

Abstract
The risk of Alzheimer’s disease increases following cerebral hypoperfusion. We studied the long-term interaction between low blood flow to the brain and Alzheimer’s disease by inducing a transient global ischemic insult in aged 3xTg-AD mice and determining the effects on AD pathology 3-months post injury. We found that global ischemia does not increase the levels of amyloid-β in these mice. However, the injury did lead to enhanced phosphorylation of the amyloid precursor protein (APP) at the Thr668 site in both the 3xTg-AD mice and wild-type controls. Furthermore, we found an increase in insoluble total tau 3-months post-injury. Together these findings further elucidate the long-term impact of cerebral hypoperfusion on Alzheimer’s disease.
Introduction
Alzheimer’s disease (AD) is the leading cause of dementia among the elderly. Evidence from human studies suggest that the pathogenesis of AD is multi-factorial, and certain co-morbid insults, such as stroke, heart attack, diabetes and traumatic brain injury may be risk factors for AD in elderly populations [3, 7, 12, 13, 18, 21–23].
Cerebral hypoperfusion is a major co-morbidity for AD. In AD models, our lab and others have reported acute post-traumatic effects of cerebral hypoperfusion on APP and BACE1 expression as well as altered APP processing [6, 8–10, 17, 24, 26, 31]. Critically, the long-lasting influence of cerebral ischemia post-injury on AD-related pathways has not yet been fully investigated in AD transgenic mice especially in older mice and in AD relevant brain regions. To elucidate the long-term effects of global ischemia on AD, we analyzed the impact of a single cerebral ischemic insult on AD-related proteins in the brains of older 3xTg-AD mice 3-months post-hypoperfusion [15].
Methods
Animals
Mouse experiments were performed in accordance with animal protocols approved by the Institutional Animal Care and Use Committee and the University of California, Irvine. 15 month old male 3xTg-AD mice [15] or non-transgenic age, strain and sex-matched mice were exposed to a 12-minute bilateral common carotid artery occlusion as previously described [28]. The mice were sacrificed and the brains collected 24 and 48-hours post injury (n=4 sham and 4 ischemic, each). 12-month old mice were sacrificed 3-months post-ischemia following perfusion with PBS (3xTg-AD n=12 sham and n=14 ischemic, Control n=4 sham 4 ischemic).
Half brains were cut sagittally, without the hindbrain, and homogenized in T-per extraction buffer as previously described [10]. Protein concentrations were determined by the Bradford method. The second half of the brains were placed in 4% paraformaldehyde for 48-hours then cut into 50 μM sections using a vibratome.
Western Blotting
Western blots were performed as previously described [10]. Western blots were normalized to GAPDH and represented as a percentage of sham. All error bars represent the standard error of the mean. We directly analyzed 4 sham animals and 4–5 ischemic animals for western blots.
Aβ ELISA
Aβ1–40 and Aβ1–42 were measured as previously described using a sensitive sandwich ELISA system [10].
Immunohistochemical analysis
Light-level immunohistochemistry was performed with an avitin-biotin immunoperoxidase technique (ABC kit; Vector Laboratories Inc, Burlingame, CA) and visualized with diaminobenzidine as previously described [10]. Fluorojade staining was performed as previously described [14].
Confocal Microscopy
Fluorescent immunolabeling was performed using free-floating sections as previously described [10]. The prevent signal bleed-through, all fluorophores were excited and scanned separately using lambda strobing.
Antibodies
Primary antibodies were used at 1:1000 unless otherwise noted and are listed below. HIF1α(Abcam Cambridge, MA), HIF1β(Abcam), HIF2α(Abcam), Synaptophysin(Sigma-Aldrich, St. Louis MO), VEGF (Abcam), GAPDH(1:10,000, Santa Cruz Biotechnology, Inc. Santa Cruz, CA), HSP90(Abcam), HSP70(Calbiochem, San Diego, CA), HSP60(Abcam), Casp3(Promega Madison, WI), HT7(1:3000, Pierce Biotechnology, Rockford, IL), AT8 (Pierce Biotechnology), AT100(Pierce Biotechnology), AT180 (Pierce Biotechnology), AT270(Pierce Biotechnology), PHF1(Kind gift of P. Davies), Thy1 (Millipore, Billerica, MA), 6E10(Signet, Dedham, MA), CT20(1:5000, Calbiochem), ADAM10(Calbiochem), ADAM17(Calbiochem), BACE1(Calbiochem), PS1(Novus, Littleton, CO), PS2(Novus), Aph1A (Novus), pAPPT668(Cell Signaling Danvers, MA), A H3(Millipore, Billerica, MA), T H3(Millipore), A H4 L8(Cell Signaling), Fe65(Millipore), MAPK(Cell Signaling), pGSK3β(Cell Signaling), tGSK3αβ(Cell Signaling), pAKT(Cell Signaling), tAKT(Cell Signaling)
Statistical Analyses
Data were analyzed by Student’s t-tests. Outliers greater than 3x the standard deviation of the mean were removed from statistical analysis. Error bars were standard error of the mean. Results were considered significant only when p<0.05.
