

Abstract
ADAR2, an RNA editing enzyme that converts specific adenosines to inosines in certain pre-mRNAs, often leading to amino acid substitutions in the encoded proteins, is mainly expressed in brain. Of all ADAR2-mediated edits, a single one in the pre-mRNA of the AMPA receptor subunit GluA2 is essential for survival. Hence, early postnatal death of mice lacking ADAR2 is averted when the critical edit is engineered into both GluA2 encoding Gria2 alleles. Adar2−/−/Gria2R/R mice display normal appearance and life span, but the general phenotypic effects of global lack of ADAR2 have remained unexplored. Here we have employed the Adar2−/−/Gria2R/R mouse line, and Gria2R/R mice as controls, to study the phenotypic consequences of loss of all ADAR2-mediated edits except the critical one in GluA2. Our extended phenotypic analysis covering ∼320 parameters identified significant changes related to absence of ADAR2 in behavior, hearing ability, allergy parameters and transcript profiles of brain.
Keywords: Gene knockout, Gene regulation, Mouse, Mouse genetics, RNA editing, Phenotyping, Systemic phenotyping
Introduction
The most common RNA modification in higher organisms is the enzymatic deamination of specific adenosines to inosine (A-to-I editing), wrought by members of the ADAR protein family (1). ADARs, short for adenosine deaminases acting on RNA, were discovered by their ability to convert in vitro in double-stranded RNA ∼50% of the adenosines to inosines, thus destabilizing the double-stranded structure (2). Occurring in worms, flies and mammals, these enigmatic enzymes are now known to switch specific adenosines to inosine in mostly primary nuclear transcripts (3–6). A-to-I editing often recodes amino acids within the gene-encoded protein, based on reading inosine as guanosine by the translational machinery, and can remove or create splice acceptor sites (7), thus altering the pattern of RNA splicing. Moreover, editing in 5′ and 3′UTRs and intronic sequence may lead to altered translational efficiency, mRNA half-life and intron splicing (5, 8, 9). Mammalian miRNAs are also ADAR targets, thus changing the target range of miRNAs and the translational efficiency of miRNA targets in the cellular mRNA pools (9).
Mammalian ADARs form a small family of three closely related proteins, whose functions in the various tissues they are expressed in are poorly understood. Mice gene-targeted for global deletion of functional Adar2 alleles succumb to severe epilepsy within 3 weeks of life (8). The ADAR2-mediated edits were mostly found in pre-mRNAs for different ligand-gated and voltage-gated ion channels and G protein-coupled receptors of the nervous system and occur in imperfect double-stranded structures formed by exonic sequence and upstream or downstream intronic sequence (8, 10). ADAR2-targeted pre-mRNAs often contain several independent edits, thus expanding the protein sequence encoded by the respective genes, with the different subpopulations of unedited and edited versions exhibiting altered functional properties. This has led to the notion that ADAR2 action may fine-tune the properties of the diverse proteins affected (4).
While the extent of A-to-I editing for different positions in diverse mammalian mRNAs varies from low (<10%) to medium (10–40%) (8), there is one ADAR2-targeted position, which always occurs fully edited (11). This is the Q/R site, located in the pore-forming segment M2 of GluA2 (GluR-B, GluR2), one of four subunits of AMPA receptors (12), which mediate most of the fast excitatory synaptic transmission in the brain. The glutamine (Q) codon CAG for this site within GluA2 pre-mRNA is efficiently converted into a CIG codon for arginine (R), such that >98% of all GluA2 contains arginine in the Q/R site, with the critical functional consequence that AMPA receptor channels carrying this subunit are Ca2+ impermeable. Gene targeting experiments in the mouse demonstrated that under-editing the Q/R site leads to early postnatal death (8, 13, 14), whereas the gene-targeted substitution of an arginine for glutamine codon for the Q/R site within both Gria2 alleles results in Gria2R/R mice with normal life span and robust appearance in absence of functional Adar2 alleles (8, 15). These experiments identified the Q/R site within GluA2 as the only ADAR2 edit essential for survival.
Gria2R/R mice non-conditionally lacking ADAR2 have normal cage behavior. However, the widespread tissue expression of ADAR2, predicting additional unknown ADAR2 targets (6, 16), would foreshadow phenotypic consequences that might be revealed in appropriate phenotypic examinations of Adar2−/−/Gria2R/R mice. Thus, to gain further insights into the physiological relevance of ADAR2 edits excepting the Q/R site (Gen/Arg site), we systematically analyzed the phenotype of the double mutant mouse line, employing Gria2R/R mice as controls, in the German Mouse Clinic (GMC)2 by determining ∼320 parameters (17). This comprehensive analysis indeed identified several phenotypic alterations caused by loss of ADAR2, although the RNA targets of ADAR2 that might underlie the phenotypes were not revealed. Analysis of known editing sites in mRNAs suggests that editing by other ADAR family members might extenuate the effects of the global ADAR2 knock-out.
EXPERIMENTAL PROCEDURES
Adar2−/−/Gria2R/R Mutant Mice
Phenotypic analysis of Adar2−/−/Gria2R/R mice was performed in the GMC as recently described (17), with Gria2R/R mice serving as controls. Heterozygous Adar2+/− and homozygous Gria2R/R mice (8), which had been backcrossed to C57BL/6J seven times, were first interbred, and the resulting double heterozygous mice were intercrossed to generate double mutant Adar2−/−/Gria2R/R mice.
