

Abstract
Autophagy is a self-digestion pathway essential for maintaining cellular homeostasis and cell survival and for degrading intracellular pathogens. Human immunodeficiency virus-1 (HIV-1) may utilize autophagy for replication as the autophagy-related protein-7 (ATG-7), microtubule-associated protein 1 light chain 3, ATG-12, and ATG-16L2 are required for productive HIV-1 infection; however, the effects of autophagy induction on HIV-1 infection are unknown. HIV-1-infected individuals have lower levels of 1α,25-dihydroxycholecalciferol, the hormonally active form of vitamin D, than uninfected individuals. with the lowest concentrations found in persons with AIDS. Using human macrophages and RNA interference for ATG-5 and Beclin-1 and chemical inhibition of phosphatidylinositol 3-kinase, we have found that physiologically relevant concentrations of 1α,25-dihydroxycholecalciferol induce autophagy in human macrophages through a phosphatidylinositol 3-kinase-, ATG-5-, and Beclin-1-dependent mechanism that significantly inhibits HIV-1 replication in a dose-dependent manner. We also show that the inhibition of basal autophagy inhibits HIV-1 replication. Furthermore, although 1α,25-dihydroxycholecalciferol induces the secretion of human cathelicidin, at the concentrations produced in vitro, cathelicidin does not trigger autophagy. Our findings support an important role for autophagy during HIV-1 infection and provide new insights into novel approaches to prevent and treat HIV-1 infection and related opportunistic infections.
Keywords: Autophagy, HIV, Macrophage, Phosphatidylinositol 3-Kinase, Vitamin D, ATG5, Apoptosis, Beclin, Cathelicidin, Cell death
Introduction
Human immunodeficiency virus type-1 (HIV-1)2 is a global health problem that at current estimates has infected 60 million people and caused 25 million deaths worldwide. Currently, there are an estimated 33 million people living with HIV-1, 2 million of whom are children (1). Despite the immune defense mechanisms that the host deploys against HIV-1 and improved antiretroviral therapies, the virus persists in long-lived cells including macrophages and dendritic cells.
One-third of HIV-1-infected individuals are co-infected with Mycobacterium tuberculosis, a leading cause of death among people living with HIV-1. Studies have suggested that 1α,25-dihydroxycholecalciferol (1,25D3), the hormonally active form of vitamin D3, exerts anti-mycobacterial effects in human macrophages in vitro (2) through phosphatidylinositol 3-kinase (PI3K) (3) and macroautophagy (4). Few publications have examined the relationship between vitamin D3 and HIV-1 disease progression and survival. However, the few studies published have shown that HIV-1-infected individuals have lower 1,25D3 levels than uninfected individuals (5–9) with the lowest concentrations found in persons with AIDS (7). Moreover, depressed levels of 1,25D3 are correlated with significantly lower CD4+ T cell counts and higher tumor necrosis factor levels (6). Interestingly, consumption of foods rich in 25-hydroxycholecalciferol (25D3), the inactive precursor for 1,25D3, correlates with an increase in CD4+ T cell counts in HIV-1-infected individuals (10). Furthermore, women with low levels of 25D3 have an increased risk of HIV-1 disease progression (11), with infants born to HIV-1-infected mothers with low 25D3 levels having an increased risk of acquiring HIV-1 in utero, intrapartum, and through breastfeeding while also having a higher mortality rate (12).
Macroautophagy (herein referred to as autophagy) is a trafficking pathway whereby cytoplasmic constituents such as subcellular organelles and microbial pathogens are engulfed by autophagosomes, which fuse with lysosomes, forming autolysosomes, degrading the engulfed component. As an obligatory intracellular parasite, HIV-1 survival is dependent upon its ability to exploit host cell machinery for replication and dissemination and to circumvent cellular processes that prevent its growth. During infection, HIV-1 down-regulates Beclin-1 and microtubule-associated protein 1 light chain 3B-II (LC3B-II), reducing both basal autophagy and the numbers of autophagosomes per cell (13–15). However, silencing of autophagy proteins inhibits HIV-1 infection of HeLa cells (16). Interestingly, the HIV-1 envelope glycoprotein expressed on the surface of infected cells induces cell death with autophagy in uninfected bystander CD4+ T cells (17–19). Therefore, we investigated the effect of 1,25D3-mediated autophagy on productive HIV-1 infection of macrophages. We demonstrate that 1,25D3 induces autophagy in macrophages that inhibits HIV-1 replication.
EXPERIMENTAL PROCEDURES
Cells and Reagents
Monocyte-derived macrophages were obtained by culturing monocytes isolated from the peripheral blood mononuclear cells of HIV-1 seronegative donors (with approval from the University of California San Diego Institutional Review Board) in RPMI 1640 (Invitrogen) supplemented with 10% (v/v) heat-inactivated fetal bovine serum (FBS; Gemini Bio-Products) and 10 ng/ml macrophage colony stimulating factor (R&D Systems) for 10 days at 37 °C, 5% CO2. MonoMac1 cells were purchased from Deutsche Sammlung von Mikroorganismen und Zellkulturen and cultured in RPMI 1640 supplemented with 10% (v/v) FBS, 2 mm l-glutamine, 100 μm nonessential amino acids and 1 mm sodium pyruvate at 37 °C, 5% CO2. TZM-bl cells were obtained from the AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, National Institutes of Health from Dr. John C. Kappes and Tranzyme Inc. (20) and cultured in Dulbecco's modified Eagle's medium supplemented with 10% (v/v) FBS at 37 °C, 5% CO2.
1,25D3, wortmannin, pepstatin A, bafilomycin A1, SID 26681509, interferon α (IFNα), staurosporine, and rapamycin were all purchased from Sigma. Maraviroc was purchased from Toronto Research Chemicals. Wortmannin and bafilomycin A1 were used at 100 nm, SID 26681509 was at 50 nm, maraviroc was at 10 nm, and pepstatin A was at 10 μg/ml with pretreatment for 1 h before the addition of 1,25D3 or rapamycin. LL-37 and the scrambled control peptide were purchased from AnaSpec. Secreted LL-37 was quantified using a LL-37 enzyme-linked immunosorbent assay (ELISA; Hycult Biotech). (4-[3-(4-iodophenyl)-2-(4-nitrophenyl)-2H-5-tetrazolio]-1,3-benzene disulfonate (WST-1) and the lactate dehydrogenase release assay were purchased from Roche Applied Science and used according to the manufacturer's directions. The single-stranded DNA (ssDNA) ELISA was purchased from Millipore and used as previously described (21). LysoTracker Red DND-99 was purchased from Invitrogen and used according to the manufacturer's directions.
HIV-1
HIV-1Ba-L was obtained through the AIDS Research and Reference Reagent Program from Dr. Suzanne Gartner and Dr. Robert Gallo (22, 23). Virus stocks were prepared as previously described (24). The 50% tissue culture infective dose (TCID50) was determined using the Spearman-Kärber method (25) using the Alliance HIV-1 p24 antigen ELISA (PerkinElmer Life Sciences). Cells were infected with 105 TCID50 HIV-1Ba-L per 5 × 105 cells for 3 h after pretreatment for 4 h with 1,25D3 or rapamycin unless otherwise stated. Virus binding was assessed by incubating 106 macrophages with 300 ng p24 at 37 °C for 3 h. Cells were then washed extensively with Dulbecco's phosphate-buffered saline at 4 °C to remove unbound virions and lysed in 120 μl of CelLytic M (Sigma) supplemented with protease inhibitors (Thermo Scientific). Viral content was assessed by monitoring p24 by ELISA. Similar experimental conditions were used to estimate the process of viral entry, with the exception that an additional step of trypsinization for 5 min at 37 °C was performed after 5 h to remove non-internalized virions. The cells were then washed once with RPMI supplemented with 10% FBS followed by 3 times with Dulbecco's phosphate-buffered saline before lysis. HIV-1 productive infection of TZM-bl cells 48 h post-HIV-1 exposure was detected using the β-Gal Staining Set (Roche Applied Science).
Immunoblotting
Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (14C10), LC3B (D11), PI3KC3 (D9A5), Beclin-1, and ATG5 antibodies were obtained from Cell Signaling, HIV-1 Nef (3D12) antibody was from Abcam, and β-actin antibody (AC-74) was from Sigma. Cell lysates were prepared using CelLytic M supplemented with protease inhibitors (all from Sigma). For co-immunoprecipitation, 50 μg anti-PI3KC3 was immobilized in a coupling gel, then 50 μg of cell lysates were incubated with the antibody-immobilized coupling gel using the ProFound-Co-Immunoprecipitation kit (Thermo Scientific). For immunoblot analyses, cell lysates were resolved using 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol-buffered 4–12% polyacrylamide gel (Invitrogen) and transferred to nitrocellulose membranes (Thermo Scientific) followed by detection with the WesternBreeze chemiluminescence kit (Invitrogen) as described previously (24, 25). Relative densities of the target bands compared with the β-actin or GAPDH bands were analyzed using ImageJ (NIH).