Results
Induction of ischemic changes in an Alzheimer’s disease model
To understand the long-term effects of a single ischemic episode on AD neuropathology, we induced a transient ischemic insult (12-minutes) by bilateral common carotid artery occlusion (BCAO) on 15-month old 3xTg-AD mice (Fig. 1A). At this age, the 3xTg-AD mice start developing Aβ plaques and tau neurofibrillary tangles. To confirm that a 12-minute BCAO was sufficient to induce an ischemic infarct, mice were sacrificed 24-hours post-injury and histology performed for degenerating cells using fluorojade. We observed extensive fluorojade-positive cells throughout the hippocampus and within the cortex of the ischemia-affected mice (Fig. 1B), whereas no fluorojade-positive cells were detected in the sham-treated mice (data not shown). These results confirm that a 12-minute BCAO induces an ischemic infarct.
Figure 1. Acute ischemic response has subsided 3-months post-injury.
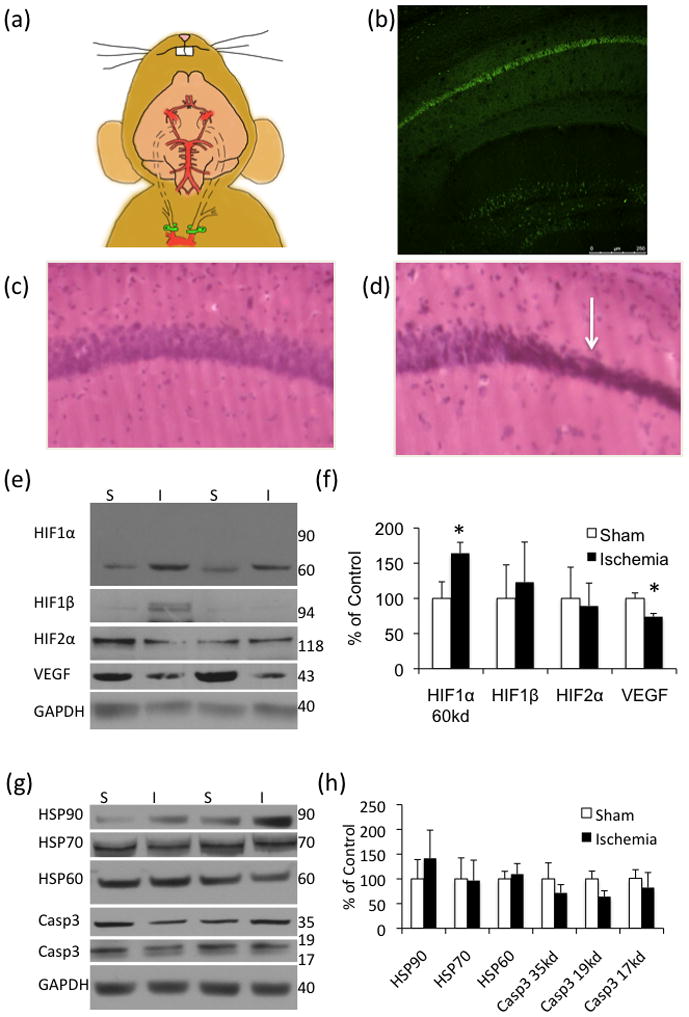
(a) Schematic of the bilateral occlusion of the common carotid artery utilized in the 3xTg-AD mice. Clamps are highlighted in green. (b) 24-hours post-ischemia there is abundant fluorojade staining in the hippocampus (n=4 sham, 4 ischemia). (c–d) H&E stain of sham and ischemic tissue, respectively. (e–f) Hypoxia inducible factors were mostly unchanged 3-months post-injury although vascular endothelial growth factor (VEGF) was significantly decreased in the ischemia-treated mice (n=12 sham, 14 ischemia). (g–h) Analysis by western blot of heat shock proteins (HSP) and caspase3 levels (casp) revealed no differences between ischemia-and sham-treated mice 3-months post injury (n=4 sham, 5 ischemia).
To assess the long-term effects of this infarct on AD neuropathology, a cohort of male 12 month old 3xTg-AD mice were survived for 3-months post-ischemia. We first looked at cell death and morphological changes at this time-point using an H&E stain. We found pyknotic cells within the CA1 of the mice that had been exposed to ischemia but not in the sham mice (Fig. 1C–D).
