

Abstract
The emergence of oral disease-modifying therapies in multiple sclerosis (MS) will have a significant impact on the evolving scenario of immunomodulatory treatments in MS where current therapies are all injectable. Reducing relapses in trials translates for individuals with MS into a therapeutic aim of stopping future events. Thus the possible absence of any perceived benefits to the individual together with the long disease course, variable outcome, and a younger age group affected in MS makes side effects the major issue. The use of disease-modifying therapies as a whole needs to be placed in the context of a widening therapeutic indication where the use of these therapies is being justified at an increasingly early stage and in pre-MS syndromes such as clinically isolated and radiologically isolated syndromes where no fixed disability is likely to have accumulated. The five oral therapies discussed (cladribine, fingolimod, laquinimod, BG-12, and teriflunomide) have just completed Phase III studies and some have just been licensed. New oral drugs for MS need to be placed within this evolving marketplace where ease of delivery together with efficacy and side effects needs to be balanced against the known issues but also the known long-term safety of standard injectables.
Keywords: multiple sclerosis, immunomodulatory treatment, cladribine, fingolimod, laquinimod, BG-12, teriflunomide
Introduction to management issues in treatment of MS – new developments with oral agents
Multiple sclerosis (MS) is the most common central nervous system (CNS) demyelinating disease worldwide. The disease incidence ranges from 2 to 10 per 100,000 population per year and has a prevalence of 100 to 150 per 100,000 in the UK, making MS the most common non-traumatic acquired cause of disability in young adults.1,2
Interferon beta (IFN-β), the first therapy in randomized trials to reduce the number of relapses that people with MS experience, has revolutionized the management of the condition, introducing the concept of disease-modifying treatment (DMT). However, as yet, DMTs have not influenced the long-term prognosis3 and their role in the management of MS remains under challenge in some health systems.4
The confirmation that relapses can be reduced by therapy has driven attempts to diagnose the condition earlier and has also paved the way for other DMTs, including glatiramer acetate (GA)5 and natalizumab.6 Until recently all available therapies were injectable, a key disadvantage both for patients in terms of side effects and for health services in terms of cost, but, based on results of large randomized controlled double-blind trials, a number of oral therapies are becoming available. Their position in management of MS is yet to be determined but will emerge during the next few years as their use becomes more widespread.7
In this article, we will introduce the evolving scenario of immunomodulatory treatments, starting with an overview of injectable therapies. We will then review five new oral immunomodulatory agents that have been recently investigated in clinical trials, with a main focus on fingolimod and cladribine, the two oral drugs that are at the most advanced stage of development and that have already, or soon will have been, submitted for marketing authorization in several countries, and that have already been approved in some countries (at the time of writing, the US for fingolimod and Russia and Australia for cladribine).
When does relapsing-remitting MS start?
The development of DMTs that have effects on relapses has probably exerted some role in the refinement of the diagnostic criteria for MS in an attempt to identify the condition at an early stage – “McDonald criteria” MS.8 Subsequent evidence that early treatment with potent immunotherapy can stop and even reverse the accumulation of disability9,10 together with evidence that relapses occurring within the first 2 years have some influence on prognosis,11 provides further support to the rationale of early treatment of MS. Identifying disease activity earlier has led to the emergence of the “clinically isolated syndrome” (CIS)12 and the “radiologically isolated syndrome” (RIS)13 as potential predictors of relapsing-remitting (RR) MS where therapeutic intervention may have a role (Figure 1). This approach together with “rapidly evolving severe” (RES) MS, a subgroup identified by some funders,14 identifies 5 clinical scenarios that offer a potential target for a therapy aimed at reducing relapses (Table 1).
Figure 1.

Attempts to identify disease activity earlier than clinically definite relapsing-remitting MS (RRMS) has resulted in the identification of “McDonald Criteria” MS, clinically isolated syndrome (CIS), and radiologically isolated syndrome (RIS) based around MRI.
Abbreviation: SPMS, secondary progressive MS.
Table 1.
Pre-MS and MS subtypes as potential targets for oral therapies
| RRMS subtype | Definition | Risk of second episode/ARR over 2 years |
|---|---|---|
| Pre-MS | ||
| RIS13 | Inflammatory lesions on MRI in the absence of clinical symptoms indicative of a related neurological episode RIS could underlie the subsequent development of RRMS or PPMS | 31% (52/166)169 |
| CIS12 | A single inflammatory neurological event. Subdivided based on number of MRI lesions present initially MRI: <three lesions on MRI there is a <10% risk of MS at 15 years. If ≥three lesions the risk of MS is 50% at 15 years | <3 MRI lesions: 10% ≥3 MRI lesions: 45%170 |
| MS | ||
| “McDonald Criteria” MS8 | Single neurological episode with MRI evidence of dissemination in time and space | 41%171 |
| Clinically definite MS | Two relapses disseminated in time and space. Group 1: CDMS from 1996, Group 2: CDMS from 2008 |
Group 1: 1.2 relapses/year Group 2: 0.5 relapses/year21 |
| RES RRMS14 | Two clinically significant relapses in one year and one or more gadolinium-enhancing lesions on brain MRI or a significant increase in T2 lesion load | 1.46 relapses/2 year172 |
Note: Risk of second episode and ARR are based on trial placebo data.
Abbreviations: ARR, annualized relapse rate; CIS, clinically isolated syndrome; RES, rapidly evolving severe; RIS, radiologically isolated syndrome; RRMS, relapsing-remitting multiple sclerosis.
Relapse-modifying therapy – treat early or never?
The drive behind identifying the disease early is the understanding that “relapse-modifying” therapy initiated in the progressive phase does not stop progression.15 Therefore treatment early in the disease can have significant beneficial long-term benefits, but if treatment is delayed, though entailing the same risk, and no matter how small, will have no benefit (Figure 2).
Figure 2.

Resetting immune tolerance can prevent subsequent disability (A); however evidence is emerging that beyond a certain point during the course of MS, a “relapse-suppressive therapy” is not effective (B).
Much of the work in the field of “immune resetting” has been undertaken on the evaluation of aggressive therapies where their effects need to be balanced against our lack of ability to predict outcome with a disease course that can have a median length of 50 years, depending upon the population.16 The onset in the majority can be relatively benign, but disability can still accumulate progressively after many years in 43% to 89% of people with MS. However, 10% to 15% will never experience any disability and treating all will expose a significant minority to therapy that is not required.16 The relationship of CIS and RIS with final disability currently is even less clear.
Though early relapse rates have a limited predictive ability in MS,11 other features that predict severity such as B-cell follicle-like structures have been found only at post-mortem.17 Otherwise, the onset of early disability is unquestionably significant, but once a certain level is reached relapse-modifying therapy does not prevent progressive deterioration.18,19 Thus there is limited potential to initiate a successful therapy based on reducing relapses.
Therefore the clinical drive is to identify those at risk of poor outcomes early in the course of disease, given the potential need for a durable therapeutic effect, with its associated risks, for many years. However the reality is that our current knowledge of MS prognosis and of both long-term efficacy and safety of DMTs is limited and treatment needs be considered with equipoise.
A changing study population
An increasingly acknowledged problem of incorporating the results of new studies into all types of MS therapy is the issue of a changing population who enter trials. As therapies have been introduced into practice and people with MS have started them, those who did not are either viewed by themselves and/or their doctor as benign cases or have issues, up to now, with injectables. As a result the relapse rate evident in trials has declined over time,20 presenting challenges for clinical trial design21 (Figure 3).
Figure 3.

Annualized relapse rates observed in the 26 trials identified in the systematic review by Nicholas et al21 plotted against the year in which the paper was published. The dashed line shows the fitted trend line.
Adapted from Nicholas et al.21 Copyright © 2011, SAGE Publications. http://msj.sagepub.com
This trend has two major implications for the clinical management of MS. Firstly this problem has been identified by funders and has led to the identification of a RES subgroup of RRMS (Table 1). Actually, the criteria for RES define a population that formed a substantial subgroup of those who took part in early randomized trials of DMTs in MS. The second issue is applicability of trial outcomes across the early onset subtypes with a genuine need to identify the benefits/risk balance in subjects commenced on any therapy (Table 1).
Summary
Injectable relapse-modifying therapies for MS are well established in the management of MS. The development of oral therapies needs to be placed in the context of a number of ongoing patient management issues in MS. Beyond clinically definite RRMS there are a number of areas where their use may be applied but this involves extrapolation of trial data and assumptions about the role of immune interventions (Table 2). The acceptability of the risks of therapy compared with the potential benefits will vary in different countries according to different standards of practice, health systems, and applicable policies. The challenge for clinicians, scientists, and regulatory authorities is how to best use the current knowledge about distinct disease subtypes and natural history studies to deliver the optimal benefit for people with MS.
Table 2.
Using data from placebo groups over 2 years based on 100 subjects in each group to predict the number of expected relapses
| Efficacy | Relapse reduction | 30% | 30% | 50% | 70% |
| Example therapy | IFN-β, GA | terifluonamide | cladribine/fingolimod | natalizumab/mitoxantrone | |
| Side effects (%) | serious | 0 | 0.1 | 0.8/0.8 | 0.1/0.7 |
| ongoing | 80% | minimal | minimal | minimal | |
| Expected relapses with therapy over 2 years | |||||
| Demyelination syndrome subtype (expected relapses in 2 year period) | Rapidly evolving severe (300) | 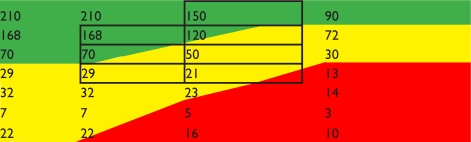 |
|||
| Clinically definite – group 1 (240) | |||||
| Clinically definite – group 2 (100) | |||||
| McDonald (41) | |||||
| CIS ≥ 3 MRI lesions (45) | |||||
| <3 MRI lesions (10) | |||||
| RIS (31) | |||||
Notes: Using trial efficacy data the relapses that will occur as result of therapy can be predicted. Acceptability is highlighted: green, acceptable in most health systems; orange: acceptable in US/parts of Europe; red: not acceptable in health systems due to side effects; grey outline: groups in trials that have been tested.
Abbreviations: CIS, clinically isolated syndrome; RIS, radiologically isolated syndrome; GA, glatiramer acetate.
