

Abstract
Oxidative stress-related damage to the DNA macromolecule produces a multitude of lesions that are implicated in mutagenesis, carcinogenesis, reproductive cell death and aging. Many of these lesions have been studied and characterized by various techniques. Of the techniques that are available, the comet assay, HPLC-EC, GC-MS, HPLC-MS and especially HPLC-MS/MS remain the most widely used and have provided invaluable information on these lesions. However, accurate measurement of DNA damage has been a matter of debate. In particular, there have been reports of artifactual oxidation leading to erroneously high damage estimates. Further, most of these techniques measure the end product of a sequence of events and thus provide only limited information on the initial radical mechanism. We report here a qualitative measurement of DNA damage induced by a Cu(II)-H2O2 oxidizing system using immuno spin-trapping (IST) with EPR, MS and MS/MS. The radical generated is trapped by DMPO immediately upon formation. The DMPO adduct formed is initially EPR active but subsequently is oxidized to the stable nitrone, which can then be detected by IST and further characterized by MS and MS/MS.
Keywords: Spin trapping; DNA radicals; Copper; nucleosides; cells; ESR; IST; LC-MS/MS, detect; identify
Introduction
DNA is continuously exposed to exogenous and endogenous mutagens including reactive oxygen and nitrogen species that could alter its integrity [1-3]. Oxidatively generated DNA damage is an inevitable consequence of cellular metabolism. Injury to this macromolecule can have severe biological consequences including mutation, cell death, carcinogenesis and aging [4]. DNA lesions include strand breaks, DNA-protein or DNA-DNA crosslinks, abasic sites, and modified bases. Despite the abundance of oxidatively generated DNA damage, the products exist in a much larger background (105-106) of unaltered nucleosides which themselves may be prone to oxidation during sample preparation and analysis [5].
Major efforts have been devoted in the last several years to the development of accurate assays aimed at measuring oxidative base damage [6-11]. Several analytical methods have been developed to quantify modified bases in a research field that remains very challenging. Two different types of approaches have been developed for monitoring different damage to DNA. First, indirect approaches such as the alkaline comet assay [12] or the alkaline elution technique [13] are very sensitive methods for measuring DNA strand breaks [9,14]. The alkaline single gel electrophoresis assay, particularly in its basal version, is not able to measure defined types of DNA damage since strand breaks thus measured may consist of frank nicks together with abasic sites and several alkali-labile oxidized bases. However, the use of repair enzymes prior to the comet assay analysis allows the detection of classes of damage including oxidized pyrimidine bases and purine base modifications [15]. In spite of all of the above, these approaches still do not detect radicals, but only products thought to be formed from radicals. These methods are very sensitive, but they are inherently not specific [16] to free radical chemistry in that the free radical nature of the strand break is only inferred as in the case of radiation damage. A second approach, which requires the extraction of DNA, is very useful for studying oxidative base lesions. It uses GC-MS and HPLC-EC to measure 8-oxo-7,8-dihydro-2′-deoxyguanosine (8-oxodGuo), one of the most widely studied DNA-damage adducts. However, with GC-MS, the artifactual generation of oxidized bases during the derivatization steps, as well as the lack of stability of several oxidized base modifications, prevented accurate, valid measurements. These methods cannot detect the primary radical species and can also cause artifactual oxidation during workup, thus leading to erroneously high damage estimates. The levels of 8-oxodGuo reported in the literature are highly variable [17]. HPLC-EC is currently a more popular method for the detection of 8-oxodGuo, but this method has also been criticized on the grounds that the variability of the assay is unacceptable [5,18-20]. Its application in the oxidative detection mode is restricted to only a few electroactive DNA lesions having a low oxidation potential such as 8-oxodGuo. In addition to the lack of versatility, the assay suffers from insufficient sensitivity [9], which hinders the accurate measurement of low levels of DNA lesions.
A significant improvement in the measurement of oxidatively damaged DNA has been obtained by the use of HPLC coupled to MS and MS/MS. This assay combines the efficiency of HPLC separation with the sensitivity of mass spectrometry [9-11]. Consequently, this method has been gaining prominence and has the potential to overcome many of the limitations outlined earlier. However, HPLC-MS, which is about 50-fold less sensitive than HPLC-MS/MS, is not suitable for measuring the frequency of the lesions within the range of a few modifications per 107 to 109 normal nucleosides [11]. HPLC-MS/MS is now considered to be the gold standard for the purpose of studying oxidatively damaged DNA. It should be added that even HPLC-MS3 is necessary for the detection of modified nucleosides whose frequency is around a few lesions per 109 nucleosides [10,11].
Some immunological assays [21,22] have also been designed for the measurement of oxidative DNA base damage, but they have been plagued by a lack of specificity. Several attempts have been made to use antibodies raised against 8-oxodGuo, but it has been difficult to obtain a highly specific antibody. 8-oxodGuo differs from dGuo by a single oxygen atom, thereby challenging the specificity of the antibody. The method's applications have been limited by cross reactivity of the antibodies with normal DNA bases and other abundant biological constituents [23].
The study of primary radicals is critical to understanding DNA damage. However, the detection of these primary transients is difficult due to their short lifetimes. To address this difficulty, a method developed in our laboratory, immuno-spin trapping (IST), combines the specificity of spin trapping with the sensitivity of an antigen-antibody-based assay [24,25]. This method has been very successful in detecting radicals in a number of macromolecules such as proteins [26-32] and in DNA [33-36]. IST allows sensitive, reliable and economical detection of radical-generated damage. Its major limitation is that the radical is not identified by this procedure. Thus, an analytical question that is often posed is, “What is ELISA detecting? ” [23]. Because the detection of DNA is limited to dot blots and ELISA assays, the identification of a radical as DNA-derived and not a contaminant (protein-derived) depends on the absolute purity of the DNA as achieved by traditional DNA purification methods. The possible structural identification of DNA radicals detected by ELISA in previous work [33-36] is further complicated by the limited investigation of DNA radical adducts by ESR or the corresponding DNA nitrone adducts by MS. The use of LC/MS to detect DMPO adducts of radicals derived from nucleosides has been reported for radicals generated photochemically from the photolysis of 5-halo-2′-deoxyuridines, 5-thiophenylmethyl-2′-deoxyuridine and thymidine (which are well known photo precursors of nucleoside free radicals) with menadione bisulfite as the photosensitizer, and a new uridine radical was detected in RNA [37].