Hypoxia inducible factor-1α (HIF1α) is a transcription factor that rapidly accumulates in response to low oxygen levels, forms a heterodimer with HIF1β and initiates the transcription of a number of genes, including vascular endothelial growth factor (VEGF) [27]. We were unable to detect full length HIF1α at the predicted 90–120kDa weight (Fig. 1E–F). However, we found an altered lower molecular weight HIF1α band, which may represent the accumulation of a breakdown product of HIF1α (Fig. 1E–F). We did not find a difference in HIF1β or HIF2α. Additionally, we found a decrease in VEGF in the ischemia-exposed mice 3-months post injury (Fig. 1E–F). Other injury markers, including heat shock proteins (HSP’s) and caspase3, did not differ between the sham- and ischemia-treated mice at this time-point, thus no ongoing cell death processes were apparent at this time-point (Fig. 1G–H).
Insoluble tau is increased 3-months post-ischemia
To determine the long-term impact of a transient ischemic insult on AD-related proteins, we analyzed total tau levels and specific phosphotau epitopes in the 3xTg-AD mice (Fig. 2A–B). Notably, we found that there was a significant decrease in total tau, detected by the antibody HT7, consistent with our previous finding that mild, oligemic, hypoperfusion injury significantly decreased total tau up to 3-weeks post-injury [10]. In addition, we found that tau phosphorylated at Thr181 (AT270) was significantly decreased 3-months post injury (Fig. 2A–B). No differences were observed in phospho-tau at both Ser202 and Thr205 (AT8) or S396 andS404 (PHF1 antibody) (Fig. 2A–B).
Figure 2. Ischemia produces long-term changes in tau solubility in the brain.
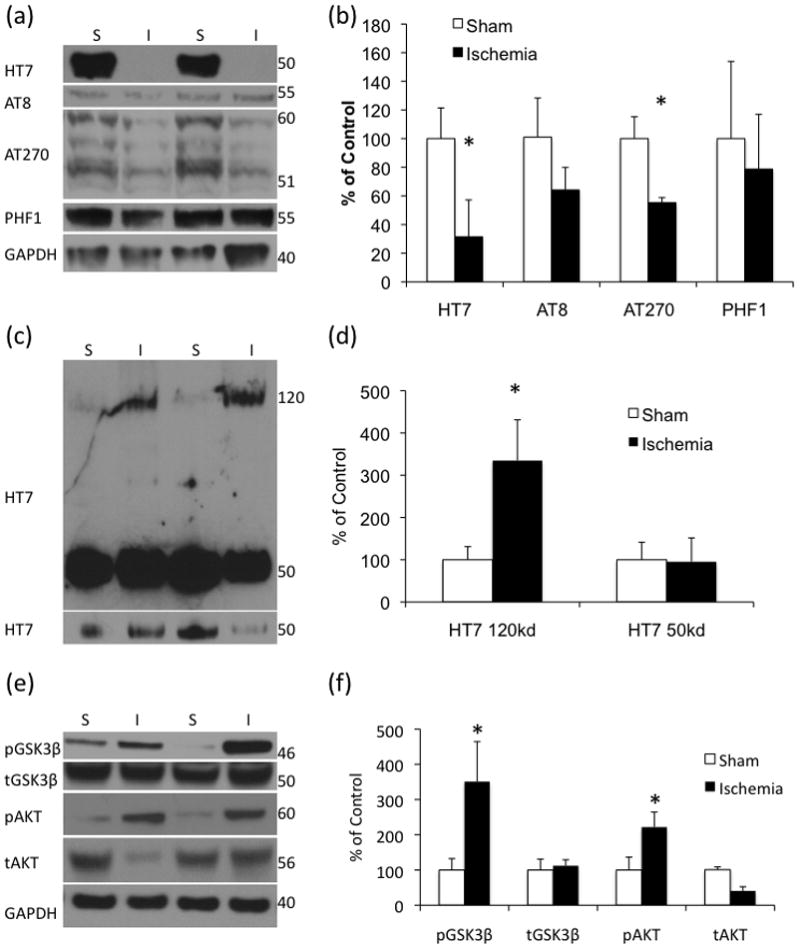
(a–b) Western blot analysis of soluble levels of total and phospho-tau’s reveal long-lasting decreases in total tau as well as the Thr181 phospho-tau. (c–d) Western blot analysis of formic-acid-insoluble tissue from 3xTg-AD mice 3-months post-injury revealed a significant increase in a high molecular weight total tau. (e–f) Active AKT, phosphoAKT and inactive phosphoGSK3β are significantly increased. (n=4 sham, 5 ischemia)
To determine if the decrease we observed in soluble tau was caused by a shift to the insoluble fraction, we analyzed detergent-insoluble tau levels, but did not find a change in tau at the expected molecular weight for aggregates broken down into monomers by the formic acid (Fig. 2C–D). However, we did find a significant increase in a high molecular weight, insoluble HT7-reactive tau band, which may represent a tightly aggregated form of insoluble tau such as tau associated with neurofibrillary tangles, or a post-translationally modified tau (Fig. 2C–D).
