

Abstract
Lambdoid phage 21 has the prototype pinholin-SAR endolysin lysis system, which is widely distributed among phages. Its prototype pinholin, S2168, triggers at an allele-specific time to form small, heptameric lesions, or pinholes, in the cytoplasmic membrane, thus initiating lysis. S2168 has two transmembrane domains, TMD1 and TMD2. Only TMD2 is required for the formation of pinholes, whereas TMD1 acts as an inhibitor of TMD2 and must be externalized to the periplasm in the lytic pathway. Previously we provided evidence that S2168 first accumulates as inactive dimers with both transmembrane domains embedded in the bilayer. Here we analyze an extensive collection of S21 mutants to identify residues and domains critical to the function and regulation of the pinholin. Evidence is presented indicating that, within the inactive dimer, TMD1 acts in trans as an inhibitor of the lethal function of TMD2. A wide range of phenotypes, from absolute lysis-defectives to accelerated lysis triggering are observed for mutations mapping to each topological domain. The pattern of phenotypes allows the generation of a model for the structure of the inactive dimer. The model identifies the faces of the two transmembrane domains involved in intramolecular and intermolecular interactions, as well as interaction with the lipid.
Keywords: bacteriophage, holin, lysis, SAR domain, membrane protein
Introduction
In phage lysis, canonical holins, like the lambda S105 and T4 T proteins, terminate the infection cycle by suddenly “triggering” to form very large holes in the cytoplasmic membrane at the end of an allele-specific period of accumulation in the bilayer (Young & White, 2008, Dewey et al., 2009). These lesions allow the non-specific escape of the phage endolysin, fully folded and enzymatically functional, across the bilayer, leading to lysis within seconds. Recently, a second type of holin, designated as a pinholin, has been discovered (Park et al., 2007). Pinholins get their name from the fact that they form small holes that simply depolarize the membrane, rather than allowing the non-specific escape of a folded protein. To effect lysis, pinholins require SAR endolysins, which have an N-terminal transmembrane domain (TMD) that engages the sec translocon and results in export of the endolysin in an inactive, membrane-tethered form (Xu et al., 2004, Sun et al., 2009). When the pinholins trigger to depolarize the membrane, these membrane-tethered enzymes are released from the bilayer and re-fold to the active state, resulting in destruction of the murein and lysis. For both pinholins and canonical holins, the molecular basis for the allele-specific timing of the triggering event is unknown. However, they share a common characteristic: they both can be prematurely triggered to lethal hole-formation by artificial depolarization of the membrane with energy poisons and uncouplers (Xu et al., 2004, Park et al., 2006).
The prototype pinholin is S2168, the holin of the lambdoid phage 21. S2168 is one of two products of S21, the first gene in the phage 21 lysis cassette (Fig. 1A). The pathway to pinhole formation begins with the holin protein in an inactive form, with both of its two transmembrane domains (TMDs) in the bilayer (Fig. 1B, C) (Park et al., 2006). To proceed towards pinhole formation, TMD1 (Fig. 1C, D) must exit from the membrane, an event which happens spontaneously in a time-dependent fashion after insertion of S2168 into the membrane. This change in membrane topology can be accelerated if the membrane is artificially depolarized with an uncoupler. In contrast, the topology change is completely blocked for a lysis-defective allele, irsS2168, which has the irs epitope, containing two positively charged residues, at the N-terminus of its protein product (Fig. 1B, D). The irsS2168 allele is dominant and cross-linking studies indicate that its product dimerizes with S2168 protein (Pang et al., 2009). Thus it has the properties of an antiholin, a heterogeneous class of holin-specific inhibitors encoded by many bacteriophages(Wang et al., 2000, Young & Wang, 2006), either as alternate product of the holin gene (Bläsi et al., 1990) or an unlinked gene (Ramanculov & Young, 2001, Tran et al., 2005). In fact, S21 produces a second polypeptide, S2171, by virtue of translational starts three codons upstream of the start used for the S2168 holin (Fig. 1A) (Barenboim et al., 1999). S2171 has weak antiholin character, in that membrane escape of TMD1 is delayed but not blocked, presumably because it has only a single positively charged residue at its N-terminus (Fig. 1A, B, D). Taken together, these results suggested a model in which the products of S21 first populate an inactive dimer stage, where TMD1 is retained in the membrane (Fig. 1C). In the subsequent pathway to pinhole formation, the pinholins in the inactive dimer are first converted to an “activated” state, with TMD1 externalized, ultimately leading to an oligomer within which the heptameric pinhole forms. Evidence for the activated dimer is indirect, in that cysteine substitutions in TMD1 can lead to the periplasmic formation of intermolecular disulfide linkages in an orientation-specific manner (Park et al., 2006). Periplasmic interaction of TMD1 is not required for pinhole formation, because a mutant in which all of TMD1 has been deleted retains robust triggering ability.
Figure 1.

A. Phage 21 lysis cassette
Phage 21 lysis genes S, R, Rz, Rz1 are located downstream of the late gene promoter, indicated by the arrow. S21 gene has a dual-start motif, which encodes both a holin S2168 (translated from the Met4 start codon), and an antiholin S2171 (translated from codon Met1) (Barenboim et al., 1999, Park et al., 2006). Both start codons are bold and underlined.
B. The membrane topology of S2168 (left), S2171 (middle), and irsS2168 (right).
TMD1: white box; TMD2: black box. The TMD1 of S2168 is initially inserted in the membrane but later released into the periplasm (Park et al., 2006) (see panel C). The externalization of TMD1 is delayed in the context of S2171 and completely blocked in the context of irsS2168, which has the irs-tag (RYIRS) fused to the N-terminus of S2168.
C. Model of S2168 hole formation pathway (top-down view from periplasm).
The two TMDs (green: TMD1; purple: TMD2) in a single S2168 molecule are boxed. Initially, both TMDs of S2168 are inserted in the membrane in an inactive dimer form. When TMD1 is released, the inactive dimer is converted into an activated dimer form. Activated dimers then aggregate and nucleate to form heptameric pinholes. (Modified from Pang et al. (2009), with permission.)
D. The distribution of mutations on the S21 reading frame.
The S21 reading frame is shown, with TMD1 and TMD2 indicated by the double-line underscore. The residues deleted in S2168ΔTMD1 are shaded in gray. The position and sequence of the irs epitope in irsS2168 is shown as a box below the sequence. Individual S2168 mutants in the collection are shown above the amino acid sequence. Orange pound signs and underlined mutations represent nonsense and missense mutations obtained by EMS mutagenesis and selection, respectively; all others were obtained by site-directed mutagenesis. Asterisks mark the position of ochre nonsense mutations, Color code: black, no significant change in triggering time compared to the parental; red, accelerating triggering time by >15 m; blue, delayed triggering by >10 m; green, triggering abolished. Highlighted mutants have reduced or undetectable level of protein.