Phenotyping in GMC
Phenotypic analysis of 38 Adar2−/−/Gria2R/R mice was performed in the GMC (17–19), with 40 Gria2R/R mice serving as controls. For parallel analysis, in two pipelines in the GMC, the animals were divided into two groups. Almost all screens analyzed nine to ten mice of each sex and phenotype. A reduced number of animals were measured in the lung function (n = 6) and molecular phenotyping screen (n = 4). In the primary screen, >320 parameters were measured, including allergy, behavior, clinical chemistry, cardiovascular analysis, dysmorphology, bone and cartilage, energy metabolism, eye and vision, immunology, lung function, molecular phenotyping, neurology, nociception, steroid metabolism, and pathology.
Sequencing of Edited Codons in Select ADAR Targets
RT-PCR products of selected sequences containing editing recoded codons were amplified from brain RNA of four control and four ADAR2 KO mice with the following forward and reverse primer sets: Cyfip2, 5′-GGATCACCCAGTGTCTTCACCAGC-3′ and 5′-GAACTTGTCTGTGGGGTGAACCAG-3′; Kcna1, 5′-GGCACGGAGATAGCTGAGCAG-3′ and 5′-GATCTCCATGTACTCAGACTTGCTG-3′; Blcap, 5′-CAGCCAGAGAGCACAGCGGCTCAGC-3′ and 5′-GAATGTGGTACCCGCAGGACTG-3′; Gabra3, 5′-GCTTGGCCATGTTGTTGGGACAGAG-3′ and 5′-AGCACTGGGAGCAGCAGCAGAC-3′; Flna, 5′-GTTGAAGGCCCCAGCAAGGTGAG-3′ and 5′-GCACTCCTCCAGAGCTCCTGAG-3′; Cadp2, 5′-CAGCATGGAAATGGGCCAAGAGC-3′ and 5′-ACCCTCACTCACTGACGCTGTAGC-3′; and Flnb, 5′-CCCAGCTGAGTTCAGCATCTGGAC-3′ and 5′-CTTGACATCGATCGTGTGGATGC-3′. Gel-purified amplicons were DNA sequenced to determine the editing extent at the individual codons.
Statistical Analysis of Data
If not otherwise stated, data were analyzed by ANOVA or Student's t test for two independent samples with equal variances. Except as in the overall test with ANOVA, data were analyzed separately for male and female mice by Student's two-sample t tests. Differences were assigned statistically significant at a significance level of α = 0.05.
RESULTS
A comprehensive phenotypic analysis of the Adar2−/−/Gria2R/R mutant mice (henceforth referred to as ADAR2 KO mice) and Gria2R/R littermates as controls was performed in the GMC (17) to identify possible phenotypic changes in all organ systems from global ADAR2 KO.
Morphological and Histological Examination of ADAR2 KO Mice
The dysmorphology and pathology screens investigated morphological abnormalities in different organ systems of ADAR2 KO relative to control mice. The external inspection and whole-body x-ray analysis did not reveal genotype-specific differences (data not shown).
Neurons of the cortex and hippocampus as well as the distribution and morphology of the Purkinje cells and neurons in the granular and molecular layer of the cerebellum were examined in detail in the pathology screen. The macro- and microscopic examination of brain and all other organs stained with H&E revealed no genotype-specific alterations. Additionally, the determination of heart weight in relation to body weight or tibia length was without pathological findings (data not shown).
Immunological, Hematological, and Clinical Chemical Plasma Parameters of ADAR2 KO Mice Are within Normal Physiological Range of C57BL/6J Mice
Some of the clinical chemical parameters were changed in the ADAR2 KO relative to control mice at a statistically significant level (p < 0.05, ANOVA) in blood samples taken at two independent time points (14 and 17 weeks of age) (supplemental Table S1). For example, decreased plasma protein and glucose concentrations were reproducibly detected in blood samples of the ADAR2 KO mice relative to controls (Fig. 1, A and B). The concentration of free fatty acids in blood plasma was increased slightly in ADAR2 KO mice (+34%; Fig. 1C). Concerning hematological parameters, a lower mean white blood cell count was observed in the ADAR2 KO mice of both sexes in comparison with controls (Fig. 1D). Despite the reproducibility and statistical significance of these changes, the clinical parameters in the ADAR2 KO mice were in the normal physiological range of the C57BL/6J mouse strain and were therefore not considered as physiopathological findings. Moreover, the changes in the white blood cell count did not go together with changes in the leukocyte subset proportions (see below). The glucose tolerance test (between 15 to 120 min after glucose injection) and plasma lipid and glucose parameters of fasted mice revealed no differences between genotypes (supplemental Table S1 and data not shown).
FIGURE 1.