Fluorescence Microscopy
Cells were fixed and permeabilized in Dulbecco's phosphate-buffered saline supplemented with 4.5% (w/v) paraformaldehyde and 0.1% (v/v) saponin for 30 min, washed, then probed with mouse anti-Gag-p17 (2D11) (Abcam) and rabbit anti-LC3B (D11) for 30 min. Cells were then probed with a goat anti-mouse Alexa Fluor 488 and goat anti-rabbit Alexa Fluor 555-conjugated antibodies for 30 min (Invitrogen), washed, and counterstained with Hoechst 33342. Labeled cells were visualized using an Olympus IX71 inverted fluorescence microscope equipped with a 100×/1.4 NA oil differential interference Plan Apochromat objective (Nikon Instruments) and a X-Cite 120Q light source (EXFO). The filters used were: for Hoechst 33342, band pass 377/50, dichroic mirror beam splitter 409, band pass 447/60; for Alexa Fluor 488, band pass 473/31, dichroic mirror beam splitter 495, band pass 520/35; for Alexa Fluor 555, band pass 525/40, dichroic mirror beam splitter 555, band pass 585/40. Images were acquired in RGB and combined at 72 dots per inch, 24-bit depth with an Olympus DP72 digital camera head equipped with a 1.45-million pixel charge-coupled device and a Bayer color filter using the DP72 Image Acquisition Interface (Olympus). As the background number of puncta was relatively low, population analysis was performed as previously described (26, 27). Briefly, cells were classified as (a) cells with diffuse LC3B fluorescence, (b) cells with ≥1 dot/cell but <10 dots/cell, or (c) cells with numerous LC3 puncta (≥10 dots/cell). The cutoff of ≥10 dots/cell was based upon the 95% confidence level calculated using the upper tail of Student's t distribution of puncta counts from 100 macrophages each from three independent donors incubated in full medium for 4 h using the equation (28)
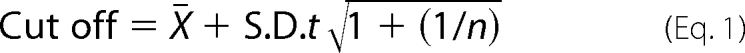 |
where X̄ is the mean of independent donor LC3B puncta counts, S.D. is the standard deviation, n is the number of independent controls, t is the (1 − α)th percentile of the one-tailed t-distribution with v = n − 1 degrees of freedom (29).
shRNA Transduction
MISSION small hairpin (shRNA) lentiviral particles were obtained from Sigma. Lentiviral transduction of MonoMac1 cells with particles for shRNAs targeting Beclin-1 (SHCLNV-NM_003766), ATG5 (SHCLNV-NM_004849), or scrambled non-target negative control (Scr, SHC002V) was performed according to the manufacturer's protocol.
Flow Cytometry
FACS data were collected using a FACSCalibur flow cytometer and analyzed using CellQuest Pro (both from BD Biosciences). Cell surface protein expression of CCR5 and CD4 was performed using fluorescently labeled monoclonal antibodies for CCR5, CD4, and CD14 (allophycocyanin-CCR5, peridin-chlorophyll protein-CD4, and fluorescein isothiocyanate-CD14) followed by treatment with Cytofix (all BD Biosciences). Intracellular LL-37 expression was assessed using 2 μm monensin (EMD Chemicals)-treated macrophages incubated with 1,25D3. Cells were fixed and permeabilized using Cytofix/Cytoperm (BD Biosciences), then probed with goat anti-LL-37 antibodies (Santa Cruz Biotechnology) followed by Alexa Fluor 647 conjugated donkey anti-goat antibodies (Invitrogen). Intracellular staining of endogenous saponin-resistant LC3B was performed as previously described (30) using rabbit anti-LC3B (D11) and allophycocyanin-conjugated mouse anti-rabbit antibodies (BD Biosciences). The Click-IT EdU (5-ethynyl-2′-deoxyuridine) Alexa Fluor 488 kit was purchased from Invitrogen and used according to the manufacturer's instructions.
Real-time PCR
Strong-stop HIV-1 DNA quantification was measured by real-time PCR using the LightCycler System and the FastStart DNA Master SYBR Green I (all Roche Applied Science). Primers and run conditions were as previously described (31). All results are expressed as the ratio between the target gene and the RNA polymerase II and normalized so that HIV-1 LTR in untreated cells equals 1.00.
Statistics
Unpaired, two-tailed, Student's t tests, α = 0.05 were used to assess whether the means of two normally distributed groups differed significantly.
RESULTS
1,25D3 Induces Autophagy in Human Macrophages
EB1089, a vitamin D3 analog, induces autophagy in MCF-7 cells (32) and human monocytes (4) and promotes autophagic responses in human myeloid leukemia cells through the induction of PI3K class III (PI3KC3) and Beclin-1 (33). The physical interaction of these two proteins forms the PI3KC3 kinase complex, which is essential for the induction of autophagosome formation at the vesicle elongation step. Therefore, we assessed the ability of physiologically relevant concentrations of 1,25D3 to induce autophagy in human macrophages. We observed the enhanced expression of Beclin-1 (Fig. 1A) and PI3KC3 (Fig. 1B) after incubation with 50–100 pm 1,25D3 for 4 h. Co-immunoprecipitation followed by immunoblotting also demonstrated that 1,25D3 induced Beclin-1 binding to PI3KC3 (Fig. 1C), forming the PI3KC3 kinase complex.
FIGURE 1.

1,25D3 induces autophagy in human macrophages. A, macrophages were left untreated or were incubated with 1,25D3, rapamycin (Rapa) or starved by incubation in Earle's balanced salt solution for 4 h, lysed, then subjected to immunoblotting with antibody to Beclin-1 and β-actin. B, shown is an immunoblot using antibody to PI3KC3 and β-actin. C, shown is an immunoblot of co-immunoprecipitation with PI3KC3-specific antibody using antibody to Beclin-1 and antibody to PI3KC3 as loading control. D, shown are immunoblots of LC3B isoforms using antibody to LC3B or β-actin using macrophages pretreated with 100 nm wortmannin, 10 μg/ml pepstatin A, or vehicle control before 1,25D3 or rapamycin treatment. E, shown is a flow cytometry analysis of saponin-resistant LC3B-II in macrophages after 1,25D3 and/or pepstatin A treatment for 4 h. Representative dot blots with the percentage of cells displaying saponin-resistant LC3B-II from three donors are shown. Ab, antibody. F, representative fluorescence microscopy images of macrophages from three donors after 1,25D3 or rapamycin treatment for 4 h are shown. Cells were fixed and permeabilized, then stained with antibody to LC3B (red) and Hoechst 33342 (blue). LC3B puncta are indicated by the white arrows. When puncta were not present, LC3B staining gives rise to a diffuse cytoplasmic pattern. The number of puncta per cell is indicated as the mean ± S.D. Scale bars indicate 10 μm. G, percentage of cells with LC3B puncta after designated treatments for 4 h is shown. H, quantification of cells with LC3B puncta after designated treatments at the time points indicated. Results are the means ± S.E. of three independent experiments performed in triplicate. †, p ≥ 0.05; *, p < 0.001 compared with untreated control.
The second established molecular marker for the induction of autophagy that we measured was the degree of LC3B lipidation (26). During autophagy, cytosolic LC3B-I is converted to LC3B-II by a ubiquitin-like system that involves autophagy-related protein-7 (ATG7), ATG3, and the ATG5-ATG12 complex. The ATG5-ATG12 complex ligates LC3B-II to the nascent autophagosome membrane through phosphatidylethanolamine with the LC3B-II associated with the inner membrane degraded after fusion of the autophagosome with lysosomes. Therefore, the conversion of LC3B-I to LC3B-II and its turnover is an indicator of autophagy induction and flux (26). We detected an increase in LC3B-II after 1,25D3 stimulation for 4 h similar to that observed with rapamycin, an inducer of autophagy through inhibition of the mammalian target of rapamycin (mTOR) complex 1 (mTORC1). Inhibition of PI3KC3, which is involved at the vesicle elongation step of autophagy, by pretreatment with wortmannin (34) inhibited the lipidation of LC3B-I by 1,25D3 (Fig. 1D). The accumulation of LC3B-II was increased in the presence of the lysosomal protease inhibitor pepstatin A (Fig. 1D), indicative of autophagic flux (35).
When autophagosomes are formed, LC3B redistributes from a soluble diffuse cytosolic pattern to an insoluble autophagosome-associated vacuolar pattern (30, 36). We, therefore, quantified the autophagosome-associated LC3B-II in human macrophages using saponin resistance with flow cytometry (30). Staining for endogenous LC3B in saponin-washed macrophages, we observed that the percentage of cells containing a saponin-resistant fraction significantly increased with dose of 1,25D3 (p < 0.05; Fig. 1E). Co-treatment with pepstatin A significantly increased the percentage of cells containing a saponin-resistant fraction upon treatment with 100 pm 1,25D3 indicative of autophagic flux (p = 0.047; Fig. 1E). We then visually tracked the autophagic response to 1,25D3 using fluorescence microscopy (Fig. 1F) and observed that the number of cells displaying LC3B puncta after 1,25D3 treatment was concentration-dependent with 1000 pm 1,25D3 sufficient to induce the maximal response (Fig. 1G). The lowest concentration of 1,25D3 that triggered a significant percentage of cells to contain numerous LC3B puncta was 50 pm (p < 0.001; Fig. 1G). Importantly, the observed autophagosome accumulation was not accompanied by pyknosis, karyorrhexis, or plasma membrane blebbing, all markers for apoptosis (Fig. 1F). Incubating macrophages with 1,25D3 for longer periods of time increased the number of cells displaying numerous LC3B puncta, although this increase was only significant for 50 and 100 pm 1,25D3 and the rapamycin treatments by 48 h (p < 0.01; Fig. 1H).