The structure of the pinhole in the membrane has been interrogated with cysteine-accessibility studies, using a library of cysteine-substitutions at positions throughout the pinholin sequence, with cross-linking experiments using the amine-specific reagent DSP (dithiobis[succinimidyl propionate]), and with computational approaches using coarse-grained simulated annealing of the TMD2 sequence (Pang et al., 2009). These efforts led to a heptameric model for the minimal pinhole, in which seven TMD2 domains oriented at 31° to the plane of the membrane form a left-handed helical bundle around a channel of ~1.5 nm diameter.
These recent results provide a much more comprehensive picture of the functional and structural aspects of the S21 pinholin than is available for any of the previously studied holins. Here we report an extensive mutational analysis of S21and discuss the results in terms of the model for the structure of the inactive dimer that is proposed to be the first significant intermediate in the pinhole formation pathway.
Results
TMD1 inhibits TMD2 pinhole formation in both cis and trans
Previously, we provided evidence that TMD1 is externalized to the periplasm by showing that the cysteine-substitution protein, S2168S16C, forms disulfide-linked dimers during the pathway to pinhole formation (Park et al., 2006). We wondered whether the functional inhibition exerted by irsS2168 also extended to the topological change of the inhibited pinholin. To answer this question, we assessed disulfide-dimer formation by S2168S16C in the presence of the antiholin allele (Fig. 2A, B). No change in dimer formation was observed, although the lethal function of the cysteine-substitution protein was completely inhibited by irsS2168. This indicates that irsS2168 does not inhibit the externalization of S2168 TMD1 and suggests that the inhibition effect must be due to the interaction of the membrane-embedded irs-tagged TMD1 of irsS2168 with TMD2 of S2168. Coupled with the previous results (Park et al., 2006), these data indicate that membrane-embedded TMD1 could interact with TMD2 both intermolecularly and intramolecularly (Fig. 2C). The strong lysis-timing and survival phenotypes available in this system suggested that a genetic analysis could identify the helix-helix interfaces involved.
Figure 2. irsS2168 does not inhibit the externalization of S2168 TMD1.
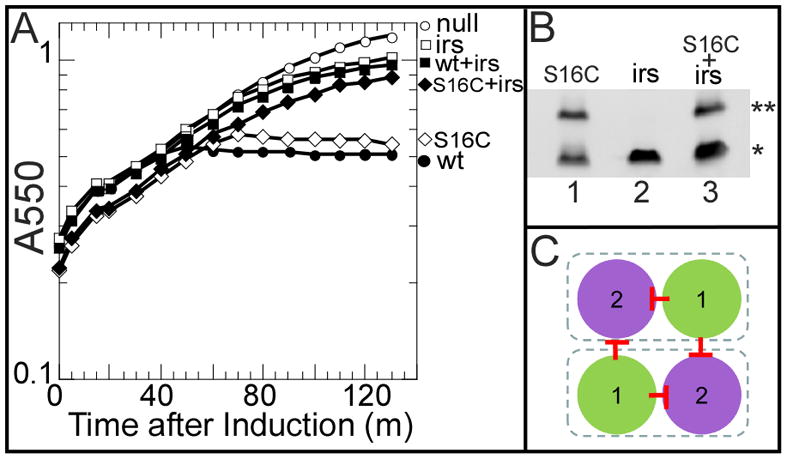
A. Triggering of S2168 alleles. Cultures were induced at t=0 and monitored for growth as A550. Open circles, prophage λQ21Δ(SRRzRz1)21, no plasmid. Closed circles, prophage λS2168, no plasmid. Open squares, prophage λQ21Δ(SRRzRz1)21, plasmid pirsS2168*. Closed squares, prophage λS2168, plasmid pirsS2168*. Open diamond, prophage λS2168S16C, no plasmid. Closed diamond, prophage λS2168S16C, plasmid pirsS2168*.
B. Periplasmic disulfide-bond formation with S2168S16C. Cultures were induced and precipitated by TCA at 1 h after induction. Samples were resuspended in sample loading buffer without reducing agent and analyzed by SDS-PAGE and western blotting. The primary antibody detects both the S2168 variants and irsS2168 since the epitope is at the S21 C-terminus. Double and single asterisks indicate the position of dimer and monomer, individually. Lane 1, prophage λS2168S16C, no plasmid. Lane 2, prophage λQ21Δ(SRRzRz1)21, plasmid pirsS2168*. Lane 3, prophage λS2168S16C, plasmid pirsS2168*.
C. Top-down view of the inactive dimer. Same scheme as Fig. 1C. TMD1 interacts with TMD2 both inter- and intra- molecularly, depicted by the red stop arrows.
Mutants in each topological domain of S2168 have altered triggering phenotypes
An extensive library of S2168 missense alleles was generated by two methods (Table 1). First, loss of function mutants were obtained by selecting for a defect in the lethal pinhole formation after EMS mutagenesis (Table 2). In this selection, 39 mutants were isolated with missense or nonsense mutations in the S2168 gene, which resolved into 7 unique nonsense and 13 unique missense mutations (Table 1, asterisk entries; Table 2; Fig. 1D). There are 10 nonsense mutations accessible by transition mutation from the parental S2168 base sequence. The most distal of these, R65op, is likely to retain lethality, based on the analysis of the C-terminal mutations (see below). Thus 7 of 9 (78%) possible nonsense mutations were recovered in the selection for loss of lethality. These considerations and the degree of repeats of the missense alleles isolated (Table 2) indicate that the selection was near saturation, in terms of mutations accessible by a single base change. Other than the extremely short N-terminal cytoplasmic domain, which in S2168 consists of only 3 residues, each of the other four topological domains (TMD1, loop, TMD2 and C-terminal cytoplasmic tail) had at least one position where non-lethal mutations were selected (Table 2; Fig. 1D). It should be noted that the plasmid context used for the selection was a hybrid, with S21 inserted in the place of S in the λ lysis cassette, but the phenotypic analysis was done with these alleles re-created in the context of the complete phage 21 lysis cassette. In a few cases (T15I, S16F, A38T, E62K), the mutant alleles recovered lethal function, albeit significantly delayed, as observed for lambda S mutants selected in a similar plasmid-based system (Raab et al., 1986). As before, this is likely because the expression level in the selection for loss of lethality was somewhat lower than that attained in the induced prophage and the cognate late gene expression context.