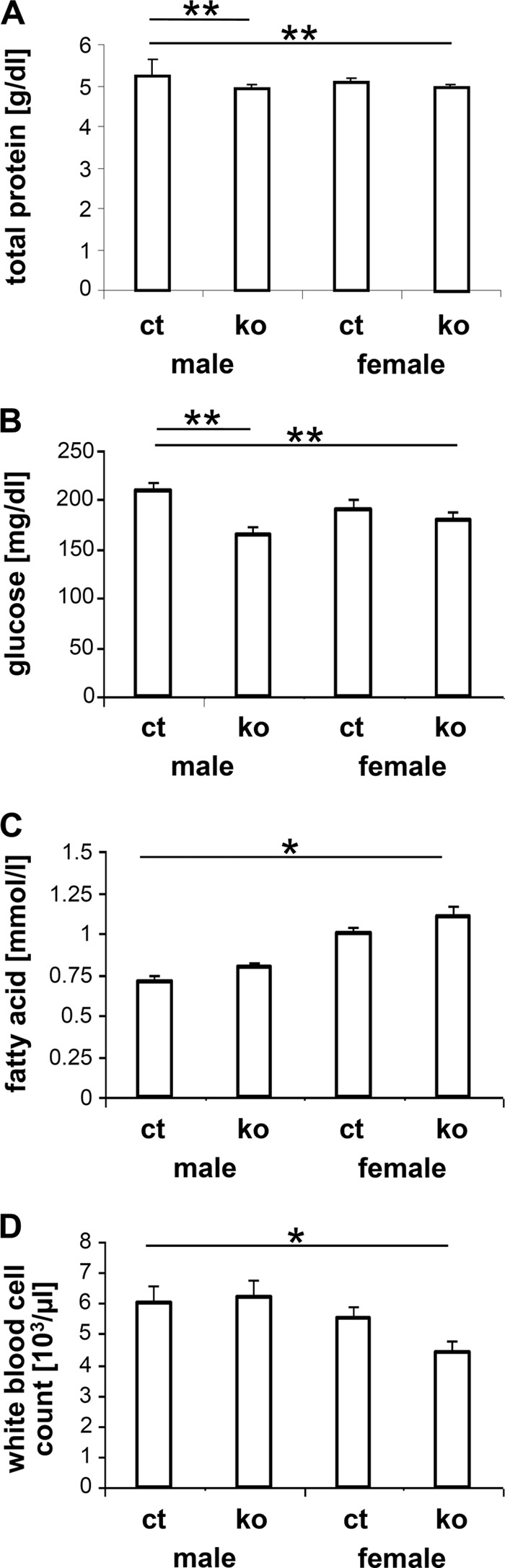
Changes in blood parameters: measurement of total protein (A), glucose (B), and fatty acid (C) concentrations and white blood cell count (D) in blood samples of ADAR2 KO (ko) and control (ct) mice. The data of total protein and glucose levels were represented as mean value of two measurements (14- and 17-week-old animals). Significance indicated by p values (n = 8–10). *, p < 0.05; **, p < 0.01.
In the immunology screen, leukocyte populations and antibody isotype levels in peripheral blood were measured by flow cytometry and Luminex bead array technology (18). No differences in leukocyte subsets, such as T cells (CD3+, CD4+, CD8+), natural killer cells, B cells, granulocytes, and antibody levels (IgM, IgG1, IgG2, IgG3, IgA, and rheumatoid factor) were revealed between ADAR2 KO and control mice (supplemental Table S2).
Increased IgE Levels in Female ADAR2 KO Mice
An involvement of ADAR2-mediated RNA editing in the regulation of total serum IgE levels has been suggested (20). The analysis of total IgE plasma levels by ELISA (18) in ADAR2 KO mice at the age of 14 weeks revealed statistically significant differences in females between mutants and controls (p = 0.012; +183.2%; Fig. 2). The same tendency was observed at the age of 17 weeks, but without reaching significance (p = 0.58; +63.7%). Male mutants showed a trend toward higher total IgE plasma levels at both time points (p = 0.67; +51.7%; Fig. 2). These data were reproduced in a second independent cohort (data not shown). The median concentration of total IgE was higher in female than male mice in both mutants and controls, which is common for many inbred mouse strains (21–23).
FIGURE 2.

Box plot showing total plasma IgE levels in ADAR2 KO (ko) and control mice (ct). Increased total IgE levels in ADAR2 KO mice were observed prominently in females. IgE levels in plasma samples at the age of 14 (A) and 17 (B) weeks were changed in ADAR2 mutant mice. Significance indicated by p values using t test and Mann-Whitney Rank Sum Test (n = 9–10). *, p < 0.05.
Lack of Evidence for Hormonal Changes in ADAR2 KO Mice
So far, a correlation of RNA editing with steroid metabolism is unknown. The steroid screen of the GMC measures key steroids such as dehydroepiandrosterone and testosterone in blood plasma. Besides sex-associated differences, no changes in the levels of these two hormones were detected between ADAR2 KO and control mice (data not shown). Atrial natriuretic peptide is a cardiac hormone used as clinical prognostic marker for cardiovascular disorders and heart failure. Its plasma concentration showed no difference between ADAR2 KO mice and controls (data not shown).
In summary, the analysis of a broad spectrum of blood parameters identified relevant changes in the total plasma IgE levels of female ADAR2 KO mice. The same tendency was found in male KO mice. Commonly, female mice are more susceptible for immunoglobulin alterations, leading to significant changes rather in females than in males.
Heart and Lung Appear Functionally Normal in ADAR2 KO Mice
ADAR2 expression has also been detected in adult heart and lung (24), but the effects of the ADAR2 KO on these organs have not been analyzed. Here, we examined basic cardiovascular functions in ADAR2 KO and control mice by noninvasive blood pressure measurement, determining systolic and diastolic pressure, mean arterial pressure, and pulse rate. A slight but statistically significant decrease in the heart rate was found in ADAR2 KO mice in comparison with controls (ANOVA, p = 0.057, −5.1%; data not shown). Because no additional cardiovascular parameters such as heart weight and atrial natriuretic peptide plasma levels were altered in ADAR2 KO mice, the reduced pulse rate is most probably secondary and without relevance for normal cardiovascular function.
