

Abstract
Conserved bacteriophage ATP-based DNA translocation motors consist of a multimeric packaging terminase docked onto a unique procapsid vertex containing a portal ring. DNA is translocated into the empty procapsid through the portal ring channel to high density. In vivo the T4 phage packaging motor deals with Y- or X-structures in the replicative concatemer substrate by employing a portal-bound Holliday junction resolvase that trims and releases these DNA roadblocks to packaging. Here using dye-labeled packaging anchored 3.7-kb Y-DNAs or linear DNAs, we demonstrate FRET between the dye-labeled substrates and GFP portal-containing procapsids and between GFP portal and single dye-labeled terminases. We show using FRET-fluorescence correlation spectroscopy that purified T4 gp49 endonuclease VII resolvase can release DNA compression in vitro in prohead portal packaging motor anchored and arrested Y-DNA substrates. In addition, using active terminases labeled at the N- and C-terminal ends with a single dye molecule, we show by FRET distance of the N-terminal GFP-labeled portal protein containing prohead at 6.9 nm from the N terminus and at 5.7 nm from the C terminus of the terminase. Packaging with a C-terminal fluorescent terminase on a GFP portal prohead, FRET shows a reduction in distance to the GFP portal of 0.6 nm in the arrested Y-DNA as compared with linear DNA; the reduction is reversed by resolvase treatment. Conformational changes in both the motor proteins and the DNA substrate itself that are associated with the power stroke of the motor are consistent with a proposed linear motor employing a terminal-to-portal DNA grip-and-release mechanism.
Keywords: DNA Enzymes, DNA Viruses, Fluorescence Resonance Energy Transfer (FRET), Molecular Motors, Viral Protein, DNA Packaging, DNA Translocase, Bacteriophage Terminase, Procapsid Portal
Introduction
The nucleic acid packaging machinery of double-stranded DNA and RNA bacteriophages and of herpesviruses and adenoviruses fills empty preassembled capsid precursors by broadly conserved ATP-driven motor mechanisms (1–3). In most of the large tailed double-stranded DNA bacteriophages, a prohead is filled to near liquid crystalline DNA density (∼500 mg/ml) from a replicative genomic concatemer in vivo by a powerful packasome motor (4). The motor consists of a multimeric terminase enzyme docked onto a unique capsid vertex containing the DNA entrance and exit channel-containing portal dodecamer. In conjunction with high ATP consumption, the terminase-portal motor translocates DNA through the prohead portal, and the terminase also cuts the genomic concatemer to form the mature viral particle DNA ends upon receiving a headful signal from the portal, hence the name terminase (5, 6).
The essential components of the packasome motor appear to share many structural and mechanistic features among intensively studied double-stranded DNA phages T4, lambda, P22, SPP1, Phi29, and others (7). Accordingly, a number of general mechanisms for DNA translocation have been proposed, although some can now apparently be eliminated by more recent experiments employing a variety of genetic, biochemical, biophysical, and structural approaches. Thus, genetic and biophysical evidence suggests that the DNA molecular motor is unlikely to operate by a once favored F1F0ATPase-like rotary mechanism (8, 9). Rather the portal is now thought to be unable to rotate but to operate as part of a linear motor that can apply high translocation force by terminase-ATPase consumption of ATP (10, 11). One linear motor mechanism has been proposed for a phage T4 DNA compression motor. The model proposes that a portal DNA grip-and-release driven by the linear force of the conformational change of the terminase to portal interaction drives DNA into the prohead by a DNA compression motor stroke (12, 13). Another linear model for the T4 motor proposes that a conformational change in the terminase pentamer situated on the portal, reflecting tense and relaxed terminase crystal structures, imparts a linear translocation force (14). Both models thus propose that major conformational change in the motor proteins is responsible for the linear motor power stroke that drives translocation. In contrast, a quite different single protein-based hexameric helicase-like motor translocase that packages single-stranded RNA into the Phi6 procapsid is proposed to act by a confined linear motor arm motion within the body of the hexamer rather than by a large scale conformational change in the motor protein (3).
Although detailed structural information is now available for a number of the DNA and the RNA motor proteins, most proposed mechanisms remain hypothetical in the absence of any direct measure of dynamic changes in the motor during translocation. In this work we have applied fluorescence techniques to study the efficient phage T4 motor that is composed of a single terminase multimer (gp17) bound to the portal dodecamer (gp20) of the prohead. This two protein-based packasome motor employs pure gp17 terminase and proheads to package short linear DNAs of any sequence at high (20 to near 100%) efficiency in vitro (15). Departures from the duplex structure, such as found in Y-DNAs or linear DNAs with nicks, represent a potent block to continued translocation of short DNAs (<200 bp) that have been shown to be released from the packaging motor (12). However, when joined to a prohead-anchoring DNA leader of substantial kb length, the packaging arrested Y-DNAs accumulate in the portal (13). In fact, it is known that the T4 motor must deal with Y- or X-structures that are present in high concentration in the replicative concatemer in vivo. It does this by employing a portal-bound Holliday junction resolvase enzyme, gp49 that acts to release the DNA roadblock by trimming the Y- or X-structures (16). In the absence of active gp49 resolvase in vivo, mature phages are not assembled, and arrested packaging into proheads leads to the accumulation of partially filled heads that cannot attach a tail and that are seen to accumulate in the infected host (17, 18). These partially filled heads can, however, be rescued into active full phage particles by supplying at low temperature a temperature-sensitive reversible gp49 enzyme activity.
In this work we show that purified gp49 resolvase can release compression or “crunching” in in vitro arrested Y-DNA structures from prohead portals. The Y-DNA structures that contain FRET dye pairs allow conformational changes in the DNA substrate that are catalyzed by the packaging motor working together with the resolvase to be measured. In addition, we show using active terminase labeled with a single peptide-specific dye molecule FRET to both a GFP-labeled portal prohead and to a single specific sequence dye-labeled packaging substrate. By employing these reagents, we can measure by FRET-fluorescence correlation spectroscopy (FRET-FCS)2 changes in dye-to-dye distances during packaging. We are thus able to address critically dynamic features of the packaging motor and assess their correspondence to features expected by proposed hypothetical packaging mechanisms.