A number of kinases are altered following ischemia and during AD. In our model, we found a significant increase in phosphorylated AKT (Fig. 2E–F), a kinase involved in GSK3β inactivation, which, in turn is a known tau-kinase. Consistent with an increase in phosphorylated AKT, we found an increase in GSK3β phosphorylated at ser9 – an inhibitory phosphorylation (Fig. 2E–F). Hence, ischemia induces reductions in GSK3β activity at this time-point, and may partially explain the reductions seen in tau phosphorylation.
Ischemia induces long-term alterations in APP phosphorylation
We next analyzed the impact of ischemia on Aβ40 and Aβ42 3-months post-injury. We found no significant difference between ischemia and sham treated mice in any Aβ species by ELISA (Fig. 3E–F). To determine if the long delay between injury and sacrifice accounted for the lack of significant difference between the sham and ischemia treated mice we performed ELISAs on tissue 24-hours (Fig. 3A–B) and 48-hours post-surgery (Fig. 3C–D). We found no significant differences between sham and ischemia treated mice at any time point post injury except for a significant decrease in soluble Aβ42 48-hours post ischemia (Fig 3).
Figure 3. Ischemia does not increase Aβ levels in older 3xTg-AD mice.

ELISA was used to measure soluble and insoluble Aβ40 and Aβ42. All results were normalized to protein concentration. (a–b) 24-hours after injury, ischemia does not lead to an alteration in Aβ40 or 42 (n=4 sham, 4 ischemia). (c–d) 48-hours after injury, ischemia does not lead to an increase in Aβ40 or 42 (n=4 sham, 4 ischemia). (e–f) 3-months post-injury there is no significant alteration in Aβ (n=12 sham, n=14 ischemia).
We stained tissue from mice 3-months following ischemia for Aβ-like species (Fig. 4A) and found that by crude measurement, overall staining in the CA1, there was no significant difference in staining in the ischemia-treated tissue (Fig. 4B). We did not find any notable differences between the sham- and the ischemia-treated mice in thioflavin staining (Fig. 4C).
Figure 4. Ischemia alters APP C-terminal fragments.
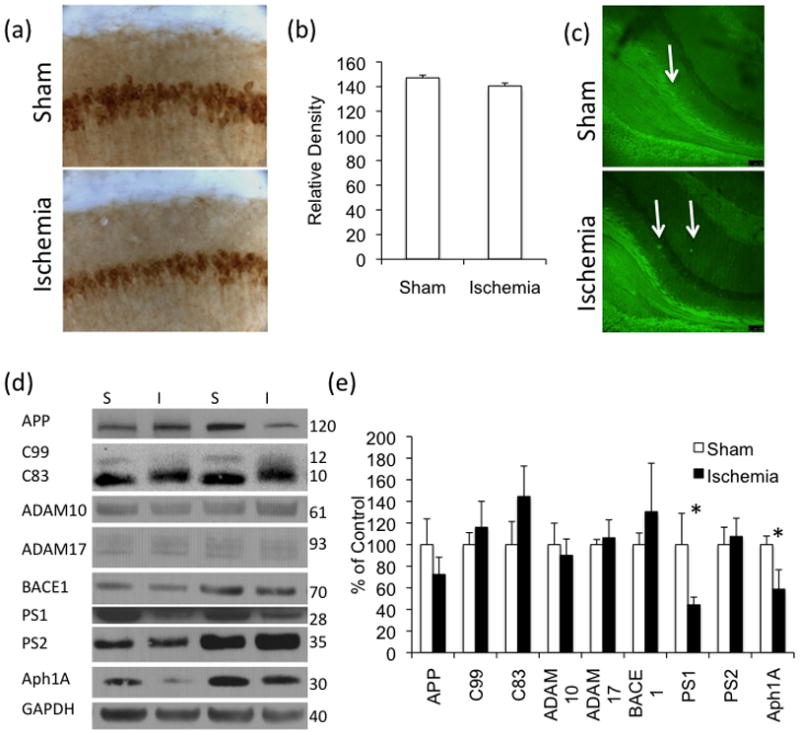
(a–b) Aβ-like staining, by 6E10 antibody, is not altered by immunohistochemistry 3-months post-ischemia. Quantification was performed using ImageJ and selecting an elliptical area of the CA1 (c) We did not find a difference in thioflavin staining. Arrows indicate areas of high thioflavin staining. (d–e) Western blot analysis of APP and APP-related proteins revealed a weight shift in the APP C-terminal fragment C83, but no changes in ADAM10, ADAM17 or BACE-1. We found a significant decrease in the γ-secretase components PS1 and Aph1 but not in PS2 (n=4 sham, 5 ischemia).