In our present work, coupling of the very sensitive IST with ESR, HPLC, MS and MS/MS provides a very useful technique for determining the primary DNA radical damage rather than the stable end product that forms after a possibly complex sequence of events. The initial DNA spin-trapped radical adducts are enzymatically digested to 2′-deoxyribonucleosides. The resulting digestion mixture is separated by reverse phase liquid chromatography and analyzed by MS and MS/MS, which allows the unequivocal identification of the radical trapped by the spin trap. Furthermore, because additional nitrone adduct formation is impossible once the DNA is separated from DMPO (even by dilution), artifactual DNA oxidation during extraction and subsequent hydrolysis cannot lead to nitrone adduct formation. Dilution of DMPO by 100-fold is sufficient to prevent artifactual nitrone formation because at DMPO concentration below 1mM, DNA radicals will decay before they can be trapped. No attempt has been made to quantitate DNA-DMPO adducts, which is beyond the scope of the present study.
Here we have studied the damage to DNA induced by a Cu(II)-H2O2 oxidizing system with the combined use of ESR, IST, HPLC, MS and MS/MS. These data presented here were reproduced with the Fe(II)- H2O2 system (the more classical Fenton hydroxyl radical generating system) as well. The hydroxyl radical reacts at diffusion-controlled rates with virtually any macromolecule including DNA. Using this combined approach, we have detected and identified a nitrone adduct on the 2′-deoxyadenosine moiety in nucleosides, pure DNA and cellular DNA.
Presentations and discussions have been limited to Cu(II)–H2O2-induced, oxidatively generated DNA damage, which is pathophysiologically important because copper associates with DNA bases in the nuclei of mammalian cells [38]. Copper-mediated ROS damage assumes significance due to the increased evidence of elevated levels of copper in tumor growth and angiogenesis [39-41]. There have been several reports of copper concentration being significantly higher in cancer patients [39-41], whereas the concentration of other elements like zinc, iron and selenium were significantly lower [39]. Copper chelation or copper depletion is under intense investigation for therapeutic purposes [39,40].
Experimental Procedures
Reagents
2′- Deoxycytidine (dCyd), 2′-deoxyguanosine (dGuo), thymidine (dThd), 2′-deoxyadenosine (dAdo) and inosine were purchased from MP Biomedicals (Irvine, CA). Calf thymus DNA, nuclease P1 (from Penicillium citrinum), 7-deazaadenosine and 3-deazaadenosine were obtained from Sigma Aldrich (St. Louis, MO). Snake venom phosphodiesterase was obtained from Worthington Chemicals (Freehold, NJ). Cupric chloride was purchased from Alfa Aesar (Ward Hill, MA). Calf intestinal alkaline phosphatase was purchased from Invitrogen. The 2′-deoxyadenosine isotopes (15N5) and (13C1015N5) were purchased from Cambridge Isotope Laboratories (Andover, MA). The spin trap DMPO was purchased from Alexis Biochemicals (San Diego, CA), purified twice by vacuum distillation at room temperature, and stored under argon at −80 °C. The DMPO concentration was measured at 228 nm assuming a molar absorption co-efficient of 7,800 M−1 cm−1. Hydrogen peroxide was obtained from Fisher Scientific Company (Fairlawn, NJ). The hydrogen peroxide concentration was verified using UV absorption at 240 nm (ε240nm = 43.6 M−1 cm−1). All buffers used were treated with Chelex 100 ion exchange resin (Bio-Rad Laboratories, Hercules, CA) to avoid transition metal-catalyzed reactions.
Chemical Reactions
Production of the DNA-nitrone adducts in nucleosides and calf thymus DNA
Typically, reaction mixtures contained 2.5-5 mM nucleoside, 300 μM CuCl2, 100 μM H2O2 and 100 mM DMPO in 100 mM chelex-treated phosphate buffer (pH 7.4) and were incubated for 1 hr at 37°C. For 2′-deoxyguanosine experiments, a saturated solution of the nucleoside was used. Calf thymus DNA (250 μg/mL) was reacted with CuCl2 (300 μM), H2O2 (100 μM) and 100 mM DMPO in chelex-treated 100 mM phosphate buffer (pH-7.4) and incubated at 37° C for 1 hr. For ELISA, the nitrone DNA reaction mixtures were diluted to 5 μg/mL of DNA in PBS. For MS analyses, standard nucleoside samples were diluted 200-300-fold with H2O just prior to analysis.
Production of the DNA-nitrone adducts in Raw-264.7 cells
The mouse macrophage cell line Raw 264.7 was grown and maintained in Dulbecco's modified eagle medium (DMEM) supplemented with 10% heat-inactivated fetal bovine serum, penicillin and streptomycin. Upon reaching 80% confluence, cells were counted and seeded at the needed density for treatment. Exponentially growing Raw cells (1-3 million/well) were seeded into 12 well-plates and treated with 100 μM CuCl2/100 μM H2O2 in the presence of DMPO for 12-15 hrs. at 37 °C in a humidified 5% CO2 environment. After this treatment, cell viability was determined using the trypan blue exclusion assay. Cells were found to be 80-90 % viable at this time point. The cells were pelleted and frozen at −80°C until DNA extraction and analysis of the DMPO-nitrone adducts.
DNA extraction and digestion procedure
Cell pellets stored at −80°C were re-suspended in digestion buffer with proteinase K, and DNA extraction was carried out as described previously [33,34]. The extraction procedure preserves the nitrone adduct covalently-bound to the DNA [33,34]. DNA concentration and purity were measured from the absorbance at 260 and 280 nm. Pure DNA will exhibit an A260/A280 ratio between 1.8 and 2. For digestion of both calf thymus and cell-derived DNA to the nucleosides, the DNA was suspended in 10 mM Tris buffer (pH 8.8) and 20 mM MgCl2 with nuclease P1 (2U), snake venom phosphodiesterase (2U) and calf intestinal alkaline phosphatase (2U) and incubated for 5-7 and 24 hrs. The hydrolyzed nucleosides were compared to the standard nucleosides by HPLC using both UV (λ = 254 nm) and MS detection. Just prior to MS analysis, the digested DNA samples were diluted 10-fold with water.
Immunoprecipitation of DNA-DMPO nitrone adducts from DNA
Immunoprecipitation of DNA-DMPO nitrone adducts was carried out with the Seize X Mammalian Immunoprecipitation Kit (Pierce, Rockford, IL) with some modifications. The samples were pre-cleared (1 hr at room temperature) with 200 μl of protein G slurry (50%). The purified DNA was incubated overnight with 25 μg of monoclonal anti-DMPO antibody, and the antigen-antibody mixture was then incubated overnight with the Protein G slurry. Immune complexes were eluted with elution buffer according to the manufacturer's instructions and collected in Effendorf tubes with 20 μl Tris buffer (pH 9.6).