Although we observed an increase in autophagic markers in the absence of visible pyknosis, karyorrhexis, or plasma membrane blebbing, it was important to confirm that the cells were not undergoing cell death at the physiological concentrations being used, as the induction of excessive autophagy can cause cell death in mammalian cells in experimental systems in vitro (37, 38). Therefore, we initially assayed for plasma membrane breakdown (as a sign of cytotoxicity) using the lactate dehydrogenase assay combined with the WST-1 assay that measures the activity of the mitochondrial respiratory chain (as an indicator of viable cells). We observed that after 96 h at physiological concentrations, 1,25D3 exhibited no cytotoxic effects and that 1,25D3 only became cytotoxic at 500 pm or above (p < 0.05; Fig. 2A). Moreover, we also assessed the dose-dependent effect of 1,25D3 on ssDNA accumulation at the same time point based on the selective thermal denaturation of apoptotic ssDNA using low heat and formamide and subsequent detection using a monoclonal antibody, a more specific indicator of apoptosis than terminal deoxynucleotidyltransferase dUTP nick end labeling (TUNEL) (39). We observed the dose-dependent increase in the number of cells exhibiting the accumulation of ssDNA that became significant at 500 pm 1,25D3 (p = 0.0005; Fig. 2B). As we were using rapamycin as a control for the induction of autophagy, we also assessed its cytotoxic effects and observed that at 100–200 nm, rapamycin was neither significantly cytotoxic as measured by lactate dehydrogenase release (p > 0.05) or WST-1 conversion (p > 0.2; Fig. 2C) or induced significant numbers of cells to accumulate ssDNA (p > 0.2; Fig. 2D). We then assessed the effect of 1,25D3 and rapamycin on EdU incorporation and observed no significant effect at the physiologically relevant concentrations tested (p > 0.2; Fig. 2E).
FIGURE 2.

Physiological concentrations of 1,25D3 are not cytotoxic. Macrophages were incubated with increasing concentrations of 1,25D3 for 96 h. A, aliquots of supernatant taken before the addition of WST-1 were tested for lactate dehydrogenase (LDH) spectrophotometrically using the LDHPLUS assay (black squares). For the last hour cells were incubated with WST-1, and the reduction of the WST-1 reagent to its formazan product was monitored spectrophotometrically (black circles). B, quantification of the number of cells with apoptotic ssDNA using the ssDNA ELISA 96 h after 1,25D3 treatment is shown. C, lactate dehydrogenase (black squares) and WST-1 (black circles) assay after 96 h of incubation of macrophages with increasing concentrations of rapamycin is shown. D, quantification of the number of cells with apoptotic ssDNA using the ssDNA ELISA 96 h after rapamycin treatment is shown. E, effect of 1,25D3 and rapamycin on the uptake of EdU by macrophages is shown. Cells were incubated with 1,25D3 or rapamycin for 78 h, after which cells were incubated a further 18 h in the presence of 10 μm EdU. Cells were harvested, fixed, permeabilized, then probed for EdU incorporation and analyzed for the percentage of EdU-positive cells by flow cytometry. Physiological concentrations of 1,25D3 and rapamycin had no cytotoxic effects. All results are the means ± S.E. of three donors performed in triplicate. †, p ≥ 0.05; *, p < 0.001 compared with untreated control cells.
We next investigated to what extent conventional autophagy mediators are required for 1,25D3-induced autophagosome formation. Pretreatment with wortmannin, which suppresses autophagy regardless of nutrient status (40), inhibited autophagosome formation mediated by 1,25D3 with no cytotoxic effects (Fig. 3A). Although 3-methyladenine is widely used as an autophagy inhibitor (41), we specifically did not use it in these experiments because recent studies have shown that it actually promotes autophagy under nutrient-rich conditions (40). RNA interference of ATG5 and Beclin-1 (Fig. 3B) resulted in the reduction in autophagosome formation after 1,25D3 treatment (Fig. 3C and 3D). Therefore, several known components of autophagosome formation, PI3K, ATG5, and Beclin-1, are essential for 1,25D3-mediated autophagy.
FIGURE 3.

1,25D3-induced autophagy requires PI3K, ATG5, and Beclin-1. A, macrophages were incubated with wortmannin or vehicle control then treated with 100 pm 1,25D3 for 4 h. Left, quantification of the percentage of LC3B-positive autophagosome-forming cells is shown. Middle, cell supernatants were tested spectrophotometrically for lactate dehydrogenase (LDH) release as a measure of plasma membrane rupture. Right, for the last hour cells were incubated with WST-1, and the reduction of the WST-1 reagent to its formazan product was monitored spectrophotometrically. B, MonoMac1 cells were transfected with scrambled shRNA (S), Beclin-1-specific (B2–B5), or ATG5-specific (A1–A4) shRNA. Top, shown is an immunoblot performed with antibodies to Beclin-1, ATG5, and β-actin. Bottom, quantification of silencing effect on protein content is shown. C, shown are representative microscopy images of LC3B staining in cells transfected with scrambled (S), ATG5 (A2), or Beclin-1 (B3) shRNA treated with 1,25D3 or rapamycin for 4 h. Cells were stained with antibody to LC3B (red) and Hoechst 33342 (blue). Scale bars indicate 10 μm. D, shown is quantification of the percentage of LC3B-positive autophagosome-forming cells transfected with scrambled (S), ATG5 (A2), or Beclin-1 (B3) shRNA and treated with 1,25D3 or rapamycin. Results are the means ± S.E. of three independent experiments performed in triplicate. †, p ≥ 0.05; *, p < 0.001 compared with untreated control cells.
1,25D3 Inhibits HIV-1 Replication in Human Macrophages
We then determined whether 1,25D3 and rapamycin influence HIV-1 infection and replication in macrophages by comparing the extent to which 1,25D3 and rapamycin pre-treatment influenced p24 antigen accumulation in the supernatants of macrophages that were subsequently infected with HIV-1. Rapamycin, at achievable in vivo plasma concentrations, inhibited HIV-1 replication by >95% over 10 days (p < 0.001; Fig. 4A). 1,25D3 induced a dose-dependent inhibition of HIV-1 replication with 50 pm being the minimum concentration required to significantly inhibit HIV-1 at 10 days post-infection (79% reduction; p = 0.01; Fig. 4B). This effect was not enhanced by further increasing the 1,25D3 concentration (Fig. 4B). Importantly, the 1,25D3 and rapamycin treatments had no significant effects on either cell viability (WST-1 assay) or ssDNA accumulation, a specific marker for apoptotic cells (39) at day 10 (p > 0.05; Fig. 4C).
FIGURE 4.
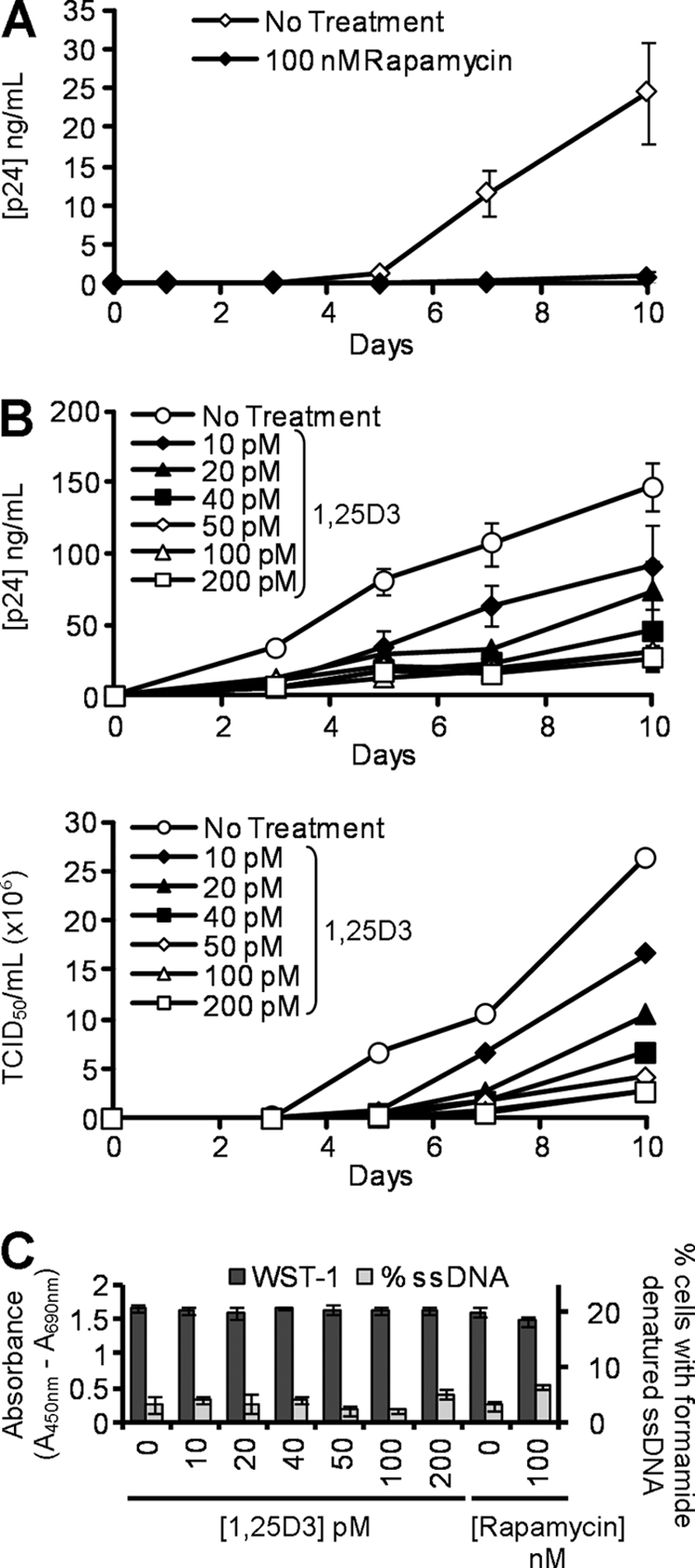
1,25D3 and rapamycin inhibit HIV-1 replication. A, macrophages were incubated with rapamycin before exposure to HIV-1Ba-L. ELISAs were performed for HIV-1 p24 antigen release over 10 days. B, macrophages were incubated with increasing concentrations of 1,25D3 before exposure to HIV-1Ba-L. Top, ELISAs were performed for HIV-1 p24 antigen over 10 days. Bottom, TCID50 is calculated from the supernatants of infected cells. 50 pm 1,25D3 was the minimum concentration required to significantly inhibit HIV-1 replication (p = 0.01). C, after 10 days of HIV-1 infection, cells were incubated with WST-1 for 1 h, and the reduction of the WST-1 reagent to its formazan product was monitored spectrophotometrically (dark gray histograms). Cells were then fixed and permeabilized, and the percentage of cells with apoptotic ssDNA was quantified by ELISA. At all concentrations tested, neither 1,25D3 nor rapamycin induced apoptotic ssDNA formation or reduced cell viability by day 10 of HIV-1 infection. Results are the means ± S.E. of three independent experiments performed in triplicate.