To further explore the impact of ischemia on APP processing, we examined steady-state levels of APP and the α-secretases ADAM17, ADAM10 and the β-secretase BACE1, but found no differences in these proteins (Fig. 4D–E). Interestingly, we found a significant decrease in the protein levels of the γ-secretase components presenlin1 (PS1) and Aph1A, but not presenilin2 (PS2).
Although we found no change in the levels of C83 or C99, we did observe an upward protein shift in the APP cleavage fragment C83 in the ischemia-exposed mice (Fig. 4D–E), which likely represents a post translational modification. APP can be phosphorylated at Thr668 which alters the conformation of APP, interfering with its binding to the scaffold/adaptor protein, Fe65, thereby decreasing its ability to activate the transcription of a number of target genes [1, 2, 16, 25]. Using a specific antibody, we analyzed Thr668-phosphorylated APP 3-months following ischemia (Fig. 5A–B). We did not find a significant difference in the 98-kDa band of phospho-APP Thr668. We found a reduction in a 188-kDa band in the mice exposed to ischemia, and a significant increase in the 120-kDa band (Fig. 5A–B). Notably, we found a increase in a 100-kDa band of APP Thr668 in control mice suggesting that this increase in APP phosphorylated at Thr668 occurs ubiquitously following ischemia and is not constrained to those with AD pathology present (Fig. 5D–E). Notably, we also found a similar pattern in the mice sacrificed 24-hours post-ischemia, with a significant increase in 120-kDa band of pAPP and a significant decrease in the levels of the 90-kDa band (Fig. 5F–G). At the 24-hour time point we did not find a significant change in the highest molecular weight band (Fig. 5F–G).
Figure 5. Ischemia induces long-term changes in APP phosphorylation and altered histone acetylation.
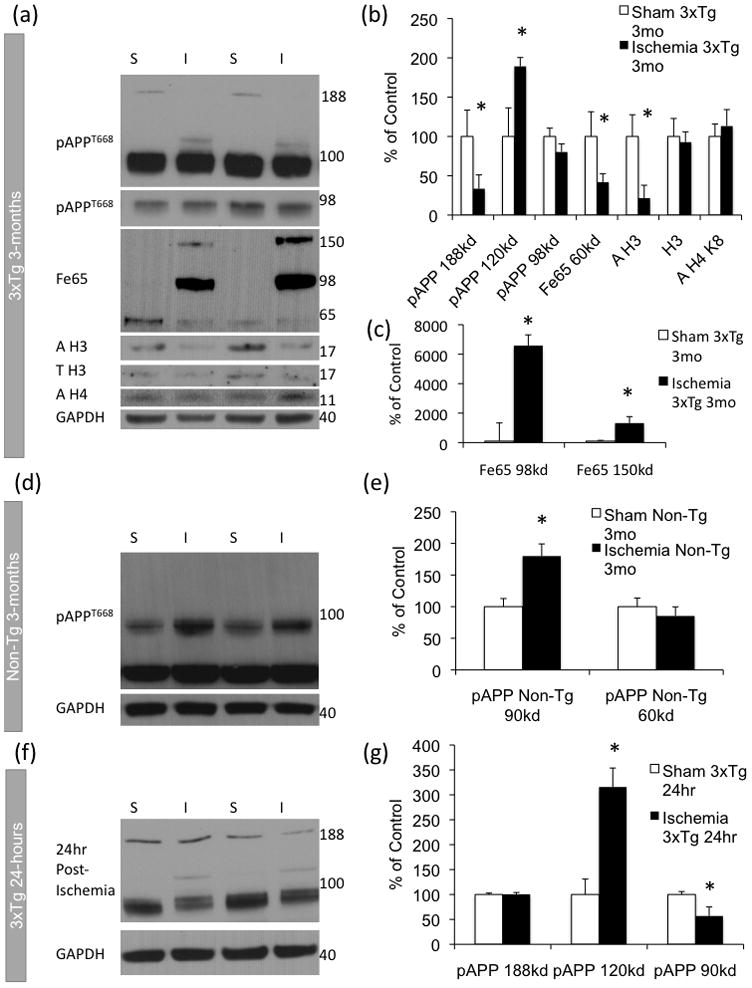
(a–b) Western blot analysis revealed a decrease in 180kDa APPT668 post-ischemia and an increase in 120kDa APPT668 in 3xTg-AD mice. Histone H3 acetylation was decreased in ischemia treated 3xTg-AD mice. We found a significant decrease in 60kd Fe65 and an increase in two higher molecular weight bands of Fe65, quantification included separately due to the scale(c). (d–e) APPT668 was increased in non-transgenic (Non-Tg) mice 3-months post ischemia. (f–g) 120kDa APPT668 was increased and 90kDa APPT668 was decreased in 3xTg-AD mice 24-hours post-ischemia.