Table 1.
S2168 mutants.
| loc1 | allele2 | full-length3 | Δ(TMD1)4 | irsS21685 | |||
|---|---|---|---|---|---|---|---|
| T6 | Prot7 | T6 | Prot7 | alone8 | +wt9 | ||
| wt | 0 | 1 | 0 | 1 | NL | NL | |
| TMD1 | S8L | 0 | 0.2 | NL | NL | ||
| G10L | +40 | ||||||
| G10Q | +40 | 0.2 | |||||
| A12L | −35 | NL | 70 | ||||
| G14Q | −35 | 30 | 70 | ||||
| G14T | 0 | ||||||
| G14L | +40 | ||||||
| G14C | −35 | NL | NL | ||||
| T15I* | +40 | ||||||
| T15L | +40 | ||||||
| T15C | −35 | NL | NL | ||||
| S16F* | +30 | ||||||
| S16C | 0 | ||||||
| A17L | +60 | ||||||
| A17Q | −35 | 60 | 40 | ||||
| S19L | NL | ||||||
| S19C | NL | ||||||
| G21L | NL | ||||||
| G21Q | −35 | 110 | 80 | ||||
| F24L | +40 | ||||||
| F24C | −20 | ||||||
| Loop | D29N* | NL | 0 | ||||
| D29V* | NL | +40 | |||||
| Q30C | +40 | +20 | 0.4 | ||||
| V31C | NL | +10 | |||||
| S32C | NL | −40 | |||||
| P33L* | NL | NL | |||||
| P33S* | NL | 0 | |||||
| S34C | NL | +10 | |||||
| TMD2 | W36C | NL | NL | 0.01 | |||
| A37V* | NL | NL | |||||
| A37T | +30 | −30 | |||||
| A37C | +40 | +30 | |||||
| A38T* | +50 | −20 | |||||
| G40L | NL | NL | 0.4 | ||||
| G40T | NL | NL | |||||
| V41C | +40 | −10 | |||||
| G43L | NL | NL | |||||
| G43E* | NL | NL | 0.2 | ||||
| G43R* | NL | 0 | |||||
| G43C | +40 | +50 | |||||
| S44L | NL | +30 | 0.3 | ||||
| S44T | −35 | NL | 90 | NL | |||
| S44N | −35 | −45 | NL | NL | |||
| S44C | NL | NL | 0.4 | ||||
| L45C | +10 | +30 | 0.2 | ||||
| V46C | 0 | −10 | |||||
| V46L | +10 | 0 | |||||
| L47C | +60 | +20 | |||||
| L47F | +50 | 0 | |||||
| G48L | NL | NL | |||||
| G48T | NL | +20 | |||||
| G48C | NL | NL | |||||
| F49C | −10 | −10 | |||||
| F49L | 0 | 0 | |||||
| L50C | +30 | +30 | 0.2 | ||||
| T51I* | NL | NL | |||||
| T51C | +20 | NL | |||||
| Y52C | 0 | NL | 0.4 | ||||
| Y52F | +10 | −10 | |||||
| T54I* | NL | NL | |||||
| T54C | NL | NL | |||||
| tail | I60och10 | NL | nt | NL | nt | ||
| E62och | +40 | nt | +60 | nt | |||
| E62K* | +40 | NL | |||||
| D63och | 0 | nt | +30 | nt | |||
| D63C | 0 | 0 | |||||
| R65och | 0 | nt | +30 | nt | |||
| A67C | 0 | 0 | |||||
Topological domain
Mutant alleles. Asterisk: mutants generated by EMS mutagenesis.
relative to parental S2168
relative to parental S2168ΔTMD1
relative to parental irsS2168
change in triggering time of each mutant (m) relative to the parental allele (50 m after induction for both S2168 and S2168ΔTMD1). NL: non-lethal.
The protein accumulation level of each mutant. Only mutants with significantly lower amount of protein accumulated than wildtype are shown. The number indicates the protein amount as a ratio to the wildtype. nt, not tested.
The triggering time of each irsS2168 mutant (m). The parental irsS2168 is NL (non-lethal).
The triggering time of each irsS2168 mutant in the presence of S2168 expressed from prophage λS2168. The wt irsS2168 inhibits the triggering of S2168 and is thus labeled as NL.
Ochre stop codon substitution of that residue.
Table 2.
Independent S2168 lysis-defective alleles recovered from EMS mutagenesis
| codon | amino acid change | codon change | isolates | ||
|---|---|---|---|---|---|
| 15 | Thr | Ile | ACA | ATA | 4 |
| 16 | Ser | Phe | TCT | TTT | 1 |
| 23 | Trp | End | TGG | TGA | 2 |
| 26 | Gln | End | CAG | TAG | 1 |
| 27 | Trp | End | TGG | TAG | 1 |
| 27 | Trp | End | TGG | TGA | 3 |
| 29 | Asp | Asn | GAT | AAT | 4 |
| 29 | Asp | Val | GAT | GTT | 1 |
| 30 | Gln | End | CAG | TAG | 1 |
| 33 | Pro | Leu | CCG | CTG | 1 |
| 33 | Pro | Ser | CCG | TCG | 1 |
| 36 | Trp | End | TGG | TAG | 1 |
| 36 | Trp | End | TGG | TGA | 2 |
| 37 | Ala | Val | GCT | GTT | 1 |
| 38 | Ala | Thr | GCG | ACG | 3 |
| 43 | Gly | Glu | GGA | GAA | 4 |
| 43 | Gly | Arg | GGA | AGA | 1 |
| 51 | Thr | Ile | ACT | ATT | 5 |
| 54 | Thr | Ile | ACA | ATA | 1 |
| 62 | Glu | Lys | GAA | AAA | 1 |
Many other mutants were obtained by site-directed mutagenesis, including a number constructed for Cys-substitution experiments aimed at probing the lumenal residues of the pinhole (Pang et al., 2009). The mutant alleles were subjected to phenotypic analysis, including triggering time and accumulation of the mutant protein (Table 1).
In overview, three mutant triggering phenotypes were observed: non-lethal (no triggering after induction), delayed, and early, summarized in Table 1 and Fig. 1D. For delayed or non-lethal mutants, only those with normal levels of protein accumulation are included in the analysis below.