To understand how 1,25D3 affects HIV-1 replication, we examined sequential steps of HIV-1 replication. Treatment of cells with 1,25D3 did not significantly affect the expression of either CD4 or CCR5, which are required for HIV-1 binding and entry into cells (Fig. 5A). Consistent with this finding, binding of HIV-1 to 1,25D3-treated cells, as measured by ELISA of cell-associated p24 Gag protein, was similar to that of untreated cells (Fig. 5B). To assay for virus entry, we examined the quantity of intracellular p24 Gag (trypsin-resistant) associated with cells exposed to virus for 5 h. Both untreated cells and 1,25D3-treated cells displayed the same intracellular p24 concentration over 5 h (p > 0.35; Fig. 5C). Treatment of cells with the CCR5 antagonist maraviroc provided a measurement of nonspecific background in these assays.
FIGURE 5.

1,25D3 does not affect HIV-1 entry or release. A, macrophages were left untreated (solid gray histogram) or treated with 100 pm 1,25D3 (dashed line) or 100 nm rapamycin (solid line). After 4 h, cells were harvested and stained for surface CD4 and CCR5. B, macrophages treated with 1,25D3, rapamycin, or maraviroc (M) for 4 h were infected with replication competent HIV-1Ba-L. Binding was measured at 3 h postinfection by washing cells extensively, then lysing and analyzing p24 by ELISA. C, entry was measured at 5 h postinfection by washing cells extensively, then trypsinization, lysing, and subsequently analyzing intracellular p24 by ELISA. D, DNA was extracted from cells at 8 h postinfection for PCR analysis of pre-integration strong-stop HIV-1 DNA. RNA polymerase II was amplified as a control. Results are expressed as the ratio between the target gene and the RNA polymerase II and normalized so that HIV-1 LTR in untreated cells equals 1.00. E, macrophages lysed at 5 h postinfection were subjected to immunoblotting for both Nef and GAPDH. F, percentage of TZM-bl cells were productively infected with HIV-1Ba-L after 4 h treatment with 1,25D3 or rapamycin. G, macrophages were incubated with 1 ng p24 antigen from the 10-day aliquots of cell-free supernatants post-1,25D3 or rapamycin treatment for 3 h then cultured for 10 days with ELISA performed for HIV-1 p24 antigen. H, macrophages were incubated with 100 pm 1,25D3 at different time points with respect to infection with HIV-1Ba-L. ELISAs were performed for HIV-1 p24 antigen over 10 days. I, 20-day-old HIV-1-infected macrophages were incubated with rapamycin or 1,25D3 with ELISA performed for HIV-1 p24 antigen over 3 days. J, shown is the percentage of cells with apoptotic ssDNA combined with p24 release after 4 h of treatment with rapamycin or 1,25D3. All data are the means ± S.E. of three independent experiments performed in triplicate. *, p < 0.001 compared with untreated control cells.
The previous results suggested that 1,25D3 treatment had no effect on HIV-1 binding or entry. Therefore, we measured viral entry by real-time PCR. Macrophages were treated with 1,25D3, rapamycin, or vehicle control for 4 h before infection with HIV-1Ba-L for 3 h. After an additional 5-h culture in fresh media, samples were analyzed by real-time PCR for the presence and quantity of strong-stop HIV-1 DNA (with LTR R/U5 primers), an early product of reverse transcription. Physiological concentrations of 1,25D3 had no effect on HIV-1Ba-L infection (p = 0.8), whereas 100 nm rapamycin treatment significantly inhibited HIV-1Ba-L infection by 67% (p < 0.0001; Fig. 5D). As Nef is the first viral product synthesized during the HIV life cycle, we also analyzed the translation of Nef at 5 h by immunoblotting and observed no difference between cells treated with the vehicle control or with 1,25D3 (Fig. 5E). We then assessed for productive HIV-1 infection by assaying for Tat activity using TZM-bl cells. We pretreated TZM-bl cells with increasing concentrations of 1,25D3 or rapamycin for 4 h then exposed them to HIV-1. We found that at the concentrations tested 1,25D3 had no significant inhibitory effect on HIV-1 productive infection as measured by Tat activity (p > 0.47; Fig. 5F). Conversely, rapamycin displayed a dose-dependent inhibitory effect that only became significant at 400 nm (p = 0.025; Fig. 5F).
We then assessed whether the inhibition of HIV-1 infection was due to 1,25D3-inducing the production of replication-incompetent viral particles; uninfected macrophages were incubated with 1 ng of p24 antigen from the day-10 aliquots of the cell-free supernatants post-1,25D3 pretreatment for 3 h then cultured for 10 days. No difference in HIV-1-replicative fitness was observed post-1,25D3 treatment (Fig. 5G). Next, we examined the effect of 100 pm 1,25D3 on HIV-1 replication at early time points post-infection by treating macrophage cultures that had been exposed to HIV-1BaL with 100 pm 1,25D3 at −4 h, at the time of infection and at 3, 5, and 7 days post-infection. At each time point, 7 days post-infection excepted, 1,25D3 significantly suppressed HIV-1 replication (p < 0.034; Fig. 5H) in the absence of toxic effects.
We also tested the effects of rapamycin and 1,25D3 on HIV-1-infected macrophages by comparing the extent to which 1,25D3 and rapamycin treatment influenced the release of p24 antigen from 20 day-old HIV-1-infected macrophage cultures. At achievable in vivo plasma concentrations, neither rapamycin nor 1,25D3 induced the significant release of p24 antigen into the supernatant over 4 h (Figs. 5I and 5J). However, both rapamycin and high concentrations of 1,25D3 significantly inhibited HIV-1 replication over 72 h (Fig. 5I). Conversely, although 50 pm 1,25D3 inhibited the release of p24 over 72 h compared with the vehicle control, the inhibition was not significant, and 10 pm 1,25D3 had no effect on HIV-1 p24 release (Fig. 5I). Interestingly, both 50 and 25 μm rapamycin induced the release of significant levels of p24 antigen after just 4 h that was accompanied by significant levels of apoptosis (Fig. 5J).
1,25D3-mediated Autophagy Inhibits HIV-1 Replication in Human Macrophages
Previous studies have shown that, in primary macrophages, HIV-1 assembles in late endosomes of the recycling pathway (42) and co-localizes with LC3B. Therefore, to assess the intersection of autophagy with HIV-1 replication and the role of autophagy in 1,25D3-mediated inhibition of HIV-1, we examined the relative distribution of HIV-1 and LC3B. In the absence of 1,25D3 stimulation, the HIV-1 Gag p17-specific antibody showed some colocalization with the autophagosome marker LC3B and almost no colocalization with acidified lysosomes (Fig. 6A). However, upon 1,25D3 treatment, the number of p17 puncta that colocalized with LC3B or LysoTracker increased significantly (Fig. 6B).
FIGURE 6.

LC3B and acidic vacuoles colocalize with HIV-1 Gag. Macrophages were treated with 100 pm 1,25D3 or vehicle control (EtOH) then infected with HIV-1Ba-L for 10 d. A, cells were fixed, permeabilized, and probed for LC3B, Gag-p17, and Hoechst 33342 (blue). Green dots indicate Gag-p17, and red dots indicate LC3B positive structures. B, after 10 days, cells were incubated with Lysotracker Red for 4 h, then fixed, permeabilized, and probed for Gag-p17. Green dots indicate Gag-p17, and red dots indicate acidic vacuoles. Scale bars indicate 10 μm.