To examine whether previous exposure to ischemia altered the relationship between phosphorylated APP and Fe65, we analyzed steady state levels of Fe65 by western blot in the 3xTg-AD mice 3-months post-injury. We observed a significant increase in the 98-kDa and 150-kDa molecular weight band of Fe65 in the ischemia-treated mice possibly due to altered binding following the phosphorylation of APP (Fig. 5A–B). Because evidence suggests that APP cleavage fragment AICD and Fe65 form a multimeric complex with the histone acetyltransferase Tip60 [2,], we examined acetylation of selected histones in the ischemia- and sham-treated mice. We found a significant decrease in the acetylation of histone H3 in the post-ischemic mice, suggesting long-term altered chromatin modifications following ischemia (Fig. 5A–B). We did not find changes in steady-state levels of total histone H3 or in histone H4 acetylated at lysine 8 (Fig. 5A–B).
Discussion
Unlike some previous studies we did not find a significant increase in Aβ. We believe that this discrepancy is due to the age of the mice used in this study, 12–15 months old, compared to the commonly used 2–8 month olds [4, 24, 29, 30]. Notably, there have been previous studies that evaluated the impact of ischemia on AD, and did not find changes in AD-related proteins. A study using double APP/PS1 transgenic mice did not find significant differences in Aβ deposition 2-months after a 45-minute common carotid artery occlusion [20]. More recently, a study found that there was a decrease in total APP levels in the cortex 7- and 30-days post-hypoperfusion [6].
Moreover, we found that there was a decrease in total tau and the AT270 phospho-tau epitope of tau as well as alterations in the kinases pAKT and GSK3β 3-months following injury. This finding not only extends our previous findings that an oligemic insult resulted in decreased tau for up to 3-weeks post-injury [10], but also correlates with published clinical data, showing that patients who have suffered from a hypoperfusion injury often present with a lower Braak score than “pure” AD patients [5, 11, 19].
In this study, we present evidence that ischemia produces long-lasting impacts on AD related-pathology. Our finding of increased high-molecular-weight insoluble tau 3-months post-injury highlights the potential of this type of injury to induce AD-pathology related aggregation. Furthermore, the altered APP phosphorylation at Thr668 and decreased histone3 acetylation underlines the complex nature of the long-term molecular relationship between ischemic injury and Alzheimer’s disease.
Acknowledgments
This work was supported by grants from the NIH AG-021982 to FML and NIH 1F31NS063650-01A1 to MAK. We thank Mr. David Cheng for assistance with the mice.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Ando K, Iijima KI, Elliott JI, Kirino Y, Suzuki T. Phosphorylation-dependent regulation of the interaction of amyloid precursor protein with Fe65 affects the production of beta-amyloid. The Journal of biological chemistry. 2001;276:40353–40361. doi: 10.1074/jbc.M104059200. [DOI] [PubMed] [Google Scholar]
- 2.Cao X, Sudhof TC. A transcriptionally [correction of transcriptively] active complex of APP with Fe65 and histone acetyltransferase Tip60. Science (New York, NY. 2001;293:115–120. doi: 10.1126/science.1058783. [DOI] [PubMed] [Google Scholar]
- 3.Doraiswamy PM, Leon J, Cummings JL, Marin D, Neumann PJ. Prevalence and impact of medical comorbidity in Alzheimer’s disease. The journals of gerontology. 2002;57:M173–177. doi: 10.1093/gerona/57.3.m173. [DOI] [PubMed] [Google Scholar]
- 4.Gentile MT, Poulet R, Di Pardo A, Cifelli G, Maffei A, Vecchione C, Passarelli F, Landolfi A, Carullo P, Lembo G. Beta-amyloid deposition in brain is enhanced in mouse models of arterial hypertension. Neurobiology of aging. 2009;30:222–228. doi: 10.1016/j.neurobiolaging.2007.06.005. [DOI] [PubMed] [Google Scholar]