EXPERIMENTAL PROCEDURES
Fluorescent DNA Construction
Y-DNAs were constructed from HPLC-purified dye-labeled oligonucleotides (Integrated DNA Technologies, Inc.) by annealing complementary single strands, followed by gel electrophoresis and purification from gel slices by extraction of the dye containing duplex DNAs (Qiaex II kit isolation; Qiagen). The TxY-DNA (see Fig. 1B) was prepared as described previously from two 90-mers (13). For the double dye-labeled 11- and 16-bp Y-DNA preparation, four oligonucleotides were synthesized. The Cy3 (C3) was joined by amino six carbon linkers of Cy3 to two different internal T residues of the 90-base oligonucleotide (Y1L), and Alexa 488 (Ax) dye was joined by three carbon linkers of Ax to the 3′-terminal C of the 33-base oligonucleotide (Y1 Rb); the two other oligonucleotides of 16 bases (Y1 Rc) and 41 bases (YI Ra) were unlabeled (see Fig. 1D, panel iv). Y1 Ra, Rb, and Rc together formed the complementary strand (YI R) for the Y1L oligonucleotide (see Fig. 1D, panels iii and iv). Unlabeled Y-DNA used for FRET-FCS (see Figs. 7 and 8) was prepared by annealing 90-base-long YIL and YIR oligonucleotides. Unlabeled Y-DNA used for resolvase sequence-specific cutting assay (see Fig. 4B) was prepared by annealing 90-base-long YIL and YIR oligonucleotides without purification from incomplete synthesis products to serve as sequencing ladders. Single dye-labeled Y-DNA was prepared using the 30-base oligonucleotide Y1La, 5′-phosphorylated 60-base oligonucleotide Y1Lb, 5′-phosphorylated 32-base oligonucleotide Y1Rb, and Ax dye-labeled at the 5′-terminal C of the 58-base oligonucleotide Y1Ra (see Fig. 1D, panel ii). The double dye AxC3Y-DNAs, single dye AxY-DNAs, and unlabeled Y-DNAs (90 bases each) were phosphorylated by T4 polynucleotide kinase (New England Biolabs), and the blunt ends were ligated to pBR322 DNA linearized with EcoRV, followed by PstI digestion and gel extraction to yield the Y-DNAs with the 3.7-kb purified pBR322 (New England Biolabs) derived leaders (Fig. 1C).
FIGURE 1.

Y-DNA packaging substrates with a 3.7-kb leader sequence complexed with gp17 terminase-ATPase stall in proximity to the prohead portal. A, diagram of the N-terminal GFP portal-containing procapsid with anchored, stalled 3.7-kb TxY-DNA. B, the structure of the 90-bp TxY-DNA and its sequence. YIL and YIR represent the two strands of the Y-DNA. C, construction of the 3.7-kb leader TxY-DNA. D, schematic of different Y-DNA modifications with the same basic sequence used in various experiments without any fluorophore: (panel i) with Alexa 488 at the junction (panel ii), with Cy3 dye and Alexa 488 in the complementary strand at 11-bp distance (panel iii), with Cy3 dye and Alexa 488 in the complementary strand at 16-bp distance (panel iv). Ra, Rb, and Rc oligonucleotides together (panel iv), form the complement to the YIL strand of the double dye-labeled Y-DNAs. The Y-DNA portal interaction studies were carried out by assaying packaging activity by FCS and nuclease assay using either the wild type or GFP-gp20 proheads and 90-base-long Y-DNA ligated to a 3.7-kb leader sequence as substrate in the reaction mixture.
FIGURE 7.

The N- and C-terminal regions of the gp17 protein interact with the stalled Y-DNA. Packaging was carried out using NT-ReAsH or CT-ReAsH-terminase, wild type proheads, and 3.7-kb Alexa 488 Y-DNA. A and D, different FRET values were observed for CT-ReAsH (A) and NT-ReAsH (D) terminases with 3.7-kb Alexa 488 Y-DNA. B and E, autocorrelation plots of the Ax-green channel are shown. C and F, nuclease assays were carried out following FCS-FRET measurements confirming the packaging of 3.7-kb AxY-DNA by CT-ReAsH terminase (C) as well as the NT-ReAsH terminase (F).
FIGURE 8.

Changes in conformational states of the terminase and prohead portal during packaging of linear and Y-DNA substrates. Packaging was carried out using GFP-gp20 proheads, CT-ReAsH terminase, and unlabeled linear (5-kb PstI-digested pL16 DNA) or unlabeled 3.7-kb Y-DNA. Different FRET values were observed between CT-ReAsH terminase and GFP-gp20 fusion with the linear (A) and Y-DNAs (B). FRET was measured with the 3.7-kb Y-DNA after resolvase treatment for 30 min (C).
FIGURE 4.

T4 Endo VII resolves the Y-DNA structure. A, gel mobility shift assays were carried out with and without resolvase treatment of the Y-DNA, and samples were analyzed on agarose gel. Molecular weight markers are indicated by arrows. B, for the analysis of cleavage sites at the nucleotide level in Y-DNA structures, either strand of the Y-DNA (Y2L/Y2R) was 5′ end-labeled with [γ-32P]ATP and hybridized with unlabeled complementary strand. The Y-DNAs with labeled Y2R strand (Y2L/Y2R*) and with labeled Y2L strand (Y2L*/Y2R) were treated with 50 and 500 units of resolvase and were run along with the untreated Y-DNA samples after boiling on 8% denaturing polyacrylamide gel and were visualized by autoradiography. The markers are shown with arrows with the number of bases indicated. The dotted line shows the DNA band corresponding to the 40-base marker. C, schematic representation of the Y-DNA sequence showing resolvase cleavage sites. The cleavage sites are represented by filled arrows of varying sizes to illustrate the differences in cutting efficiencies.
DNA Packaging Components
Unmodified wild type empty large procapsids (elps) were prepared from the T4 procapsid producing mutant-infected bacteria. Procapsid purification employed differential centrifugation, glycerol gradient ultracentrifugation, and FPLC-DEAE chromatography as described previously (15). Portal fusion protein GFP-gp20 containing procapsids were produced by supplying GFP-gp20 in trans from an expression vector to terminase, and portal-deficient phage mutant-infected bacteria as described previously (10). The same purification as of wild type procapsids was followed and then characterized for packaging activity by FCS and nuclease assay as described previously (19). GFP-gp20 proheads fluorescence was measured by Typhoon imager 9400 (GE Healthcare) (see Fig. 2D) on 0.8% agarose gel using excitation and emission wavelengths 489/509 nm as well as in a Picoquant MicroTime 200 confocal microscope (see Fig. 3A) with excitation at 470 nm. The wild type and both N- and C-terminal tetra cysteine-tagged gp17 terminase large subunit were purified from the chitin-binding domain-intein fusion protein as described previously (20). DNA packaging was carried out as described previously, except that PEG 20,000 was added to 2% (w/v) final concentration to the packaging assay mixture for FCS measurements (15).
FIGURE 2.