Overview of the standard injectables for MS treatment
Four major classes of standard injectables are used in the treatment of MS. These include IFN-β, GA, natalizumab, and mitoxantrone. Other less commonly used therapies include intravenous immunoglobulin and the oral agent azathioprine (reviewed by Nicholas and Chataway).22
Interferon-β (IFN-β)
Mode of action
IFN-β is a type I interferon that has anti-viral and anti-inflammatory characteristics. The mode of action is to inhibit T-cell activation and reduce the blood–brain barrier (BBB) permeability to inflammatory cells.23 Two types of IFN-β are available (IFN-β1b and IFN-β1a). Their main difference is in their pharmacokinetics and pharmacodynamics.24
Efficacy
In RRMS (two relapses in the previous 2 or 3 years) IFN-β reduces relapse rates by ∼30% compared with placebo25 as well as reducing lesion accumulation on the magnetic resonance imaging (MRI) scan.26 In CIS, three studies have found that IFN-β significantly reduces the risk of a second event and therefore of conversion to clinically definite MS.27–29
Side effects
Few serious adverse events are associated with IFN-β25 though there is a risk of hepatic injury.30 Early flu-like symptoms occur in 50% of people but injection site reactions occur with subcutaneous administration in 80% of people. There is evidence that neutralizing antibodies to IFN-β that can develop after 1 year of therapy and is one cause of reduction in efficacy of the drug.31 Unfortunately, the parenteral mode of administration is a leading cause for compliance and tolerability problems in many patients.32,33
Glatiramer acetate (GA)
Mode of action
GA is a random combination of four amino acids (alanine, lysine, glutamate, and tyrosine)34 which has a structural similarity to myelin basic protein. GA is an immune modulator, working, it is thought, by shifting the T helper (Th)1 lymphocytes in MS patients towards a predominance of Th2 phenotype. It represents the first successful use of an agent that ameliorates MS autoimmune responses by altering signals through the T cell receptor.35
Efficacy
GA, subcutaneously administered on a daily basis to patients with RRMS, significantly reduced relapse rates by ∼30% over 2 years compared with placebo5,36 and prevented the appearance of new lesions detectable by MRI.36,37 Comparator studies against IFN-β have shown no difference.38,39 GA also significantly reduces the time to conversion to clinically definite MS from CIS.40
Side effects
There are no major adverse reactions but a self-limiting allergic-type reaction (flushing, chest tightness, and anxiety) can occur in 15% of people taking GA.36 Local injection site reactions are increased compared with placebo, occurring in up to 80% of people.5
Natalizumab
Mode of action
Natalizumab is a monoclonal antibody to the α4 subunit of α4β1 integrin (VLA-4), a protein found on the surface of lymphocytes. α4β1 integrins interact with the vascular-cell adhesion molecule 1 enabling adhesion of lymphocytes to the vascular endothelium. Its main mode of action is to prevent the transmigration of inflammatory lymphocytes across the BBB into the CNS.41
Efficacy
Clinical trials confirmed that natalizumab produced a 68% reduction in annualized relapse rate, 92% reduction in gadolinium enhancing lesions in MRI, and a 42% reduction in disability progression rate over 2 years compared with placebo.6 Natalizumab reduced relapse rates by 53% in combination with IFN-β1a compared with IFN-β1a monotherapy.42 To our knowledge natalizumab has not been studied in CIS populations.
Side effects
After obtaining its license in 2005 natalizumab was withdrawn when three fatal progressive multifocal leukoencephalopathy (PML) cases were associated with combination therapy with IFN-β1a. It was subsequently reapproved as monotherapy for MS after safety re-evaluation.43 The estimated risk of PML with natalizumab is 1 in 1000 (0.1%).44 Neutralizing antibodies, which cause loss of drug efficacy, have been identified in about 6% of patients.45
Mitoxantrone
Mode of action
Mitoxantrone is an anthrecenedione immunosuppressive cytotoxic agent that acts on a number of possible pathways, including inhibition of T-cell activation, suppression of T-cell, B-cell and macrophage proliferation, impaired antigen presentation, prevention of macrophage mediated demyelination, and reduction of pro-inflammatory cytokines.46
Efficacy
Clinical trials have supported the use of mitoxantrone for the treatment of worsening RRMS, progressive relapsing MS, and secondary progressive (SP) MS.47–49 Mitoxantrone significantly reduced relapse rates per year at 2 years by 69% and significantly reduced the proportion of people with sustained disease progression at 2 years.47
Side effects
The major risks are dose-related cardiotoxicity and leukemia. The risk of congestive heart failure exceeds 0.20% with a mean cumulative dose of (60.5 mg/m2); in addition 2.2% of patients showed an asymptomatic reduction in left ventricular ejection fraction.50 The risk of leukemia varies but has been found to be as high as 0.67%.51 Over a quarter of women taking mitoxantrone developed amenorrhea compared with none taking placebo, 8% of whom were still experiencing this at the end of the trial. Nausea and vomiting, alopecia, and urinary tract infections were more frequent in people taking mitoxantrone.
Using the standard injectables
Recommendations for starting injectable therapies in RRMS vary worldwide. In the UK, despite evidence, injectables are not usually commenced until the development of clinically definite RRMS, whereas in the US they are used in RIS. Generally IFN-β or GA is commenced as first-line medication. Treatment failure would indicate transferring either to the alternative first-line therapy or to second-line treatments such as natalizumab if the patient has definite RRMS (or RES RRMS in the UK) or mitoxantrone where progression may be starting. However there is no fixed therapeutic pathway and with licensing of natalizumab in the UK it can be prescribed as first-line pharmacotherapy if patients fulfill the relevant criteria.14
Pharmacodynamics and pharmacokinetics of the new oral agents
Cladribine
Mode of action
Cladribine is a synthetic purine nucleoside analog that produces selective lymphocyte depletion through inhibition of cellular DNA synthesis and repair.52,53 Lymphocyte damage arises from the accumulation of cladribine active triphosphate deoxynucleotide, produced by high levels of deoxycytidine kinase, and poorly metabolized by 5′-nucleotidase;54 furthermore cladribine metabolites are resistant to degradation by adenosine deaminase.55
The peculiar balance of enzymes required for the metabolism of purine nucleoside determines cladribine’s specificity for particular immune cell types. Hence, cladribine affects both B and CD4 and CD8 T lymphocytes, with minor effects on natural killer, null, and innate immune cells.53,56 Cladribine also impairs the migratory capacity into the CNS via the BBB and within the CNS of CD4 and CD8 T lymphocytes and, to a lesser extent, CD14 monocytes,57 Cladribine also reduces the production of the proinflammatory cytokine interleukin (IL)-2 and chemokines.58–60
Pharmacokinetics
Cladribine is a pro-drug that needs intracellular phosphorylation to activate nucleotides, and it is the intracellular concentration of these metabolites, which is several hundred-fold higher than that of cladribine in plasma, that is important. There is no correlation between the plasma concentration of cladribine and that of the intracellular metabolites, however, much of the pharmacokinetic data on cladribine was derived after subcutaneous and intravenous administration (2-chloro-2′-deoxyadenosine). After subcutaneous administration the cerebrospinal fluid concentration is 25% of that in plasma in healthy subjects, whereas in patients with meningeal disease, cladribine concentration in the cerebrospinal fluid exceeds that of plasma. This is relevant in MS because BBB breakdown in active disease phases will result in higher CNS bioavailablity.61 The distribution of intravenous cladribine in patients with hematological malignancies showed a plasma protein binding of 25% (21.1% in healthy controls) and a volume of distribution of 6.57 L/kg.62
The oral preparation used in RRMS has a new dosage form (with 2-hydroxypropyl- β-cyclodextrin; HPβCD) containing 3 mg or 10 mg of cladribine. Absorption of this preparation has a peak in 0.5 to 1.5 hours, being slightly delayed when administered with a high-fat meal. After administration of 10 mg tablet, the bioavailability is 39% to 43% and 28.5% is eliminated via the renal route, with a renal clearance of 29.9 L/h.63
Pretreatment with cladribine increases the intracellular accumulation of the active metabolite of cytarabine, cytosine arabinoside 5′-triphosphate, by 36% to 40%, which supports the use of an annual, short-course dosing regimen.61
Fingolimod
Mode of action
Fingolimod is the first of a new class of drugs that inhibits immune cell migration by interacting with sphingosine 1-phosphate (S1P) receptors. S1P binds to S1P receptors predominantly on lymphocytes, signaling for them to exit lymph nodes and enter the circulation.64 S1P also regulates diverse cellular functions such as survival and proliferation.65 S1P receptors belong to a G protein–coupled receptor family (S1P1-S1P5) which are targeted by fingolimod–phosphate (fingolimod-P) 66 an active metabolite that arises from the reversible phosphorylation of fingolimod by sphingosine kinase.67
With initial dosing of fingolimod, there is agonism of S1P receptors and transient signaling.68 However, with continued fingolimod dosing, functional antagonism occurs with internalization of S1P receptors.69 Nevertheless, S1P1 receptors activated by fingolimod-P retain signaling activity for hours in spite of a quantitative internalization. The ultimate effect, however, is to prevent the egress of lymphocytes from lymphoid organs, including self-reactive circulating lymphocytes, but lymphocytes persist in central lymph nodes, maintaining a reactivity in systemic infection.64,68,70 Besides in lymphocytes, the presence of S1P1 receptors in endothelial cells is reported to contribute to the egress prevention of lymphocytes.71–73
Pharmacokinetics
After oral administration (1.25 mg) in man there is a median 0.5 hours before fingolimod appears in blood at quantifiable levels. An initial absorption “shoulder” occurs in 4 hours and the peak of 1.1 ng/mL is reached 12 hours after dosing.74,75 Its absorption is not affected by food, regardless of the fat content.76 The area under the concentration-time curve extrapolated to infinite time (AUC) is 85% ± 6% and the absolute oral bioavailability is 94%. Fingolimod and fingolimod-P have a red blood cell distribution respectively of 86% and <17% and are both highly protein bound, >99.7% with an extensive volume of distribution of 1200 ± 260 L. Steady state blood concentrations are reached after 2 months of daily dosing and during maintenance dosing, the conversion between fingolimod and fingolimod-P yields a stable blood level ratio of the two moieties.
The biotransformation of the fingolimod metabolites occurs through three pathways.
One is the reversible phosphorylation to fingolimod-P mainly through the action of sphingosine kinase type 2 with a lesser contribution from sphingosine kinase type 1. Reverse dephosphorylation to fingolimod occurs through the action of the lipid phosphate phosphohydrolases (LPP)1a and LPP3 and to a lesser extent by the specific sphingosine 1-phosphate phosphatase SPP1. The second pathway is the fingolimod oxidative biotransformation, through cytochrome (CYP)4F2 hydroxylation, resulting in the M12 metabolite, followed by M1, M2, M3, and M4. The third pathway involves transformation by the dihydroceramide synthase to form the nonpolar metabolites M29, M30, M27, and M28.77
The steady-state oral clearance of fingolimod averages 10.8 L/h and its half-life is 6.1 ± 1 days. After an oral administration, about 81% of the dose is slowly excreted in the urine as inactive metabolites, and after 34 days 89% of the administered dose can be recovered.74,75
BG-12
Mode of action
The exact mechanism of action by which the oral formulation of dimethylfumarate (DMF) and monomethylfumarate (MMF), its primary metabolite, exert their effects in MS is still unclear. In vitro experiments indicate three potential pathways of action. First, BG-12 has been found able to switch the T-helper response from Th1 to Th2 phenotype,78 both increasing the Th2 driving molecules, such as IL-4, IL-10, and IL-5,79–82 and reducing the Th1 driving molecules, IL-6, IL-1β and tumor necrosis factor (TNF) alpha,81,83–87 The second pathway involves the activation of the nuclear factor E2-related factor 2 (Nrf2) transcriptional pathway,88 which acts on one side as an oxidative stress modulator by the induction of phase II detoxification genes.87,89,90 Treatment of cultured astroglia and microglia with fumaric acid esters enhances the activity of phase II detoxification enzyme NAD(P)H:quinone oxidoreductase-1 (NQO-1) mRNA, and increases the cellular glutathione content of activated mixed glial cells.86 On the other side the Nrf2 has neuroprotective capabilities such as inhibition of excitotoxic and oxidative neuronal damage,91–95 BBB protection,96 and regulation of myelin maintenance.97 Through the third pathway, DMF almost completely inhibits the TNF-induced CD62E, responsible for the accumulation of blood leukocytes at sites of inflammation by mediating the adhesion of cells to the vascular lining.84 These data suggest that BG-12 could have dual neuroprotective and anti-inflammatory effects.
Pharmacokinetics
After oral intake, DMF is rapidly hydrolyzed by esterases to its metabolite MMF. Completely absorbed in the small intestine,86 it can interact with immune cells in the blood circulation.83 MMF, the most bioactive metabolite,98 is further metabolized in the citrate cycle to carbon dioxide and water and finally eliminated mainly though breathing, while only small amounts of intact MMF are excreted through urine or feces. The half-life of DMF and MMF are about 12 minutes and 36 hours, respectively. The peak concentration of MMF in human serum is reached 5 to 6 hours after oral intake. There is no evidence for a cytochrome P450-dependent metabolism in the liver.99
Laquinimod
Mode of action
Laquinimod is a new synthetic immunoregulator derived from roquinimex but is pharmacologically and chemically distinct from its progenitor; the clinical Phase III trial of roquinimex was stopped due to side effects.100–102 Chemical modifications of the roquinimex structure have given laquinimod a favorable pharmacology/toxicology profile. In addition to being a more potent disease inhibitor,103 with better tolerability, the effects on the immune system have become increasingly clear.