TMD1 mutants suggest its orientation in the inactive dimer
Since TMD1 is not required for lethal function but must be removed from the bilayer to allow fruitful oligomerization by TMD2, any mutation in TMD1 affecting the triggering time is most likely affecting its ability to exit the membrane. Presumably, such changes would be due either to changing its inter- or intramolecular affinity for TMD2, or changing its hydrophobicity, or both. The clearest example of such effects comes from mutations at Ala17, where changes to the more hydrophilic Gln and the more hydrophobic Leu had the expected opposite effects on triggering time (Fig. 3A, Table 1): dramatically early triggering for the former and drastically delayed for the latter. Similar patterns were observed at Gly14 and Gly21 (Table 1). The simplest interpretation is that these positions face the lipid (Fig. 4A), a notion supported by the fact that the early-triggering mutants G14Q, A17Q and G21Q all restored lytic function in the context of irsS2168 (Fig. 3B, Table 1).
Figure 3. Triggering phenotypes of TMD1 mutants.

Same procedure as in Fig. 2A.
A. A17 mutant phenotypes mirror hydrophobicity. Each culture contained prophage λQ21Δ(SRRzRz1)21, with no plasmid (open circles), or plasmids pS2168 (closed circles), pS2168A17Q (open squares), or pS2168A17L (closed squares).
B. Hydrophilic changes suppress the triggering defect of irsS2168. Each culture contained prophage λQ21Δ(SRRzRz1)21 with no plasmid (open circles), or plasmids: pirsS2168* (closed circles), pirsS2168*G14Q (open squares), pirsS2168*A17Q (open diamonds), or pS2168G21Q (open triangles).
C. A12L confers early triggering and reduces trans inhibition by irsS2168. Circles, prophage λQ21Δ(SRRzRz1)21 with either pS2168 (closed), or pS2168A12L (open). Dashed line with squares, prophage λQ21Δ(SRRzRz1)21 with either pirsS2168* (open), or pirsS2168*A12L (closed). Triangles, prophage λS2168 with either pirsS2168* (open), or pirsS2168*A12L (closed).
Figure 4. Mapping TMD1 mutations.
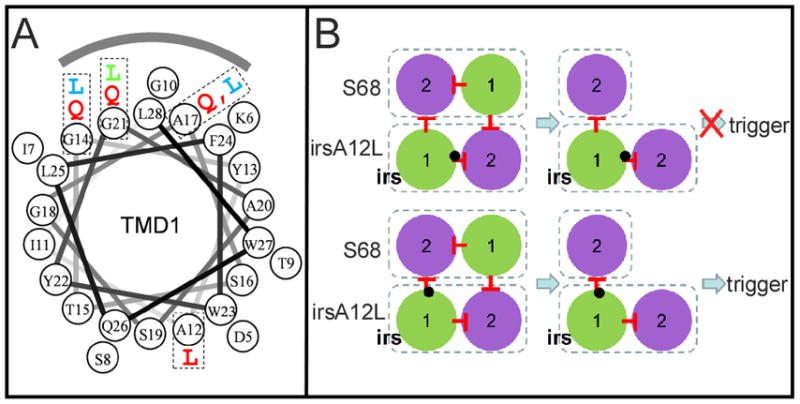
A. Helical wheel projection of S2168 TMD1. Mutations used to analyze the orientation of TMD1 in the inactive dimer are labeled with the same color code as Fig. 1D. The shaded arc indicates the face interacting with lipid in the inactive dimer.
B. The effect of the position of A12L on the antiholin function of irsS2168. Upper, A12L mutation (black dot) in the context of irsS2168 is shown either affecting the cis (intramolecular; upper diagram) or the trans (intermolecular; lower diagram) interaction with TMD1. In the former, inhibition of triggering should be maintained, but not in the latter.
In contrast, an Ala →Leu change at position 12 on the opposite face of TMD1, although increasing hydrophobicity, dramatically advanced triggering (Fig. 3C, 4A), suggesting that the A12L mutation destabilizes a TMD1– TMD2 interaction. We reasoned that if it was the cis (intramolecular) interaction that was affected, then A12L would not affect the negative dominance in the context of irsS2168 (Fig. 4B, upper). However, the opposite was observed, with irsS2168A12L clearly compromised in its antiholin character (Fig. 3C). This suggests that A12L disrupts the trans (intermolecular) TMD1–TMD2 interface (Fig. 4B, lower).
Substitutions with cysteine at five positions in TMD1 generated a wide range of phenotypes: dramatic advance of the triggering time (G14C, T15C), small advance or no change (S16C, F24C), and abolition of triggering (S19C) (Table 1). These changes clearly cannot be interpreted in terms of a significant alteration in hydrophobic character. The phenotypes are complicated by the fact that, after extraction from the bilayer, TMD1 undergoes homotypic interactions that, in the case of S16C, can lead to disulfide-bond-linked homodimer formation (Park et al., 2006). To assess the degree of covalent dimerization, cells were harvested by TCA (trichloroacetic acid) precipitation at the time of triggering (or at 60 m for the non-triggering allele S19C). Western blot analysis of these samples prepared in the absence of reducing agent revealed that the two very early lysis mutants, G14C and T15C, and the S16C mutant have a substantial fraction of the pinholin in covalently linked dimer state at the time of triggering (Fig. 5A). When DTT (dithiothreitol) was added to the cultures immediately after induction, disulfide bond formation was reduced or eliminated and the triggering times of all three mutants were significantly delayed, indicating that in all three cases disulfide bond formation contributes positively to pinhole formation.
Figure 5.