Immuno-spin trapping analysis by ELISA
A standard ELISA procedure in 96 well plates (Greiner Labortechnik, Frickenhausen, Germany) was used as described in detail [34]. Briefly, DNA solutions were diluted to 5 μg/mL in 1× PBS. 25 μL of the DNA solution and 25 μL of Reacti-Bind DNA coating solution were placed in each well of the plate and the plate incubated for 2-4 hrs at 37 °C. The plates were washed once with washing buffer (1×PBS containing 0.05% non-fat dry milk and 0.1% Tween-20) and blocked with blocking buffer (1×PBS containing 3% non - fat dry milk) for 2 hr at 37 ° C or overnight at 4 ° C. After washing once with the washing buffer, the rabbit anti-DMPO serum (diluted 1:10,000 in the wash buffer) was added and incubated at 37 °C for 60 min. After 3 more washes, 100 μL of the secondary antibody (HRP- conjugated), diluted 1:10,000 in the wash buffer, were added and incubated for 60 min. After 3 washes, the antigen-antibody complexes were developed using Immobilon chemiluminescence substrate (Millipore Corp., Billerica, MA), and the light emitted was recorded as arbitrary light units using X-Fluor software (Tecan US, Research Triangle Park, NC).
ESR spin-trapping experiments
Upon mixing, the reaction mixture was immediately transferred to a flat cell and the ESR scan initiated within 1 min after starting the reaction. ESR spectra were obtained with an ELEXSYS E500 ESR spectrometer (Bruker Biospin, Billerica, MA) equipped with an ER4122SHQ cavity operating at 9.76 GHz and at room temperature. The ESR spectrometer settings were as follows: scan range, 100 G; modulation frequency, 100 kHz; modulation amplitude, 1.0 G; microwave power, 20 mW; receiver gain, 2 × 104; time constant, 327 ms; and conversion time, 327 ms.
Mass spectrometry analysis
Ion trap analysis
On-line HPLC-ESI/MS-MS measurements were carried out using an Agilent 1100 HPLC system (Palo Alto, CA) with a photo diode array detector. Separations were performed on a Waters C18 column (3 μm, 2.1×150 mm). The elution was achieved at a flow rate of 200 μL/min using a gradient mode of acetonitrile in 5 mM ammonium acetate from 0% - 40 % in 30 min. The HPLC was coupled to a Finnigan LCQ ion trap mass spectrometer (West Palm Beach, FL) equipped with an electrospray ionization probe and Finnigan Xcalibur software. The column eluent was split between the UV and the mass detectors and analyzed in the positive ion mode. This configuration was used for the UV (λ = 254 nm) and mass detection of standard and DNA digested nucleosides only.
Q-TOF analysis
LC/ESI/MS/MS analyses were performed using a Waters Q-Tof Premier hybrid mass spectrometer equipped with a nanoAcquity UPLC system (Milford, MA). LC analyses were performed as follows: 5 μL injections were loaded onto a Waters Symmetry™ C18 trapping column (20 mm × 180 μm id), followed by a 100 mm × 100 μm id Waters Atlantis™ dC18 column. Specifically, separations were performed by first trapping for 10 minutes at a flow rate of 5 μL/min, followed by linear gradients of 2-40% (acetonitrile/0.1% FA v/v) over 40 min and 40-95% over 5 min at a flow rate of 300 nL/min. The Q-Tof instrument is equipped with a nano-lockspray source, thereby resulting in a calibration with a measured mass accuracy of 5 ppm or less. Capillary and cone voltages were set to ~4 kV and 45 V, respectively. Fragmentation of oligonucleotides was accomplished using collision energy of 25 V. Data processing and MS/MS interpretation were performed manually using MassLynx software (Milford, MA).
Results
Free nucleosides as targets of oxidation: Nucleoside-DMPO adduct
ESR spin trapping
ESR studies were carried out for each of the 2′-deoxyribonucleosides (2′-deoxyguanosine, 2′-deoxyadenosine, thymidine and 2′-deoxycytidine). When 5 mM (or saturated, for deoxyguanosine) solutions of the nucleosides were reacted with CuCl2 (300 μM), H2O2 (100 μM) and 100 mM (DMPO), a radical adduct formation was observed only with 2′-deoxyadenosine. Under our experimental conditions, 2′-deoxyadenosine produced an ESR-detectable DMPO/dAdo· radical adduct (Fig. 1A) that was inhibited in the presence of the copper chelator DTPA (Fig. 1B). The presence of all the components was required for adduct formation. In the absence of the 2′-deoxyadenosine, a DMPO/·OH adduct was detected as a result of the spin trapping of the generated ·OH [42].
Fig 1. Formation of the DMPO/dAdo radical adduct from the reaction of dAdo with CuCl2-H2O2 in the presence of DMPO. All components of the reaction must be present for the adduct formation.

(A). Typically nucleosides (300μM) were reacted with CuCl2 (300μM), H2O2 (100μM) and DMPO (100mM) and spectra recorded immediately on mixing. Instrumental conditions: modulation amplitude1.0G; time constant, 327ms; receiver gain, 2 ×104; and microwave power, 20mW, (B) in the presence of the metal chelator DTPA, adduct formation was not observed, (C-F) are the various control experiments where one of the components is not present. Details are discussed in the results section.
The DMPO/·OH adduct was suppressed when either Cu(II) or H2O2 was absent from the system [42]. Without 2′-deoxyadenosine in the reaction mixture, background nitroxide species were formed which were subtracted from the species formed in the presence of dAdo. The spectrum consists of three species, one with a nitrogen-centered adduct with aN1=15.4, aN2=2.6, and , implying the formation of a radical adduct formed by the dAdo-derived nitrogen radical reacting with the spin trap DMPO. The spectrum also contains two non-2′-deoxyadenosine-derived components: the DMPO/·OH adduct with aN=14.9 and and one minor species with aN=14.6 and aH=1.0 attributed to DMPO ring opening [43]. The simulated spectrum fits well with the experimental spectrum and is the sum of the three species above (Fig. 2). Similar DMPO radical adduct formation was observed with adenine and 5′-AMP (adenosine 5′-monophosphate). In experiments with inosine, where the exocyclic NH2 is replaced by a keto group, adduct formation was not detected (data not shown). Because of the absence of a substrate-derived radical from inosine and the hyperfine splitting constant observed, the 2′-deoxyadenosine radical was assigned to the exocyclic NH2 according to the literature [44].No significant adduct formation was detected from any other nucleosides. When we did similar experiments with nitrogen-saturated solutions, similar results were obtained. ESR experiments using the Fe(II)-H2O2oxidizing system were also studied and similar data were obtained (data are presented in the supplemental section).
Fig 2. Composite ESR spectra of A (all components of reaction mixture present)-C (dAdo absent). Computer simulation and the deconvolution of the ESR spectrum.