Initial evidence of its immunomodulatory effects demonstrate that laquinimod biases the CD4+ phenotype in favor of the Th2/Th3 cytokine production IL-4, IL-10, and transforming growth factor-b and inhibits disease development and infiltration of inflammatory cells, CD4+ T cells, and macrophages, into the CNS.104,105 Another effect on the immune system is the suppression of major histocompatibility class II antigen presentation and down-regulation of epitope spreading.106
The effect of laquinimod treatment is not dependent on endogenous IFN-β for its disease inhibition in chronic experimental autoimmune encephalomyelitis (EAE), but combined with exogenous IFN-β for treatment of chronic EAE a significant synergistic inhibitory effect was seen.107 Lymphocytes from laquinimod-treated animals displayed reduced VLA-4-mediated adhesiveness, without suppression of spontaneous and antigen-induced T-cell proliferation;108 laquinimod reduces chemotaxis and adhesion molecules through the suppression of the nuclear factor kappa-light-chain-enhancer of activated B cells (NFκB) pathway that interact with the apoptotic activation of immuno-competent cells.106
Pharmacokinetics
Laquinimod has high oral bioavailability, small volume of distribution, and a low total clearance rate. The peak plasma concentration is reached within 1 hour of oral administration. Laquinimod is metabolized as a low affinity substrate for CYP3A4 in liver microsomes. Four hydroxylated and two dealkylated products are cleared primarily through the urine and are not metabolically active. Less than 5% of laquinimod is eliminated unchanged in urine or feces. Strong specific inhibitors of the CYP3A4 enzyme such as ketoconazole can slow the elimination of laquinimod.109
Teriflunomide
Mode of action
Teriflunomide is a reversible non-competitive inhibitor of dihydroorotate dehydrogenase (DHODH),110,111 an integral mitochondrial membrane protein essential for pyrimidine synthesis.112,113 Inhibiting DHODH prevents clonal expansion of B and T cells and antibody production.114–116 Consistent with its tyrosine kinase inhibitory properties, further immunomodulatory effects have been reported, such as the block of immunoglobulin (Ig) G1 production from IgM isotype switching after IL-4 receptor stimulation;117 inhibition of calcium mobilization in lymphoma cells stimulated by anti-CD3 antibody, IL-2 production, IL-2 receptor expression on human T lymphocytes, and tyrosine phosphorylation in CTLL-4 cells stimulated by IL-2;118 and diminished activation of inducible nitric oxide synthase in astrocytes.119
Pharmacokinetics
The pharmacokinetics of teriflunomide, appear to be linear over the therapeutic dosage range. Mean plasma half-life is 15 to 18 days and the bioavailability is nearly 100%, with total plasma clearance of 0.30 mL/kg/hour. It is extensively protein bound (>99%) primarily to albumin, with an almost constant portion (0.5%) of free teriflunomide, and cleared via several metabolic pathways, including biliary and urinary excretion.120
Clinical efficacy, tolerability, and safety of the new oral agents
Cladribine
Clinical efficacy
Parenteral cladribine has been studied in progressive and relapsing MS in three randomized, double-blind, placebo-controlled trials. The first, a 2-year crossover study evaluating the clinical and radiological efficacy of intravenous cladribine in 51 chronic progressive MS subjects, started in 1992. In the first year the treatment arm was given four monthly courses of 0.10 mg/kg/day for 7 days, a total of 2.8 mg/kg of cladribine. In the second year patients treated with placebo in the first year were given a half dosage of cladribine in three successive monthly courses of 0.10 mg, 0.05 mg, and 0.05 mg/kg/day for 7 days. This study showed that cladribine could temporarily stabilize clinical progression, with a possible dose-dependent effect. There was a reduction in enhancing lesions on MRI (absent at 1 year in 22 out of 24 treated versus 12 out of 24 placebo subjects, P < 0.001), but no significant change in T2 lesion load.121 The second study was an 18-month study in 52 RRMS patients who received placebo or cladribine at 0.07 mg/kg/day by subcutaneous injection for five consecutive days as six monthly courses achieving a total dose of 2.1 mg/kg. Patients were followed up for further 12 months. Those receiving cladribine had a significant reduction in a combined measure of the severity and frequency of relapses compared with patients receiving placebo from months 7 to 12, which was maintained at month 18. There was a significant reduction in enhancing lesions on MRI at 6 months after treatment that was maintained at 12 and 18 months compared with placebo.122 In the third study 159 patients with progressive MS (30% primary, 70% secondary) received placebo or subcutaneous injection of cladribine at 0.07 mg/kg/day for five consecutive days every four weeks for either two or six cycles (total dose, 0.7 mg/kg or 2.1 mg/kg, respectively), followed by placebo, for a total of eight cycles. This study confirmed cladribine’s ability to suppress enhancing lesions on MRI. The higher dosage also reduced the accumulation of T2 lesions, but both doses failed to affect disability progression.56
In the light of these results, three trials were designed in order to assess the effectiveness of oral cladribine in RRMS. In the first, CLARITY (CLAdRIbine Tablets Treating MS OrallY), 1326 patients were enrolled in a 1:1:1 ratio to receive one of two cumulative doses of cladribine tablets (either 3.5 mg or 5.25 mg/kg body weight) or matching placebo, given in two or four short courses for the first 48 weeks, then in two short courses starting at week 48 and week 52 (for a total of 8 to 20 days per year). During the 96-week study, patients receiving 3.5 and 5.25 mg/kg showed, respectively, versus placebo, a relative reduction in annualized relapse rate of 57.6% and 54.5%, a higher relapse-free rate (odds ratio: 2.53 and 2.43), an increased time to first relapse (hazard ratio [HR] 0.44 and 0.46), a lower risk of 3-month sustained progression of disability (HR 0.67 and 0.69), and a relative reduction of enhancing lesions of 85.7% and 87.9% and of active T2 lesions of 73.4% and 76.9%.23 This study has been extended for 2 years, so that patients originally randomized to placebo in the CLARITY study will all receive two courses of oral cladribine per year. Patients in either of the cladribine groups will be randomized 2:1 to receive two courses of cladribine per year or placebo. Patients will maintain the same treatment for both years of the extension study.124 Two further studies are still running. ONWARD (Oral cladribine added oN to Rebif new formulation in patients With Active Relapsing Disease) is a 96-week, double-blind, placebo-controlled, Phase IIb study, in which oral cladribine is added to IFN-β1a (subcutaneous three times weekly, 44 μg).125 The primary end-point is the safety and tolerability of oral cladribine compared with placebo in patients with active MS (RRMS and SPMS with superimposed relapses). Clinical and MRI end-points are secondary outcome measures in this study. ORACLE (ORAl CLadribine in Early MS) is a randomized, double-blind, placebo-controlled study of 600 subjects with CIS and positive MRI brain scans. The aim is to assess the effect of early treatment with oral cladribine in delaying the development of definite MS. The design provides for three arms (1:1:1 ratio) receiving one of two different dosage regimens of cladribine tablets or matching placebo: in the first year two or four treatment cycles are scheduled, with each cycle consisting of once daily administration for four to five consecutive days; in the second year, two treatment cycles are to be administered to all patient groups. Patients will be treated for 96 weeks or up to the time when they experience a second attack, in which case they would be offered an open-label treatment with IFN-β1a 44 μg three times a week for a 96-week maintenance treatment period. Patients who do not convert to clinically definite MS within the initial 96-week period of the study will be eligible to enrol in a 96-week long-term follow-up treatment period.126
Tolerability and safety
A review of adverse events in all MS subjects enrolled in cladribine studies,63 both for oral and parental preparations, found adverse events were increased in cladribine-treated groups. Neoplasms and serious adverse events were reported, respectively, in 3.7% and 12.9% of all cladribine-treated subjects compared with 1.7% and 8.1% of placebo-treated subjects. Eighteen common adverse events were found to have at least 2% greater incidence within cladribine-treated than placebo-treated subjects: headache (25.8% vs 19.4%); lymphopenia (21.3% vs 1.7%); upper respiratory tract infection (15.9% vs 11.3%); nasopharyngitis (13.7% vs 11.5%); nausea (13.4% vs 11.1%); fatigue (10.0% vs 7.9%); back pain (9.3% vs 7.1%); depression (7.8% vs 4.0%); leucopenia (5.9% vs 0.6%); pyrexia (5.7% vs 2.7%); injection site bruising (4.5% vs 1.7%); rash (4.4% vs 1.5%); MS (3.9% vs 1.0%); alopecia (3.3% vs 1.3%); lymphocyte count decreased (3.5% vs 0.0%); muscular weakness (3.2% vs 1.0%); and herpes zoster (2.9% vs 0.2%).
Lymphopenia was a dose-dependent side effect of cladribine, occurring in 6.1% at lower doses (0 to 1.75 mg/kg); 15.2% for doses from 1.75 to 3.5 mg/kg; 20.6% for doses from 3.5 to 5.25 mg/kg; and 28.4% at higher doses (5.25 to 7.0 mg/kg).
In the CLARITY study the reported adverse events were more frequent in the cladribine groups and included lymphocytopenia (21.6% in the 3.5 mg group and 31.5% in the 5.25 mg group, vs 1.8% in placebo patients), herpes zoster (eight and twelve patients, respectively, vs none in placebo patients), and one patient who was treated with cladribine had reactivation of latent tuberculosis and died.123 A concern with using cladribine is the risk of malignancies, which were elevated in the cladribine-treated groups in the CLARITY study whereas none occurred in the placebo group, which is atypical in a study of this size.123 Cladribine has been used extensively in hairy cell leukemia (HCL) where it is still unclear whether the higher risk of subsequent malignancy is related to the use of cladribine or hairy cell leukemia itself.127 These concerns will be addressed more fully when the results of ONWARD and ORACLE become available.
Fingolimod
Clinical efficacy
Fingolimod has been recently licensed for use in RRMS by the US Food and Drug Administration (FDA).128 Evidence of its effectiveness is based on studies in animal models,70,129,130 Phase II trials131–133 and in particular two Phase III trials.
In the first,134 1272 subjects with RRMS were enrolled to receive daily oral fingolimod 0.5 mg, 1.25 mg, or placebo. Over 24 months of 1033 subjects who completed the study, those receiving placebo, 0.5, or 1.25 mg of fingolimod showed an annualized relapse rate of 0.40, 0.18, and 0.16, respectively, and a cumulative probability of disability progression, confirmed after 3 months, of 24.1%, 17.7%, and 16.6%, respectively. Fingolimod significantly reduced the risk of disability progression over the 24-month period at doses of 0.5 mg and 1.25 mg with an HR of 0.70 and 0.68, respectively, versus placebo. Enhancing lesions, lesion load, and brain atrophy on MRI were also significantly reduced over the 24-month period in treated versus placebo patients.
In the second Phase III study,135 which compared the efficacy of oral fingolimod with intramuscular IFN-β1a, 1292 subjects with RRMS were enrolled to receive daily oral fingolimod 0.5 mg, 1.25 mg, or IFN-β1a 30 μg weekly. Notably, subjects were allowed to have failed IFN-β prior to enrolment and thus could be re-exposed to a therapy they had failed – IFN-β. Over 12 months 1153 subjects completed the study. Patients receiving IFN-β1a, fingolimod 0.5 mg, or 1.25 mg showed an annualized relapse rate of 0.33, 0.16, and 0.20, respectively, but no significant differences were seen in disability progression. Enhancing lesions, lesion load, and brain atrophy on MRI were also significantly reduced over the 12-month period in fingolimod versus IFN-β1a patients.