As autophagy proteins are required for efficient HIV-1 replication in HeLa cells (16), to determine whether 1,25D3 affects HIV-1 replication in macrophages through autophagy, we examined whether the inhibition of sequential steps of the autophagy pathway inhibits HIV-1 replication. Therefore, we initially assessed the effect of the inhibition of basal autophagy on HIV-1 replication in macrophages by comparing the extent to which wortmannin and Beclin-1 and ATG5 silencing influenced p24 accumulation in the supernatants of HIV-1-infected cells. Importantly, wortmannin had no effect on HIV-1 infection over 7 days (p = 0.68; Fig. 7A) but significantly inhibited HIV-1 by day 10 (p < 0.001; data not shown). We then infected the shRNA-transduced cells silenced for Beclin-1 and ATG5 and observed a significant reduction in p24 antigen concentration over 10 days (Fig. 7B).
FIGURE 7.

1,25D3-mediated induction of autophagy inhibits HIV-1 replication. A, macrophages were incubated with wortmannin or vehicle control before treatment with 50–100 pm 1,25D3 and infection with HIV-1Ba-L. ELISA performed for HIV-1 p24 antigen release over 7 days. B, shown is quantification of p24 antigen secreted by HIV-1-infected ATG5 shRNA (A2) and Beclin-1 shRNA (B3) transduced cells compared with nonspecific scrambled shRNA-transduced cells. C, ATG5 shRNA (A2), Beclin-1 shRNA (B3), and nonspecific scrambled shRNA (S)-transduced cells were incubated with 0–100 pm 1,25D3 or 0–200 nm rapamycin then infected with HIV-1Ba-L. ELISAs were performed for HIV-1 p24 antigen over 10 days. D, macrophages were incubated with bafilomycin A1, SID 26681509, or vehicle control before treatment with 50–100 pm 1,25D3 and infection with HIV-1Ba-L. ELISAs were performed for HIV-1 p24 antigen release over 7 days. Results are the means ± S.E. of three independent experiments performed in triplicate.
To assess whether 1,25D3-induced autophagy contributes to the observed inhibition of HIV-1 replication by 1,25D3, we initially compared the extent to which wortmannin influences 1,25D3-mediated inhibition of HIV-1 replication. The p24 antigen levels in the supernatants of wortmannin and 1,25D3 co-treated cells were similar to those in the wortmannin only-treated cells (p > 0.13; Fig. 7A), suggesting that the inhibition of PI3K reversed the 1,25D3-mediated inhibition of HIV-1 replication. We then assessed the effect of silencing Beclin-1 and ATG5. In the scrambled control RNAi treated cells, 25, 50, and 100 pm 1,25D3 inhibited HIV-1 p24 levels by 80, 89, and 91%, respectively, by day 10 post-infection (p < 0.0004; Fig. 7C). Beclin-1 silencing reduced the inhibitory effect of 25 and 50 pm 1,25D3 to 20 and 37%, respectively, which were not significantly different to the untreated cells (p > 0.07; Fig. 7C). However, 100 pm 1,25D3 maintained a significant inhibitory effect on HIV-1, although this was reduced to just 54% inhibition by day 10 (p = 0.03). ATG5 silencing had a more pronounced effect, with 25 pm 1,25D3 actually increasing HIV-1 p24 levels 20% by day 10, although this was not significant (p = 0.08; Fig. 7C). Conversely, the inhibitory effect of 50 and 100 pm 1,25D3 was reduced to just 14% (p = 0.05) and 32% (p = 0.07), respectively, which were not significantly different to the untreated cells. We then assessed the effect of silencing Beclin-1 and ATG5 on the rapamycin-induced inhibition of HIV-1 replication and again observed a reduction in the observed inhibition, although at no point did this reduction become significant (p > 0.05; Fig. 7C), suggesting that the rapamycin-mediated inhibition of HIV-1, although partially through autophagy, is also through another mechanism.
We next investigated whether autophagosome acidification is required for the autophagic inhibition of HIV-1. Macrophages were treated with bafilomycin A1, an inhibitor of the vacuolar H+ ATPase and, thus, autophagosome-lysosome fusion, and subsequently infected with HIV-1. In the absence of 1,25D3, bafilomycin A1 increased HIV-1 production over 7 days, although this was not significant. In the presence of 1,25D3, bafilomycin A1 reversed the 1,25D3-mediated inhibition (Fig. 7D), suggesting that the acidic pH of autophagolysosomes is required for the autophagy-mediated control of HIV-1.
We then investigated whether lysosomal hydrolases are important for 1,25D3-mediated inhibition of HIV-1 through autophagy. Using SID 26681509, a novel thiocarbazate specific inhibitor of the lysosome hydrolase cathepsin L, we observed that, in the absence of 1,25D3, there was no net inhibition of HIV-1 (Fig. 7D). Moreover, in the presence of 1,25D3, SID 26681509 abrogated the HIV-1 inhibition (Fig. 7D). Together, these data indicate that the 1,25D3-mediated induction of autophagy in macrophages is responsible for the inhibition of HIV-1 replication.
Human Cathelicidin (LL-37) Does Not Trigger Autophagy at Low Concentrations
A recent study using RNA interference for the human cationic antimicrobial protein 18 (hCAP18), from which the active antimicrobial peptide cathelicidin (LL-37) is cleaved extracellularly from the C terminus, suggested that 1,25D3 induced autophagy in human monocytes is via a cathelicidin-dependent mechanism (4). Moreover, LL-37 inhibits HIV-1 infection of peripheral blood mononuclear cells (43). Therefore, we investigated whether the LL-37 secreted from 1,25D3-treated macrophages (Fig. 8, A and B) influences autophagy and HIV-1 replication in macrophages. To determine whether the LL-37 elicited by 1,25D3 is sufficient to induce autophagy, macrophages were treated with the greatest concentration of LL-37 measured. No significant increase in the redistribution of LC3B from diffuse to punctate staining was observed (p = 0.31; Fig. 8C).
FIGURE 8.

1,25D3-induced cathelicidin does not induce autophagy or inhibit HIV-1. A, macrophages were treated with 1,25D3 and secreted LL-37 measured by ELISA. B, shown is a flow cytometry analysis of LL-37 in macrophages left unstimulated (gray histograms) or stimulated with 2000 units/ml IFNα (blue histograms), 100 pm 1,25D3 (green histograms), or 200 nm rapamycin (red histograms). Left, representative flow cytometry histograms are shown. Right, mean fluorescence change is shown. C, macrophages were left untreated or treated with LL-37, fixed, permeabilized, then probed for LC3B and analyzed by fluorescence microscopy. Scale bars indicate 10 μm. Left, representative microscopy images are shown. Right, quantification of cells with LC3B puncta is shown. D, the percentage of TZM-bl cells productively infected with HIV-1Ba-L after pretreatment with increasing concentrations of LL-37 or scrambled control peptide is shown. E, macrophages were incubated with varying concentrations of LL-37 or a scrambled control peptide (CP) for 4 h before exposure to HIV-1Ba-L. ELISAs were performed for HIV-1 p24 antigen release over time. Results are the means ± S.E. of three independent experiments performed in triplicate.
To assess whether LL-37 inhibits productive HIV-1 infection, we pretreated TZM-bl cells with LL-37 for 4 h then exposed them to HIV-1. LL-37 inhibited HIV-1 productive infection in a dose-dependent manner (Fig. 8D), with no inhibitory effect observed at the concentrations elicited by 1,25D3-treated macrophages (p > 0.7; Fig. 8D). We then examined the effect of LL-37 on HIV-1 replication in macrophages and observed that the concentrations elicited by macrophages in response to 1,25D3 in vitro induced the dose-dependent augmentation of HIV-1 replication (Fig. 8E). Conversely, at levels of LL-37 higher than those produced by macrophages in vitro in response to physiological concentrations of 1,25D3, we observed the inhibition of HIV-1 (p < 0.05; Fig. 8E).
DISCUSSION
Our results demonstrate that physiological concentrations of 1,25D3 induce autophagy in human macrophages and that autophagy is required for the 1,25D3-mediated inhibition of HIV-1 replication. To our knowledge these results are the first to link the induction of autophagy with the inhibition of HIV-1 replication in macrophages.
Studies of 1,25D3 as an autophagy inducer have focused on immortalized cell lines (32, 33, 44) and its potential role as an anti-oncogenic drug as it induces cell death via a caspase-independent mechanism (33) at supraphysiological concentrations (32, 45). These concentrations are associated in patients with hypercalcemia and hypercalciuria that result from the increased intestinal absorption of calcium combined with the calcium-immobilizing effects of vitamin D3. Therefore, 1,25D3 or vitamin D3 analogs are being pursued as anti-cancer therapies only in combination with glucocorticoids or anti-mitotic agents (46). Importantly, we observed no decrease in cell viability or increase in apoptotic cells resulting from physiologically relevant concentrations of 1,25D3 that significantly inhibited HIV-1 through autophagy.
