cell fresh weight)) as indicated under “Experimental Procedures.” The data are the mean and confidence interval (95%) for four independent experiments, with several replicates per experiment. B, immunodetection of His tag PMA2, PMA2, and PMA4. Microsomal membrane fractions (10 μg of protein) were analyzed by SDS-PAGE and Western blotting using antibodies against His-tagged PMA2 (top), PMA2 (middle), and PMA4 (bottom).
DISCUSSION
A well known documented mechanism of regulation of the H+-ATPase is its activation by phosphorylation of its penultimate residue, a Thr, and the subsequent binding of 14-3-3 proteins. Recently, several studies in Arabidopsis thaliana, O. sativa, and N. plumbaginifolia have led to the identification of new phosphorylation sites in the C-terminal region of the H+-ATPase (for review, see Ref, 1). Two of these, corresponding to PMA2 Thr-931 (14) and to PMA2 Ser-938 (14) or AHA2 931 (15), have been shown to negatively regulate the enzyme by impairing the binding of 14-3-3 proteins.
In this study, we described the identification of PMA2 Thr-889 phosphorylated in the plant and its molecular and biochemical characterization in both yeast and plant cell expression systems. Expression of the PMA2-Thr889Ala mutant did not allow yeast growth, whereas mutation of this residue to Asp resulted in improved growth and increased H+-ATPase activity when compared with wild-type PMA2, suggesting that Thr-889 phosphorylation is involved in enzyme activation. Surprisingly, in contrast to what would be expected from the generally accepted regulation model, the PMA2-Thr889Asp mutation resulted in a reduction in phosphorylated Thr-955 and a more drastic reduction in bound 14-3-3 proteins. Activation of the enzyme thus does not depend on high levels of Thr-955 phosphorylation and 14-3-3 protein binding. However, this does not mean that Thr-955 phosphorylation and/or 14-3-3 protein binding are dispensable. Indeed, no yeast growth was observed when T889D was combined with the T955A mutation, which prevents phosphorylation of this residue and subsequent 14-3-3 protein binding (9). Additional information was provided by the double mutant of PMA2-Thr889Asp and PMA2-Ser938Asp, a mutation that allows Thr-955 phosphorylation, but not 14-3-3 protein binding, and so prevents yeast growth (14). This combination allowed better growth than wild-type PMA2, strongly suggesting that the mechanism of activation involving phosphorylation of Thr-889 is indeed independent of 14-3-3 protein binding. However, the PMA2-Thr889Asp-Ser938Asp mutant grew more slowly than the PMA2-Thr889Asp mutant, indicating that 14-3-3 protein binding generates additional activation.
Expression of the Thr-889 mutants in N. tabacum BY2 cells led to similar observations, although the reduction in 14-3-3 protein binding with the PMA2-Thr889Asp-Ser938Asp mutant was less severe than in yeast. However, we should bear in mind that in transgenic BY2 cells, the endogenous wild-type PMA2 is still present and, because H+-ATPases are known to dimerize, wild-type PMA2 can be co-purified with the tagged isoform and thus contribute to 14-3-3 protein binding.
In N. tabacum BY2 cells, expression of the PMA2-Thr889Asp (PMA2-T889D) isoform did not result in increased external acidification, most probably because the contribution of the ectopic PMA2 was too low when compared with the endogenous PMA2, PMA4, and possibly other unknown PMA isoforms. The reason might be that the ectopic expression of H+-ATPase in N. tabacum BY2 cells might be detrimental, and thus, only transgenic lines with low expression were selected. This is in agreement with the observation that transgenic lines selected for the expression of ectopic PMA2 or PMA4 (30) expressed these isoforms less and less over time, necessitating the repetition of the transformation.
PMA2 Thr-889 is well conserved among H+-ATPases. The corresponding residue is also phosphorylated in the OSA3 rice isoform (20), belonging, like PMA2, to subfamily I of the H+-ATPases, and in the AHA2 Arabidopsis isoform (16, 21), belonging to subfamily II. Phosphorylation of AHA2 Thr-881 is decreased in Arabidopsis cells treated with the bacterial elicitor flagellin (21) and is increased in Arabidopsis seedlings supplied with sucrose (16). Niittylä et al. (16) suggested that AHA2 Thr-881 phosphorylation is independent of phosphorylation of the penultimate residue and hence 14-3-3 protein binding. Here, using phospho-specific antibodies and purified mutated PMA2 isoforms, we were able to demonstrate that upon Thr-889 phosphorylation, Thr-955 must also be phosphorylated for enzyme activation, whereas 14-3-3 protein binding is not required. This is the first mechanism for H+-ATPase activation by phosphorylation that has been shown not to be strictly dependent on 14-3-3 protein binding as previously an increase in enzyme activity has been correlated with increased binding of 14-3-3 proteins to the enzyme C terminus.