Tolerability and safety
In the placebo-controlled fingolimod study the incidence of lower respiratory tract infections was higher in the fingolimod than the placebo group, infections occurring in 9.6% patients receiving 0.5 mg and 11.4% patients receiving 1.25 mg of fingolimod versus 6.0% patients receiving placebo. The fingolimod group also had a dose-dependent slowing of the heart rate and a higher incidence of atrioventricular conduction block at the time of the first dose. A higher occurrence of macular edema was also observed, which resolved with discontinuation of the drug, as well as the frequent elevations in liver-enzyme levels.
In the comparator study versus IFN-β, 2 fatal cases of herpes infection occurred. One was a case of herpes simplex encephalitis who was originally given a course of intravenous methylprednisolone for an MS relapse and the second was a case of disseminated primary varicella zoster infection in a patient with a negative baseline varicella zoster antibody titer who was exposed to a child with chicken pox during an 8-day course of corticosteroids (intravenous and then oral methylprednisolone) for a MS relapse.
BG-12
Clinical efficacy
In light of its mode of action and effectiveness in psoriasis, a pilot study investigated the safety and efficacy of oral DMF and ethylhydrogen fumarate in MS patients.82 This was a prospective, open-label study conducted in 10 patients with RRMS. The study medication was titrated over 9 weeks up to a dose of 240 mg three times daily. The study medication significantly reduced the mean number of enhancing lesions after 22 and 70 weeks of treatment. A Phase IIb double-blind, placebo-controlled, dose-ranging, multinational study was conducted involving 257 RRMS patients randomized 1:1:1:1 ratio to receive oral DMF 120 mg once daily, 120 mg three times daily, 240 mg three times daily, or placebo for 24 weeks.136 During the subsequent 24-week safety extension period all patients received DMF, those receiving placebo starting 240 mg three times daily. The 240 mg three times daily arm met the primary end-point, with a significant reduction in the total number of enhancing lesions by 69% as well as the number of new or enlarging T2-hyperintense lesions and the number of new T1-hypointense lesions compared with placebo.
Two Phase III studies are underway aiming to determine the safety and efficacy of oral fumarate in RRMS. The first, DEFINE (Determination of the Efficacy and safety of oral Fumarate IN rElapsing-remitting MS), is a double-blind study in which 1011 patients have been randomized 1:1:1 to receive oral DMF 240 mg three times a day, twice a day, or placebo, completed in January 2011. The second, CONFIRM (COmparator and aN oral Fumarate In Relapsing-remitting MS), is a study comparing two dosages of oral DMF and subcutaneous GA 20 mg once daily, 1232 patients affected by RRMS have been enrolled to receive 1:1:1:1 oral DMF 240 mg three times daily, twice daily, GA, or placebo. Both trials have a treatment period of 96 weeks. After 1 year of treatment patients who experience sustained disability progression or one relapse after 24 weeks in DEFINE and two in CONFIRM can optionally switch to any MS therapy as open-label rescue therapy.
Tolerability and safety
In the pilot, open-label study82 the most common adverse events reported were gastrointestinal symptoms (diarrhea, cramps, nausea) and flushing, which were experienced by almost all patients but decreased after the first 6 weeks of the treatment. In the much larger Phase IIb study,136 the common adverse events were flushing, headache, nasopharyngitis and nausea. No difference in the frequency of infections was seen between all groups. Although only recently studied in MS, the use of this class of drugs in psoriasis has shown them to be generally well tolerated and safe agents for up to 14 years.137
Laquinimod
Clinical efficacy
Laquinimod is effective in experimental studies using animal MS models99,100,103 and has shown neuroprotective capacity.105,138,139
In the Phase II study140 209 RRMS patients were randomized to receive two different doses of laquinimod, 0.1 mg or 0.3 mg, compared with placebo, as three daily tablets for 24 weeks. In this multicenter, double-blind, randomized trial, patients receiving laquinimod 0.3 mg had a significant 44% reduction in mean cumulative number of active lesions versus placebo. No differences were found in relapses and disability between the three groups.
In the double-blind, placebo-controlled Phase IIb study,141 306 patients affected by RRMS with at least one enhancing lesion on the screening MRI were enrolled to receive oral laquinimod 0.3 mg, 0.6 mg, or placebo, in a 1:1:1 ratio, for 36 weeks. The results showed a 40.4% reduction of enhancing lesions in the last 4 months MRI scans in the group receiving the higher dosage of laquinimod versus placebo.
Two Phase III trials are being conducted. The first, ALLEGRO (Assessment of oral laquinimod in preventing progression of MS), is a double-blind study that recruited 1000 patients with RRMS who were randomized to receive laquinimod 0.6 mg once a day or placebo for 24 months. The study demonstrated a significant reduction in annualized relapse rate compared with placebo and showed a significant reduction in disability progression, though the extent of these effects are not yet published.142 The second Phase III randomized, double-blind, parallel-group, placebo-controlled study, BRAVO (Benefit-Risk assessment of AVOnex and laquinimod), aims to compare the effect of daily oral laquinimod 0.6 mg with placebo and with IFN-β1a (Avonex) administered intramuscularly 30 μg weekly. Recruitment was completed in June 2009 and the results are awaited.
Tolerability and safety
In the Phase II study140 there was no significant difference in adverse events between the groups. Two serious adverse events, breast cancer and acute tonsillitis, were reported in the laquinimod higher dosage group, as well as a mild and transient increase in liver function tests. In addition a small dose-related elevation of erythrocyte sedimentation rate was also noted. In the Phase IIb randomized controlled trial,141 again transient and dose-dependent increases in liver enzymes were noted. A case of Budd–Chiari syndrome occurred after 1 month of the higher laquinimod dosage exposure in a patient with underlying hypercoagulability (heterozygosis for factor V Leiden mutation). Chest pain occurred in four patients receiving 0.6 mg and in two patients receiving laquinimod 0.3 mg. Arthralgia was reported by two patients in the 0.6 mg group and by four in the laquinimod 0.3 mg group. Viral infections, herpes simplex, and herpes zoster were more frequent in 0.3 mg laquinimod arm.
Teriflunomide
Clinical efficacy
Preclinical testing of teriflunomide has been found to be effective in an EAE-MS model.143 The initial results of a clinical trial in MS have been recently presented.144–147 TEMSO (Teriflunomide in Reducing the Frequency of Relapses and Accumulation of Disability in Patients With Multiple Sclerosis) is a Phase III 2-year randomized, double-blind, placebo-controlled study that included 1088 relapsing MS patients. Patients were randomized to placebo or teriflunomide, 7 mg or 14 mg, once daily. Both doses of teriflunomide significantly reduced annualized relapse rate by 31% versus placebo. The 12-week sustained risk of disability progression was also significantly reduced by 30% for the 14 mg and 24% for the 7 mg dose. Teriflunomide also significantly reduced the lesion load at MRI by 39% and 67% at the 7 and 14 mg doses relative to placebo.
TENERE (Comparing the Effectiveness and Safety of Teriflunomide and Interferon Beta-1a in Patients With Relapsing Multiple Sclerosis) is a further randomized, parallel-group, single-blind study comparing the effectiveness and safety of the two doses of teriflunomide (7 and 14 mg) versus IFN-β1a 44 μg three times a week in 300 patients with RRMS. The objective is to make a head-to-head comparison over 1 year treatment with “time to failure” as primary endpoint, defined as the first occurrence of relapse or permanent study treatment discontinuation for any cause, whichever comes first. This study is expected to be completed by the end of 2011. TOPIC is an international, multi-center, randomized, double-blind, placebo-controlled, parallel group study evaluating the efficacy and safety of 2 years’ treatment with teriflunomide once daily at 7 mg and 14 mg versus placebo in approximately 780 CIS patients. The primary outcome of this study is the reduction of time to conversion to MS. This study is expected to be completed in 2015.
Tolerability and safety
In the recently presented preliminary results of TEMSO, no serious opportunistic infections occurred in patients treated with teriflunomide, and there was no difference in the serious infections between the groups, occurring in 2.2% of placebo, and in 1.6% of the 7 mg and 2.5% of the 14 mg groups.144–147
Patient-focused perspectives
A major issue with therapies in MS is that it is not clear to date whether they benefit subjects in the longer term and whether they fulfil the patients’ perception of benefit. The current medical aim of therapy is to prevent relapses with the ultimate intention of preventing progressive neurological deterioration that may or may not occur in the future. This means that therapies may have no beneficial effects to the subject other than a future “lack of event”.
In terms of baseline neurological symptoms therapies such as GA and IFN-β have shown a beneficial affect on health-related quality of life (HR-QOL) in some subjects.148,149 This has to be balanced against a discontinuation rate of 28% to 41%,150 principally due to adverse effects (52%), physician-assessed disease progression (40%), and patient perceived ineffectiveness (20%). Similar to the findings of a Cochrane review of IFN-β1a in randomized controlled trials25 adherence to therapies including GA and IFN-β is about 75% over an average of 31 months. Adherent patients had shorter disease length and shorter duration of treatment together with a better quality of life.151 Subjects failing to respond to treatment and dropping out would explain why those who adhere long-term to standard therapies such as IFN-β can apparently benefit from therapy in open studies.152 More intense therapies such as natalizumab, especially when subjects have rapidly evolving severe RRMS, have a favorable affect on disability153 as well as more extensive benefits on HR-QOL.154
As therapies are being initiated earlier, as in CIS or even RIS stages, however, subjects will have no ongoing neurological symptoms or impairment. Thus any side effects that affect QOL will have an impact on their continued use. For the current injectable therapies in MS to be beneficial they need to be used long term, and this is proving a problem in RRMS. In CIS and RIS they will need to be used for much longer in essentially asymptomatic subjects, and thus one might expect higher rates of dropout. Some of these issues will arise as a result of long-term administration of any therapy, including oral therapies.
Drug administration
Therapies must be tolerated in terms of the modality of drug administration. This means that injectables are at an immediate disadvantage compared with oral administration. Subcutaneous and intramuscular injections are a discomfort, can be painful, and are currently rejected by some patients on that basis alone. IFN-β and GA commonly cause skin site reactions that can be irritating only but can also cause scarring of tissue, leading to lipoatrophy and lipohypertrophy that can make continued administration difficult.155 Very occasionally, injections, usually as a result of poor injection technique, cause skin infections that require treatment with antibiotics. Natalizumab requires intravenous administration and entails a risk of allergic reactions that require admission to hospital for a short period with implications for funders and for resource allocation.
The oral therapies have immediate benefits over injectables in terms of administration; however, subjects prefer oral administration but only if efficacy is not compromised.156 Fingolimod is a daily therapy. However, cladribine is administered only every 6 months, and thus has further potential advantages.
Day-to-day
The injectable IFN-β can cause transient flu-like symptoms. However, in some cases side effects can be ongoing. These include muscle aches and spasticity in 15% of subjects157 and low mood has been reported,158 whereas GA has minimal day-to-day side effects. These side effects are an issue in the longer term. Nevertheless, it is not clear how day-to-day side effects contribute to the 52% of side-effect dropouts as opposed to drug administration issues.150
Oral therapies emerging in 2011 have few day-to-day issues, though they are yet to be extensively used. Fingolimod can cause nausea and headaches but is reported to be well tolerated.135 No significant day-to-day issues are associated with use of cladribine.123
Risk of infections
Therapies that impair the immune system function can put the user at risk of infections. Infection themselves represent a significant issue for people with MS, not only by requiring treatment themselves, but also by affecting MS symptoms in ways that often mimic a relapse of MS.159 In MS, urinary dysfunction increases as disability increases,160 making urinary tract infections a major problem as disability accumulates.