GFP-gene 20 fusion portals form small heat labile esp proheads at 28 °C and heat stable elp proheads at 32 °C. A, GFP-gp20 proheads produced at 28 °C were predominantly esps and at 32 °C elps as assessed for heat stability by release of the major capsid protein gp23 into the gel by not heating (−) or heating (+) prohead samples prior to 10% SDS-PAGE electrophoresis followed by Coomassie staining. B and C, Western blot analyses of wild type proheads and GFP-gp20 proheads were done using gp20 antibody (B) and GFP antibody (C). Molecular weight markers are indicated by arrows on the right sides of the gels and blots. D, fluorescent GFP-gp20 proheads were analyzed by running the unboiled prohead samples on native agarose gels and visualizing the unstained gel on Typhoon imager.
FIGURE 3.

TxY-DNA stalls in close proximity to GFP-containing portal. A, FRET-FCS analysis of the TxY-DNA with 3.7-kb leader sequence and the GFP-gene 20 fusion protein portal in the packaging mixture revealed a ∼5-nm separation by FRET. B, nuclease assays were carried out following FCS measurements confirming the packaging of the TxY-DNA with leader. Arrows indicate markers in bp.
Cloning of Tetra Cysteine Tag at the N and C Termini of the gp17 Gene
The tetra cysteine peptide FLNCCPGCCMEP (21) was added to both the N and C termini of the wild type gp17 gene in the pTYB2 vector (20). The oligonucleotides used for addition of peptide to the N-terminal (NT) and C-terminal (CT) ends of the gp17 gene are: forward, NT-5′-CCG CGG TTT CTG AAC TGC TGC CCG GGC TGC TGC ATG GGA CCG CCG CGG-3′, and reverse, NT-5′-GGC GCC AAA GAC TTG ACG ACG GGC CCG ACG ACG TAC CCT GGC GGC GCC-3′; and forward, CT-5′-CCG GGT TTC TGA ACT GCT GCC CGG GCT GCT GCA TGG GAC CGC-3′, and reverse, CT-5′-CCG GGC GGT TCC ATG CAG CAG CCC GGG CAG CAG TTC AGA AAC-3′. gp17-pTYB2 was digested with SacII and XmaI for addition of the oligonucleotides at the NT and CT, respectively. The digested vector was treated with shrimp alkaline phosphatase and ligated with the T4 polynucleotide kinase-phosphorylated oligonucleotides. The ligation mix was transformed into competent DH10B Escherichia coli cells, and colonies were screened by PCR followed by sequence confirmation of peptide additions.
In Vitro Labeling with ReAsH-Ethanedithiol (EDT2)
The labeling of the purified tetra-Cys gp17 (NT) and gp17 tetra-Cys (CT) with ReAsH-EDT2 was accomplished following the manufacturer's instructions (Invitrogen). Protein and dye were mixed in the ratio of 1:2 in terms of micromolar concentration in the Q buffer (50 mm Tris-HCl, pH 8.0, 100 mm NaCl, 0.1 mm EDTA, 5 mm MgCl2, 0.2 mm ZnCl2, and 1 mm ATP containing 10% glycerol) and incubated on ice for 1 h. The labeled protein was diluted to 300 μl in 1× BAL buffer (supplied with the kit) and purified using G-25 Sephadex spin column. Diluted labeled complex was concentrated using Amicon Ultra centrifugal filters of 10,000 molecular weight cut-off. The purified complex was stable for several days at 4 °C. ReAsH fluorescence was measured by Typhoon imager (see Fig. 6B) on 10% native polyacrylamide gel using excitation and emission wavelengths of 593 and 608 nm, as well as in a Variant Cary Eclipse fluorescence spectrophotometer (see Fig. 6A).
FIGURE 6.

ReAsH-EDT2 labeled N- and C-terminal tetra cysteine-tagged gp17 proteins are active in DNA packaging. A and B, both the NT and CT tetra cysteine-tagged gp17 proteins were labeled with ReAsH EDT2 dye, and fluorescence was measured in a Picoquant MicroTime 200 confocal microscope (A) and Typhoon imager (B). C, the labeled and unlabeled CT and NT terminases were run on native-PAGE followed by Coomassie staining as a loading control, and unstained gel was analyzed in Typhoon imager. D and E, nuclease assays were carried out to check the activities of the unlabeled (D) and the labeled CT and NT terminases (E) using 5-kb PstI linearized pL16 DNA as a substrate and wild type proheads in the reaction mixtures. Wild type terminase was used as a positive control in the assays.
DNA Packaging Assays
A nuclease protection assay measured the amount of packaged DNA by agarose gel electrophoresis after incubation at 37 °C for 30 min with pancreatic DNase, followed by release of the packaged procapsid DNA by incubation at 65 °C for 30 min in the presence of EDTA, proteinase K, and 1% SDS as described previously (15). The packaged ethidium bromide-stained DNAs were quantitated using UVP software to integrate gel DNA band density in comparison with known quantities of standard DNA ladders (Fermentas). For nuclease assay of packaging following FCS measurements, the sample was removed from the microscope observation stage and processed by the nuclease assay described above to determine the packaging that had been achieved during the FCS measurements. Packaging was assayed in addition by change in diffusability of the added dye-labeled DNA as compared with the added DNA without packaging (omitting either terminase or procapsids). Employing FCS and FRET-FCS measurements at room temperature, FCS and FRET-FCS assays of packaging were carried out as described previously (12, 22). FCS measurement of packaging after resolvase treatment employed a 30-min incubation with resolvase at room temperature assay (addition of 500 units of resolvase in 1 μl to the 16-μl packaging mixture) followed by FRET-FCS.