- 5.Goulding JM, Signorini DF, Chatterjee S, Nicoll JA, Stewart J, Morris R, Lammie GA. Inverse relation between Braak stage and cerebrovascular pathology in Alzheimer predominant dementia. Journal of neurology, neurosurgery, and psychiatry. 1999;67:654–657. doi: 10.1136/jnnp.67.5.654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hiltunen M, Makinen P, Peraniemi S, Sivenius J, van Groen T, Soininen H, Jolkkonen J. Focal cerebral ischemia in rats alters APP processing and expression of Abeta peptide degrading enzymes in the thalamus. Neurobiology of disease. 2009;35:103–113. doi: 10.1016/j.nbd.2009.04.009. [DOI] [PubMed] [Google Scholar]
- 7.Hofman A, Ott A, Breteler MM, Bots ML, Slooter AJ, van Harskamp F, van Duijn CN, Van Broeckhoven C, Grobbee DE. Atherosclerosis, apolipoprotein E, and prevalence of dementia and Alzheimer’s disease in the Rotterdam Study. Lancet. 1997;349:151–154. doi: 10.1016/S0140-6736(96)09328-2. [DOI] [PubMed] [Google Scholar]
- 8.Itoh T, Satou T, Nishida S, Tsubaki M, Hashimoto S, Ito H. Expression of amyloid precursor protein after rat traumatic brain injury. Neurol Res. 2009;31:103–109. doi: 10.1179/016164108X323771. [DOI] [PubMed] [Google Scholar]
- 9.Jellinger KA. Traumatic brain injury as a risk factor for Alzheimer’s disease. Journal of neurology, neurosurgery, and psychiatry. 2004;75:511–512. [PMC free article] [PubMed] [Google Scholar]
- 10.Koike MA, Green KN, Blurton-Jones M, Laferla FM. Oligemic Hypoperfusion Differentially Affects Tau and Amyloid-{beta} The American journal of pathology. 2010 Jul;177(1):300–10. doi: 10.2353/ajpath.2010.090750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lee JH, Olichney JM, Hansen LA, Hofstetter CR, Thal LJ. Small concomitant vascular lesions do not influence rates of cognitive decline in patients with Alzheimer disease. Archives of neurology. 2000;57:1474–1479. doi: 10.1001/archneur.57.10.1474. [DOI] [PubMed] [Google Scholar]
- 12.Li ZG, Zhang W, Sima AA. Alzheimer-like changes in rat models of spontaneous diabetes. Diabetes. 2007;56:1817–1824. doi: 10.2337/db07-0171. [DOI] [PubMed] [Google Scholar]
- 13.Marklund N, Blennow K, Zetterberg H, Ronne-Engstrom E, Enblad P, Hillered L. Monitoring of brain interstitial total tau and beta amyloid proteins by microdialysis in patients with traumatic brain injury. J Neurosurg. 2009;110:1227–1237. doi: 10.3171/2008.9.JNS08584. [DOI] [PubMed] [Google Scholar]
- 14.McLin JP, Steward O. Comparison of seizure phenotype and neurodegeneration induced by systemic kainic acid in inbred, outbred, and hybrid mouse strains. The European journal of neuroscience. 2006;24:2191–2202. doi: 10.1111/j.1460-9568.2006.05111.x. [DOI] [PubMed] [Google Scholar]
- 15.Oddo S, Caccamo A, Shepherd JD, Murphy MP, Golde TE, Kayed R, Metherate R, Mattson MP, Akbari Y, LaFerla FM. Triple-transgenic model of Alzheimer’s disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron. 2003;39:409–421. doi: 10.1016/s0896-6273(03)00434-3. [DOI] [PubMed] [Google Scholar]
- 16.Pardossi-Piquard R, Petit A, Kawarai T, Sunyach C, Alves da Costa C, Vincent B, Ring S, D’Adamio L, Shen J, Muller U, St George Hyslop P, Checler F. Presenilin-dependent transcriptional control of the Abeta-degrading enzyme neprilysin by intracellular domains of betaAPP and APLP. Neuron. 2005;46:541–554. doi: 10.1016/j.neuron.2005.04.008. [DOI] [PubMed] [Google Scholar]
- 17.Pluta R. Role of ischemic blood-brain barrier on amyloid plaques development in Alzheimer’s disease brain. Current neurovascular research. 2007;4:121–129. doi: 10.2174/156720207780637207. [DOI] [PubMed] [Google Scholar]
- 18.Purandare N, Burns A. Cerebral emboli in the genesis of dementia. Journal of the neurological sciences. 2009;283:17–20. doi: 10.1016/j.jns.2009.02.306. [DOI] [PubMed] [Google Scholar]