There is minimal risk of infections with IFN-β and GA. The original reports found no increased risk of infections with natalizumab use. However, the emergence of PML and other opportunistic infections has demonstrated that there is impaired immunity in subjects on this therapy and has suggested that rigorous long-term follow-up is appropriate.161
Oral therapies such as fingolimod and cladribine have increased rates of infections. Fingolimod increased lower respiratory tract infections compared with placebo134 and produced more serious herpes virus infections, even though this was not replicated in the comparator trial.135 Cladribine increased the risk of infections overall and of herpetic infections in particular.123 These issues are a concern as the schemes of administration do not require subjects to automatically attend clinical follow-up frequently, and thus infections could become more serious if not managed proactively.
Considering the mechanism of action of the available drugs (IFN-β, GA, and natalizumab) and the new oral therapies (fingolimod, cladribine, terifluonamide, laquinimod, and BG12), it would be expected that there may be some similarity in side effects between drug groups (Figure 4). Natalizumab, fingolimod, and cladribine potentially reduce lymphocyte numbers at the site of action whereas terifluonamide, laquinimod and BG12 are more similar to IFN-β/GA. The current limited knowledge of side effects does seem to support this distinction (Table 2).
Figure 4.

Simplified overview of postulated mechanism of action of standard injectables and new oral therapies. Fingolimod and cladribine reduce available circulating lymphocytes by blockade of lymphocyte egress from lymph nodes (fingolimod), by cytotoxicity and depletion (cladribine). Natalizumab acts by blockade of adhesion and transmigration across the blood–brain barrier. All three drugs (fingolimod, cladribine, and natalizumab), indirectly (fingolimod and cladribine) or directly (natalizumab), reduce the number of lymphocyte in the central nervous system (CNS). In contrast, IFN-β and glatiramer acetate (GA) shift the balance of lymphocytes and modulate cytokine secretion. The newer oral therapies, terifluonamide, laquinimod, and BG-12, do not reduce lymphocytes in the CNS and appear to act in a similar way to IFN-β and GA by modulating cytokines and lymphocyte activation. None of these injectable (IFN-β and GA) and oral (terifluonamide, laquinimod, and BG-12) drugs appear to significantly reduce the numbers of circulating lymphocytes, suggesting an immunomodulatory rather than an immunosuppressive profile.
Life-threatening adverse events
Major risks from therapy also need to be considered as standard therapies have proven to be safe over 15+ years. IFN-β has few risks apart from liver damage, which is rare and can be attenuated by following monitoring guidelines,30 and GA has no major risks.
Natalizumab can cause reactivation of the JC virus causing PML.162 This life-threatening condition was initially fatal in all cases but can now be managed by using plasma exchange to remove natalizumab, allowing the host immune system to reconstitute and to clear the virus, although at a risk of inducing the immune reconstitution syndrome (IRIS).162 An emerging risk-management strategy is evolving around the finding that those who have never had the JC virus infection can not get PML unless they develop a new infection. The development of an assay to test for the presence of JC virus means that the risk of this complication can be stratified.163 The risk of malignancy with natalizumab is not elevated; although initial links were made with melanoma this has not being confirmed in large populations.164
Oral therapies have shown evidence of life-threatening adverse events in randomized controlled trials. In the placebo-controlled fingolimod study, two subjects died due to disseminated herpes infections134 and in the comparator study there was an increased frequency of localized skin cancers in the fingolimod-treated group.135 Cladribine was associated with an increased risk of malignancy with none in the placebo group and 10 (1.1%) in the treated group. Notably, five were benign uterine leiomyomas, the significance of which is as yet unclear.123
These issues with oral therapies hinted at in the randomized controlled trials (Table 2) require further information to be collected post-licensing to quantify the existence and extent of any association. In the light of the prolonged disease course in MS, long-term side effects need to be quantified in order to optimize treatment pathways.
Pregnancy
This is a significant consideration as MS presents more commonly in childbearing women than any other group. It is a key issue for young women considering disease-modifying therapy, as MS itself has no effect on pregnancy outcomes and pregnancy can have a beneficial effect on the disease, though it has to be balanced against deterioration after delivery.165 Of the standard therapies, GA has not caused major concerns in pregnancy. IFN-β is associated with an abortive effect at 40 times the recommended dose.166 GA can be continued in pregnancy (FDA category B) but it is currently recommended that IFN-β be halted prior to pregnancy or if the patient becomes pregnant (FDA category C). Recent work in 88 pregnancies exposed to IFN-β showed no evidence of any fetal complications, malformations, or developmental abnormalities.165 Natalizumab has shown evidence of fetotoxicity in animal studies. Without controlled data in pregnancy it is recommended only when benefit outweighs risk (FDA category C). However cohort studies have confirmed that subjects can have a successful pregnancy after natalizumab.153
Animal data have revealed evidence that at doses equivalent to the recommended dose in humans, cladribine can be teratogenetic, embryotoxic, and fetotoxic, can increase resorptions and reduce litter size, and can cause fetal death. As a result cladribine has been assigned to pregnancy category D by the FDA. There are no anecdotal or controlled data from human pregnancy and there is no evidence of teratogenicity in humans. However other drugs that inhibit DNA synthesis have been teratogenic in humans.
The S1P1 receptor blocked by fingolimod is involved in vascular formation, and consistent with this fingolimod has been found to produce vascular defects in one animal species, including persistent truncus arteriosus and ventricular septal defect.
Fifty-two pregnancies have been reported in fingolimod clinical trials in MS as well as three pregnancies in the renal transplant population.167 The incidence of spontaneous abortion was consistent with expected rates. A single malformation has been reported, unilateral congenital postero-medial bowing of the tibia, but it was not clearly related to fingolimod. However, given the findings in animals, the use of fingolimod in women who are or may become pregnant should be considered only if benefit is deemed to exceed potential risk to the fetus. Women considering pregnancy should stop fingolimod for at least 2 months.
Place in therapy in comparison with conventional treatment options
The efficacy of oral therapies in reducing relapse rates places them a step above the standard injectables, IFN-β and GA, but below natalizumab and mitoxantrone.
However, there are some concerns about the RRMS populations in which the recent oral trials were conducted, which were different from those of the IFN-β/GA studies in terms of baseline relapse rate (Figure 3). This is notable in that the extent of relapse reduction in natalizumab comparator studies showed a reduction in relapse rates of 45% by adding natalizumab (IFN-β1a: annualized relapse rate [ARR] 0.75 vs IFN-β1a + natalizumab: ARR 0.3442), which is similar (48%) to that seen in the fingolimod comparator study (IFN-β1a: ARR 0.33 vs fingolimod 1.25 mg daily: ARR 0.16).135 However, the comparative study currently available for fingolimod,35 though it recruited a proportion of IFN-β failures, would indicate that it is more effective than IFN-β and, by implication, GA. Because comparator studies using cladribine versus IFN-β are not yet available, at this point cladribine is lacking some supportive data. However, no direct studies have yet been performed with either fingolimod or cladribine versus GA.
The key question is whether oral therapies will take the place of IFN-β/GA or mitoxantrone/natalizumab. If the decision was based solely on efficacy and mode of administration they would replace IFN-β/GA. However IFN-β/GA are safe therapies, and even though subjects may experience ongoing side effects, these medications are likely to remain first-line offerings for some time (Table 3). In subjects with no evidence of ongoing disease activity on IFN-β/GA there is no reason to proactively change therapy until more information on side effects of oral treatments becomes available. In females with MS who wish to become pregnant in future GA is currently favored, but with the knowledge that IFN-β can be stopped and pregnancy can be successful, this drug can also be used. Increasing experience of natalizumab in pregnancy might suggest that it can be used like IFN-β and stopped prior to pregnancy.
Table 3.
Effect of therapy on long-term adherence
|
Type |
Injectables |
Orals |
||||
|---|---|---|---|---|---|---|
| Therapy | IFN-β | GA | Natalizumab | Fingolimod | Cladribine | Teriflunomide |
| Administration | ++ weekly –3 times per week, sc/im |
+++ daily sc | + every 28 days, iv | – | – | – |
| Day-to-day | ++ | + | + | ± | – | – |
| Risk of infections | – | – | + | + | ± | – |
| Life threatening (malignancies/infections) | – | – | ± | + | ++ | ± |
| Pregnancy (FDA category) | C | B | C | ? | D | ? |
Notes: Administration is the problem caused by the method and frequency of drug administration. Day-to-day is the ongoing side effects of therapy. The risk of infections requires increasing levels of surveillance by physicians and patients. Life-threatening side effects include infections, malignancy, and other causes of major clinical events.
Abbreviation: GA, glatiramer acetate.
The key determinants that will affect the prescription of oral therapies in early demyelinating syndromes appear to be side effects and risks. Natalizumab is not used in the early demyelinating syndromes such as RIS, CIS, and “McDonald criteria” MS where the prognosis is unclear because of concerns about side effects. In fact serious side effects for cladribine and fingolimod appear at this point to be higher than natalizumab’s risk of PML (Table 2). Though further data are needed on long-term side effects with oral therapies, successfully stratifying risk in subjects considering natalizumab by the antibody test for JC virus could significantly reassure patients and physicians about its safety in low-risk patients. This could weaken the argument for using oral therapies, as a more effective, equally safe agent would be available, and then their only advantage would be the oral formulation. Taking into consideration the current perceived risk of therapy by licensing agencies as an indication, it would appear that orals may be recommended by some health systems in “McDonald criteria” MS. Their use does not appear justifiable in CIS or RIS at this point without direct trial evidence (Table 3).
In more aggressive MS the evidence for efficacy of oral agents may not support the delay in initiating effective therapy, given that natalizumab has a track record of efficacy, and although it has side effects they are well known and with stratification the risk profile could be significantly improved. Furthermore, competition from new emerging “immune resetting” treatments such as alemtuzumab,9 and mesenchymal or hematopoietic stem cell transplantation168 are likely to arise as potential therapies of choice in more severe, treatment-resistant forms of disease that still present inflammatory features.
Conclusion
Oral therapies will unquestionably have a major impact on the treatment of RRMS. They are attractive to both patients and health care providers in that they avoid injection-related discomfort and adverse events and reduce the costs of administering them.
Efficacy matters to patients and, together with the way of administration, oral drugs represent a strong new entry to the relapse-modifying therapy market. Further data are required on their long-term safety profile to establish their exact role in treatment of different forms of MS (early vs established, mild vs severe) and their placement in relation to the existing treatments.
Current data support their gradual introduction as an option for patients with RRMS who decline, opt out of, or fail to adequately respond to injectable treatments. Appropriate programs for monitoring adverse events are warranted.
Acknowledgments
This work was supported by the Multiple Sclerosis Trials Collaboration (Charity number 1113598; to PG, AA), the Medical Research Council (Ref. G0800679; to PM and RN) and the Italian MS society (Ref. 2008/R/16; to PM).
Footnotes
Disclosure
The authors declare no conflicts of interest in this work.