Assays of Resolvase Activity
For gel mobility shift assays, unlabeled Y-DNA (Fig. 1D, panel i) was used as substrate. The reaction mixture consisted of 1 μm Y-DNA in packaging buffer (100 mm Tris-HCl, pH 8.0, 12 mm MgCl2, 1 mm dithiothreitol, 4 mm spermidine, 10 mm ATP, 200 mm NaCl, 4 mm putrescine, and 10% polyethylene glycol; 15–20 kDa; Sigma) with and without 500 units of resolvase (T4 endonuclease VII was a gift from Prof. B. Kemper, University of Cologne). The reactions were incubated at 37 °C for 60 min and stopped by the addition of EDTA. The samples were run on 1% agarose gel (see Fig. 4A). To identify the cutting sites in the Y-DNA, the Y2L and Y2R strands of the Y-DNA were 5′ end-labeled with [γ-32P]ATP using T4 polynucleotide kinase. 1 μl (∼ 104 cpm) of the labeled strand was annealed to 2 μl of unlabeled complementary strand in the hybridization buffer (70 mm Tris-HCl, pH 7.6, 70 mm MgCl2, and 500 mm KCl). For annealing, the samples were heated at 90 °C for 2 min, and then the tubes carrying the samples were placed on the top of a 500-ml beaker with 90 °C water and cooled gradually to room temperature over more than 30 min. The samples were purified using Sephadex G-25 spin columns (GE Healthcare). For resolvase assays, the reaction mixture contained 1 μl of Y-DNA with labeled Y2R strand (Y2L/Y2R*) or with labeled Y2L strand (Y2L*/Y2R) and 50 or 500 units of resolvase in reaction buffer (10 mm Tris-HCl, pH 8.0, 10 mm MgCl2). The reactions were incubated at 37 °C for 60 min and stopped by the addition of loading dye containing formamide. The samples were incubated at 90 °C for 2 min before electrophoresis on 8% denaturing polyacrylamide gels. Cleaved products were visualized by autoradiography on Typhoon Imager (see Fig. 4B). One unit of endonuclease VII is defined as the amount of enzyme that cleaves 100 ng of cruciform DNA under standard reaction conditions within 15 min at 37 °C (31).
FCS and FRET-FCS Measurements and Analyses
FCS measurements were performed using a Picoquant MicroTime 200 confocal microscope (Picoquant microtime system coupled to an Olympus IX71 microscope). The excitation laser (λex = ∼470 nm) was reflected by a dichroic mirror (centered at 476 nm) to a high numerical aperture oil objective (100×; numerical aperture, 1.3) and focused onto the solution sample. The fluorescence signals were collected through the same objective and dichroic mirror. An additional long pass filter was used to eliminate the scattered laser light. Fluorescence responses from the donor and the acceptor molecules were separated by a beam splitter and detected by two avalanche photodiode detectors using the method of time-correlated single photon counting and the time-tagged time-resolved mode of the TimeHarp 200 board. High quality bandpass (Chroma) filters were used for recording donor and acceptor fluorescence in two separate detection channels. FCS measurements were performed in a constant detection volume. PicoQuant Symphotime software was used to generate the autocorrelation curves and in analyzing the FRET data. The autocorrelation curves were fitted with the following diffusion model. The autocorrelation function for n fluorescent species traversing a three-dimensional Gaussian volume with radius ω0 and half-axial z0 is as follows,
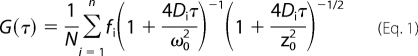 |
where τ is the lag time, N is the number of molecules in the volume, and fi is the fraction of the corresponding diffusion coefficient Di (23).
The collected single photon data were binned by 1-ms bin in each channel (donor or acceptor), which resulted in intensity-time traces and count-rate histograms. Threshold values in each channel were used to identify the single molecule bursts from the corresponding background signal level. Significant signals from adjacent intervals were binned together to get a sum photon number for each burst. Thus, fluorescence bursts were recorded simultaneously in donor and acceptor channels, and FRET efficiencies were calculated using E = nA/(nA + γnD), where nD and nA are the sums of donor counts and acceptor counts for each burst, taking into account the possible difference in the detection efficiency (γ) in two separate channels (24). The donor-to-acceptor distance (r) in terms of efficiency of energy transfer (E) and Förster distance (R0) is given by r = R0 [1/E − 1]1/6. We have used the value of R0 of 6.749 nm for the Alexa 488 (donor) and Cy3 (acceptor) pair for estimating the donor-to-acceptor distance (25). We calculated the value of R0 of 6.2 nm for the Alexa 488 (donor) to ReAsH (acceptor) pair from the spectral properties of the donor and acceptor and the donor quantum yield. For the GFP (donor) and ReAsH (acceptor) pair, R0 of 5.5 nm was calculated (23, 26). We assume that orientation factor changes in the donor-acceptor fluorophores bound to a rigid Y-DNA stem can account for at most a minor portion of the observed Förster Distance changes (22, 23).
RESULTS
GFP-Gene 20 Fusion Protein Portals Can Form Proheads with Both Empty Small Prohead (esp) and Empty Large Prohead (elp) Forms at Different Temperatures
Proheads containing GFP-gp20 (green fluorescent protein fused to the N terminus of portal protein gp20) were produced in vivo in bacteria that supply the GFP-gp20 fusion protein in trans from an expression vector. When infected with a T4 mutant lacking the normal portal protein (20amB8) as well as the small and large (16amN66 17amA465) gp16 and gp17 terminase subunits, proheads containing a mixed mutant and GFP fusion portal are assembled (10). T4 proheads with the same capsid components are found in vivo in esp and elp forms. The elps are more stable to temperature than esps and release the major capsid protein gp23 into SDS-PAGE gels only upon heating; heat-dependent gel mobility of gp23 has been shown to be diagnostic of the maturation-expansion (esp versus elp state) of the proheads by extensive structural studies (summarized in (27). As previously reported (28), the growth temperature of the infected bacterial culture strongly influences the relative proportion of esps and elps produced. However, mutant proheads containing C-terminal gp20-GFP fusion portals were shown to be locked into the esp conformation at high and low temperatures until DNA packaging drives expansion to the elp form (19). In contrast, the N-terminal GFP-gp20-containing portal proheads are comparable with wild type in producing both esps and elps depending upon the growth conditions. At temperature above 28 °C, mostly elps (>90%) and few esps (<10%) were detected, but ∼80% of the total prohead yield was esps at 28 °C temperature as determined by SDS-PAGE (Fig. 2A). Proheads with N-terminal fusion portal proteins produced in this way typically have ∼2–3 of the 12 portal subunits tagged with full-length GFP (10), consistent with Western blot analysis using gp20 and GFP antibodies (Fig. 2, B and C). GFP-gp20 proheads are fluorescent when checked on 0.8% agarose gel in Typhoon Imager (Fig. 2D), whereas the wild type proheads could not be detected on gel. Although both esp and elp proheads package DNA in vitro with comparable efficiency (19), the GFP-gp20 portal proheads produced at 28 °C or lower were generally used because of apparently greater GFP fluorescence.
FRET-FCS gp20-GFP Fusion and Tx-Y-DNA
Previously we have shown that the Y-junction portion of the 3.7-kb TxY-DNA was stalled in the vicinity of the gp20-GFP fusion, and Tx-GFP FRET (10%) was observed (13). With GFP-gp20 fusion, we observed a higher FRET value (20%) using a 3.7-kb TxY-DNA construct (Fig. 3A). Fluorescence correlation analysis of the 3.7-kb TxY-DNA in the packaging assay samples showed a decrease in the linear diffusion constant, suggesting that a substantial portion of the DNA had been packaged that was further determined by the gel-based nuclease protection assay (Fig. 3B). The diffusion coefficient of the 3.7-kb leader TxY-DNA anchored into the GFP portal was measured to be ∼2 μm2/s, comparable with previous measurements (22). Fig. 3A shows the FRET histogram when 3.7-kb TxY-DNA is packaged in the GFP portal. From this measurement, the donor-to-acceptor distance (r) is estimated to be 5 nm, given a Förster distance of 4 nm for a GFP-Tx donor-acceptor pair (29).