The lung function screen assessed spontaneous breathing in unrestrained mice at rest and during activity by whole body plethysmography and revealed no physiologically relevant alterations in ADAR2 KO mice. Although breathing patterns differed between male ADAR2 KO and control mice with respect to inspiratory time, such alterations were not evident for females (supplemental Table S3). Inspiratory time was 8.31% shorter in male ADAR2 KO mice, probably caused by somewhat higher respiratory rates. The latter fact was not statistically significant (p = 0.06). All other parameters characterizing breathing such as tidal volume, flow rates, minute ventilation, and expiratory time did not show statistically significant differences.
ADAR2 KO Mice Are Hypermetabolic
In the metabolic screen, mice were analyzed in a multichannel indirect calorimetry system, combining gas exchange measurements and the monitoring of food intake and unspecific activity in the cage. Trials were conducted at room temperature over a period of 21 h. A statistically significant increase of mean heat production between ADAR2 KO and control mice was found (Fig. 3), varying between +7% in female and +12% in male mutants. This difference was mainly due to a significant increase of energy expenditure and maximum metabolic rates, which occur soon after lights-off. The difference in minimum heat production was not statistically significant. Interestingly, locomotor activity also was increased in the first half of the dark phase, even though it did not reach significance due to high biological variability. Food consumption was increased slightly in female but not male mutants, whereas body mass and body temperature did not differ between mutants and controls (data not shown). The slight differences in overall energy expenditure could be due to increased activity levels, resulting in higher energy expenditure.
FIGURE 3.

Mean heat production calculated from gas exchange data collected over 21 h in the indirect calorimetry system plotted versus body mass. ADAR2 KO mice (ko) show a moderate increase in heat production compared with controls (ct) (n = 7).
Interestingly, of the clinical chemical parameters associated with metabolic functions, glucose was decreased in non-fasted ADAR2 KO mice, whereas e.g. triglycerides did not differ between mutants and controls (supplemental Table S1). Therefore, the changes in mean oxygen consumption remain of unclear relevance and could be secondary. In conclusion, basal metabolic functions were seemingly not affected by the mutation.
Neurological and Behavioral Screens Reveal Specific Mutant Phenotype
The neurology and behavior screens were of particular interest for the ADAR2 KO mice, given the relatively strong expression of ADAR2 in the nervous system (4) and the association of the ADAR2 loss-of-function allele with neurological dysfunctions (10). An overall assessment of basic neurological functions (modified SHIRPA protocol (25, 26)) revealed no significant changes between ADAR2 KO mice and controls. Forepaw and all-paw grip strength measurements (force meter) showed slightly increased all-paw grip strength in ADAR2 KO mice (+5.5%; data not shown). The rotarod test for the evaluation of differences in motor coordination and balance revealed no significant changes (Fig. 4A). However, the mutant mice rotated more often passively (p = 0.05; +32%; Fig. 4B), rendering the measured latencies of limited significance.
FIGURE 4.

Rotarod and open field analysis. Rotarod performance, mean latency of three trials on the acceleration rotarod (4–40 rpm/300 s; 15-min intertrail intervals) (A) and percentage of passive rotation and falling as cause for trial termination (B). C, analysis of total rearing frequency of ADAR2 KO and control in an open field as a novel environment. Mice were observed for 20 min in the open field; data are presented as mean ± S.E. per 5-min intervals. The left panel shows data of male mice, the right panel shows data of female mice. For statistics, see “Results.” ko, ADAR2 KO mice; ct, control mice (n = 10).
The open field test in the behavior screen was used to evaluate exploratory behavior, reactivity to novelty, and emotionality. In this test, the ADAR2 KO mice showed increased rearing activity during the first 5 min in the arena compared with the respective control groups (ANOVA, p < 0.05; +28.85%; Fig. 4C). Male ADAR2 KO mice reared less frequently during the last 5 min of the 20-min observation period than the male control group (p < 0.05; 28.9%; Fig. 4C). In male controls, rearing activity increased over time from the first to the last 5 min, whereas it stayed approximately at the same level in ADAR2 KO males (genotype effect on time course, p < 0.01; Fig. 4C). Other open field parameters such as forward locomotor activity or anxiety-related phenotypes were not significantly altered in ADAR2 KO mice (data not shown).
A Hearing Deficit in ADAR2 KO Mice
The functionality of sensory organs in ADAR2 KO mice was analyzed in the eye (vision), nociception (thermal pain), dysmorphology (hearing ability), and behavior screen (sensorimotor gating). Slit lamp examination and funduscopy for the analysis of the anterior and posterior segments of the eye revealed no abnormalities. In addition, laser interference biometry indicated the typical sex-specific differences of axial eye lengths. Differences between ADAR2 KO and control mice were not statistically significant (data not shown).
The nociception screen tests the responsiveness of the somatosensory system to thermal pain by means of the hotplate test identifying no differences in pain sensation between ADAR2 KO and control mice (data not shown).