At least four phosphorylation sites within the PMA2 C terminus have been clearly identified and shown to be involved in enzyme regulation. Moreover, in a recent study, a fifth residue in the PMA2 C terminus has been suggested to be phosphorylated (6); however, its phosphorylation has not been demonstrated, and only mutagenesis results support this conclusion. Nevertheless, three regions of auto-inhibition, each controlled by an important conserved threonine, have been identified (6), Thr-869 in region A, Thr-889 in region B, and the penultimate residue, Thr-955, in region C. It has been shown that for PMA2, the mimicking of phosphorylation of Thr-869 alone or together with that of Thr-889 in a yeast expression system increases enzyme activity and overrides the inhibition caused by the alanine mutation of the penultimate residue (6). Our results showed that Thr-889 phosphorylation is not by itself able to override the absence of Thr-955 phosphorylation and support the hypothesis that this is achieved by the combination of an upstream event and Thr-889 phosphorylation.
In conclusion, we have demonstrated phosphorylation of Thr-889 of PMA2. Its characterization by site-directed mutagenesis uncovers a new aspect of the regulation of H+-ATPases, that of H+-ATPase activation in the absence of 14-3-3 protein binding. Thr-889 phosphorylation can activate the PMA2 provided the penultimate residue, Thr-955, is also phosphorylated, whereas binding of 14-3-3 proteins is not required, but can cause additional activation. These results highlight the fact that H+-ATPase activation relies on different mechanisms involving phosphorylation steps and the binding of 14-3-3 proteins.
Acknowledgments
We thank Prof Claudia Oecking for providing the PMA2-Δ52 plasmid. We also thank J. Nader for excellent technical support.
This work was supported by grants from the Interuniversity Poles of Attraction Program (Belgian State, Scientific, Technical and Cultural Services) and the Belgian National Fund for Scientific Research.
- H+-ATPase
- proton pump ATPase
- PMA2
- N. tabacum plasma membrane H+-ATPase isoform 2
- AHA2
- A. thaliana H+-ATPase isoform 2
- NTA
- nitrilotriacetic acid.
REFERENCES
- 1. Duby G., Boutry M. (2009) Pflugers Arch. 457, 645–655 [DOI] [PubMed] [Google Scholar]
- 2. Palmgren M. G. (2001) Annu. Rev. Plant Physiol. Plant Mol. Biol. 52, 817–845 [DOI] [PubMed] [Google Scholar]
- 3. Arango M., Gévaudant F., Oufattole M., Boutry M. (2003) Planta 216, 355–365 [DOI] [PubMed] [Google Scholar]
- 4. Baxter I., Tchieu J., Sussman M. R., Boutry M., Palmgren M. G., Gribskov M., Harper J. F., Axelsen K. B. (2003) Plant. Physiol. 132, 618–628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Axelsen K. B., Venema K., Jahn T., Baunsgaard L., Palmgren M. G. (1999) Biochemistry 38, 7227–7234 [DOI] [PubMed] [Google Scholar]
- 6. Speth C., Jaspert N., Marcon C., Oecking C. (2010) Eur. J. Cell Biol. 89, 145–151 [DOI] [PubMed] [Google Scholar]
- 7. Fuglsang A. T., Visconti S., Drumm K., Jahn T., Stensballe A., Mattei B., Jensen O. N., Aducci P., Palmgren M. G. (1999) J. Biol. Chem. 274, 36774–36780 [DOI] [PubMed] [Google Scholar]
- 8. Kinoshita T., Shimazaki K. (1999) EMBO J. 18, 5548–5558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Maudoux O., Batoko H., Oecking C., Gevaert K., Vandekerckhove J., Boutry M., Morsomme P. (2000) J. Biol. Chem. 275, 17762–17770 [DOI] [PubMed] [Google Scholar]