T4 Endo VII Resolvase Cleavage Sites in Y-DNAs
The 90-bp Y-DNA was cleaved by the T4 Endo VII resolvase as seen by mobility shift assay. The cleaved Y-DNA runs faster than the intact Y-DNA on an agarose gel (Fig. 4A). The exact locations of the Endo VII cleavage sites were determined in the two strands (Y2L/Y2R) in the Y-DNA. For this purpose, Y-DNAs with one of the strands labeled with [γ-32P]ATP were used in the Endo VII treatment. The reaction products were separated on denaturing polyacrylamide gels and visualized by autoradiography. One major L strand cleavage product of 36 bases (b) and, for reasons that are not clear, increased minor R products of 38 and 39 bases in B form DNA were seen on the gel (Fig. 4B). The cleavage sites 3′ to a departure from B form structure are comparable with those determined by Kemper and co-workers (30, 31). The cleavage sites for both the strands are shown in the Fig. 4C. The major L strand (∼36 bases) nicking is expected to release the upper Y arm, thereby releasing the structural roadblock to packaging.
Effect of T4 Endo VII Resolvase on the Compression of 11-bp and 16-bp Y-DNAs
To examine the effects of resolvase on the molecular orientation of stalled DNA substrates, 90-bp Y-DNA packaging substrates were internally labeled with an energy transfer dye pair: a Cy3 (C3; acceptor) was positioned in the duplex Y stem either 11 or 16 bp apart from an Ax (donor) located on the complementary strand (Fig. 1D, panels iii and iv). A nick was located 3′ to the Ax attached at C, because of the three oligonucleotide construction of the Ax containing strand. These Y-DNAs were ligated to a 3.7-kb stem extension (Fig. 1C) to allow anchoring and stalling in the procapsid portal; however, the AxC residue nick cannot be sealed. FRET-FCS measurements of 3.7-kb AxC3Y-DNA substrates in buffer and following packaging are shown in Fig. 5 (A and B). Following the fluorescence measurements, packaging protection of a portion of the 3.7-kb AxC3Y-DNAs added to the fluorescence chamber was confirmed by nuclease assay (Fig. 5, D and F); in a parallel control, mixtures without proheads or terminase nuclease assay did not show protected DNA (data not shown).
FIGURE 5.

T4 Endo VII resolvase affects the compression of 11- and 16-bp Y-DNAs. A and B, FRET-FCS measurements of 3.7 -kb AxC3Y-DNAs with 11-bp (A) and 16-bp (B) interdye distances DNAs in buffer and in packaging mix before and after resolvase addition (1 μl to 16 μl packaging mixture) were carried out at room temperature. C and E, autocorrelation plots to measure diffusion of the unpackaged, packaged with and without resolvase 11-bp (C) and 16-bp (E) 3.7-kb AxC3Y-DNAs. D and F, nuclease assays were carried out following FCS-FRET measurements confirming the packaging of the 11-bp (D) and 16-bp (F) 3.7-kb AxC3Y-DNAs. The markers are shown with arrows.
In analyzing packaging by FRET-FCS, a single-species diffusion model does not exactly fit the data, which may indicate the heterogeneity of the environment in the reaction buffer. The autocorrelation plots of the packaged 3.7-kb AxC3Y-DNAs (11 or 16 bp) fitted with a double-species model are presented in Fig. 5 (C and E). The fitted parameters and errors associated with the measured diffusion coefficients are tabulated in Table 1. Correlation spectroscopy of the packaged 3.7-kb AxC3Y-DNAs revealed a portion with procapsid-like diffusion, consistent with the nuclease assay following the fluorescence measurements (Table 1). The diffusion coefficient of the anchored 3.7-kb AxC3Y-DNA (11 or 16 bp) into normal procapsids was measured to be 1–3 μm2/s. When compared with our previous findings with 10- and 14-bp 2.7-kb AxC3Y-DNAs (13), two components were observed in the case of the 16-bp spacing displaying 25 and 45% energy transfer efficiencies (Fig. 5B), whereas the 11-bp spacing showed only one broad component with 45% efficiency (Fig. 5A). In the absence of terminase and ATP, FRET efficiencies of 35 and 25% were observed with 11- and 16-bp 3.7-kb AxC3Y-DNAs, respectively (data not shown). Because the difference in the FRET values for the unpackaged and the packaged 11-bp 3.7-kb AxC3Y-DNA was only ∼10%, the two energy components seen previously could not be resolved here. However two components were seen in the case of 16-bp 3.7-kb AxC3Y-DNA, which could be attributed to the unpackaged (FRET value of 25%) and packaged (FRET value of 45%) DNAs. We attribute the high efficiency FRET to the packaged DNAs and the low efficiency component to the freely diffusing unpackaged 3.7-kb AxC3Y-DNA substrates. For 11-bp AxC3Y-DNA, in the absence of procapsids, we observed energy transfer of 35% (Fig. 5A), which resulted in a donor-to-acceptor distance (r) of 7.5 nm. For 11-bp AxC3Y-DNA when packaged into the procapsid, we observed an energy transfer of 45%. Consequently, in this case we estimated r = 6.9 nm. Thus, the change in donor-to-acceptor distance after AxC3Y-DNA (11 bp) is packaged into the procapsid is 0.6 nm, which is equivalent to ∼9% change in the interdye spacing. For 16-bp AxC3Y-DNA, in the absence of procapsid, we observed energy transfer of 20% (Fig. 5B), which corresponds to a value of r = 8.5 nm. For the 16-bp AxC3Y-DNA, we observed energy transfer of 45% when packaged into the procapsid, corresponding to r = 6.9 nm. Thus, the change in the donor-to-acceptor distance after AxC3Y-DNA (16 bp) is packaged is 1.6 nm, which is equivalent to ∼18% change in the interdye spacing. Hence, from the changes in the FRET efficiency and interdye distances, we calculate an average DNA compression of 10–20% that is associated with the packaging reaction. When resolvase was subsequently added to the packaging mix with 3.7-kb AxC3Y-DNA (11 or 16 bp) and incubation was carried out for 30 min, a decrease in the FRET efficiency was observed that could be attributed in part to the linearization and completion of packaging of the Y-DNAs by release of the upper Y arm (Fig. 4), because the lowered FRET values were similar to that of the unpackaged DNAs (Fig. 5, A and B). An increase in the fractional contribution of the free Y-DNA and a decrease in the prohead bound Y-DNA was also observed after resolvase treatment (Table 1). This is expected because of 3′ cleavage of the intact YIL strand apposed to the AxC residue Rc-Rb nick, a site also expected to be nicked by resolvase (30). This cleavage is expected to release stalled AxC3Y-DNA from the packaged 3.7-kb leader (Fig. 1D, panels iii and iv) in the prohead, thus increasing the amount of fast diffusing free Y-DNA in solution.