The spectrum obtained is a sum of three radical adducts as indicated. It consists of one component with aN1=15.4, aN2=2.6, , implying the formation of a radical adduct formed by the dAdo-derived nitrogen radical and also contains two non-2′-deoxyadenosine-derived components: the DMPO/·OH adduct with aN=14.9 and =14.9 and one minor species with aN=14.6, aH=1.0 attributed to DMPO ring opening
Mass spectrometric analysis
The four nucleoside reaction mixtures were also analyzed by LC/MS, and the corresponding (M+H) + ions in each of the reaction mixtures were observed. In the LC/MS analysis of the reaction mixture containing 2′-deoxyadenosine, an abundant ion of m/z 363.16 corresponding in mass to the DMPO-dAdo nitrone adduct of 2′-deoxyadenosine [theoretical (M+H) + = 363.31)] was observed in addition to an ion of m/z 252.09, which corresponds in mass to unmodified 2′-deoxyadenosine. Fig. 3A shows the extracted ion chromatograms of these two ions while the corresponding mass spectra at 7.0 min (m/z 252.1) and 34.6 min (m/z 363.2) are shown in Fig. 3B and C, respectively. The ion of m/z 363.16 corresponding to the DMPO-dAdo nitrone adduct was not observed in the control experiments (where one of the components was missing). The MS/MS analyses of the m/z 363.2 and m/z 252.1 ions are shown in Fig 4. The MS/MS spectrum of the unmodified dAdo (m/z 252.1) in Fig. 4A shows a base-derived BH2+ fragment ion of m/z 136.06, corresponding in mass to the loss of 2-deoxyribose (−116 Da), and sugar-derived ions labeled as S+ and SH2+ of m/z 117.06 and 119.04, respectively. The MS/MS spectrum of the protonated molecule of m/z 363.2 (DMPO-dAdo nitrone adduct, Fig 4B) shows an abundant fragment ion of m/z 247.13, a base-derived BH2+ ion corresponding in mass to the loss of the 2-deoxyribose (−116 Da). In addition, an ion corresponding to the DMPO moiety was observed at m/z 111.10. The observation of this ion demonstrates that DMPO adduct formation has occurred and that DMPO trapped the dAdo radical. The fragment ions of m/z 117.06 and 119.04 correspond in mass to the S+ and SH2+ ions. The ion of m/z 229.12 corresponds in mass to the loss of H2O from the BH2+ ion. Using a variety of labeled 2′-deoxyadenosine compounds and deazaadenosine compounds, we determined that the ions of m/z 94.07, 97.03, 137.05, and 172.06 are the result of cross-ring cleavages and rearrangements. MS nomenclatures used are in accordance with a previous publication [45]. A similar pattern of fragmentation across the base-sugar bond was observed in the MS/MS data of both the unmodified dAdo ion and the DMPO adduct of 2′-deoxyadenosine. When MS and MS/MS of inosine was carried out, no adduct formation was detected. No ions corresponding in mass to the other nucleosides plus DMPO were observed. MS and MS/MS experiments using the Fe(II)-H2O2 oxidizing system were also conducted and similar data were observed (data are presented in the supplemental section). The adduct was absent in the control without DMPO and the Fe(II) /H2O2 control. A trace amount of the adduct was formed when DMPO and one of the reactants was present in the system.
Fig 3. MS data obtained from the reaction of deoxyadenosine with CuCl2/H2O2 in the presence of DMPO.



(A) EIC of the unmodified 2′-deoxyadenosine (m/z 252.1) and the radical adduct of 2′-deoxyadenosine (m/z 363.2) (B) Corresponding mass spectra at 7 min (C) and at 34.6 min.
Fig 4. MS/MS of the ions from the reaction of dAdo with CuCl2/H2O2 in the presence of DMPO.


(A) MS/MS of the (M+H)+ with m/z 252.09 (B) and the radical adduct ion with m/z 363.16
DNA (calf thymus) as a target of oxidation: DNA-DMPO adduct
To determine whether radical adducts could be observed in DNA, the Cu(II)-H2O2 Fenton reaction was repeated using calf thymus DNA. The ESR of DNA under similar experimental conditions used for the nucleosides yielded no assignable spectrum.
Immuno-spin trapping analysis
Because ELISA is much more sensitive than ESR, the calf thymus reaction mixture was analyzed by ELISA using the anti-DMPO antibody. A DNA-derived DMPO radical adduct was obtained, indicating the presence of a radical intermediate trapped by DMPO. Production of DNA nitrone adducts required the presence of all components of the reaction mixture (Fig. 5).
Fig 5. Immunological detection of the DNA-derived radical adduct from the reaction of calf thymus DNA with CuCl2 /H2O2 in the presence of DMPO.

Reaction mixtures were incubated at 37°C for 1 hr and then analyzed as described in experimental procedures with IST detection. No significant adduct formation was observed in the control experiments where one of the components was omitted. All components of the reaction must be present for DNA-adduct formation.
Mass spectrometric analysis
In order to determine the nature of the DMPO adducts which had occurred in the calf thymus DNA subjected to oxidation by Cu(II)-H2O2 Fenton chemistry, we performed LC/MS and MS/MS analyses of digested samples of both reacted and control DNA. DNA was digested as described in the experimental procedure section. Comparison of the HPLC (λ = 254nm) and MS detection confirmed the release of the nucleosides from DNA (data not shown). From the LC/MS and MS/MS analyses, ions corresponding to 2′-deoxyadenosine and DMPO-modified 2′-deoxyadenosine were observed. No ions corresponding in mass to the other nucleosides plus DMPO were observed. The extracted ion chromatograms corresponding in mass to the dAdo (m/z 252.1) and the DMPO-dAdo nitrone adduct (m/z 363.2) are shown in Fig. 6A (panels 3 and 4). The ion corresponding to the DMPO adduct was not observed in the unreacted control DNA subjected to enzymatic digestion (Fig. 6A, panels 1 and 2). No DMPO-adduct formation was observed from any other control samples. The corresponding mass spectra for the peaks at 6.78 min and 33.93 min were observed at the same retention times as in the 2′-deoxyadenosine reaction mixture (Fig. 3A). The corresponding MS/MS fragmentation spectra of the ion of m/z 252.1 and 363.2 are shown in Fig. 6B. For the unmodified 2′-deoxyadenosine, fragments corresponding to cleavage between the base and the sugar were observed (BH2+ and S+). Comparison of the MS/MS analysis of the ion of m/z 363.2 from the calf thymus DNA mixture (Fig. 6B) with the 2′-deoxyadenosine reaction mixture (Fig. 4B) confirmed its identity. The observation of the ion corresponding to DMPO (111.09 Da) demonstrated that DMPO adduct formation had occurred on 2′-deoxyadenosine. The loss of the 2-deoxyribose sugar moiety (−116 Da) indicated that a DMPO-nucleoside adduct was formed.