- 10. Svennelid F., Olsson A., Piotrowski M., Rosenquist M., Ottman C., Larsson C., Oecking C., Sommarin M. (1999) Plant Cell 11, 2379–2391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kanczewska J., Marco S., Vandermeeren C., Maudoux O., Rigaud J. L., Boutry M. (2005) Proc. Natl. Acad. Sci. U.S.A. 102, 11675–11680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ottmann C., Marco S., Jaspert N., Marcon C., Schauer N., Weyand M., Vandermeeren C., Duby G., Boutry M., Wittinghofer A., Rigaud J. L., Oecking C. (2007) Mol. Cell 25, 427–440 [DOI] [PubMed] [Google Scholar]
- 13. Ekberg K., Palmgren M. G., Veierskov B., Buch-Pedersen M. J. (2010) J. Biol. Chem. 285, 7344–7350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Duby G., Poreba W., Piotrowiak D., Bobik K., Derua R., Waelkens E., Boutry M. (2009) J. Biol. Chem. 284, 4213–4221 [DOI] [PubMed] [Google Scholar]
- 15. Fuglsang A. T., Guo Y., Cuin T. A., Qiu Q., Song C., Kristiansen K. A., Bych K., Schulz A., Shabala S., Schumaker K. S., Palmgren M. G., Zhu J. K. (2007) Plant Cell 19, 1617–1634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Niittylä T., Fuglsang A. T., Palmgren M. G., Frommer W. B., Schulze W. X. (2007) Mol. Cell Proteomics. 6, 1711–1726 [DOI] [PubMed] [Google Scholar]
- 17. Nühse T. S., Stensballe A., Jensen O. N., Peck S. C. (2003) Mol. Cell Proteomics. 2, 1234–1243 [DOI] [PubMed] [Google Scholar]
- 18. Ookura T., Komatsu S., Kawamura Y., Kasamo K. (2005) Jarq-Jpn. Agr. Res. Q. 39, 99–104 [Google Scholar]
- 19. Sugiyama N., Nakagami H., Mochida K., Daudi A., Tomita M., Shirasu K., Ishihama Y. (2008) Mol. Syst. Biol. 4, 193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Whiteman S. A., Nühse T. S., Ashford D. A., Sanders D., Maathuis F. J. (2008) Plant J. 56, 146–156 [DOI] [PubMed] [Google Scholar]
- 21. Nühse T. S., Bottrill A. R., Jones A. M., Peck S. C. (2007) Plant. J. 51, 931–940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. de Kerchove d'Exaerde A., Supply P., Dufour J. P., Bogaerts P., Thinés D., Goffeau A., Boutry M. (1995) J. Biol. Chem. 270, 23828–23837 [DOI] [PubMed] [Google Scholar]
- 23. Treco D. A., Lundblad V. (2001) in Current Protocols in Molecular Biology, pp. 13.1.1–13.1.7, John Wiley & Sons, Inc., New York: [DOI] [PubMed] [Google Scholar]
- 24. Morsomme P., Dambly S., Maudoux O., Boutry M. (1998) J. Biol. Chem. 273, 34837–34842 [DOI] [PubMed] [Google Scholar]
- 25. Goderis I. J., De Bolle M. F., François I. E., Wouters P. F., Broekaert W. F., Cammue B. P. (2002) Plant. Mol. Biol. 50, 17–27 [DOI] [PubMed] [Google Scholar]
- 26. Zhao R. M., Moriau L., Boutry M. (1999) Plant Sci. 149, 157–165 [Google Scholar]
- 27. Nagata T., Nemoto Y., Hasezawa S. (1992) Int. Rev. Cytol. 132, 1–30 [Google Scholar]
- 28. van der Fits L., Deakin E. A., Hoge J. H., Memelink J. (2000) Plant. Mol. Biol. 43, 495–502 [DOI] [PubMed] [Google Scholar]
- 29. Laemmli U. K. (1970) Nature 227, 680–685 [DOI] [PubMed] [Google Scholar]
- 30. Bobik K., Duby G., Nizet Y., Vandermeeren C., Stiernet P., Kanczewska J., Boutry M. (2010) Plant. J. 62, 291–301 [DOI] [PubMed] [Google Scholar]
- 31. Woloszynska M., Kanczewska J., Drabkin A., Maudoux O., Dambly S., Boutry M. (2003) Ann. N.Y. Acad. Sci. 986, 198–203 [DOI] [PubMed] [Google Scholar]
- 32. Luo H., Morsomme P., Boutry M. (1999) Plant. Physiol. 119, 627–634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Olsson A., Svennelid F., Ek B., Sommarin M., Larsson C. (1998) Plant. Physiol. 118, 551–555 [DOI] [PMC free article] [PubMed] [Google Scholar]