The envelope glycoproteins, gp120 and gp41, activate protein kinase C and Akt, upstream effectors of the mTORC1, that trigger autophagy in uninfected cells (17, 19, 47). Moreover, the G-U-rich long terminal repeat region of the HIV-1 genome is a ligand for Toll-like receptor 8 found in monocytes (48) that triggers downstream signaling via Toll-interleukin-1 receptor-domains activating the interferon regulatory factor, nuclear factor κ-light-chain enhancer of activated B cells, and mitogen-activated protein kinase pathways leading to the induction of autophagy (49). As an obligatory intracellular parasite, HIV-1 survival is dependent upon its ability to exploit the host cell machinery for replication and dissemination and to circumvent cellular processes that prevent its growth. One such mechanism is that, upon infection, HIV-1 down-regulates IL-1 receptor-associated kinase 4, which is essential for virtually all Toll-like receptor signaling (50). Recent studies have shown that HIV-1 down-regulates autophagy in productively infected CD4+ T cells, U937 cells (15) and myeloid dendritic cells through an Env-dependent manner (14) and that HIV-1 is undetectable in highly autophagic macrophages (13). Moreover, in myeloid cells, HIV-1 Gag co-localizes with LC3B puncta (Fig. 6) and CD9 (51). These data combined with our observations that pharmacological inhibitors of autophagy and RNA interference of autophagy proteins inhibits HIV-1 replication in macrophages are consistent with the hypothesis that HIV-1 is assembled on endocytic membranes that intersect with the autophagy pathway (42, 52). However, our data obtained using wortmannin appears to be in conflict with a previous study that showed that PI3K inhibition blocked HIV infection of macrophages within 24 h (53). That study used LY 294002, which has recently been shown to inhibit a large number of other kinases as well as ATP-binding proteins (34). Therefore, the rapidity of HIV-1 inhibition observed in that study may be due to the nonspecific inhibition of other kinases involved during HIV-1 replication. By triggering autophagy, physiologically relevant concentrations of 1,25D3 overcomes the HIV-1-mediated down-regulation of autophagy and inhibits viral replication through an autophagy-dependent mechanism without inducing apoptosis or necrosis even though the autophagic response is similar to that observed in cancer cells (33). We also show that blocking 1,25D3-triggered autophagy with pharmacological inhibitors or RNA interference decreases the inhibition of HIV-1 replication. Therefore, this study demonstrates, to our knowledge for the first time, that 1,25D3 inhibits HIV-1 replication through the induction of autophagy.
In this study we also show that blocking rapamycin-triggered autophagy with pharmacological inhibitors or RNA interference also decreases the inhibition of HIV-1 replication. Therefore, although it is known that rapamycin inhibits HIV-1 replication through the down-regulation of CCR5 and the inhibition of HIV-1 mRNA synthesis (54, 55), we demonstrate the novel finding that rapamycin also inhibits HIV-1 replication at least in part through the induction of autophagy. A recent publication observed that 50 μg/ml (54.7 μm) rapamycin treatment of HIV-1-infected cells induced higher yields of p24 antigen into the supernatant than from rapamycin-untreated cells and attributed this to the induction of autophagy (51). However, rapamycin is cytotoxic at concentrations above 2 μm and induces apoptosis (56) (Fig. 2), which may lead to the secretion of HIV-1 p24 antigen even in the absence of lactate dehydrogenase release, a marker for secondary necrosis. Moreover, in vivo concentrations of therapeutically administered rapamycin are 500–5000-fold lower, at which concentrations we and others observed the inhibition of HIV-1 replication (Fig. 3) and the induction of autophagy (15, 57) (Fig. 1). Furthermore, rapamycin is now being used in HIV-1-infected transplantation recipients for its potential antiretroviral activity as well as for its immunosuppressive effects (58).
We also investigated the effect of extracellular LL-37 on HIV-1 replication. We observed that although HIV-1 is inhibited at LL-37 concentrations found in the plasma of healthy individuals, at the concentrations found to be elicited by macrophages in response to physiological concentrations of 1,25D3 in vitro and in the absence of 1,25D3, LL-37 is unable to induce autophagy and actually enhances HIV-1 replication. Interestingly, similar concentrations of LL-37 are found in cervicovaginal secretions where higher base-line genital levels are associated with increased acquisition of HIV-1, independent of the behavioral and immune correlates of HIV-1 acquisition (59). It is possible that endogenous hCAP18 may be more relevant than the secreted LL-37, as Yuk et al. (4) clearly showed that hCAP18 is upstream of Beclin-1 and ATG5 induction post-supraphysiological levels of 1,25D3 treatment (20 nm). However, we observed no role for exogenous LL-37 in 1,25D3-mediated autophagy at physiological concentrations (12.5–100 pm).
Vitamin D3 deficiency is conservatively defined as <50 nm 25D3, which is the estimated mean concentration of 25D3 present in people worldwide (60). A number of studies have found that the concentrations of vitamin D3 metabolites in HIV-1-infected persons are lower than that of uninfected controls (5, 6, 8, 9) with individuals with the lowest concentrations having an increased risk for HIV-1 disease progression (11). Moreover, a history of AIDS-defining events is associated with lower 1,25D3 concentrations (7). The major source of vitamin D3 is through the endogenous photochemical conversion of 7-dehydrocholesterol in the skin to previtamin D3 by ultraviolet B light exposure, which then undergoes a 1,7-sigmatropic hydrogen transfer forming cholecalciferol. This is then transferred from the skin by the vitamin D-binding protein and is subsequently hydroxylated by 25-hydroxylase CYP2R1 in hepatocytes to form 25D3 in a poorly regulated manner. Lesser amounts of vitamin D3 metabolites are also consumed through fortified dairy products and oily fish. Vitamin D3 status, therefore, is largely dependent upon the availability of cholecalciferol. Why HIV-1-infected individuals tend to have lower levels of 1,25D3 and/or 25D3 is largely unknown but is thought to be related to inadequate renal 1α-hydroxylation mediated by pro-inflammatory cytokines and/or antiretroviral drugs (6, 7, 61). The effects of HIV-1 viral products on 1,25D3 and/or 25D3 synthesis have not yet been evaluated. Four genes contribute to the variability of serum 25D3 concentrations: 7-dehydrocholesterol reductase (involved in cholesterol synthesis and the availability of 7-dehydrocholesterol in the skin), 25-hydroxylase CYP2R1 and CYP24A1 (degrades and recycles vitamin D3), and GC, which encodes for the vitamin d-binding protein. Genetic variations at these loci were recently identified in individuals who have a substantially increased risk of vitamin D insufficiency (62). Studies to assess the genetic variations at these loci and their impact on HIV-1 progression and disease status are now under way.
In summary, this study demonstrates a role for autophagy during the early phases of HIV-1 infection and a potential link between vitamin D3 and rapamycin-induced autophagy and the inhibition of HIV-1 replication in macrophages. Whether vitamin D3 supplementation will be a useful adjunct to antiretroviral treatment of HIV-1-infected persons is unknown but is testable through clinical trials. Dissecting the molecular mechanisms by which HIV-1 utilizes autophagic machinery will greatly enhance our understanding of this critical nexus in the host-virus relationship that in turn may lead to the development of novel strategies to prevent and treat HIV-1 infection and related opportunistic infections.
Acknowledgments
We thank Carol Mundy for technical assistance and Dennis Young (Flow Cytometry Core Facility, University of California San Diego) for assistance with flow cytometry.
This work was supported, in whole or in part, by National Institutes of Health Grants AI084573 and AI068632 (NIAID; to the International Maternal Pediatric Adolescent AIDS Clinical Trials Group).
- HIV
- human immunodeficiency virus
- 1,25D3
- 1α,25-dihydroxycholecalciferol
- 25D3
- 25-hydroxycholecalciferol
- LC3B
- light chain 3B
- WST-1
- 4-[3-(4-iodophenyl)-2-(4-nitrophenyl)-2H-5-tetrazolio]-1,3-benzene disulfonate
- ssDNA
- single-stranded DNA
- TCID50
- 50% tissue culture infective dose
- EdU
- 5-ethynyl-2′-deoxyuridine
- hCAP18
- human cationic antimicrobial protein 18.