Fig 6. MS data obtained from the reaction of calf thymus DNA with CuCl2-H2O2 in the presence of DMPO.


(A) EIC of m/z 252.1 (unmodified 2′-deoxyadenosine) and of m/z 363.2 (DMPO-2′-deoxyadenosine) Panels 1 and 2 show the EIC obtained from the LC/MS of the control DNA (no DMPO present). Note the absence of the adduct ion. Panels 3 and 4 show the EIC's obtained from the DNA subjected to oxidation in the presence of DMPO. Note the presence of the adduct ion. Inset, shows the HPLC chromatogram, of the standard nucleosides and released nucleosides from the DNA digestion.(B) MS/MS of the ion of m/z 252.09 and m/z 363.16.
DNA (cellular) as a target of oxidation: DNA-DMPO adduct
To determine whether DNA radical adducts could be formed in cells, Raw cells 264.7 were treated with CuCl2 and H2O2 in the presence of DMPO. Cellular DNA was extracted following the treatment and analyzed initially by immuno-spin trapping (data not shown).
Immuno-spin trapping analysis
Production of DNA nitrone adducts required the presence of all components of the reaction mixture in the cells. A DNA-derived DMPO radical adduct was obtained, indicating the presence of a radical intermediate.
Mass spectrometric analysis
To enrich the extracted DNA samples for DMPO adducts, the DNA was digested and then subjected to immunoprecipitation using the anti-DMPO antibody. The samples were then analyzed by LC/MS. Fig. 7A shows the extracted ion chromatograms for the ions corresponding in mass to the dAdo (m/z 252.1) and the DMPO-dAdo nitrone adducts (m/z 363.2). The ion of m/z 363.2 was not observed in the LC/MS analysis of the control cells (data not shown). The MS/MS data for these two ions are shown in Fig. 7B. The fragmentation spectra of these ions are comparable to the fragmentation spectra of the same ions obtained from the adenosine reaction mixture (Fig. 4). The observation of the ion of m/z 111.08 corresponded in mass to DMPO (−111 Da), thus demonstrating that DMPO adduct formation has occurred. The loss of the deoxyribose sugar moiety suggested that the DMPO-nucleoside adduct formation occurred in the cellular DNA, similar to the calf thymus DNA.
Fig 7. MS data obtained from the reaction of RAW 264.7 cells treated with CuCl2/H2O2 in the presence of DMPO.


(A) EIC of unmodified 2′-deoxyadenosine of m/z 252.1 and m/z 363.2 from cellular DNA. (B) MS/MS of the ion of m/z 252.09 and m/z 363.16
Discussion
Oxidative processes in DNA may lead to several types of DNA modifications including chain breaks, abasic sites, DNA-protein crosslinks, purine-reactive aldehyde adducts and oxidized DNA bases [7,46,47]. As discussed in earlier reviews [5,9-11,17], considerable effort has been devoted to the development of reliable assays aimed at measuring oxidative damage in DNA. It has been unambiguously shown that unrepaired oxidized base lesions can either cause mutations or block DNA replication [48,49]. The measurement of oxidized bases and nucleosides in DNA may be used to gain insights into the nature and importance of the chemical reactions that modify DNA. A significant improvement for the measurement of oxidative damage to DNA has been obtained by the use of HPLC coupled to MS and MS/MS.
The objective of our present work was to combine the very sensitive IST technique, which has been shown to be a quick method for DNA damage detection [34,35], with ESR, MS and MS/MS to identify spin-trapped DNA adducts. We first studied standard nucleosides in an effort to detect and identify the exact location of the radical trapped by the spin trap and previously not structurally identified. Next, calf thymus and cellular DNA were reacted with hydroxyl radicals generated by the Cu(II)-H2O2 oxidizing system. Due to the presence of high DMPO concentrations, the radicals thus generated are trapped by DMPO immediately upon formation to generate DMPO radical adducts. Conversely, during the extraction and digestion steps the DMPO concentration is very low, which eliminates the generation of artifacts. While the DNA-DMPO adducts are initially EPR active, they are subsequently oxidized to more stable nitrone adducts. These stable adducts can then be detected by immuno-spin trapping [25,33,34] and further characterized by mass spectrometry. Based on MS analyses, ions corresponding in mass to 2′-deoxyadenosine and a DMPO adduct of 2′-deoxyadenosine were observed. In addition, MS/MS analyses allowed for the confirmation of the DMPO-adenosine adduct (Fig. 4B, 6B and 7B). Specifically, MS/MS analysis of the ion at m/z 363.2 (corresponding in mass to the DMPO-dAdo nitrone adduct) produced a fragment ion of m/z 247.11, thereby indicating the loss of the deoxyribose group (−116 Da). The loss of the sugar moiety suggests that the deoxyribose moiety is intact and, thus, is not the site of DMPO attachment. In addition, the observation of the ion of m/z 111.11 demonstrates that the DMPO adduct formation has occurred. The detection of the neutral loss of 116 Da due to deoxyribose is very specific, and ensures that only compounds derived from 2′-deoxyribonucleosides are detected [37] and excludes that the trapped radical is on the sugar group. MS and MS/MS of related compounds confirmed the exocyclic nitrogen as the site of radical formation.
In this paper, we report for the first time the characterization of a DNA-DMPO-nitrone adduct from a DNA radical generated via the Cu(II)-H2O2 oxidation chemistry. The LC-MS/MS analysis of the nucleoside-DMPO nitrone adducts is made possible due to the higher stability of the nitrones relative to radical adducts. In this work, an adenosine-DMPO adduct was characterized. It is probable that other nucleoside-derived radicals are formed, but in our studies using ESR and MS, only one adduct was detected with DMPO spin trapping. Although sugar radicals are formed [50, 51], we did not detect any DMPO-sugar adduct. Our data clearly suggest the formation of the DMPO-dAdo nitrone adduct in nucleoside, calf thymus and in cellular DNA. ESR experiments strongly suggest the formation of the radical on the exocyclic NH2 (radical 1 below). This aminyl radical arises from the dehydration of the hydroxyl radical adduct at C4 of the purine moiety of the 2′-deoxyadenosine. MS and MS/MS of related compounds were carried out extensively to rule out detectable radical formation at any other sites of dAdo. 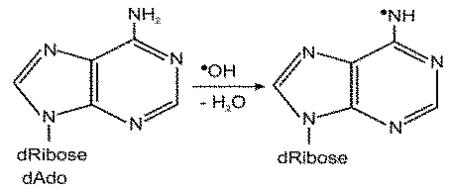 The efficiency of DMPO trapping of this radical may be due to the more sterically accessible exocyclic NH2-derived adduct and/or product stability (the exocyclic adduct has an intact aromatic ring). This trapped radical may assume significance in DNA as this redox–neutral radical may be relatively stable [52], leading to A(adenine)-mediated hole migration to adjacent bases[53]. It has been predicted by hole-trapping researchers that the adenine radical cation contributes to the hole-transfer (HT) process through A/T sequences and exists as a real chemical intermediate [54-58]. One-electron oxidation of DNA results in migration and localization of the electron loss center to guanine [53]. Our data clearly indicate that an adenosine radical intermediate is indeed formed.