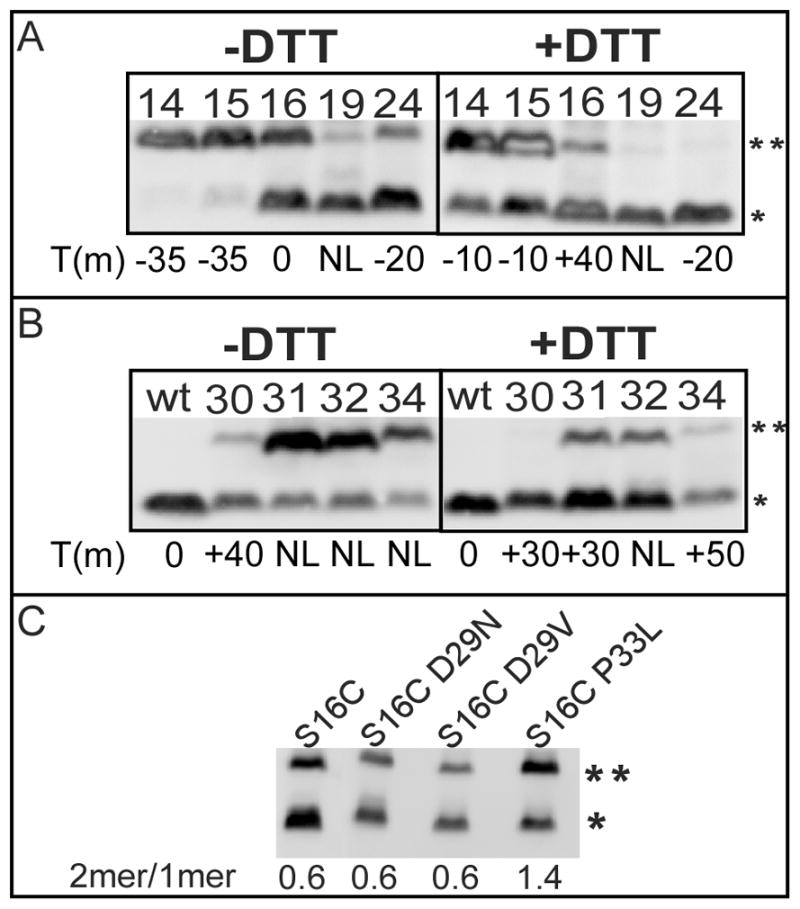
A, B. The effect of DTT on the disulfide-bond formation of S2168 cysteine mutants
Cultures carrying prophage λQ21Δ(SRRzRz1)21 and derivatives of plasmid pS2168 were thermally induced and precipitated by TCA either at the time of triggering or, for the delayed or non-lethal mutants, at the time when S2168S16C triggers. Samples were analyzed by SDS-PAGE and western blotting without reducing agent. Right panels show samples from cultures treated with 1 mM DTT at the time of induction. Numbers on each lane indicate residue positions of Cys substitutions. Asterisks indicate the position of monomer and dimers, as in Fig 2B. The triggering time of each mutant relative to that of wt S2168, in minutes, is indicated at the bottom. A, Cys substitutions in TMD1. B, Cys substitutions in the connecting loop.
C. The effect of each mutation on the externalization of S2168 TMD1.
Same procedure as in Fig 2B, except that cells carried prophage λQ21Δ(SRRzRz1)21 with derivatives of plasmid pS2168 encoding mutations on the S2168 gene as indicated on top of each lane. Asterisks indicate the position of monomer and dimers, as in Fig 2B. The dimer to monomer ratio of each mutant is illustrated on the bottom.
Mutations in the periplasmic loop
The periplasmic loop was significantly over-represented among the mutants selected for loss of lethality after EMS mutagenesis (Fig. 1D, Tables 1, 2). At D29, the only possible G to A transition, resulting in D29N, was among the non-lethal isolates selected, as well as the presumably much rarer D29V, which requires an A to T transversion. Both of the missense mutations accessible by a C to T transition mutation from P33 (P33L, P33S) were also selected. Moreover, every site-directed change made in the loop residues, all of which resulted in Cys substitutions, caused a non-lethal or severely delayed lysis phenotype (Fig. 1D, Table 1). However, in these cases, the formation of the disulfide bond appears to be irrelevant to the triggering defect, since DTT treatment reduced or eliminated the formation of the linkages but did not affect the triggering time significantly (Fig. 5B). The simplest explanation for these defects would be that changes in the loop would compromise externalization of TMD1, in which case the defective alleles should regain lethality in the ΔTMD1 context. However, neither of the D29 mutants nor P33L had a detectable defect in terms of externalization of TMD1, as indicated by the effect of mutation on the dimer to monomer ratio of S2168S16C (Fig. 5C). Moreover, D29V significantly delayed the triggering and P33L retained the non-lethal character in the S2168ΔTMD1 context (Table 1). These results suggest that these mutations exerted their effects at stages beyond TMD1 export.
Analysis of mutants in TMD2 suggests faces for TMD1 interaction
Phenotypic analysis of mutations in TMD2 (Fig. 1D, Table 1) is more difficult because each change could affect the release of TMD1, by interfering with the TMD1–TMD2 interaction or TMD2-TMD2 interactions downstream in the pathway to pinhole formation. To identify the residues involved in the TMD1– TMD2 interface in the inactive dimer form, the effect of each mutation was tested both in the context of the full-length and TMD1 deletion alleles. Mutations in five positions (A37T, A38T, V41C, L47F, G48T; Fig. 6A; Table 1) were found to affect the triggering time of S2168 and S2168ΔTMD1 differently, without affecting accumulation of either, and in each case, the lethal function was improved, either recovering lethality or triggering earlier, in the ΔTMD1 context (Table 1). This indicates that the original defect in each case was due to the retention of TMD1 in the bilayer. It follows that these positions, occupying a wide arc on the helical surface of TMD2, are likely to be involved in a TMD1–TMD2 interface (Fig. 6A), either intramolecular or intermolecular, and that the mutations increase the affinity of TMD2 for TMD1.
Figure 6. Orientation map of the inactive dimer.

A. Helical wheel projection of TMD2.
Positions where mutations exhibit accelerated lethal function in the ΔTMD1 context, and thus predicted to interact with TMD1, are colored in orange. V46 and F49, colored in green, exhibit phenotypic insensitivity and thus are likely to be facing the lipid, indicated by the shaded arc. Hydrophilic and weakly hydrophobic residues are indicated by asterisk.
B. The orientations of TMD1 and TMD2 in the inactive dimer.
Same scheme as in Fig. 1C, but, in addition, residue positions are indicated in white circles. Residues predicted to face lipid and involved in TMD1–TMD2 interactions are colored green and black, respectively. The blue arc inside TMD2 represents the extent of the hydrophilic and weakly hydrophobic residues that eventually face the lumen of the pinhole. ID1, 2 = inactive forms 1 and 2; AD = putative activated dimer.
In comparison with the analysis of TMD1 mutations (see above), there is less clear evidence specifying a lipid-interacting face of TMD2. However, replacing residues V46 or F49 with either C or L has no effect on the triggering time of either S2168 or S2168ΔTMD1 (Table 1). The tolerance of these two positions to major changes in side chain architecture suggests that they may not be involved in protein- protein interactions and may thus face the lipid (Fig. 6A).