REFERENCES
- 1. Joint United Nations Programme on HIV/AIDS (2008) Report on the Global HIV/AIDS Epidemic 2008, Executive Summary, Joint United Nations Programme on HIV/AIDS, Geneva, Switzerland [Google Scholar]
- 2. Martineau A. R., Honecker F. U., Wilkinson R. J., Griffiths C. J. (2007) J. Steroid Biochem. Mol. Biol. 103, 793–798 [DOI] [PubMed] [Google Scholar]
- 3. Sly L. M., Lopez M., Nauseef W. M., Reiner N. E. (2001) J. Biol. Chem. 276, 35482–35493 [DOI] [PubMed] [Google Scholar]
- 4. Yuk J. M., Shin D. M., Lee H. M., Yang C. S., Jin H. S., Kim K. K., Lee Z. W., Lee S. H., Kim J. M., Jo E. K. (2009) Cell Host. Microbe 6, 231–243 [DOI] [PubMed] [Google Scholar]
- 5. Haug C., Müller F., Aukrust P., Frøland S. S. (1994) J. Infect. Dis. 169, 889–893 [DOI] [PubMed] [Google Scholar]
- 6. Haug C. J., Aukrust P., Haug E., Mørkrid L., Müller F., Frøland S. S. (1998) J. Clin. Endocrinol. Metab. 83, 3832–3838 [DOI] [PubMed] [Google Scholar]
- 7. Mueller N. J., Fux C. A., Ledergerber B., Elzi L., Schmid P., Dang T., Magenta L., Calmy A., Vergopoulos A., Bischoff-Ferrari H. A. (2010) AIDS 24, 1127–1134 [DOI] [PubMed] [Google Scholar]
- 8. Teichmann J., Stephan E., Discher T., Lange U., Federlin K., Stracke H., Friese G., Lohmeyer J., Bretzel R. G. (2000) Metabolism 49, 1134–1139 [DOI] [PubMed] [Google Scholar]
- 9. Teichmann J., Stephan E., Lange U., Discher T., Friese G., Lohmeyer J., Stracke H., Bretzel R. G. (2003) J. Infect. 46, 221–227 [DOI] [PubMed] [Google Scholar]
- 10. de Luis D. A., Bachiller P., Aller R., de Luis J., Izaola O., Terroba M. C., Cuellar L., González Sagrado M. (2002) Nutr. Hosp. 17, 285–289 [PubMed] [Google Scholar]
- 11. Mehta S., Giovannucci E., Mugusi F. M., Spiegelman D., Aboud S., Hertzmark E., Msamanga G. I., Hunter D., Fawzi W. W. (2010) PLoS ONE 5, e8770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Mehta S., Hunter D. J., Mugusi F. M., Spiegelman D., Manji K. P., Giovannucci E. L., Hertzmark E., Msamanga G. I., Fawzi W. W. (2009) J. Infect. Dis. 200, 1022–1030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Espert L., Varbanov M., Robert-Hebmann V., Sagnier S., Robbins I., Sanchez F., Lafont V., Biard-Piechaczyk M. (2009) PLoS ONE 4, e5787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Blanchet F. P., Moris A., Nikolic D. S., Lehmann M., Cardinaud S., Stalder R., Garcia E., Dinkins C., Leuba F., Wu L., Schwartz O., Deretic V., Piguet V. (2010) Immunity 32, 654–669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zhou D., Spector S. A. (2008) AIDS 22, 695–699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Brass A. L., Dykxhoorn D. M., Benita Y., Yan N., Engelman A., Xavier R. J., Lieberman J., Elledge S. J. (2008) Science 319, 921–926 [DOI] [PubMed] [Google Scholar]
- 17. Denizot M., Varbanov M., Espert L., Robert-Hebmann V., Sagnier S., Garcia E., Curriu M., Mamoun R., Blanco J., Biard-Piechaczyk M. (2008) Autophagy 4, 998–1008 [DOI] [PubMed] [Google Scholar]
- 18. Espert L., Denizot M., Grimaldi M., Robert-Hebmann V., Gay B., Varbanov M., Codogno P., Biard-Piechaczyk M. (2006) Med. Sci. (Paris) 22, 677–678 [DOI] [PubMed] [Google Scholar]
- 19. Espert L., Denizot M., Grimaldi M., Robert-Hebmann V., Gay B., Varbanov M., Codogno P., Biard-Piechaczyk M. (2006) J. Clin. Invest. 116, 2161–2172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Platt E. J., Wehrly K., Kuhmann S. E., Chesebro B., Kabat D. (1998) J. Virol. 72, 2855–2864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Campbell G. R., Watkins J. D., Loret E. P., Spector S. A. (2011) AIDS Res. Hum. Retroviruses, in press [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Gartner S., Markovits P., Markovitz D. M., Kaplan M. H., Gallo R. C., Popovic M. (1986) Science 233, 215–219 [DOI] [PubMed] [Google Scholar]
- 23. Popovic M., Gartner S., Read-Connole E., Beaver B., Reitz M. (October 27–29, 1988) in Retroviruses of Human AIDS and Related Animal Diseases, Colloque Des Cent Gardes (Girard M., Valette L. eds) pp. 21–27, Fondation Marcel Mérieux, Pasteur Vaccins, Marnes-La-Coquette, Paris [Google Scholar]
- 24. Campbell G. R., Loret E. P., Spector S. A. (2010) J. Biol. Chem. 285, 1681–1691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Japour A. J., Mayers D. L., Johnson V. A., Kuritzkes D. R., Beckett L. A., Arduino J. M., Lane J., Black R. J., Reichelderfer P. S., D'Aquila R. T., Crumpacker C. S. The RV-43 Study Group, and The AIDS Clinical Trial Group Virology Committee Resistance Working Group (1993) Antimicrob. Agents Chemother. 37, 1095–1101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Klionsky D. J., Abeliovich H., Agostinis P., Agrawal D. K., Aliev G., Askew D. S., Baba M., Baehrecke E. H., Bahr B. A., Ballabio A., Bamber B. A., Bassham D. C., Bergamini E., Bi X., Biard-Piechaczyk M., Blum J. S., Bredesen D. E., Brodsky J. L., Brumell J. H., Brunk U. T., Bursch W., Camougrand N., Cebollero E., Cecconi F., Chen Y., Chin L. S., Choi A., Chu C. T., Chung J., Clarke P. G., Clark R. S., Clarke S. G., Clavé C., Cleveland J. L., Codogno P., Colombo M. I., Coto-Montes A., Cregg J. M., Cuervo A. M., Debnath J., Demarchi F., Dennis P. B., Dennis P. A., Deretic V., Devenish R. J., Di Sano F., Dice J. F., Difiglia M., Dinesh-Kumar S., Distelhorst C. W., Djavaheri-Mergny M., Dorsey F. C., Dröge W., Dron M., Dunn W. A., Jr., Duszenko M., Eissa N. T., Elazar Z., Esclatine A., Eskelinen E. L., Fésüs L., Finley K. D., Fuentes J. M., Fueyo J., Fujisaki K., Galliot B., Gao F. B., Gewirtz D. A., Gibson S. B., Gohla A., Goldberg A. L., Gonzalez R., González-Estévez C., Gorski S., Gottlieb R. A., Häussinger D., He Y. W., Heidenreich K., Hill J. A., Høyer-Hansen M., Hu X., Huang W. P., Iwasaki A., Jäättelä M., Jackson W. T., Jiang X., Jin S., Johansen T., Jung J. U., Kadowaki M., Kang C., Kelekar A., Kessel D. H., Kiel J. A., Kim H. P., Kimchi A., Kinsella T. J., Kiselyov K., Kitamoto K., Knecht E., Komatsu M., Kominami E., Kondo S., Kovács A. L., Kroemer G., Kuan C. Y., Kumar R., Kundu M., Landry J., Laporte M., Le W., Lei H. Y., Lenardo M. J., Levine B., Lieberman A., Lim K. L., Lin F. C., Liou W., Liu L. F., Lopez-Berestein G., López-Otín C., Lu B., Macleod K. F., Malorni W., Martinet W., Matsuoka K., Mautner J., Meijer A. J., Meléndez A., Michels P., Miotto G., Mistiaen W. P., Mizushima N., Mograbi B., Monastyrska I., Moore M. N., Moreira P. I., Moriyasu Y., Motyl T., Münz C., Murphy L. O., Naqvi N. I., Neufeld T. P., Nishino I., Nixon R. A., Noda T., Nürnberg B., Ogawa M., Oleinick N. L., Olsen L. J., Ozpolat B., Paglin S., Palmer G. E., Papassideri I., Parkes M., Perlmutter D. H., Perry G., Piacentini M., Pinkas-Kramarski R., Prescott M., Proikas-Cezanne T., Raben N., Rami A., Reggiori F., Rohrer B., Rubinsztein D. C., Ryan K. M., Sadoshima J., Sakagami H., Sakai Y., Sandri M., Sasakawa C., Sass M., Schneider C., Seglen P. O., Seleverstov O., Settleman J., Shacka J. J., Shapiro I. M., Sibirny A., Silva-Zacarin E. C., Simon H. U., Simone C., Simonsen A., Smith M. A., Spanel-Borowski K., Srinivas V., Steeves M., Stenmark H., Stromhaug P. E., Subauste C. S., Sugimoto S., Sulzer D., Suzuki T., Swanson M. S., Tabas I., Takeshita F., Talbot N. J., Tallóczy Z., Tanaka K., Tanaka K., Tanida I., Taylor G. S., Taylor J. P., Terman A., Tettamanti G., Thompson C. B., Thumm M., Tolkovsky A. M., Tooze S. A., Truant R., Tumanovska L. V., Uchiyama Y., Ueno T., Uzcátegui N. L., van der Klei I., Vaquero E. C., Vellai T., Vogel M. W., Wang H. G., Webster P., Wiley J. W., Xi Z., Xiao G., Yahalom J., Yang J. M., Yap G., Yin X. M., Yoshimori T., Yu L., Yue Z., Yuzaki M., Zabirnyk O., Zheng X., Zhu X., Deter R. L. (2008) Autophagy 4, 151–175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Brady N. R., Hamacher-Brady A., Yuan H., Gottlieb R. A. (2007) FEBS J. 274, 3184–3197 [DOI] [PubMed] [Google Scholar]