Hearing ability was assessed by a click box test producing a short 20 kHz sound and observing the Preyer's reflex. This test identified a significant difference between male ADAR2 KO and control mice at the age of 9 weeks (ANOVA, p = 0.001; Fig. 5A). Although most of the male mutant mice showed either no or a strongly reduced Preyer's reflex (mean score, 1.22), the male controls had a normal hearing reaction (mean score, 2.8). We observed a slightly reduced Preyer's reflex (mean score 2.0) in female ADAR2 KO mice compared with the controls (mean score, 2.3; Fig. 5A).
FIGURE 5.

Sensory perception (n = 10). A, click box test: reaction of 9-week-old ADAR2 KO (ko) and control (ct) mice to a 20-kHz sound classified into six categories: 0, no reaction; 1, no Preyer's reflex; 2, retarded reaction; 3, Preyer's reflex; 4, strong reaction; 5, particularly strong reaction. B, sensorimotor gating in ADAR2 KO mice measured by % PPI of the acoustic startle reflex at a startle pulse intensity of 110 db and prepulse intensities of 67, 69, 73, and 81 db. Mean value represents the mean PPI value of all four prepulse intensities. C, acoustic startle responses (ASR) measured at background noise (NS), and sound pressure intensities of 70–120 db. Data are presented as means + S.E., significant differences between mutants and controls are indicated by asterisks. *, p < 0.05; **, p < 0.01; ***, p < 0.001. Left panels show data of male mice, and right panels show data of female mice.
In addition, prepulse inhibition (PPI) of the acoustic startle reflex was used to assess sensorimotor gating. A significant PPI deficit was observed in male ADAR2 KO mice at the 67 db intensity as well as in the mean value of all measurements (Fig. 5B). The analysis of acoustic startle reflex clearly indicated reduced startle reactivity at higher sound pressure levels in the ADAR2 KO mice (Fig. 5C). Overall, these data suggest reduced hearing ability at both high (click box test) and broadband (acoustic startle reflex) frequencies in ADAR2 KO mice compared with controls and an additional sensorimotor gating prepulse inhibition deficit in male mutants.
Transcript Profiling Analysis
Transcript profiling of brain, comparing four male ADAR2 KO versus control mice (27), identified 80 significantly overexpressed genes in the ADAR2 KO mice (Fig. 6).
FIGURE 6.

Transcript profiling of brain from four ADAR2 KO mice: heat plots of significantly regulated genes from eight DNA microarray experiments of mutant versus control mice. One dye-flip pair represents two experimental replicates of each analyzed mouse (n = 4). Official gene symbols and names are given. The scale bar encodes the ratio of the fold induction; red is up-regulated, and green is down-regulated in ADAR2 KO mice.
The two DNA probes for Adar2 on our microarray locate to the 5′ part of the Adar2 allele upstream of the mutated exon 4 (15). The transcriptional down-regulation of the mutated allele of Adar2 reaching <10% of the level in wild-type mice (15) is in accordance with the observed down-regulation of Adar2 in all four analyzed mutant mice. However, Adar2 down-regulation did not reach statistical significance due to interindividual variability (Fig. 7). We specifically also examined the expression levels of the ADAR gene family member, Adar1, as well as of the glutamate receptor genes, Gria1 (encoding GluA1), Gria2 (GluA2), Gria3 (GluA3), and Gria4 (GluA4), most of which are known ADAR2 targets (15). None of these genes was transcriptionally regulated in brains of ADAR2 KO mice (Fig. 7).
FIGURE 7.

Transcript profiles of selected ADAR and glutamate receptor genes (Gria1-4) in brains from ADAR2 KO mice: heat plots of gene expression patterns from eight DNA microarray experiments of mutant versus control mice. One dye-flip pair represents two experimental replicates of each analyzed mouse (n = 4). The scale bar encodes the ratio of the fold induction. Red is up-regulated, and green is down-regulated in ADAR2 KO mice.
The 80 significantly up-regulated genes were categorized into biological functions using the Ingenuity Pathway Analysis software. In particular, nucleic acid metabolism, cellular growth, hematopoiesis, lipid, carbohydrate and amino acid metabolism, immune response, cancer, and cell-to-cell signaling were among the statistically overrepresented functional annotations (significance level of α = 0.05).
Changes in A-to-I Editing Levels in mRNA Codons
We compared the extent of RNA editing in codons of potential ADAR2 targets (6, 28) in the brain of adult ADAR2 KO and control mice, selecting positions edited to >20% in the controls. ADAR2 ablation mostly decreased but rarely abolished the extent of editing (8), indicating that other ADAR family members can also edit ADAR2 targets (supplemental Table S4).
DISCUSSION
We employed Adar2−/−/Gria2R/R mice to search for phenotypic changes that might be expected from absence of editing of all known and unknown ADAR2 substrates, except the “exon-edited” Q/R site provided by the Gria2R/R alleles. Indeed, systematic phenotyping in the GMC revealed several phenotypic differences between ADAR2 KO and control mice, indicating that respective normal physiological functions require ADAR2.
The phenotypic analysis indicated no effects of the lack of ADAR2 on morphological, immunological, hormonal, and pathological parameters, or on sensory organs for vision and nociception. Changes found for clinical chemical and hematological parameters in the ADAR2 KO mice were within the range of normal physiological variability of the C57BL/6J strain and therefore not considered as a mutant phenotype. The alterations identified in the cardiovascular and lung function as well as in mean oxygen consumption of ADAR2 KO mice were interpreted as secondary effects without relevance for the function of the respective organ systems.