Mutations in the C-terminal cytoplasmic domain
A nearly universal feature of class I and II holins is the presence of a short cytoplasmic C-terminus, usually very hydrophilic and carrying predominantly positively charged residues (Young & Wang, 2006). S2168 is no exception, with a 13 residue cytoplasmic tail bearing 9 charged residues, six of which are Arg or Lys (Fig. 1D). Table 1 shows that most of this domain is dispensable, since a stop codon at D63, eliminating 9 of the 13 residues, has no effect on lysis timing. Stops earlier in the reading frame severely damage or ablate lethal function, but, since the antibody used is specific for the C-terminal oligopeptide sequence, it is not known whether protein localization or accumulation is compromised. Finally, E62K, which increases the number of basic residues by one and the overall predicted positive charge by two, is a lysis-delay allele in the full-length context and is non-lethal in the deletion context (Table 1). Similar inhibitory phenotypes have been observed for analogous mutants in the C-terminal cytoplasmic domain of the canonical class I holin, lambda S105 (Bläsi et al., 1999), suggesting a regulatory role for this domain in both types of holin.
Discussion
The temporally-regulated lethal function of phage holins is not only an important fundamental process in its own right, as the most common cytocidal event in the biosphere, but is also arguably one of the simplest temporally scheduled processes in biology. Holin triggering occurs with remarkable precision at an allele-specific time on a 10 – 100 minute time scale after the onset of holin gene expression (Young & Wang, 2006). The recently discovered pinholin class of holins is a particularly attractive system for dissecting this fundamental process, because of the small size of the pinholin, only 68 residues, the interesting dynamic membrane topology of its TMD1, and the rather simple nature of the lethal pinhole, a heptamer of TMD2 (Pang et al., 2009). Here we analyze the cellular and molecular phenotypes of an extensive collection of S21 mutants in an effort to dissect the pathway to pinhole formation and to provide a genetic basis for future biochemical, biophysical and structural investigation.
The results presented here allow two specific advances in our understanding of the S21 pathway, allowing us to discern the orientations of the two TMDs in the inactive dimer and revealing the existence of a second class of inactive dimer, with only 1 TMD1 in the membrane. The findings also suggest refinements of the model for S21 that may apply to holin function in general.
The structure of the inactive dimer
Previously, we demonstrated that TMD1 is non-essential for pinhole formation and, in the growing population of pinholin proteins, is progressively externalized during the pathway to triggering (Park et al., 2006). Moreover, triggering induced by addition of energy poisons is associated with quantitative release of TMD1s from the bilayer. In contrast, the irsS2168 variant, which cannot externalize its TMD1, is non-lethal, does not oligomerize beyond the dimer stage (Pang et al., 2009), and is negative-dominant, hetero-dimerizing with and blocking the lethal function of the parental pinholin. These observations indicated that TMD1 is an intrinsic inhibitor of TMD2 oligomerization and suggested that an inactive dimer, with TMD1 still in the membrane, is part of the normal pathway to triggering. Here we further show that the irsS2168 dominance is imposed without preventing the externalization of TMD1 of the functional pinholin with which it dimerizes (Fig. 2A, B). This result has several implications. First, mutations in TMD1 that affect lysis timing are likely to do so by affecting the release of TMD1 from the bilayer, either by altering interactions with TMD2 or by altering the intrinsic hydrophobicity of TMD1 and thus its stability in the membrane. Second, TMD1 can externalize spontaneously without coordinating its topological change with its sister TMD1 in the inactive dimer (see below). Finally, and most importantly, the fact that dimers with only one TMD1 in the membrane do not oligomerize or progress to pinhole formation indicates that TMD1 must make trans (i.e., intermolecular) inhibitory contacts with TMD2 in the inactive dimer. Moreover, careful examination of the patterns of phenotypic variance in the three possible contexts (the full-length holin form (S2168), the strong antiholin form (irsS2168) and the ΔTMD1 form (S2168ΔTMD1)) has allowed us to establish an orientation map for the TMDs in the inactive dimer (Fig. 6B). Residues important for assigning the orientation are: residues G14, A17 and G21 in TMD1, suggested to face the lipid; A12 in TMD1, suggested to face TMD2 in trans, and residues A37T, A38T, V41C, L47F, and G48T, which are indicated to interact with TMD1. Actually, we do not have direct evidence for a cis (intramolecular) interaction between TMD1 and TMD2, although S2168 protein must start as a monomer in the membrane, during which time presumably a cis interaction would maintain the holin in an inactive form. Moreover, the length of the arc defined by the five TMD2 mutations that have TMD1-sensitive phenotypes suggests that at least some of them, likely including positions 37 and 47, must occupy the intramolecular interface.
Although the orientation model is low resolution, it should be testable experimentally, either with biochemistry, using cysteine-scanning and copper-phenanthroline-mediated disulfide bond formation (Gründling et al., 2000, Sun & Kaback, 1997), with genetics, by isolating intragenic suppressors of the lethal and/or lytic phenotypes, or, perhaps, with structural studies, if the TMD1–TMD2 interaction can be covalently locked with disulfide bonds and thus survive the removal of the membrane by detergent.
The role of dimer formation in holin function
Both the λ holin S105, and the prototype pinholin S2168, have an antiholin form with a short N-terminal extension carrying an extra positively charged residue that affects topology of TMD1. In the case of λ, the TMD1 of the antiholin form S107 is inhibited from entering the bilayer, whereas that of S2171 is retarded in its ability to escape from it. In both cases, the properties of the antiholin form have been made discernible by studying mutant alleles more strongly blocked from making the topological transition than the native antiholin; i.e., a deletion of TMD1 in λ S (White et al., 2010), and the irs-tag allele in S21. In both of these cases, the enhanced antiholins are capable of an absolute block in the lethal function and in the formation of dimers with the holin form. It is critical to note that in neither case is antiholin inhibition the fundamental basis of the temporal regulation of triggering. Even in the absence of the antiholin form, holin-mediated timing retains its fundamental characteristics: long-term accumulation of the holin without affect on membrane integrity, sudden triggering associated with lethality and depolarization of the membrane, and sensitivity to premature triggering by energy poisons (Gründling et al., 2001, Park et al., 2006) The simplest notion, by inference, is that antiholin-mediated inhibition or retardation of triggering reflects the stabilization of a normal dimer intermediate in the pathway to lethal function. Support for this idea is provided by the existence of multiple S lysis-defective alleles that are blocked at the dimer stage (Gründling et al., 2000). Similarly, many S21 non-lethal mutants are blocked at the dimer stage (T. Pang and R. Young, unpublished). However, until kinetic evidence from in vivo or in vitro experiments demonstrating a precursor-product relationship between dimers and the lethal holes is available, alternative scenarios in which both antiholin inhibition and mutational inactivation lead to off-pathway, “dead-end” dimers are also possible. Nevertheless, a potential rationale for the existence of dimers as a normal intermediate in hole formation may be inferred from consideration of S2168. According to the orientation map of the inactive dimer generated here, the relatively hydrophilic residues of TMD2 that ultimately face the lumen of the pinhole are sequestered against TMD1 in both cis and trans (Fig. 6A, B). Moreover, evidence provided here suggests that, in the normal pathway, there is a second form of the inactive dimer, in which only one TMD1 remains in the bilayer. The proposed orientation map would require the least rotational re-arrangement to allow continued sequestration of the future lumenal face, in this case by sequestration against the remaining embedded TMD1 and the other TMD2 (Fig. 6B). By this reasoning, the putative “activated” dimer, produced by the externalization of both TMD1s, would be much less able to sequester the lumenal residues, thus providing a driving force for oligomerization.