TABLE 1.
Diffusion coefficients and respective fractional contributions for different constructs of Y-DNA under various packaging conditions
D1 and D2 are the diffusion coefficients of unpackaged and packaged DNAs. The values are given as averages ± S.E. Fractional contributions are shown as percentage values.
| Sample | D1 |
D2 |
||
|---|---|---|---|---|
| Value | Contribution | Value | Contribution | |
| μm2/s | % | μm2/s | % | |
| 3.7-kb AxCyY-DNA 11-bp | 99 ± 8 | |||
| 3.7-kb AxCyY-DNA 11-bp packaged | 3.5 ± 1 | 29 | 94 ± 8 | 71 |
| 3.7-kb AxCyY-DNA 11-bp packaged + resolvase | 3.4 ± 1 | 22 | 90 ± 6 | 78 |
| 3.7-kb AxCyY-DNA 16-bp | 90 ± 5 | |||
| 3.7-kb AxCyY-DNA 16-bp packaged | 2 ± 0.6 | 26 | 90 ± 7 | 74 |
| 3.7-kb AxCyY-DNA 16-bp packaged + resolvase | 1.5 ± 0.5 | 9 | 90 ± 5 | 91 |
Changes in Conformational States of the Terminase and Prohead Portal during Packaging of Linear and Y-DNA Substrates
To label the terminase with a single fluorescent dye at a known position, a ReAsH-binding tetra cysteine peptide FLNCCPGCCMEP was added to either the C terminus or the N terminus of the wild type gp17 gene in pTYB2 vector (20, 21). Both proteins were labeled with ReAsH-EDT2 dye, and fluorescence was measured (Fig. 6A). Labeled proteins were also seen on gel in Typhoon Imager (Fig. 6B) using the red filter, whereas the unlabeled proteins could not be detected. The Coomassie-stained gel shows that equal amounts of the proteins were loaded (Fig. 6C). Both the NT and CT tetra cysteine-tagged proteins were active before as well as after the labeling as determined by nuclease assay (Fig. 6, D and E). To look at the DNA-terminase interaction, packaging was carried out using NT-ReAsH or CT-ReAsH terminase, wild type proheads, and 3.7-kb Alexa 488 Y-DNA. Different FRET values were observed for CT-ReAsH and NT-ReAsH terminases with 3.7-kb Alexa 488 Y-DNA. The FRET efficiency was 35% with CT-ReAsH terminase, which corresponds to a donor-to-acceptor distance (r) of 6.8 nm (Fig. 7A), and 20% with NT-ReAsH terminase, which corresponds to donor-to-acceptor distance (r) of 7.8 nm (Fig. 7D). The autocorrelation plots of the packaged 3.7-kb Alexa 488 Y-DNA fitted with a double-species model are presented in (Fig. 7, B and E). The diffusion coefficient of the 3.7-kb AxC3Y-DNA anchored into normal procapsids was measured to be ∼4–7 μm2/s. Packaging of the 3.7-kb Alexa 488 Y-DNA by the labeled terminases was confirmed by nuclease assays (Fig. 7, C and F).
To look at the portal terminase interaction, packaging was carried out using GFP-gp20 proheads, CT-ReAsH terminase and unlabeled linear (5-kb PstI-digested pL16 DNA), or unlabeled 3.7-kb Y-DNA (Fig. 1D, panel i). FRET was observed between the ReAsH-labeled terminases and the gp20-GFP fusion. A FRET value of 20% was observed between NT-ReAsH terminase and GFP-gp20 fusion, resulting in a donor-to-acceptor distance (r) of 6.9 nm (data not shown).
However, higher and strikingly different FRET values were observed between CT-ReAsH terminase and GFP-gp20 fusion when different DNAs were used (the linear and Y-DNAs.) A substantially higher FRET value of 60% was observed when 3.7-kb Y-DNA was used in packaging, resulting in a donor-to-acceptor distance (r) of 5.1 nm, whereas 45% FRET was seen when packaging linear DNA, which corresponds to a value of r = 5.7 nm (Fig. 8, A and B). To confirm this result, resolvase was added to the packaging mix containing 3.7-kb Y-DNA following packaging and incubated for 30 min with FRET observed before and after resolvase treatment. The FRET efficiency decreased from 60 to 45% after resolvase treatment, which was similar to the FRET observed with the linear DNA, suggesting linearization and completion of packaging of the 3.7-kb Y-DNA by the resolvase aided terminase (Fig. 8, B and C).
DISCUSSION
The major findings of our study of the bacteriophage T4 packaging motor are that (i) in vitro the purified Endo VII resolvase mimics its action in vivo to release and complete packaging of entrapped Y-DNA substrates, not previously shown to be the case in vitro; (ii) using specific dye-labeled substrates, Endo VII resolvase action is correlated with release of compression from the Y-DNA substrates; (iii) resolvase release of trapped Y-DNAs is also correlated with increased distance between the terminase and portal; and (iv) the correlated protein and DNA conformational changes are most likely interpreted as reflecting relaxed and tense states of the motor in the arrested (Y-DNA) versus translocating states (linear) of DNA.