A puzzling observation was an increase in IgE levels predominantly in female ADAR2 KO mice. RNA editing by activation-induced deaminase has been reported to be crucial for the class-switching mechanism of distinct immunoglobulins. Missing RNA editing resulted in hyper-IgM and a lack of IgG, IgA, and IgE (20). Changes of total IgE plasma levels in ADAR2 KO mice suggest potential ADAR2 targets that alter IgE plasma levels. In contrast to results described by Noguchi and colleagues (20), increased IgE levels upon ADAR2 KO affected mostly females. It will be of interest to analyze the allergy and IgE phenotype of ADAR2 KO mice under conditions of allergic sensitization and airway challenge using a model allergen.
An impaired Preyer's reflex was identified in ADAR2 KO mice. This mutant phenotype could be caused by functional deficits of the cochlea or reduction in motor recruitment or both. The alterations observed in PPI indicate a change in sensorimotor integration, hence changes in brain function, which might be caused by differences in any of several neurochemical systems (29). The 5-HT2C serotonin receptor, a known ADAR2 substrate (30), modulates sensory responses in C. elegans by acting directly on sensory neurons (31). Changes in this receptor signaling may be related to the PPI deficit identified in the ADAR2 KO mice. Interestingly, these findings are in accordance with an analogous PPI deficit in schizophrenic patients (32), showing altered editing of the 5-HT2C receptor pre-mRNA in the prefrontal cortex (33). An altered 5-HT2C receptor function might contribute to the hearing deficit in the ADAR2 KO mice, as this receptor is expressed in both the cochlea (34) and inferior colliculus (35).
The hearing deficit in mutant mice might correlate with the up-regulation of Rgs3 (regulator of G-protein signaling 3) gene observed in the brains of ADAR2 KO mice. Rgs3, a negative regulator of signal transduction, plays an important role in response to sensory stimuli, as its loss in sensory neurons leads to defective responses to external stimuli (36). A compensatory effect might be suggested for the up-regulation of Rgs3 in the brains of ADAR2 KO mice, thus reducing G-protein signaling in sensory neurons and contributing to the reduced hearing ability of the ADAR2 KO mice.
In addition, transcriptome analysis indicated pronounced changes in the expression levels of many brain transcripts, none of which is known to undergo RNA editing (6). Several up-regulated genes are involved in neuroprotection (Cdk6, Col8a1, Dnajb6, Hdac9, and Id3) and repair processes in astrocytes (Itsn, Rgs3, and Clint1), whereas others are associated with synaptic trafficking, which might be of interest in correlation with the important role of glutamate receptor channels in synaptic plasticity (37). For example, Dlgap2, a member of the scaffolding protein family SAPAP, is involved in activity-mediated modification of postsynaptic densities (38). Its overexpression was described in hippocampus of patients with Fragile X syndrome, a mental retardation disease, and also in a cognate mouse model (39).
In previous analyses common targets of RNA editing were identified that are involved in nervous system functions (11, 40, 41), and Li et al. (6) found an enrichment of target sites associated with synaptic functions, cell trafficking, and membrane functions. Several other identified sites are implicated in human brain-related diseases. Similar functional annotations were also present within our data set of regulated genes identified in the brain of ADAR2 KO mice. Therefore, these genes are potential candidate targets for ADAR2-mediated editing.
Although ADAR2 activity has been best characterized as selective A-to-I editing in pre-mRNAs, mostly in the nervous system, recent studies have identified noncoding RNAs such as miRNAs as ADAR2 targets, and ADAR2 may also participate in A-to-I editing the numerous repetitive RNA sequences in intronic and untranslated sequences in primary gene transcripts (9). Moreover, ADAR2 may have a biological role in processes other than A-to-I editing, resulting for instance from binding to double-stranded RNA stretches and from interacting with other proteins (42). These additional ADAR2 functions should also be considered when interpreting a phenotype associated with genetically induced absence of ADAR2.
In summary, the comprehensive phenotypic analysis of the Adar2−/−/Gria2R/R mice revealed several specific mutant phenotypes. The changes found in behavior, hearing ability, allergy parameters, and gene expression profiles in the brain of mutant mice now provide a rational basis for targeted studies of ADAR2 functions and for the identification of new functional ADAR2 targets.
Supplementary Material
Acknowledgments
We thank R. Seeliger, M. Backs, S. Bothur, S. Geiβler, M. Grandl, T. Halex, S. Holthaus, E. Holupirek, M. Kugler, A. Langer, K. Laube, J. Müller, E. Samson, F. Schleicher, D. Schmidt, W. Schneider, AE. Schwarz, B. Sperling, W. Stettinger, L. Thumann, S. Wittich, and C. Zeller as well as the GMC animal caretaker team for expert technical help. We greatly appreciate the help of Tonia Ludwig for statistical analysis.
This work was supported by the Bundesministerium für Bildung und Forschung (BMBF) Nationales Genomforschungsnetz (NGFN-Plus) Grants 01GS0850, 01GS0851, 01GS0852, 01GS0868, 01GS0869, 01GS0853, and 01GS0854) and by European Union Grant EUMODIC LSHG-2006037188 (German Mouse Clinic).

The on-line version of this article (available at http://www.jbc.org) contains supplemental Tables S1–S4.