Influence of covalent dimers on pinholin timing
For the mutants G14C, T15C and S16C, inhibiting disulfide bond formation in the periplasm with DTT retards triggering. Three interpretations are possible, keeping in perspective the fact that TMD1 is non-essential for hole formation. One possibility is that TMD1 externalization is reversible and thus formation of the disulfide bond traps TMD1 in the periplasm. This seems unlikely, since the sec localization machinery, especially SRP (Signal Recognition Particle), is not available from the periplasm (Luirink et al., 2005). A simpler interpretation is that the covalent linkage stabilizes the activated dimer by simply eliminating potential dissociation of the TMD2s. Alternatively, the disulfide linkage between the perplasmic TMD1s might indirectly simply favor an orientation of TMD2 that facilitates progress down the pathway. Distinguishing between these general rationales will require an in vitro system to assess the monomer-dimer partition in a lipid bilayer.
Experimental procedures
Bacterial strains, plasmids, media and culture growth
Mutant selections and constructions were done in E.coli XL1-Blue cells (Smith & Young, 1998) carrying plasmid pS2168(RRzRz1)λ, which has the λ S gene and its Shine-Dalgarno sequence of the plasmid pS105 replaced by the S2168 gene and its upstream Shine-Dalgarno sequence. E.coli strain MG1655lacIq1tonA::Tn10 (Guyer et al., 1981) carrying plasmid pQ, which carries the gene for the λ late gene activator, Q, under Plac/ara-1 control, has been described (Gründling et al., 2001). Other E.coli strains, and plasmids bearing wildtype S2168, S2168ΔTMD1 and irsS2168 alleles have also been described previously (Pang et al., 2009). These plasmids have the entire phage 21 lysis cassette under its native late promoter. In each case, the phage 21 lysis genes R21, Rz21 and Rz121 are inactivated by nonsense mutations. The lysogen MDS12ΔtonA(λQ21Δ(SRRzRz1)21) carrying plasmid pS2168 or its derivatives was used for phenotypic analysis of the wildtype or mutant S2168 alleles. For S2168ΔTMD1 alleles, the E.coli strain MG1655lacIq1tonA::Tn10 carrying the compatible plasmids pQ and pS2168ΔTMD1 or its derivatives were used to express the wildtype or mutant S2168ΔTMD1 alleles. To test the function of irsS2168 mutant alleles, derivatives of plasmid pirsS2168* were used, which has the promoter and the entire lysis cassette region identical to pS2168, except that it has codons encoding the irs-epitope (RYIRS) fused to the N-terminus of S2168 gene, and it carries AmpR, instead of KanR. They were transformed into either the lysogen MDS12ΔtonA(λQ21Δ(SRRzRz1)21), or MDS12ΔtonA(λS2168) which provides a chromosomal copy of S2168.
Cultures were grown in standard Luria- Bertani (LB) media supplemented with antibiotics where needed: kanamycin (40 μg/ml), ampicillin (100 μg/ml), and chloramphenicol (10 μg/ml).
Plasmids carried by lysogens were induced by shifting the culture into 42°C for 15 m. Plasmid pQ was induced by 1 mM isopropyl-β-D-thiogalactopyranoside (IPTG) and 0.2% arabinose. Lysis profiles were obtained by monitoring A550 after thermal or IPTG/arabinose inductions, as described previously (Smith & Young, 1998, Tran et al., 2005). When needed, 1mM DTT was added into the culture at the time of induction.
EMS mutagenesis and screen for the S2168 lysis defective mutants
EMS mutagenesis was performed by the procedure of Miller (1992) using the recA− strain XL1-blue carrying monomeric pS2168(RRzRz1)λ. After exposure to EMS, cells were washed and plated on LB agar to measure cell viability. The degree of mutagenesis was estimated by the frequency of rifampicin-resistance mutants after outgrowth of the EMS-treated cells, as described by Miller (1992).
After overnight growth of the EMS-treated cells, plasmid DNA was recovered and transformed into MG1655lacIq1tonA::Tn10 (Guyer et al., 1981) carrying the plasmid pQ. A portion of the transformant pool was inoculated into 25 ml LB medium supplemented with Amp and the culture was grown to A550 ~0.2 and induced with IPTG and arabinose for 2 h. After induction, the cells were collected by centrifugation and plated on LB-Amp plates without inducer. Surviving colonies were inoculated into individual cultures, induced as above and then treated with 1% chloroform. Cultures that continued to grow after induction but underwent lysis after chloroform addition were presumed to have expressed the downstream λR endolysin gene and were likely to harbor a lysis-defective S2168 allele. The pS2168(RRzRz1)λ plasmid was recovered from these cultures and was re-transformed into fresh MG1655lacIq1tonA::Tn10 cells to confirm that the lysis defective phenotype segregated with the plasmid. All the candidate plasmids that passed this final test were subjected to DNA sequence analysis.
Each of the EMS mutations was re-created in plasmid pS2168. Each plasmid was transformed into lysogen MDS12ΔtonA(λQ21Δ(SRRzRz1)21), and the effect on the triggering time of S2168 was tested.
Site-directed mutagenesis, and general DNA manipulations
Procedures for plasmid DNA isolation, QuikChange (Stratagene) mutagenesis, cloning steps and DNA sequencing have been described (Smith et al., 1998). Oligonucleotides were obtained from Integrated DNA Technologies, Coralville, IA, and were used without further purification. All enzymes were purchased from New England Biolabs, with the exception of Pfu polymerase, which was from Stratagene. Automated fluorescent sequencing was performed at the Laboratory for Plant Genomic Technologies at Texas A&M University.