References
- 1.Ford HL, Gerry E, Johnson M, et al. A prospective study of the incidence, prevalence and mortality of multiple sclerosis in Leeds. J Neurol. 2002;249:260–265. doi: 10.1007/s004150200002. [DOI] [PubMed] [Google Scholar]
- 2.Sloka JS, Pryse-Phillips WE, Stefanelli M. Incidence and prevalence of multiple sclerosis in Newfoundland and Labrador. Can J Neurol Sci. 2005;32:37–42. doi: 10.1017/s0317167100016851. [DOI] [PubMed] [Google Scholar]
- 3.Ebers GC, Traboulsee A, Li D, et al. Investigators of the 16-year Long-Term Follow-Up Study Analysis of clinical outcomes according to original treatment groups 16 years after the pivotal IFNB-1b trial. J Neurol Neurosurg Psychiatry. 2010;81:907–912. doi: 10.1136/jnnp.2009.204123. [DOI] [PubMed] [Google Scholar]
- 4.McCabe C, Chilcott J, Claxton K, et al. Continuing the multiple sclerosis risk sharing scheme is unjustified. BMJ. 2010;340:c1786. doi: 10.1136/bmj.c1786. [DOI] [PubMed] [Google Scholar]
- 5.Munari L, Lovati R, Boiko A. The Cochrane Library. 4. Chichester, UK: John Wiley & Sons, Ltd; 2006. Therapy with glatiramer acetate for multiple sclerosis. [DOI] [PubMed] [Google Scholar]
- 6.Polman CH, O’Connor PW, Havrdova E, et al. A randomized, placebo-controlled trial of natalizumab for relapsing multiple sclerosis. N Engl J Med. 2006;354:899–910. doi: 10.1056/NEJMoa044397. [DOI] [PubMed] [Google Scholar]
- 7.Gold R. Oral therapies for multiple sclerosis: a review of agents in Phase III development or recently approved. CNS Drugs. 2011;25:37–52. doi: 10.2165/11539820-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 8.Polman CH, Reingold SC, Edan G, et al. Diagnostic criteria for multiple sclerosis: 2005 revisions to the “McDonald Criteria”. Ann Neurol. 2005;58:840–846. doi: 10.1002/ana.20703. [DOI] [PubMed] [Google Scholar]
- 9.CAMMS223 Trial Investigators Alemtuzumab vs interferon beta-1a in early multiple sclerosis. N Engl J Med. 2008;359:1786–1801. doi: 10.1056/NEJMoa0802670. [DOI] [PubMed] [Google Scholar]
- 10.Burt RK, Loh Y, Cohen B, et al. Autologous non-myeloablative haemopoietic stem cell transplantation in relapsing-remitting multiple sclerosis: a phase I/II study. Lancet Neurol. 2009;8(3):244–253. doi: 10.1016/S1474-4422(09)70017-1. Erratum in: Lancet Neurol. 2009;8:309. [DOI] [PubMed] [Google Scholar]
- 11.Scalfari A, Neuhaus A, Degenhardt A, et al. The natural history of multiple sclerosis: a geographically based study 10: relapses and long-term disability. Brain. 2010;133:1914–1929. doi: 10.1093/brain/awq118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fisniku LK, Brex PA, Altmann DR, et al. Disability and T2 MRI lesions: a 20-year follow-up of patients with relapse onset of multiple sclerosis. Brain. 2008;131:808–817. doi: 10.1093/brain/awm329. [DOI] [PubMed] [Google Scholar]
- 13.Okuda DT, Mowry EM, Beheshtian A, et al. Incidental MRI anomalies suggestive of multiple sclerosis: the radiologically isolated syndrome. Neurology. 2009;72:800–805. doi: 10.1212/01.wnl.0000335764.14513.1a. Erratum in: Neurology. 2009;72:1284. [DOI] [PubMed] [Google Scholar]
- 14.National Institute for Health and Clinical Excellence (NICE) Natalizumab for adults with highly active relapsing-remitting multiple sclerosis. NICE technology appraisal guidance 12, Issue date: August 2007. Available at: http://www.nice.org.uk/nicemedia/live/11822/36136/36136.pdf. Accessed January 1, 2011.
- 15.Jones JL, Coles AJ. New treatment strategies in multiple sclerosis. Exp Neurol. 2010;225:34–39. doi: 10.1016/j.expneurol.2010.06.003. [DOI] [PubMed] [Google Scholar]
- 16.Degenhardt A, Ramagopalan SV, Scalfari A, et al. Clinical prognostic factors in multiple sclerosis: a natural history review. Nat Rev Neurol. 2009;5:672–682. doi: 10.1038/nrneurol.2009.178. [DOI] [PubMed] [Google Scholar]
- 17.Magliozzi R, Howell O, Vora A, et al. Meningeal B-cell follicles in secondary progressive multiple sclerosis associated with early onset of disease and severe cortical pathology. Brain. 2007;130:1089–1104. doi: 10.1093/brain/awm038. [DOI] [PubMed] [Google Scholar]
- 18.Coles AJ, Wing MG, Molyneux P, et al. Monoclonal antibody treatment exposes three mechanisms underlying the clinical course of multiple sclerosis. Ann Neurol. 1999;46:296–304. doi: 10.1002/1531-8249(199909)46:3<296::aid-ana4>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- 19.Burt RK, Cohen BA, Russell E, et al. Hematopoietic stem cell transplantation for progressive multiple sclerosis: failure of a total body irradiation-based conditioning regimen to prevent disease progression in patients with high disability scores. Blood. 2003;102:2373–2378. doi: 10.1182/blood-2003-03-0877. [DOI] [PubMed] [Google Scholar]
- 20.Inusah S, Sormani MP, Cofield SS, et al. Assessing changes in relapse rates in multiple sclerosis. Mult Scler. 2010;16:1414–1421. doi: 10.1177/1352458510379246. [DOI] [PubMed] [Google Scholar]
- 21.Nicholas R, Straube S, Schmidli H, et al. Trends in annualized relapse rates in relapsing remitting multiple sclerosis and consequences for clinical trial design. Mult Scler. 2011 doi: 10.1177/1352458511406309. In press. [DOI] [PubMed] [Google Scholar]
- 22.Nicholas R, Chataway J. Multiple Sclerosis: Clinical Evidence Handbook. London, UK: BMJ Publishing Group Ltd; 2007. [Google Scholar]
- 23.Dhib-Jalbut S. Mechanisms of interferon beta action in multiple sclerosis. Mult Scler. 1997;3:397–401. doi: 10.1177/135245859700300609. [DOI] [PubMed] [Google Scholar]
- 24.Alam J, Goelz S, Rioux P, et al. Comparative pharmacokinetics and pharmacodynamics of two recombinant human interferon beta-1a (IFN beta-1 a) products administered intramuscularly in healthy male and female volunteers. Pharm Res. 1997;14:546–549. doi: 10.1023/a:1012128406432. [DOI] [PubMed] [Google Scholar]
- 25.Rice GPA, Incorvaia B, Munari L, et al. The Cochrane Library. 4. Chichester, UK: John Wiley & Sons, Ltd; 2006. Interferon in relapsing-remitting multiple sclerosis. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jacobs LD, Cookfair DL, Rudick RA, et al. Intramuscular interferon beta-1a for disease progression in relapsing multiple sclerosis. The Multiple Sclerosis Collaborative Research Group (MSCRG) Ann Neurol. 1996;39:285–294. doi: 10.1002/ana.410390304. [DOI] [PubMed] [Google Scholar]
- 27.Jacobs LD, Beck RW, Simon JH, et al. Intramuscular interferon beta-1a therapy initiated during a first demyelinating event in multiple sclerosis. N Engl J Med. 2000;343:898–904. doi: 10.1056/NEJM200009283431301. [DOI] [PubMed] [Google Scholar]
- 28.Comi G, Fillipi M, Barkhof F, et al. Effect of early interferon treatment on conversion to definite multiple sclerosis: a randomised study. Lancet. 2001;357:1576–1582. doi: 10.1016/s0140-6736(00)04725-5. [DOI] [PubMed] [Google Scholar]
- 29.Kappos L, Polman CH, Freedman MS, et al. Treatment with interferon beta-1b delays conversion to clinically definite and McDonald MS in patients with clinically isolated syndromes. Neurol. 2006;67:1242–1249. doi: 10.1212/01.wnl.0000237641.33768.8d. [DOI] [PubMed] [Google Scholar]
- 30.US Food and Drug Administration Medwatch safety information alerts. 2006. Available at: http://www.fda.gov/Safety/MedWatch/SafetyInformation/SafetyAlertsforHumanMedicalProducts/ucm150730.htm. Accessed January 1, 2011.
- 31.Sorensen PS, Koch-Henriksen N, Ross C, et al. Appearance and disappearance of neutralizing antibodies during interferon-beta therapy. Neurology. 2005;65:33–39. doi: 10.1212/01.WNL.0000166049.51502.6A. [DOI] [PubMed] [Google Scholar]
- 32.Treadaway K, Cutter G, Salter A, et al. Factors that influence adherence with disease-modifying therapy in MS. J Neurol. 2009;256:568–576. doi: 10.1007/s00415-009-0096-y. [DOI] [PubMed] [Google Scholar]
- 33.Turner AP, Williams RM, Sloan AP, et al. Injection anxiety remains a long-term barrier to medication adherence in multiple sclerosis. Rehabil Psychol. 2009;54:116–121. doi: 10.1037/a0014460. [DOI] [PubMed] [Google Scholar]
- 34.Bornstein MB, Miller A, Slagle S, et al. A pilot trial of Cop 1 in exacerbating-remitting multiple sclerosis. N Engl J Med. 1987;317:408–414. doi: 10.1056/NEJM198708133170703. [DOI] [PubMed] [Google Scholar]
- 35.Duda PW, Schmied MC, Cook SL, et al. Glatiramer acetate (Copaxone) induces degenerate, Th2-polarized immune responses in patients with multiple sclerosis. J Clin Invest. 2000;105:967–976. doi: 10.1172/JCI8970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Johnson KP, Brooks BR, Cohen JA, et al. Copolymer 1 reduces relapse rate and improves disability in relapsing-remitting multiple sclerosis: results of a phase III multicenter, double-blind placebo-controlled trial. The Copolymer 1 Multiple Sclerosis Study Group. Neurology. 1995;45:1268–1276. doi: 10.1212/wnl.45.7.1268. [DOI] [PubMed] [Google Scholar]
- 37.Filippi M, Rovaris M, Rocca MA, et al. Glatiramer acetate reduces the proportion of new MS lesions evolving into “black holes”. Neurology. 2001;57:731–733. doi: 10.1212/wnl.57.4.731. [DOI] [PubMed] [Google Scholar]
- 38.Mikol DD, Barkhof F, Chang P, et al. Comparison of subcutaneous interferon beta-1a with glatiramer acetate in patients with relapsing multiple sclerosis: a multicentre, randomised, parallel, open-label trial. Lancet Neurol. 2008;7:903–914. doi: 10.1016/S1474-4422(08)70200-X. [DOI] [PubMed] [Google Scholar]
- 39.O’Connor P, Devonshire V. The use of disease-modifying agents in multiple sclerosis – by the Canadian Network of MS Clinics. Can J Neurol Sci. 2008;35:127–132. [PubMed] [Google Scholar]
- 40.Comi G, Martinelli V, Rodegher M, et al. PreCISe study group Effect of glatiramer acetate on conversion to clinically definite multiple sclerosis in patients with clinically isolated syndrome (PreCISe study): a randomised, double-blind, placebo-controlled trial. Lancet. 2009;374:1503–1511. doi: 10.1016/S0140-6736(09)61259-9. [DOI] [PubMed] [Google Scholar]
- 41.Steinman L. Blocking adhesion molecules as therapy for multiple sclerosis: natalizumab. Nat Rev Drug Discov. 2005;4:510–518. doi: 10.1038/nrd1752. [DOI] [PubMed] [Google Scholar]