- GMC
- German Mouse Clinic
- ANOVA
- analysis of variance
- PPI
- prepulse inhibition.
REFERENCES
- 1. Bass B. L. (2002) Annu. Rev. Biochem. 71, 817–846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bass B. L., Weintraub H. (1988) Cell 55, 1089–1098 [DOI] [PubMed] [Google Scholar]
- 3. Reenan R. A. (2001) Trends Genet 17, 53–56 [DOI] [PubMed] [Google Scholar]
- 4. Seeburg P. H. (2002) Neuron 35, 17–20 [DOI] [PubMed] [Google Scholar]
- 5. Maas S., Kawahara Y., Tamburro K. M., Nishikura K. (2006) RNA Biol. 3, 1–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Li J. B., Levanon E. Y., Yoon J. K., Aach J., Xie B., Leproust E., Zhang K., Gao Y., Church G. M. (2009) Science 324, 1210–1213 [DOI] [PubMed] [Google Scholar]
- 7. Rueter S. M., Dawson T. R., Emeson R. B. (1999) Nature 399, 75–80 [DOI] [PubMed] [Google Scholar]
- 8. Higuchi M., Maas S., Single F. N., Hartner J., Rozov A., Burnashev N., Feldmeyer D., Sprengel R., Seeburg P. H. (2000) Nature 406, 78–81 [DOI] [PubMed] [Google Scholar]
- 9. Nishikura K. (2010) Annu. Rev. Biochem. 79, 321–349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Higuchi M., Single F. N., Köhler M., Sommer B., Sprengel R., Seeburg P. H. (1993) Cell 75, 1361–1370 [DOI] [PubMed] [Google Scholar]
- 11. Sommer B., Köhler M., Sprengel R., Seeburg P. H. (1991) Cell 67, 11–19 [DOI] [PubMed] [Google Scholar]
- 12. Keinänen K., Wisden W., Sommer B., Werner P., Herb A., Verdoorn T. A., Sakmann B., Seeburg P. H. (1990) Science 249, 556–560 [DOI] [PubMed] [Google Scholar]
- 13. Brusa R., Zimmermann F., Koh D. S., Feldmeyer D., Gass P., Seeburg P. H., Sprengel R. (1995) Science 270, 1677–1680 [DOI] [PubMed] [Google Scholar]
- 14. Feldmeyer D., Kask K., Brusa R., Kornau H. C., Kolhekar R., Rozov A., Burnashev N., Jensen V., Hvalby O., Sprengel R., Seeburg P. H. (1999) Nat. Neurosci. 2, 57–64 [DOI] [PubMed] [Google Scholar]
- 15. Kask K., Zamanillo D., Rozov A., Burnashev N., Sprengel R., Seeburg P. H. (1998) Proc. Natl. Acad. Sci. U.S.A. 95, 13777–13782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zaranek A. W., Levanon E. Y., Zecharia T., Clegg T., Church G. M. (2010) PLoS Genet 6, e1000954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gailus-Durner V., Fuchs H., Becker L., Bolle I., Brielmeier M., Calzada-Wack J., Elvert R., Ehrhardt N., Dalke C., Franz T. J., Grundner-Culemann E., Hammelbacher S., Hölter S. M., Hölzlwimmer G., Horsch M., Javaheri A., Kalaydjiev S. V., Klempt M., Kling E., Kunder S., Lengger C., Lisse T., Mijalski T., Naton B., Pedersen V., Prehn C., Przemeck G., Racz I., Reinhard C., Reitmeir P., Schneider I., Schrewe A., Steinkamp R., Zybill C., Adamski J., Beckers J., Behrendt H., Favor J., Graw J., Heldmaier G., Höfler H., Ivandic B., Katus H., Kirchhof P., Klingenspor M., Klopstock T., Lengeling A., Müller W., Ohl F., Ollert M., Quintanilla-Martinez L., Schmidt J., Schulz H., Wolf E., Wurst W., Zimmer A., Busch D. H., de Angelis M. H. (2005) Nat. Methods 2, 403–404 [DOI] [PubMed] [Google Scholar]
- 18. Gailus-Durner V., Fuchs H., Adler T., Aguilar Pimentel A., Becker L., Bolle I., Calzada-Wack J., Dalke C., Ehrhardt N., Ferwagner B., Hans W., Hölter S. M., Hölzlwimmer G., Horsch M., Javaheri A., Kallnik M., Kling E., Lengger C., Mörth C., Mossbrugger I., Naton B., Prehn C., Puk O., Rathkolb B., Rozman J., Schrewe A., Thiele F., Adamski J., Aigner B., Behrendt H., Busch D. H., Favor J., Graw J., Heldmaier G., Ivandic B., Katus H., Klingenspor M., Klopstock T., Kremmer E., Ollert M., Quintanilla-Martinez L., Schulz H., Wolf E., Wurst W., de Angelis M. H. (2009) Methods Mol. Biol. 530, 463–509 [DOI] [PubMed] [Google Scholar]
- 19. Fuchs H., Gailus-Durner V., Adler T., Aguilar-Pimentel J. A., Becker L., Calzada-Wack J., Da Silva-Buttkus P., Neff F., Götz A., Hans W., Hölter S. M., Horsch M., Kastenmüller G., Kemter E., Lengger C., Maier H., Matloka M., Möller G., Naton B., Prehn C., Puk O., Rácz I., Rathkolb B., Römisch-Margl W., Rozman J., Wang-Sattler R., Schrewe A., Stöger C., Tost M., Adamski J., Aigner B., Beckers J., Behrendt H., Busch D. H., Esposito I., Graw J., Illig T., Ivandic B., Klingenspor M., Klopstock T., Kremmer E., Mempel M., Neschen S., Ollert M., Schulz H., Suhre K., Wolf E., Wurst W., Zimmer A., Hrabě de Angelis M. (2011) Methods 53, 120–135 [DOI] [PubMed] [Google Scholar]