- 19.Riekse RG, Leverenz JB, McCormick W, Bowen JD, Teri L, Nochlin D, Simpson K, Eugenio C, Larson EB, Tsuang D. Effect of vascular lesions on cognition in Alzheimer’s disease: a community-based study. Journal of the American Geriatrics Society. 2004;52:1442–1448. doi: 10.1111/j.1532-5415.2004.52405.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sadowski M, Pankiewicz J, Scholtzova H, Li Y-s, Quartermain D, Duff K, Wisniewski T. Links between the pathology of Alzheimer’s disease and vascular dementia. Neurochemical research. 2004;29:1257–1266. doi: 10.1023/b:nere.0000023612.66691.e6. [DOI] [PubMed] [Google Scholar]
- 21.Sheng B, Cheng LF, Law CB, Li HL, Yeung KM, Lau KK. Coexisting cerebral infarction in Alzheimer’s disease is associated with fast dementia progression: applying the National Institute for Neurological Disorders and Stroke/Association Internationale pour la Recherche et l’Enseignement en Neurosciences Neuroimaging Criteria in Alzheimer’s Disease with Concomitant Cerebral Infarction. Journal of the American Geriatrics Society. 2007;55:918–922. doi: 10.1111/j.1532-5415.2007.01171.x. [DOI] [PubMed] [Google Scholar]
- 22.Skoog I, Lernfelt B, Landahl S, Palmertz B, Andreasson LA, Nilsson L, Persson G, Oden A, Svanborg A. 15-year longitudinal study of blood pressure and dementia. Lancet. 1996;347:1141–1145. doi: 10.1016/s0140-6736(96)90608-x. [DOI] [PubMed] [Google Scholar]
- 23.Snowdon DA, Greiner LH, Mortimer JA, Riley KP, Greiner PA, Markesbery WR. Brain infarction and the clinical expression of Alzheimer disease. The Nun Study. Jama. 1997;277:813–817. [PubMed] [Google Scholar]
- 24.Sun X, He G, Qing H, Zhou W, Dobie F, Cai F, Staufenbiel M, Huang LE, Song W. Hypoxia facilitates Alzheimer’s disease pathogenesis by up-regulating BACE1 gene expression. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:18727–18732. doi: 10.1073/pnas.0606298103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Suzuki T, Nakaya T. Regulation of amyloid beta-protein precursor by phosphorylation and protein interactions. The Journal of biological chemistry. 2008;283:29633–29637. doi: 10.1074/jbc.R800003200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.van Groen T, Puurunen K, Maki HM, Sivenius J, Jolkkonen J. Transformation of diffuse beta-amyloid precursor protein and beta-amyloid deposits to plaques in the thalamus after transient occlusion of the middle cerebral artery in rats. Stroke; a journal of cerebral circulation. 2005;36:1551–1556. doi: 10.1161/01.STR.0000169933.88903.cf. [DOI] [PubMed] [Google Scholar]
- 27.Wang GL, Jiang BH, Rue EA, Semenza GL. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proceedings of the National Academy of Sciences of the United States of America. 1995;92:5510–5514. doi: 10.1073/pnas.92.12.5510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Webster CM, Kelly S, Koike MA, Chock VY, Giffard RG, Yenari MA. Inflammation and NFκB activation is decreased by hypothermia following global cerebral ischemia. Neurobiology of disease. 2009;33:301–312. doi: 10.1016/j.nbd.2008.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ye JT, Pi RB, Mao XX, Chen XH, Qin J, Xu SW, Liu PQ. Alterations in mRNA expression of BACE1, cathepsin B, and glutaminyl cyclase in mice ischemic brain. Neuroreport. 2009;20:1456–1460. doi: 10.1097/WNR.0b013e328332024a. [DOI] [PubMed] [Google Scholar]
- 30.Zhang X, Zhou K, Wang R, Cui J, Lipton SA, Liao F-F, Xu H, Zhang Y-w. Hypoxia-inducible factor 1alpha (HIF-1alpha)-mediated hypoxia increases BACE1 expression and beta-amyloid generation. The Journal of biological chemistry. 2007;282:10873–10880. doi: 10.1074/jbc.M608856200. [DOI] [PubMed] [Google Scholar]
- 31.Zhiyou C, Yong Y, Shanquan S, Jun Z, Liangguo H, Ling Y, Jieying L. Upregulation of BACE1 and beta-amyloid protein mediated by chronic cerebral hypoperfusion contributes to cognitive impairment and pathogenesis of Alzheimer’s disease. Neurochemical research. 2009;34:1226–1235. doi: 10.1007/s11064-008-9899-y. [DOI] [PubMed] [Google Scholar]