- 42.Rudick RA, Stuart WH, Calabresi PA, et al. Natalizumab plus interferon beta-1a for relapsing multiple sclerosis. N Engl J Med. 2006;354:911–923. doi: 10.1056/NEJMoa044396. [DOI] [PubMed] [Google Scholar]
- 43.Yousry TA, Major EO, Ryschkewitsch C, et al. Evaluation of patients treated with natalizumab for progressive multifocal leukoencephalopathy. N Engl J Med. 2006;354:924–933. doi: 10.1056/NEJMoa054693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Thompson JP, Noyes K, Dorsey ER, et al. Quantitative risk-benefit analysis of natalizumab. Neurology. 2008;71:357–364. doi: 10.1212/01.wnl.0000319648.65173.7a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Calabresi PA, Giovannoni G, Confavreux C, et al. The incidence and significance of anti-natalizumab antibodies: results from AFFIRM and SENTINEL. Neurology. 2007;69:1391–1403. doi: 10.1212/01.wnl.0000277457.17420.b5. [DOI] [PubMed] [Google Scholar]
- 46.Fox EJ. Mechanism of action of mitoxantrone. Neurology. 2004;63(12 Suppl 6):S15–S18. doi: 10.1212/wnl.63.12_suppl_6.s15. [DOI] [PubMed] [Google Scholar]
- 47.Martinelli Boneschi F, Rovaris M, Capra R, et al. The Cochrane Library. 4. Chichester, UK: John Wiley & Sons, Ltd; 2006. Mitoxantrone for multiple sclerosis. [Google Scholar]
- 48.Edan G, Miller D, Clanet M, et al. Therapeutic effect of mitoxantrone combined with methylprednisolone in multiple sclerosis: a randomised multicentre study of active disease using MRI and clinical criteria. J Neurol Neurosurg Psychiatry. 1997;62:112–118. doi: 10.1136/jnnp.62.2.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hartung HP, Gonsette R, Konig N, et al. Mitoxantrone in progressive multiple sclerosis: a placebo-controlled, double-blind, randomised, multicentre trial. Lancet. 2002;360:2018–2025. doi: 10.1016/S0140-6736(02)12023-X. [DOI] [PubMed] [Google Scholar]
- 50.Ghalie RG, Edan G, Laurent M, et al. Cardiac adverse effects associated with mitoxantrone (Novantrone) therapy in patients with MS. Neurology. 2002;59:909–913. doi: 10.1212/wnl.59.6.909. [DOI] [PubMed] [Google Scholar]
- 51.Martinelli V, Radaelli M, Straffi L, et al. Mitoxantrone: benefits and risks in multiple sclerosis patients. Neurol Sci. 2009;30(Suppl 2):S167–S170. doi: 10.1007/s10072-009-0142-7. [DOI] [PubMed] [Google Scholar]
- 52.Carson DA, Wasson DB, Taetle R, et al. Specific toxicity of 2-chlorodeoxyadenosine toward resting and proliferating human lymphocytes. Blood. 1983;62:737–743. [PubMed] [Google Scholar]
- 53.Brousil JA, Roberts RJ, Schlein AL. Cladribine: an investigational immunomodulatory agent for multiple sclerosis. Ann Pharmacother. 2006;40:1814–1821. doi: 10.1345/aph.1H037. [DOI] [PubMed] [Google Scholar]
- 54.Kawasaki H, Carrera CJ, Piro LD, et al. Relationship of deoxycytidine kinase and cytoplasmic 50-nucleotidase to the chemotherapeutic efficacy of 2-chlorodeoxyadenosine. Blood. 1993;81:597–601. [PubMed] [Google Scholar]
- 55.Carson DA, Kaye J, Seegmiller JE. Lymphospecific toxicity in adenosine deaminase deficiency and purine nucleoside phosphorylase deficiency: possible role of nucleoside kinase(s) Proc Natl Acad Sci U S A. 1977;74:5677–5681. doi: 10.1073/pnas.74.12.5677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rice GP, Filippi M, Comi G, Cladribine Study Group Cladribine and progressive MS: clinical and MRI outcomes of a multicenter controlled trial. Neurology. 2000;54:1145–1155. doi: 10.1212/wnl.54.5.1145. [DOI] [PubMed] [Google Scholar]
- 57.Kopadze T, Döbert M, Leussink VI, et al. Cladribine impedes in vitro migration of mononuclear cells: a possible implication for treating multiple sclerosis. Eur J Neurol. 2009;16:409–412. doi: 10.1111/j.1468-1331.2008.02433.x. [DOI] [PubMed] [Google Scholar]
- 58.Janiec K, Wajgt A, Kondera-Anasz Z. Effect of immunosuppressive cladribine treatment on serum leucocytes system in two-year clinical trial in patients with chronic progressive multiple sclerosis. Med Sci Monit. 2001;7:93–98. [PubMed] [Google Scholar]
- 59.Bartosik-Psujek H, Belniak E, Mitosek-Szewczyk K, et al. Interleukin-8 and RANTES levels in patients with relapsing-remitting multiple sclerosis treated with cladribine. Acta Neurol Scand. 2004;109:390–392. doi: 10.1111/j.1600-0404.2004.00259.x. [DOI] [PubMed] [Google Scholar]
- 60.Laugel B, Challier J, Siegfried C, et al. Cladribine exerts a modulatory effect of T cell activation. Mult Scler. 2008;14:S52–S53. [Google Scholar]
- 61.Liliemark J. The clinical pharmacokinetics of cladribine. Clin Pharmacokinet. 1997;32:120–131. doi: 10.2165/00003088-199732020-00003. [DOI] [PubMed] [Google Scholar]
- 62.Albertioni F, Herbgren L, Juliusson G, Liliemark J. Protein binding of 2-chloro 2′-deoxyadenosine (cladribine) in healthy subjects and in patients with leukaemia. Eur J Clin Pharmacol. 1994;46:563–564. doi: 10.1007/BF00196117. [DOI] [PubMed] [Google Scholar]
- 63.Australian Public Assessment Report for cladribine tablets. 2010 Dec; Available at: http://www.tga.gov.au/pmeds/auspar/auspar-movectro.pdf. Accessed March 19. http://www.tga.gov.au/pmeds/auspar/auspar-movectro.pdf. Accessed March 19. 2011. [Google Scholar]
- 64.Matloubian M, Lo CG, Cinamon G, et al. Lymphocyte egress from thymus and peripheral lymphoid organs is dependent on S1P receptor 1. Nature. 2004;427:355–360. doi: 10.1038/nature02284. [DOI] [PubMed] [Google Scholar]
- 65.Spiegel S, Milstien S. Sphingosine-1-phosphate: an enigmatic signalling lipid. Nat Rev Mol Cell Biol. 2003;4:397–407. doi: 10.1038/nrm1103. [DOI] [PubMed] [Google Scholar]
- 66.Cyster JG. Chemokines, sphingosine-1-phosphate, and cell migration in secondary lymphoid organs. Annu Rev Immunol. 2005;23:27–159. doi: 10.1146/annurev.immunol.23.021704.115628. [DOI] [PubMed] [Google Scholar]
- 67.Hanel P, Andreani P, Graler MH. Erythrocytes store and release sphingosine 1-phosphate in blood. FASEB J. 2007;21:1202–1209. doi: 10.1096/fj.06-7433com. [DOI] [PubMed] [Google Scholar]
- 68.Brinkmann V, Davis MD, Heise CE, et al. The immune modulator FTY720 targets sphingosine 1-phosphate receptors. J Biol Chem. 2002;277:21453–21457. doi: 10.1074/jbc.C200176200. [DOI] [PubMed] [Google Scholar]
- 69.Graler MH, Goetzl EJ. The immunosuppressant FTY720 down-regulates sphingosine 1-phosphate G-protein-coupled receptors. FASEB J. 2004;18:551–553. doi: 10.1096/fj.03-0910fje. [DOI] [PubMed] [Google Scholar]
- 70.Schwab SR, Pereira JP, Matloubian M, et al. Lymphocyte sequestration through S1P lyase inhibition and disruption of S1P gradients. Science. 2005;309:1735–1739. doi: 10.1126/science.1113640. [DOI] [PubMed] [Google Scholar]
- 71.Wei SH, Rosen H, Matheu MP, et al. Sphingosine 1-phosphate type 1 receptor agonism inhibits transendothelial migration of medullary T cells to lymphatic sinuses. Nat Immunol. 2005;6:1228–1235. doi: 10.1038/ni1269. [DOI] [PubMed] [Google Scholar]
- 72.Jo E, Sanna MG, Gonzalez-Cabrera PJ, et al. S1P1-selective in vivo-active agonists from high-throughput screening: off the-shelf chemical probes of receptor interactions, signaling, and fate. Chem Biol. 2005;12:703–715. doi: 10.1016/j.chembiol.2005.04.019. [DOI] [PubMed] [Google Scholar]
- 73.Sanna MG. Enhancement of capillary leakage and restoration of lymphocyte egress by a chiral S1P1 antagonist in vivo. Nat Chem Biol. 2006;2:434–441. doi: 10.1038/nchembio804. [DOI] [PubMed] [Google Scholar]
- 74.Kovarik JM, Schmouder RL, Slade AJ. Overview of FTY720 clinical pharmacokinetics and pharmacology. Ther Drug Monit. 2004;26:585–587. doi: 10.1097/00007691-200412000-00001. [DOI] [PubMed] [Google Scholar]
- 75.Kovarik JM, Hartmann S, Bartlett M, et al. Oral-intravenous crossover study of fingolimod pharmacokinetics, lymphocyte responses and cardiac effects. Biopharm Drug Dispos. 2007;28:97–104. doi: 10.1002/bdd.535. [DOI] [PubMed] [Google Scholar]
- 76.Schmouder RL, Choudhury S, Barilla D, et al. Prolonged, consistent oral absorption of FTY720. (Abstract) Am J Transplant. 2001;1:475. [Google Scholar]
- 77.Zollinger M, Gschwind HP, Jin Y, Sayer C, Zécri F, Hartmann S. Absorption and disposition of the sphingosine 1-phosphate receptor modulator fingolimod (FTY720) in healthy volunteers: a case of xenobiotic biotransformation following endogenous metabolic pathways. Drug Metab Dispos. 2011;39:199–207. doi: 10.1124/dmd.110.035907. [DOI] [PubMed] [Google Scholar]
- 78.Ockenfels HM, Schultewolter T, Ockenfels G, et al. The antipsoriatic agent dimethylfumarate immunomodulates T-cell cytokine secretion and inhibits cytokines of the psoriatic cytokine network. Br J Dermatol. 1998;139:390–395. doi: 10.1046/j.1365-2133.1998.02400.x. [DOI] [PubMed] [Google Scholar]
- 79.Asadullah K, Schmid H, Friedrich M, et al. Influence of monomethylfumarate on monocytic cytokine formation eplanation for adverse and therapeutic effects in psoriasis? Arch Dermatol Res. 1997;289:623–630. doi: 10.1007/s004030050251. [DOI] [PubMed] [Google Scholar]
- 80.De Jong R, Bezemer AC, Zomerdijk TP, et al. Selective stimulation of T helper 2 cytokine responses by the anti-psoriasis agent monomethylfumarate. Eur J Immunol. 1996;26:2067–2074. doi: 10.1002/eji.1830260916. [DOI] [PubMed] [Google Scholar]
- 81.Schilling S, Goelz S, Linker R, et al. Fumaric acid esters are effective in chronic experimental autoimmune encephalomyelitis and suppress macrophage infiltration. Clin Exp Immunol. 2006;145:101–107. doi: 10.1111/j.1365-2249.2006.03094.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Schimrigk S, Brune N, Hellwig K, et al. Oral fumaric acid esters for the treatment of active multiple sclerosis: an open-label, baseline controlled pilot study. Eur J Neurol. 2006;13:604–610. doi: 10.1111/j.1468-1331.2006.01292.x. [DOI] [PubMed] [Google Scholar]
- 83.Litjens NH, Rademaker M, Ravensbergen B, et al. Monotheylfumarate affects polarization of monocyte-derived dendritic cells resulting in down-regulated Th1 lymphocyte responses. Eur J Immunol. 2004;34:565–575. doi: 10.1002/eji.200324174. [DOI] [PubMed] [Google Scholar]