- 20. Noguchi E., Shibasaki M., Inudou M., Kamioka M., Yokouchi Y., Yamakawa-Kobayashi K., Hamaguchi H., Matsui A., Arinami T. (2001) J. Allergy Clin. Immunol. 108, 382–386 [DOI] [PubMed] [Google Scholar]
- 21. Alessandrini F., Jakob T., Wolf A., Wolf E., Balling R., Hrabé de Angelis M., Ring J., Behrendt H. (2001) Int. Arch Allergy Immunol. 124, 25–28 [DOI] [PubMed] [Google Scholar]
- 22. Corteling R., Trifilieff A. (2004) BMC Pharmacol. 4, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Seymour B. W., Friebertshauser K. E., Peake J. L., Pinkerton K. E., Coffman R. L., Gershwin L. J. (2002) Dev. Immunol. 9, 47–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Melcher T., Maas S., Herb A., Sprengel R., Higuchi M., Seeburg P. H. (1996) J. Biol. Chem. 271, 31795–31798 [DOI] [PubMed] [Google Scholar]
- 25. Rogers D. C., Fisher E. M., Brown S. D., Peters J., Hunter A. J., Martin J. E. (1997) Mamm. Genome 8, 711–713 [DOI] [PubMed] [Google Scholar]
- 26. Schneider I., Tirsch W. S., Faus-Kessler T., Becker L., Kling E., Busse R. L., Bender A., Feddersen B., Tritschler J., Fuchs H., Gailus-Durner V., Englmeier K. H., de Angelis M. H., Klopstock T. (2006) J. Neurosci. Methods 157, 82–90 [DOI] [PubMed] [Google Scholar]
- 27. Horsch M., Schädler S., Gailus-Durner V., Fuchs H., Meyer H., de Angelis M. H., Beckers J. (2008) Proteomics 8, 1248–1256 [DOI] [PubMed] [Google Scholar]
- 28. Kiran A., Baranov P. V. (2010) Bioinformatics 26, 1772–1776 [DOI] [PubMed] [Google Scholar]
- 29. Geyer M. A., Krebs-Thomson K., Braff D. L., Swerdlow N. R. (2001) Psychopharmacology 156, 117–154 [DOI] [PubMed] [Google Scholar]
- 30. Schmauss C., Howe J. R. (2002) Sci. STKE 2002, pe26. [DOI] [PubMed] [Google Scholar]
- 31. Chao M. Y., Komatsu H., Fukuto H. S., Dionne H. M., Hart A. C. (2004) Proc. Natl. Acad. Sci. U.S.A. 101, 15512–15517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Braff D. L., Geyer M. A., Swerdlow N. R. (2001) Psychopharmacology 156, 234–258 [DOI] [PubMed] [Google Scholar]
- 33. Sodhi M. S., Burnet P. W., Makoff A. J., Kerwin R. W., Harrison P. J. (2001) Mol. Psychiatry 6, 373–379 [DOI] [PubMed] [Google Scholar]
- 34. Oh C. K., Drescher M. J., Hatfield J. S., Drescher D. G. (1999) Brain Res. Mol. Brain Res. 70, 135–140 [DOI] [PubMed] [Google Scholar]
- 35. Holt A. G., Asako M., Lomax C. A., MacDonald J. W., Tong L., Lomax M. I., Altschuler R. A. (2005) J. Neurochem. 93, 1069–1086 [DOI] [PubMed] [Google Scholar]
- 36. Ferkey D. M., Hyde R., Haspel G., Dionne H. M., Hess H. A., Suzuki H., Schafer W. R., Koelle M. R., Hart A. C. (2007) Neuron 53, 39–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Krestel H. E., Shimshek D. R., Jensen V., Nevian T., Kim J., Geng Y., Bast T., Depaulis A., Schonig K., Schwenk F., Bujard H., Hvalby Ø., Sprengel R., Seeburg P. H. (2004) J. Neurosci. 24, 10568–10578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Dosemeci A., Jaffe H. (2010) Biochem. Biophys. Res. Commun. 391, 78–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Schütt J., Falley K., Richter D., Kreienkamp H. J., Kindler S. (2009) J. Biol. Chem. 284, 25479–25487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Levanon E. Y., Hallegger M., Kinar Y., Shemesh R., Djinovic-Carugo K., Rechavi G., Jantsch M. F., Eisenberg E. (2005) Nucleic Acids Res. 33, 1162–1168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lomeli H., Mosbacher J., Melcher T., Höger T., Geiger J. R., Kuner T., Monyer H., Higuchi M., Bach A., Seeburg P. H. (1994) Science 266, 1709–1713 [DOI] [PubMed] [Google Scholar]
- 42. Heale B. S., Keegan L. P., O'Connell M. A. (2009) Cell Cycle 8, 4011–4012 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.