- 28. Dixon W. J., Massey F. J., Jr. (1983) Introduction To Statistical Analysis, 3rd Ed., p. 92, McGraw-Hill, New York [Google Scholar]
- 29. Federighi E. T. (1959) J. Am. Stat. Assoc. 54, 683–688 [Google Scholar]
- 30. Eng K. E., Panas M. D., Karlsson Hedestam G. B., McInerney G. M. (2010) Autophagy 6, 634–641 [DOI] [PubMed] [Google Scholar]
- 31. Campbell G. R., Spector S. A. (2008) J. Biol. Chem. 283, 30745–30753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Høyer-Hansen M., Bastholm L., Mathiasen I. S., Elling F., Jäättelä M. (2005) Cell Death. Differ. 12, 1297–1309 [DOI] [PubMed] [Google Scholar]
- 33. Wang J., Lian H., Zhao Y., Kauss M. A., Spindel S. (2008) J. Biol. Chem. 283, 25596–25605 [DOI] [PubMed] [Google Scholar]
- 34. Bain J., Plater L., Elliott M., Shpiro N., Hastie C. J., McLauchlan H., Klevernic I., Arthur J. S., Alessi D. R., Cohen P. (2007) Biochem. J. 408, 297–315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Mizushima N., Yoshimori T. (2007) Autophagy 3, 542–545 [DOI] [PubMed] [Google Scholar]
- 36. Mizushima N., Yamamoto A., Hatano M., Kobayashi Y., Kabeya Y., Suzuki K., Tokuhisa T., Ohsumi Y., Yoshimori T. (2001) J. Cell Biol. 152, 657–668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Galluzzi L., Aaronson S. A., Abrams J., Alnemri E. S., Andrews D. W., Baehrecke E. H., Bazan N. G., Blagosklonny M. V., Blomgren K., Borner C., Bredesen D. E., Brenner C., Castedo M., Cidlowski J. A., Ciechanover A., Cohen G. M., De Laurenzi V., De Maria R., Deshmukh M., Dynlacht B. D., El-Deiry W. S., Flavell R. A., Fulda S., Garrido C., Golstein P., Gougeon M. L., Green D. R., Gronemeyer H., Hajnóczky G., Hardwick J. M., Hengartner M. O., Ichijo H., Jäättelä M., Kepp O., Kimchi A., Klionsky D. J., Knight R. A., Kornbluth S., Kumar S., Levine B., Lipton S. A., Lugli E., Madeo F., Malomi W., Marine J. C., Martin S. J., Medema J. P., Mehlen P., Melino G., Moll U. M., Morselli E., Nagata S., Nicholson D. W., Nicotera P., Nuñez G., Oren M., Penninger J., Pervaiz S., Peter M. E., Piacentini M., Prehn J. H., Puthalakath H., Rabinovich G. A., Rizzuto R., Rodrigues C. M., Rubinsztein D. C., Rudel T., Scorrano L., Simon H. U., Steller H., Tschopp J., Tsujimoto Y., Vandenabeele P., Vitale I., Vousden K. H., Youle R. J., Yuan J., Zhivotovsky B., Kroemer G. (2009) Cell Death Differ. 16, 1093–1107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kroemer G., Levine B. (2008) Nat. Rev. Mol. Cell Biol. 9, 1004–1010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Frankfurt O. S., Krishan A. (2001) J. Immunol. Methods 253, 133–144 [DOI] [PubMed] [Google Scholar]
- 40. Wu Y. T., Tan H. L., Shui G., Bauvy C., Huang Q., Wenk M. R., Ong C. N., Codogno P., Shen H. M. (2010) J. Biol. Chem. 285, 10850–10861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Blommaart E. F., Krause U., Schellens J. P., Vreeling-Sindelárová H., Meijer A. J. (1997) Eur. J. Biochem. 243, 240–246 [DOI] [PubMed] [Google Scholar]
- 42. Pelchen-Matthews A., Kramer B., Marsh M. (2003) J. Cell Biol. 162, 443–455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Bergman P., Walter-Jallow L., Broliden K., Agerberth B., Söderlund J. (2007) Curr. HIV Res. 5, 410–415 [DOI] [PubMed] [Google Scholar]
- 44. Demasters G., Di X., Newsham I., Shiu R., Gewirtz D. A. (2006) Mol. Cancer Ther. 5, 2786–2797 [DOI] [PubMed] [Google Scholar]
- 45. Pepper C., Thomas A., Hoy T., Milligan D., Bentley P., Fegan C. (2003) Blood 101, 2454–2460 [DOI] [PubMed] [Google Scholar]
- 46. Trump D. L., Potter D. M., Muindi J., Brufsky A., Johnson C. S. (2006) Cancer 106, 2136–2142 [DOI] [PubMed] [Google Scholar]
- 47. Missé D., Gajardo J., Oblet C., Religa A., Riquet N., Mathieu D., Yssel H., Veas F. (2005) AIDS 19, 897–905 [DOI] [PubMed] [Google Scholar]
- 48. Heil F., Hemmi H., Hochrein H., Ampenberger F., Kirschning C., Akira S., Lipford G., Wagner H., Bauer S. (2004) Science 303, 1526–1529 [DOI] [PubMed] [Google Scholar]
- 49. Delgado M. A., Elmaoued R. A., Davis A. S., Kyei G., Deretic V. (2008) EMBO J. 27, 1110–1121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Pathak S., De Souza G. A., Salte T., Wiker H. G., Asjö B. (2009) Scand. J. Immunol. 70, 264–276 [DOI] [PubMed] [Google Scholar]
- 51. Kyei G. B., Dinkins C., Davis A. S., Roberts E., Singh S. B., Dong C., Wu L., Kominami E., Ueno T., Yamamoto A., Federico M., Panganiban A., Vergne I., Deretic V. (2009) J. Cell Biol. 186, 255–268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Deneka M., Pelchen-Matthews A., Byland R., Ruiz-Mateos E., Marsh M. (2007) J. Cell Biol. 177, 329–341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. François F., Klotman M. E. (2003) J. Virol. 77, 2539–2549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Roy J., Paquette J. S., Fortin J. F., Tremblay M. J. (2002) Antimicrob. Agents. Chemother. 46, 3447–3455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Heredia A., Amoroso A., Davis C., Le N., Reardon E., Dominique J. K., Klingebiel E., Gallo R. C., Redfield R. R. (2003) Proc. Natl. Acad. Sci. U.S.A. 100, 10411–10416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Harris J., Hanrahan O., De Haro S. A. (2009) Curr. Protoc. Immunol. Chapter 14, Unit 14, 14 [DOI] [PubMed] [Google Scholar]
- 57. Mercalli A., Sordi V., Ponzoni M., Maffi P., De Taddeo F., Gatti G., Servida P., Bernardi M., Bellio L., Bertuzzi F., Secchi A., Bonifacio E., Piemonti L. (2006) Am. J. Transplant. 6, 1331–1341 [DOI] [PubMed] [Google Scholar]
- 58. Di Benedetto F., Di Sandro S., De Ruvo N., Montalti R., Ballarin R., Guerrini G. P., Spaggiari M., Guaraldi G., Gerunda G. (2010) Transplantation 89, 733–738 [DOI] [PubMed] [Google Scholar]
- 59. Levinson P., Kaul R., Kimani J., Ngugi E., Moses S., MacDonald K. S., Broliden K., Hirbod T. (2009) AIDS 23, 309–317 [DOI] [PubMed] [Google Scholar]
- 60. Hagenau T., Vest R., Gissel T. N., Poulsen C. S., Erlandsen M., Mosekilde L., Vestergaard P. (2009) Osteoporos. Int. 20, 133–140 [DOI] [PubMed] [Google Scholar]
- 61. Welz T., Childs K., Ibrahim F., Poulton M., Taylor C. B., Moniz C. F., Post F. A. (2010) AIDS 24, 1923–1928 [DOI] [PubMed] [Google Scholar]
- 62. Wang T. J., Zhang F., Richards J. B., Kestenbaum B., van Meurs J. B., Berry D., Kiel D. P., Streeten E. A., Ohlsson C., Koller D. L., Peltonen L., Cooper J. D., O'Reilly P. F., Houston D. K., Glazer N. L., Vandenput L., Peacock M., Shi J., Rivadeneira F., McCarthy M. I., Anneli P., de Boer I. H., Mangino M., Kato B., Smyth D. J., Booth S. L., Jacques P. F., Burke G. L., Goodarzi M., Cheung C. L., Wolf M., Rice K., Goltzman D., Hidiroglou N., Ladouceur M., Wareham N. J., Hocking L. J., Hart D., Arden N. K., Cooper C., Malik S., Fraser W. D., Hartikainen A. L., Zhai G., Macdonald H. M., Forouhi N. G., Loos R. J., Reid D. M., Hakim A., Dennison E., Liu Y., Power C., Stevens H. E., Jaana L., Vasan R. S., Soranzo N., Bojunga J., Psaty B. M., Lorentzon M., Foroud T., Harris T. B., Hofman A., Jansson J. O., Cauley J. A., Uitterlinden A. G., Gibson Q., Järvelin M. R., Karasik D., Siscovick D. S., Econs M. J., Kritchevsky S. B., Florez J. C., Todd J. A., Dupuis J., Hyppönen E., Spector T. D. (2010) Lancet 376, 180–188 [DOI] [PMC free article] [PubMed] [Google Scholar]