The efficiency of DMPO trapping of this radical may be due to the more sterically accessible exocyclic NH2-derived adduct and/or product stability (the exocyclic adduct has an intact aromatic ring). This trapped radical may assume significance in DNA as this redox–neutral radical may be relatively stable [52], leading to A(adenine)-mediated hole migration to adjacent bases[53]. It has been predicted by hole-trapping researchers that the adenine radical cation contributes to the hole-transfer (HT) process through A/T sequences and exists as a real chemical intermediate [54-58]. One-electron oxidation of DNA results in migration and localization of the electron loss center to guanine [53]. Our data clearly indicate that an adenosine radical intermediate is indeed formed.
In summary, with our novel method of combining IST with ESR and mass spectrometry, we have confirmed that DMPO traps a radical adduct from DNA in accordance with published ELISA studies [6, 7]. We have been able to trap and identify a DMPO adduct in cellular DNA. This methodology provides an unequivocal site-specific assignment of radical formation during DNA damage induced by the Cu(II)-H2O2 system. The Cu(II)–H2O2 oxidatively damages DNA and is pathophysiologically important [38-41]. The radical produced when calf thymus and cellular DNA were treated with this oxidizing system was observed on the adenosine (rather than on 2′-deoxyguanosine), which may be an intermediate in a sequence of events leading to the formation of 8-oxodGuo and other products such as 8-oxo-7,8-dihydro-2′-deoxyadenosine (8-oxodAdo) . The immuno-spin trapping technique, which requires only simple and inexpensive equipment, is a sensitive and reliable tool for detecting DNA damage. However, the complexity of the free radical chemistry makes it prudent that the combination of ESR, IST, LC/MS and MS/MS be used before structural and mechanistic conclusions are drawn. Although still only qualitative in nature, this method provides a means to detect primary radicals in nucleosides, DNA and in cells.
Supplementary Material
Acknowledgements
We gratefully acknowledge, Jean Corbett for her unconditional help during all stages of the project. We thank Dr. Ann Motten and Mary Mason for their invaluable help in preparing this manuscript. We are very thankful to Dr. Mike P. Waalkes and Dr. Birandra Sinha for reviewing the manuscript. We thank Chrissy Pratter for the technical help. This work has been supported by the Intramural Research Program of the National Institutes of Health and the National Institute of Environmental Health Sciences.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Lindahl T. Instability and decay of the primary structure of DNA. Nature. 1993;362:709–715. doi: 10.1038/362709a0. [DOI] [PubMed] [Google Scholar]
- 2.Ames BN, Shigenaga MK, Gold LS. DNA lesions, inducible DNA repair, and cell division: three key factors in mutagenesis and carcinogenesis. Environ. Health Perspect. 1993;101(Suppl. 5):35–44. doi: 10.1289/ehp.93101s535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cadet J, Douki T, Gasparutto D, Ravanat JL. Oxidative damage to DNA: formation, measurement and biochemical features. Mutat. Res. 2003;531:5–23. doi: 10.1016/j.mrfmmm.2003.09.001. [DOI] [PubMed] [Google Scholar]
- 4.Halliwell B, Gutteridge JMC. Free radicals in biology and medicine. Oxford Science; Oxford: 1999. [Google Scholar]
- 5.Beckman KB, Ames BN. Oxidative decay of DNA. J. Biol. Chem. 1997;272:19633–19636. doi: 10.1074/jbc.272.32.19633. [DOI] [PubMed] [Google Scholar]
- 6.Dizdaroglu M. Chemical determination of free radical-induced damage to DNA. Free Radic. Biol. Med. 1991;10:225–242. doi: 10.1016/0891-5849(91)90080-m. [DOI] [PubMed] [Google Scholar]
- 7.Cadet J, Berger M, Douki T, Ravanat JL. Oxidative damage to DNA: formation, measurement, and biological significance. Rev. Physiol. Biochem. Pharmacol. 1997;131:1–87. doi: 10.1007/3-540-61992-5_5. [DOI] [PubMed] [Google Scholar]
- 8.Cadet J, Weinfeld M. Detecting DNA damage. Anal. Chem. 1993;65:675A–682A. doi: 10.1021/ac00063a001. [DOI] [PubMed] [Google Scholar]
- 9.Cadet J, Douki T, Frelon S, Sauvaigo S, Pouget JP, Ravanat JL. Assessment of oxidative base damage to isolated and cellular DNA by HPLC-MS/MS measurement. Free Radic. Biol. Med. 2002;33:441–449. doi: 10.1016/s0891-5849(02)00820-1. [DOI] [PubMed] [Google Scholar]
- 10.Cadet J, Poulsen H. Measurement of oxidatively generated base damage in cellular DNA and urine. Free Radic. Biol. Med. 2010;48:1457–1459. doi: 10.1016/j.freeradbiomed.2010.03.004. [DOI] [PubMed] [Google Scholar]
- 11.Cadet J, Douki T, Ravanat JL. Oxidatively generated base damage to cellular DNA. Free Radic. Biol. Med. 2010;49:9–21. doi: 10.1016/j.freeradbiomed.2010.03.025. [DOI] [PubMed] [Google Scholar]
- 12.Olive PL, Banath JP, Durand RE. Heterogeneity in radiation-induced DNA damage and repair in tumor and normal cells measured using the “comet” assay. Radiat. Res. 1990;122:86–94. [PubMed] [Google Scholar]
- 13.Swenberg JA, Petzold GL, Harbach PR. In vitro DNA damage/alkaline elution assay for predicting carcinogenic potential. Biochem. Biophys. Res. Commun. 1976;72:732–738. doi: 10.1016/s0006-291x(76)80100-3. [DOI] [PubMed] [Google Scholar]
- 14.Frelon S, Douki T, Ravanat JL, Pouget JP, Tornabene C, Cadet J. High-performance liquid chromatography-tandem mass spectrometry measurement of radiation-induced base damage to isolated and cellular DNA. Chem. Res. Toxicol. 2000;13:1002–1010. doi: 10.1021/tx000085h. [DOI] [PubMed] [Google Scholar]