The FRET measured conformational changes in substrate and motor components may be interpreted more specifically from a working model of the packaging motor (Fig. 9). This model uses crystal structures of the gp17 terminase of T4 and the portal protein of SPP1 (14, 32, 33). Both the N- and C-terminal peptide regions of gp17 and the N-terminal peptide of the SPP1 portal, to which our fluorophores are specifically attached, are not seen in the crystal structures, suggesting that these portions of both proteins are relatively unstructured. Nevertheless, the following overall structural properties of the T4 and SPP1 packaging motor are known. The gp17 terminase is docked onto the prohead portal dodecamer (here displayed as a 13-mer as found in the free SPP1 portal protein crystal) as a pentamer, as shown. The N terminus of the SPP1 portal (and T4 portal) is known to be displayed outside the prohead, close to the outer portal rim (10, 33). Most of the SPP1 portal mass is beneath the rim in the wide portion interior to the prohead. This portion of the portal surrounds the DNA channel, and its C terminus lies deep within the prohead interior in a crown structure associated with the headful measuring role of the portal (32). The SPP1 and T4 portal proteins are of nearly identical sizes (501 and 524 residues, respectively) and, as part of strict headful DNA measuring devices, likely of even greater similarity in overall structure and function than is generally found to be the case among known portal dodecamers (34, 35). If the genetically (intergenic sequence specific cs-ts suppressor mutations), biochemically, and immunologically determined gp17-gp20 interaction sites in the T4 portal (gp20 residues 281–311) are painted onto the SPP1 structure, they localize to the outer exposed lip of the portal, shown in gold (36). Three gp17 interaction sites are also displayed as gold regions in the terminase, two defined genetically (residue 364 in the middle of the gp17 and residue 583 near the C terminus), with the latter site confirmed here by 5.1- or 5.7-nm (Y versus linear DNA distances) FRET between the GFP and the C-terminal ReAsH of the 610-residue-long gp17. A third more distant interaction of 6.9 nm is seen by FRET between the GFP-gp20 and the N-terminal ReAsH-labeled terminase. Although both the N and C termini of the terminase are likely to be on relatively flexible tether-like portions of the molecule, their proximity to the N terminus of the portal as judged by a statistically determined series of FRET-FCS distances is consistent with the overall structures of both proteins. Thus, the good fits of what is known of the two structures to the genetic and FRET-defined interaction regions (shown by the three arrows in Fig. 9), near to the N- and C-terminal crystal structure ends, suggests that this model is a reasonably close approximation to the actual T4 structure. The anchored Y-DNA junction point is also determined by FRET to be localized to this interaction region within 5 nm of the GFP portal fluorophores (Fig. 3). Although localized statistically by the FRET-FCS measurements in this “hybrid packasome” model, is there in fact a single precisely determined T4 structure?
FIGURE 9.

The N- and C-terminal regions of the gp17 protein interact with the portal. Diagram showing terminase portal-binding sites (in gold) inferred from genetics, biochemistry, and FRET in DNA-GFP portal-terminase complexes. The gp17 terminase is shown to dock onto the prohead portal dodecamer (here displayed as a 13-mer as found in the free SPP1 portal protein crystal) as a pentamer. gp17-gp20 interaction sites painted onto the SPP1 portal structure are shown in gold. Three GFP-portal interaction regions in gp17 at 6.9 nm (N-terminal) and 5.7 nm (C-terminal) are also displayed as gold regions; the reduced C-terminal 5.1-nm distance in arrested Y-DNA as compared with linear DNA complexes is shown. The diagram is based on Protein Data Bank structures 3CPE and 3EZK.
Our FRET measurements suggest that in fact the power stroke of the T4 motor is associated with significant conformational changes in both the DNA substrate and motor proteins (Fig. 10). The FRET-FCS measurements serve to localize over a 0.5–8-nm range distance changes among the interacting components. We find that the leader arrested Y-DNA in the portal undergoes an apparent compression or “crunching” by the motor power stroke. Reasons for preferring the DNA compression model to DNA bending—comparable FRET changes with ∼10-bp-spaced double dyes situated on the same side and opposite sides of the helix, reversible DNA compression comparable with what we infer from FRET experimentally measured and found to be within the measured force of the packaging motor, etc.—were discussed previously (13). The compression changes we measure here for the 10- and 16-bp separated double dye Y-DNAs (10–20%) are somewhat less than our previous measurements (20–25%). This difference may be explained by the fact that previously we measured the compression within two intact duplex strands, whereas the current study required synthesis of one nicked dye containing strand in the double dye duplex. Because we have determined that in short DNA substrates nicks have a powerful inhibitory effect on packaging that cause release from the motor, the reduction in a statistically determined FRET-FCS DNA compression likely relates to enhanced fluctuations in the arrested motor. Pausing and slipping of the phage T4 motor was observed in single molecule optical tweezers experiments even in the absence of Endo VII or known deviations from duplex DNA (37). How these fluctuations arise remains an open question, but good additional evidence for their occurrence in the motor is shown by the action of the Endo VII resolvase. The resolvase has been shown to sit on the T4 portal together with the terminase (16). Evidently from this position it is able to rescue stalled X- or Y-structure-containing concatemeric substrate DNAs in vivo and, as we now show, in vitro as well. Presumably there must be conformational fluctuations in the prohead anchored DNA substrate and motor proteins, fluctuating grips and releases, that allow access of the resolvase to the immobilizing structures.
FIGURE 10.

Both the motor proteins and the DNA undergo conformational changes during DNA packaging. A, schematic of FRET between different dyes within the DNA and site of action of gp49. Portal-bound Endo VII resolvase action releases the compression from the Y-DNA substrates. B, resolvase release of trapped Y-DNAs is also correlated with increased distance (∼0.6 nm) between the terminase and portal as shown by changes in FRET between the labeled terminase and portal during packaging.
Our studies are based on our prior demonstrations that affinity of the terminase to the portal is much greater in the presence of packaged DNA. A leader segment of a substantial number of kilobases is required to anchor the Y-DNA to the portal motor complex in the prohead, otherwise the short non-B form is released from the motor (12, 13). Our FRET-FCS measurements show that the terminase-to-portal distance is apparently decreased by ∼0.6 nm in the arrested Y-DNA as compared with linear DNA (Fig. 10B). The most likely explanation is that an ATP-driven power stroke of the motor moves the terminase toward the portal, a motion also correlated with compression of the DNA (Fig. 10A), and these changes are trapped in the arrested Y-DNA state. This magnitude of conformational change is consistent with a comparable structural change seen in two crystal structures of gp17 that have been hypothesized to be related to DNA translocation (14). However, there is also evidence that conformational changes in the SPP1 portal are required for DNA translocation (32). Overall, our statistical FRET-FCS studies support our proposed linear grip-and-release compression motor mechanism. Further single molecule studies using precisely localized dyes in the terminase and portal will be necessary to more precisely determine dynamic conformational structural changes in the terminase, the portal, or in both of these motor components of the prohead packasome. Single molecule detection will also be required to determine whether conformational structural changes to both the packasome proteins and the substrate that we measure in a motor-arrested Y-DNA substrate apply also to the motor as it is actively translocating linear DNA.
Acknowledgments
We thank Börries Kemper and Christian Biertumpfel for T4 Endo VII materials, A-Lien Lu-Chang for assistance with Endo VII sequencing, and Mark Oram for constructing the NT gp17 ReAsH peptide expression vector.