TCA precipitation, SDS-PAGE and western blotting
Ten percent TCA was used to immediately stop cell growth and precipitate protein as previously described (Park et al., 2006). Precipitates were resuspended in sample loading buffer with the addition of 5% β-mercaptoethanol, unless for the purpose of detecting the disulfide-bond linked dimer. SDS-PAGE and Western blotting were performed as described (Pang et al., 2009). Proteins were separated on 10% Tris-Tricine gels and transferred to 0.1 μm nitrocellulose membrane (Whatman, NJ). Antiserum against the S21 C-terminal peptide KIREDRRKAARGE was used as primary antibody (Barenboim et al., 1999). Blots were developed by using the Supersignal West Femto maximum sensitivity substrate kit (Thermo Scientific) according to the manufacturer’s instructions. Images were obtained by using the Molecular Imager Gel Doc DR system (Bio-Rad) and analyzed by software Quantity One (Bio-Rad).
Acknowledgments
This work was supported by Public Health Service Grant GM27099 to RY, by the Robert A. Welch Foundation, and by the Program for Membrane Structure and Function at Texas A&M University.
References
- Barenboim M, Chang CY, dib Hajj F, Young R. Characterization of the dual start motif of a class II holin gene. Mol Microbiol. 1999;32:715–727. doi: 10.1046/j.1365-2958.1999.01385.x. [DOI] [PubMed] [Google Scholar]
- Bläsi U, Chang CY, Zagotta MT, Nam K, Young R. The lethal λ S gene encodes its own inhibitor. EMBO Journal. 1990;9:981–989. doi: 10.1002/j.1460-2075.1990.tb08200.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bläsi U, Fraisl P, Chang CY, Zhang N, Young R. The C-terminal sequence of the lambda holin constitutes a cytoplasmic regulatory domain. J Bacteriol. 1999;181:2922–2929. doi: 10.1128/jb.181.9.2922-2929.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dewey JS, Savva CG, White RL, Vitha S, Holzenburg A, Young R. Micron-scale holes terminate the phage infection cycle. Proc Natl Acad Sci U S A. 2009;107:2219–2223. doi: 10.1073/pnas.0914030107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gründling A, Bläsi U, Young R. Genetic and biochemical analysis of dimer and oligomer interactions of the λ S holin. J Bacteriol. 2000;182:6082–6090. doi: 10.1128/jb.182.21.6082-6090.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gründling A, Manson MD, Young R. Holins kill without warning. Proc Natl Acad Sci USA. 2001;98:9348–9352. doi: 10.1073/pnas.151247598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guyer MS, Reed RR, Steitz JA, Low KB. Identification of a sex-factor-affinity site in E. coli as gamma delta. Cold Spring Harb Symp Quant Biol. 1981;45(Pt 1):135–140. doi: 10.1101/sqb.1981.045.01.022. [DOI] [PubMed] [Google Scholar]
- Luirink J, von Heijne G, Houben E, de Gier JW. Biogenesis of inner membrane proteins in Escherichia coli. Annu Rev Microbiol. 2005;59:329–355. doi: 10.1146/annurev.micro.59.030804.121246. [DOI] [PubMed] [Google Scholar]
- Miller JH. A short course in bacterial genetics: A laboratory manual and handbook for Escherichia coli and related bacteria. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, NY: 1992. [Google Scholar]
- Pang T, Savva CG, Fleming KG, Struck DK, Young R. Structure of the lethal phage pinhole. Proc Natl Acad Sci U S A. 2009;106:18966–18971. doi: 10.1073/pnas.0907941106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park T, Struck DK, Dankenbring CA, Young R. The pinholin of lambdoid phage 21: control of lysis by membrane depolarization. J Bacteriol. 2007;189:9135–9139. doi: 10.1128/JB.00847-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park T, Struck DK, Deaton JF, Young R. Topological dynamics of holins in programmed bacterial lysis. Proc Natl Acad Sci U S A. 2006;103:19713–19718. doi: 10.1073/pnas.0600943103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raab R, Neal G, Garrett J, Grimaila R, Fusselman R, Young R. Mutational analysis of bacteriophage lambda lysis gene S. J Bacteriology. 1986;167:1035–1042. doi: 10.1128/jb.167.3.1035-1042.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramanculov ER, Young R. An ancient player unmasked: T4 rI encodes a t - specific antiholin. Mol Microbiol. 2001;41:575–583. doi: 10.1046/j.1365-2958.2001.02491.x. [DOI] [PubMed] [Google Scholar]
- Smith DL, Struck DK, Scholtz JM, Young R. Purification and biochemical characterization of the lambda holin. J Bacteriol. 1998;180:2531–2540. doi: 10.1128/jb.180.9.2531-2540.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith DL, Young R. Oligohistidine tag mutagenesis of the lambda holin gene. J Bacteriol. 1998;180:4199–4211. doi: 10.1128/jb.180.16.4199-4211.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun J, Kaback HR. Proximity of periplasmic loops in the lactose permease of Escherichia coli determined by site-directed cross-linking. Biochemistry. 1997;36:11959–11965. doi: 10.1021/bi971172k. [DOI] [PubMed] [Google Scholar]
- Sun Q, Kuty GF, Arockiasamy A, Xu M, Young R, Sacchettini JC. Regulation of a muralytic enzyme by dynamic membrane topology. Nat Struct Mol Biol. 2009;16:1192–1194. doi: 10.1038/nsmb.1681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran TAT, Struck DK, Young R. Periplasmic domains define holin-antiholin interactions in T4 lysis inhibition. J Bacteriol. 2005;187:6631–6640. doi: 10.1128/JB.187.19.6631-6640.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang IN, Smith DL, Young R. Holins: the protein clocks of bacteriophage infections. Annu Rev Microbiol. 2000;54:799–825. doi: 10.1146/annurev.micro.54.1.799. [DOI] [PubMed] [Google Scholar]
- White R, Tran TA, Dankenbring CA, Deaton J, Young R. The N-terminal transmembrane domain of λ S is required for holin but not antiholin function. J Bacteriol. 2010;192:725–733. doi: 10.1128/JB.01263-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu M, Struck DK, Deaton J, Wang IN, Young R. The signal arrest-release (SAR) sequence mediates export and control of the phage P1 endolysin. Proc Natl Acad Sci USA. 2004;101:6415–6420. doi: 10.1073/pnas.0400957101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young R, Wang IN. Phage Lysis. In: Calendar R, editor. The Bacteriophages. Oxford: Oxford University Press; 2006. pp. 104–126. [Google Scholar]
- Young R, White RL. Lysis of the host by bacteriophage. In: Mahy BWJ, Van Regenmortel MHV, editors. Encyclopedia of Virology. 3. Boston: Academic Press; 2008. pp. 248–258. [Google Scholar]