- 15.Collins AR, Oscoz AA, Brunborg G, Gaivao I, Giovannelli L, Kruszewski M, Smith CC, Stetina R. The comet assay : topical issues. Mutatgenesis. 2008;23:143–151. doi: 10.1093/mutage/gem051. [DOI] [PubMed] [Google Scholar]
- 16.Collins AR, Dobson VL, Dusinska M, Kennedy G, Stetina R. The comet assay: what can it really tell us? Mutat. Res. 1997;375:183–193. doi: 10.1016/s0027-5107(97)00013-4. [DOI] [PubMed] [Google Scholar]
- 17.Poulsen HE, Weimann A, Loft S. Methods to detect DNA damage by free radicals: relation to exercise. Proc. Nutr. Soc. 1999;58:1007–1014. doi: 10.1017/s0029665199001329. [DOI] [PubMed] [Google Scholar]
- 18.Adachi S, Zeisig M, Moller L. Improvements in the analytical method for 8-hydroxydeoxyguanosine in nuclear DNA. Carcinogenesis. 1995;16:253–258. doi: 10.1093/carcin/16.2.253. [DOI] [PubMed] [Google Scholar]
- 19.Nakae D, Mizumoto Y, Kobayashi E, Noguchi O, Konishi Y. Improved genomic/nuclear DNA extraction for 8-hydroxydeoxyguanosine analysis of small amounts of rat liver tissue. Cancer Lett. 1995;97:233–239. doi: 10.1016/0304-3835(95)03980-b. [DOI] [PubMed] [Google Scholar]
- 20.Nakajima M, Takeuchi T, Morimoto K. Determination of 8-hydroxydeoxyguanosine in human cells under oxygen-free conditions. Carcinogenesis. 1996;17:787–791. doi: 10.1093/carcin/17.4.787. [DOI] [PubMed] [Google Scholar]
- 21.Musarrat J, Wani AA. Quantitative immunoanalysis of promutagenic 8-hydroxy-2′-deoxyguanosine In oxidized DNA. Carcinogenesis. 1994;15:2037–2043. doi: 10.1093/carcin/15.9.2037. [DOI] [PubMed] [Google Scholar]
- 22.Toyokuni S, Tanaka T, Hattori Y, Nishiyama Y, Yoshida A, Uchida K, Hiai H, Ochi H, Osawa T. Quantitative immunohistochemical determination of 8-hydroxy-2′-deoxyguanosine by a monoclonal antibody N45.1: its application to ferric nitrilotriacetate-induced renal carcinogenesis model. Lab. Invest. 1997;76:365–374. [PubMed] [Google Scholar]
- 23.Cooke MS. A commentary on “Urea, the most abundant component in urine, cross-reacts with a commercial 8-OH-dG ELISA kit and contributes to overestimation of urinary 8-OH-dG”. What is ELISA detecting? Free Radic. Biol. Med. 2009;47:30–31. doi: 10.1016/j.freeradbiomed.2009.04.003. [DOI] [PubMed] [Google Scholar]
- 24.Detweiler CD, Deterding LJ, Tomer KB, Chignell CF, Germolec D, Mason RP. Immunological identification of the heart myoglobin radical formed by hydrogen peroxide. Free Radic. Biol. Med. 2002;33:364–369. doi: 10.1016/s0891-5849(02)00895-x. [DOI] [PubMed] [Google Scholar]
- 25.Mason RP. Using anti-5,5-dimethyl-1-pyrroline N-oxide (anti-DMPO) to detect protein radicals in time and space with immuno-spin trapping. Free Radic. Biol. Med. 2004;36:1214–1223. doi: 10.1016/j.freeradbiomed.2004.02.077. [DOI] [PubMed] [Google Scholar]
- 26.Ramirez DC, Chen YR, Mason RP. Immunochemical detection of hemoglobin-derived radicals formed by reaction with hydrogen peroxide: involvement of a protein-tyrosyl radical. Free Radic. Biol. Med. 2003;34:830–839. doi: 10.1016/s0891-5849(02)01437-5. [DOI] [PubMed] [Google Scholar]
- 27.Ehrenshaft M, Mason RP. Protein radical formation on thyroid peroxidase during turnover as detected by immuno-spin trapping. Free Radic. Biol. Med. 2006;41:422–430. doi: 10.1016/j.freeradbiomed.2006.02.023. [DOI] [PubMed] [Google Scholar]
- 28.Deterding LJ, Ramirez DC, Dubin JR, Mason RP, Tomer KB. Identification of free radicals on hemoglobin from its self-peroxidation using mass spectrometry and immuno-spin trapping: observation of a histidinyl radical. J. Biol. Chem. 2004;279:11600–11607. doi: 10.1074/jbc.M310704200. [DOI] [PubMed] [Google Scholar]
- 29.Bhattacharjee S, Deterding LJ, Jiang J, Bonini MG, Tomer KB, Ramirez DC, Mason RP. Electron transfer between a tyrosyl radical and a cysteine residue in hemoproteins: spin trapping analysis. J. Am. Chem. Soc. 2007;129:13493–13501. doi: 10.1021/ja073349w. [DOI] [PubMed] [Google Scholar]
- 30.Bonini MG, Siraki AG, Bhattacharjee S, Mason RP. Glutathione-induced radical formation on lactoperoxidase does not correlate with the enzyme's peroxidase activity. Free Radic. Biol. Med. 2007;42:985–992. doi: 10.1016/j.freeradbiomed.2006.12.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ranguelova K, Suarez J, Magliozzo RS, Mason RP. Spin trapping investigation of peroxide- and isoniazid-induced radicals in Mycobacterium tuberculosis catalase-peroxidase. Biochemistry. 2008;47:11377–11385. doi: 10.1021/bi800952b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chatterjee S, Ehrenshaft M, Bhattacharjee S, Deterding LJ, Bonini MG, Corbett J, Kadiiska MB, Tomer KB, Mason RP. Immuno-spin trapping of a post-translational carboxypeptidase B1 radical formed by a dual role of xanthine oxidase and endothelial nitric oxide synthase in acute septic mice. Free Radic. Biol. Med. 2009;46:454–461. doi: 10.1016/j.freeradbiomed.2008.10.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ramirez DC, Gomez-Mejiba SE, Mason RP. Immuno-spin trapping of DNA radicals. Nat. Methods. 2006;3:123–127. doi: 10.1038/nmeth852. [DOI] [PubMed] [Google Scholar]