This work was supported, in whole or in part, by National Institutes of Health Grants AI11676 (to L. W. B.), HG002655 (to J. R. L.), and K25AI087968 (to K. R.).
- FRET-FCS
- fluorescence resonance energy transfer-fluorescence correlation spectroscopy
- elp
- empty large procapsid
- esp
- empty small procapsid
- NT
- N-terminal
- CT
- C-terminal
- ReAsH
- resorufin arsenical hairpin
- EDT2
- ethanedithiol
- Ax
- Alexa 488
- Endo
- endonuclease
- C3
- Cy3.
REFERENCES
- 1. Black L. W. (1989) Annu. Rev. Microbiol. 43, 267–292 [DOI] [PubMed] [Google Scholar]
- 2. Sheaffer A. K., Newcomb W. W., Gao M., Yu D., Weller S. K., Brown J. C., Tenney D. J. (2001) J. Virol. 75, 687–698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kainov D. E., Tuma R., Mancini E. J. (2006) Cell Mol. Life Sci. 63, 1095–1105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Rickgauer J. P., Fuller D. N., Grimes S., Jardine P. J., Anderson D. L., Smith D. E. (2008) Biophys. J. 94, 159–167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Rao V. B., Feiss M. (2008) Annu. Rev. Genet. 42, 647–681 [DOI] [PubMed] [Google Scholar]
- 6. Rao V. B., Black L. W. (2010) Virol J. 7, 356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Johnson J. E., Chiu W. (2007) Curr. Opin. Struct. Biol. 17, 237–243 [DOI] [PubMed] [Google Scholar]
- 8. Hendrix R. W. (1978) Proc. Natl. Acad. Sci. U.S.A. 75, 4779–4783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Simpson A. A., Tao Y., Leiman P. G., Badasso M. O., He Y., Jardine P. J., Olson N. H., Morais M. C., Grimes S., Anderson D. L., Baker T. S., Rossmann M. G. (2000) Nature 408, 745–750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Baumann R. G., Mullaney J., Black L. W. (2006) Mol. Microbiol. 61, 16–32 [DOI] [PubMed] [Google Scholar]
- 11. Hugel T., Michaelis J., Hetherington C. L., Jardine P. J., Grimes S., Walter J. M., Falk W., Anderson D. L., Bustamante C. (2007) PLoS Biol. 5, e59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Oram M., Sabanayagam C., Black L. W. (2008) J. Mol. Biol. 381, 61–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ray K., Sabanayagam C. R., Lakowicz J. R., Black L. W. (2010) Virology 398, 224–232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sun S., Kondabagil K., Draper B., Alam T. I., Bowman V. D., Zhang Z., Hegde S., Fokine A., Rossmann M. G., Rao V. B. (2008) Cell 135, 1251–1262 [DOI] [PubMed] [Google Scholar]
- 15. Black L. W., Peng G. (2006) J. Biol. Chem. 281, 25635–25643 [DOI] [PubMed] [Google Scholar]
- 16. Golz S., Kemper B. (1999) J. Mol. Biol. 285, 1131–1144 [DOI] [PubMed] [Google Scholar]
- 17. Luftig R. B., Wood W. B., Okinaka R. (1971) J. Mol. Biol. 57, 555–573 [DOI] [PubMed] [Google Scholar]
- 18. Kemper B., Brown D. T. (1976) J. Virol. 18, 1000–1015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ray K., Oram M., Ma J., Black L. W. (2009) Virology 391, 44–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Baumann R. G., Black L. W. (2003) J. Biol. Chem. 278, 4618–4627 [DOI] [PubMed] [Google Scholar]
- 21. Adams S. R., Campbell R. E., Gross L. A., Martin B. R., Walkup G. K., Yao Y., Llopis J., Tsien R. Y. (2002) J. Am. Chem. Soc. 124, 6063–6076 [DOI] [PubMed] [Google Scholar]
- 22. Sabanayagam C. R., Oram M., Lakowicz J. R., Black L. W. (2007) Biophys. J. 93, L17–L19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lakowicz J. R. (2006) Principles of Fluorescence Spectroscopy, 3rd Ed., Chapters 13 and 24, Springer-Verlag New York Inc., New York [Google Scholar]
- 24. Schuler B., Lipman E. A., Eaton W. A. (2002) Nature 419, 743–747 [DOI] [PubMed] [Google Scholar]
- 25. Elangovan M., Wallrabe H., Chen Y., Day R. N., Barroso M., Periasamy A. (2003) Methods 29, 58–73 [DOI] [PubMed] [Google Scholar]
- 26. Martin B. R., Giepmans B. N., Adams S. R., Tsien R. Y. (2005) Nat. Biotechnol. 23, 1308–1314 [DOI] [PubMed] [Google Scholar]
- 27. Black L. W., Showe M. K., Steven A. C. (1993) in Molecular Biology of Bacteriophage T4 (Karam J. D. ed.) pp. 218–258, ASM Press, Washington, D.C [Google Scholar]
- 28. Rao V. B., Black L. W. (1985) J. Mol. Biol. 185, 565–578 [DOI] [PubMed] [Google Scholar]
- 29. Duckworth B. P., Zhang Z., Hosokawa A., Distefano M. D. (2007) ChemBioChem 8, 98–105 [DOI] [PubMed] [Google Scholar]
- 30. Jensch F., Kemper B. (1986) EMBO J. 5, 181–189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Pottmeyer S., Kemper B. (1992) J. Mol. Biol. 223, 607–615 [DOI] [PubMed] [Google Scholar]
- 32. Cuervo A., Vaney M. C., Antson A. A., Tavares P., Oliveira L. (2007) J. Biol. Chem. 282, 18907–18913 [DOI] [PubMed] [Google Scholar]
- 33. Lebedev A. A., Krause M. H., Isidro A. L., Vagin A. A., Orlova E. V., Turner J., Dodson E. J., Tavares P., Antson A. A. (2007) EMBO J. 26, 1984–1994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Driedonks R. A., Caldentey J. (1983) J. Mol. Biol. 166, 341–360 [DOI] [PubMed] [Google Scholar]
- 35. Driedonks R. A., Engel A., tenHeggeler B., van Driel R. (1981) J. Mol. Biol. 152, 641–662 [DOI] [PubMed] [Google Scholar]
- 36. Lin H., Rao V. B., Black L. W. (1999) J. Mol. Biol. 289, 249–260 [DOI] [PubMed] [Google Scholar]
- 37. Fuller D. N., Raymer D. M., Kottadiel V. I., Rao V. B., Smith D. E. (2007) Proc. Natl. Acad. Sci. U.S.A. 104, 16868–16873 [DOI] [PMC free article] [PubMed] [Google Scholar]