- 34.Ramirez DC, Gomez-Mejiba SE, Mason RP. Immuno-spin trapping analyses of DNA radicals. Nat. Protoc. 2007;2:512–522. doi: 10.1038/nprot.2007.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kojima C, Ramirez DC, Tokar EJ, Himeno S, Drobna Z, Styblo M, Mason RP, Waalkes MP. Requirement of arsenic biomethylation for oxidative DNA damage. J. Natl. Cancer Inst. 2009;101:1670–1681. doi: 10.1093/jnci/djp414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ogusucu R, Rettori D, Netto LES, Augusto O. Superoxide dismutase 1-mediated production of ethanol- and DNA-derived radicals in yeasts challenged with hydrogen peroxide. Molecular insights into the genome instability of peroxiredoxin-null strains. J. Biol. Chem. 2009;284:5546–5556. doi: 10.1074/jbc.M805526200. [DOI] [PubMed] [Google Scholar]
- 37.Maurel V, Ravanat JL, Gambarelli S. Detection of reactive free radicals derived from nucleosides by liquid chromatography coupled to tandem mass spectrometry of DMPO spin trapping adducts. Rapid Commun. Mass Spectrom. 2006;20:2235–2242. doi: 10.1002/rcm.2579. [DOI] [PubMed] [Google Scholar]
- 38.Imlay JA, Chin SM, Linn S. Toxic DNA damage by hydrogen peroxide through the Fenton reaction in vivo and in vitro. Science. 1988;240:640–642. doi: 10.1126/science.2834821. [DOI] [PubMed] [Google Scholar]
- 39.Gupte A, Mumper RJ. Elevated copper and oxidative stress in cancer cells as a target for cancer treatment. Cancer Treatment Rev. 2009;35:32–46. doi: 10.1016/j.ctrv.2008.07.004. [DOI] [PubMed] [Google Scholar]
- 40.Tisato F, Marzano C, Porchia M, Pellei M, Santini C. Copper in diseases and treatments, and copper-based anticancer stategies. Med. Res. Rev. 2010;30:708–749. doi: 10.1002/med.20174. [DOI] [PubMed] [Google Scholar]
- 41.Wang F, Jiao P, Frezza M, Dou QP, Yan B. Turning tumor-promoting copper into anti-cancer weapon via high throughput chemistry. Curr. Med. Chem. 2010;17:2685–2698. doi: 10.2174/092986710791859315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hanna PM, Chamulitrat W, Mason RP. When are metal ion-dependent hydroxyl and alkoxyl radical adducts of 5,5-dimethyl-1-pyrroline N-oxide artifacts. Arch. Biochem. Biophys. 1992;296:640–644. doi: 10.1016/0003-9861(92)90620-c. [DOI] [PubMed] [Google Scholar]
- 43.Rota C, Barr DP, Martin MV, Guengerich FP, Tomasi A, Mason RP. Detection of free radicals produced from the reaction of cytochrome P-450 with linoleic acid hydroperoxide. Biochem. J. 1997;328:565–571. doi: 10.1042/bj3280565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hawkins CL, Davies MJ. Hypochlorite-induced damage to nucleosides: formation of chloramines and nitrogen-centered radicals. Chem. Res. Toxicol. 2001;14:1071–1081. doi: 10.1021/tx010071r. [DOI] [PubMed] [Google Scholar]
- 45.Crow FW, Tomer KB, Gross ML, McCloskey JA, Bergstrom DE. Fast atom bombardment combined with tandem mass spectrometry for the determination of nucleosides. Anal. Biochem. 1984;139:243–262. doi: 10.1016/0003-2697(84)90412-3. [DOI] [PubMed] [Google Scholar]
- 46.Cadet J. DNA damage caused by oxidation, deamination, ultraviolet radiation and photoexcited psoralens. IARC Sci. Publ. 1994;125:245–276. [PubMed] [Google Scholar]
- 47.Cadet J, Douki T, Ravanat JL. Artifacts associated with the measurement of oxidized DNA bases. Environ. Health Perspect. 1997;105:1034–1039. doi: 10.1289/ehp.105-1470384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Basu AK, Loechler EL, Leadon SA, Essigmann JM. Genetic effects of thymine glycol: site-specific mutagenesis and molecular modeling studies. Proc. Natl. Acad. Sci. USA. 1989;86:7677–7681. doi: 10.1073/pnas.86.20.7677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cheng KC, Cahill DS, Kasai H, Nishimura S, Loeb LA. 8-Hydroxyguanine, an abundant form of oxidative DNA damage, causes G→T and A→C substitutions. J. Biol. Chem. 1992;267:166–172. [PubMed] [Google Scholar]
- 50.Pogozelski WK, Tullius TD. Oxidative strand scission of nucleic acids: Routes initiated by hydrogen abstraction from sugar moiety. Chem. Rev. 1998;98:1089–1107. doi: 10.1021/cr960437i. [DOI] [PubMed] [Google Scholar]
- 51.Dizdaroglu M. Chemical determination of free radical-induced damage to DNA. Free Radic. Biol.Med. 1991;10:225–242. doi: 10.1016/0891-5849(91)90080-m. [DOI] [PubMed] [Google Scholar]
- 52.Ito T, Kuno S, Uchida T, Fujita S, Nishimoto S. Properties and reactivity of the adenosine radical generated by radiation-induced oxidation in aqueous solution. J. Phys. Chem. B. 2009;113:389–394. doi: 10.1021/jp801976t. [DOI] [PubMed] [Google Scholar]
- 53.Bamatraf MMM, O'Neill P, Rao BSM. OH radical-induced charge migration in oligodeoxynucleotides. J. Phys. Chem. B. 2000;104:636–642. [Google Scholar]
- 54.Williams TT, Odom DT, Barton JK. Variations in DNA charge transport with nucleotide composition and sequence. J. Am. Chem. Soc. 2000;122:9048–9049. [Google Scholar]
- 55.Henderson PT, Jones D, Hampikian G, Kan Y, Schuster GB. Long-distance charge transport in duplex DNA: The phonon-assisted polaron-like hopping mechanism. Proc. Natl. Acad. Sci. USA. 1999;96:8353–8358. doi: 10.1073/pnas.96.15.8353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Giese B. Long-distance electron transfer through DNA. Annu. Rev. Biochem. 2002;71:51–70. doi: 10.1146/annurev.biochem.71.083101.134037. [DOI] [PubMed] [Google Scholar]
- 57.Bixon M, Jortner J. Charge transport in DNA via thermally induced hopping. J. Am. Chem. Soc. 2001;123:12556–12567. doi: 10.1021/ja010018p. [DOI] [PubMed] [Google Scholar]
- 58.Dohno C, Ogawa A, Nakatani K, Saito I. Hole trapping at N6-cyclopropyldeoxyadenosine suggests a direct contribution of adenine bases to hole transport through DNA. J. Am. Chem. Soc. 2003;125:10154–10155. doi: 10.1021/ja035887o. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.