- 84.Loewe R, Pillinger M, de Martin R, et al. Dimethylfumarate inhibits tumor-necrosis-factor-induced CD62E expression in an NF-kappa B-dependent manner. J Invest Dermatol. 2001;117:1363–1368. doi: 10.1046/j.0022-202x.2001.01576.x. [DOI] [PubMed] [Google Scholar]
- 85.Loewe R, Holnthoner W, Groger M, et al. Dimethylfumarate inhibits TNF-induced nuclear entry of NF-kappa B/p65 in human endothelial cells. J Immunol. 2002;168:4781–4787. doi: 10.4049/jimmunol.168.9.4781. [DOI] [PubMed] [Google Scholar]
- 86.Wierinckx A, Breve J, Mercier D, et al. Detoxification enzyme inducers modify cytokine production in rat mixed glial cells. J Neuroimmunol. 2005;166:132–143. doi: 10.1016/j.jneuroim.2005.05.013. [DOI] [PubMed] [Google Scholar]
- 87.Chen XL, Dodd G, Thomas S, et al. Activation of Nrf2/ARE pathway protects endothelial cells from oxidant injury and inhibits inflammatory gene expression. Am J Physiol Heart Circ Physiol. 2006;290:H1862–H1870. doi: 10.1152/ajpheart.00651.2005. [DOI] [PubMed] [Google Scholar]
- 88.Lukashev M, Zeng W, Ryan S, et al. Activation of Nrf2 and modulation of disease progression in EAE models by BG00012 (Dimethyl Fumarate) suggests a novel mechanism of action combining anti-inflammatory and neuroprotective modalities. Mult Scler. 2007;13:S149. [Google Scholar]
- 89.Itoh K, Chiba T, Takahashi S, et al. An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem Biophys Res Commun. 1997;236:313–322. doi: 10.1006/bbrc.1997.6943. [DOI] [PubMed] [Google Scholar]
- 90.Venugopal R, Jaiswal AK. Nrf2 and Nrf1 in association with Jun proteins regulate antioxidant response element-mediated expression and coordinated induction of genes encoding detoxifying enzymes. Oncogene. 1998;17:3145–3156. doi: 10.1038/sj.onc.1202237. [DOI] [PubMed] [Google Scholar]
- 91.Calabrese V, Ravagna A, Colombrita C, et al. Acetylcarnitine induces heme oxygenase in rat astrocytes and protects against oxidative stress: involvement of the transcription factor Nrf2. J Neurosci Res. 2005;79:509–521. doi: 10.1002/jnr.20386. [DOI] [PubMed] [Google Scholar]
- 92.Li J, Johnson D, Calkins M, et al. Stabilization of Nrf2 by tBHQ confers protection against oxidative stress-induced cell death in human neural stem cells. Toxicol Sci. 2005;83:313–328. doi: 10.1093/toxsci/kfi027. [DOI] [PubMed] [Google Scholar]
- 93.Satoh T, Okamato SI, Cui J, et al. Activation of the Keap1/Nrf2 pathway for neuroprotection by electrophillic phase II inducers. Proc Natl Acad Sci. 2006;103:768–773. doi: 10.1073/pnas.0505723102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Shih AY, Li P, Murphy TH. A small-molecule-inducible Nrf2-mediated antioxidant response provides effective prophylaxis against cerebral ischemia in vivo. J Neurosci. 2005;25:10321–10335. doi: 10.1523/JNEUROSCI.4014-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Vargas MR, Pehar M, Cassina P, et al. Fibroblast growth factor-1 induces heme oxygenase-1 via nuclear factor erythroid 2-related factor 2 (Nrf2) in spinal cord astrocytes: consequences for motor neuron survival. J Biol Chem. 2005;27:25571–25579. doi: 10.1074/jbc.M501920200. [DOI] [PubMed] [Google Scholar]
- 96.Zhao J, Moore AN, Redell JB, et al. Enhancing expression of Nrf2-driven genes protects the blood brain barrier after brain injury. J Neurosci. 2007;27:10240–10248. doi: 10.1523/JNEUROSCI.1683-07.2007. 19; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Hubbs AF, Benkovic SA, Miller DB, et al. Vacuolar leukoencephalopathy with widespread astrogliosis in mice lacking transcription factor Nrf2. Am J Pathol. 2007;170:2068–2076. doi: 10.2353/ajpath.2007.060898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Nibbering PH, Thio B, Zomerdijk TP, et al. Effects of monomethylfumarate on human granulocytes. J Invest Dermatol. 1993;101:37–42. doi: 10.1111/1523-1747.ep12358715. [DOI] [PubMed] [Google Scholar]
- 99.Mrowietz U, Christophers E, Altmeyer P. Treatment of severe psoriasis with fumaric acid esters: scientific background and guidelines for therapeutic use. The German Fumaric Acid Ester Consensus Conference. Br J Dermatol. 1999;141:424–429. doi: 10.1046/j.1365-2133.1999.03034.x. [DOI] [PubMed] [Google Scholar]
- 100.Noseworthy JH, Wolinsky JS, Lublin FD, et al. North American Linomide Investigators Linomide in relapsing and secondary progressive MS. Part I: trial design and clinical results. Neurology. 2000;54:1726–1733. doi: 10.1212/wnl.54.9.1726. [DOI] [PubMed] [Google Scholar]
- 101.Wolinsky JS, Narayana PA, Noseworthy JH, et al. MRI analysis center of the University of Texas–Houston, health science center, and the North American Linomide investigators Linomide in relapsing and secondary progressive MS. Part II: MRI results. Neurology. 2000;54:1734–1741. doi: 10.1212/wnl.54.9.1734. [DOI] [PubMed] [Google Scholar]
- 102.Tan IL, Lycklama à Nijeholt GJ, Polman CH, et al. 95 Line 146/147 Study Group Linomide in the treatment of multiple sclerosis: MRI results from prematurely terminated phase-III trails. Mult Scler. 2000;6:99–104. doi: 10.1177/135245850000600208. [DOI] [PubMed] [Google Scholar]
- 103.Brunmark C, Runström A, Ohlsson L, et al. The new orally active immunoregulator laquinimod (ABR-215062) effectively inhibits development and relapses of experimental autoimmune encephalomyelitis. J Neuroimmunol. 2002;130:163–172. doi: 10.1016/s0165-5728(02)00225-4. [DOI] [PubMed] [Google Scholar]
- 104.Yang JS, Xu LY, Xiao BG, et al. Laquinimod (ABR-215062) suppresses the development of experimental autoimmune encephalomyelitis, modulates the Th1/Th2 balance and induces the Th3 cytokine TGF-beta in Lewis rats. J Neuroimmunol. 2004;156:3–9. doi: 10.1016/j.jneuroim.2004.02.016. [DOI] [PubMed] [Google Scholar]
- 105.Wegner C, Pfortner R, Bruck W. Laquinimod rescue therapy in mice with experimental autoimmune encephalomyelitis. Mult Scler. 2010;16:S41–S196. Abstract P251. [Google Scholar]
- 106.Gurevich M, Gritzman T, Orbach R, et al. Laquinimod suppress antigen presentation in relapsing-remitting multiple sclerosis: in-vitro high-throughput gene expression study. J Neuroimmunol. 2010;221:87–94. doi: 10.1016/j.jneuroim.2010.02.010. [DOI] [PubMed] [Google Scholar]
- 107.Runstrom A, Leanderson T, Ohlsson L, et al. Inhibition of the development of chronic experimental autoimmune encephalomyelitis by laquinimod (ABR-215062) in IFN-beta k.o. and wild type mice. J Neuroimmunol. 2006;173:69–78. doi: 10.1016/j.jneuroim.2005.11.023. [DOI] [PubMed] [Google Scholar]
- 108.Wegner C, Stadelmann C, Pförtner R, et al. Laquinimod interferes with migratory capacity of T cells and reduces IL-17 levels, inflammatory demyelination and acute axonal damage in mice with experimental autoimmune encephalomyelitis. J Neuroimmunol. 2010;227:133–143. doi: 10.1016/j.jneuroim.2010.07.009. [DOI] [PubMed] [Google Scholar]
- 109.Preiningerova J. Oral laquinimod therapy in relapsing multiple sclerosis. Expert Opin Investig Drugs. 2009;18:985–989. doi: 10.1517/13543780903044944. [DOI] [PubMed] [Google Scholar]
- 110.Fox RI, Herrmann ML, Frangou CG, et al. Short analytical review: mechanism of action for leflunomide in rheumatoid arthritis. Clin Immunol. 1999;93:198–208. doi: 10.1006/clim.1999.4777. [DOI] [PubMed] [Google Scholar]
- 111.Ullrich A, Knecht K, Fries M, et al. Recombinant expression of Nterminal truncated mutants of the membrane bound mouse, rat, and human flavozyme dihydroorotate dehydrogenase. Eur J Biochem. 2001;268:1861–1868. [PubMed] [Google Scholar]
- 112.Bruneau J-M, Yea CM, Spinella-Jaegle S, et al. Purification of human dihydro-orotate dehydrogenase and its inhibition by A771726, the active metabolite of leflunomide. Biochem J. 1998;336:299–303. doi: 10.1042/bj3360299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Greene S, Watanabe K, Braatz-Trulson J, et al. Inhibition of dihydro-orotate dehydrogenase by the immunosuppressive agent leflunomide. Biochem Pharmacol. 1995;50:861–867. doi: 10.1016/0006-2952(95)00255-x. [DOI] [PubMed] [Google Scholar]
- 114.Cherwinski HM, Cohn RG, Cheung P, et al. The immunosuppressant leflunomide inhibits lymphocyte proliferation by inhibiting pyrimidine biosynthesis. J Pharmacol Exp Therap. 1995;275:1043–1049. [PubMed] [Google Scholar]
- 115.Ruckemann K, Fairbanks LD, Carrey EA, et al. Leflunomide inhibits pyrimidine de novo synthesis in mitogen-stimulated T-lymphocytes from healthy humans. J Biol Chem. 1998;273:21682–21691. doi: 10.1074/jbc.273.34.21682. [DOI] [PubMed] [Google Scholar]
- 116.Siemasko KF, Chong AS, Williams JW, et al. Regulation of B cell function by the immunosuppressive agent leflunomide. Transplant. 1996;27:635–642. doi: 10.1097/00007890-199602270-00020. [DOI] [PubMed] [Google Scholar]
- 117.Siemasko K, Chong AS, Jack HM, et al. Inhibition of JAK3 and STAT6 tyrosine phosphorylation by the immunosuppressive drug leflunomide leads to a block in IgG1 production. J Immunol. 1998;160:1581–1588. [PubMed] [Google Scholar]
- 118.Xu X, Williams JW, Bremer EG, et al. Inhibition of protein tyrosine phosphorylation in T cells by a novel immunosuppressive agent, leflunomide. J Biol Chem. 1995;270:12398–12403. doi: 10.1074/jbc.270.21.12398. [DOI] [PubMed] [Google Scholar]
- 119.Miljkovic D, Samardzic T, Mostarica SM, et al. Leflunomide inhibits activation of inducible nitric oxide synthase in rat astrocytes. Brain Res. 2001;889:331–338. doi: 10.1016/s0006-8993(00)03181-4. [DOI] [PubMed] [Google Scholar]
- 120.Mladenovic V, Domljan Z, Rozman B, et al. Safety and effectiveness of leflunomide in the treatment of patients with active rheumatoid arthritis. Results of a randomized, placebo-controlled, phase II study. Arthritis Rheum. 1995;38:1595–1603. doi: 10.1002/art.1780381111. [DOI] [PubMed] [Google Scholar]
- 121.Beutler E, Sipe JC, Romine JS, et al. The treatment of chronic progressive multiple sclerosis with cladribine. Proc Natl Acad Sci U S A. 1996;93:1716–1720. doi: 10.1073/pnas.93.4.1716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Romine JS, Sipe JC, Koziol JA, et al. A double-blind, placebo-controlled, randomized trial of cladribine in relapsing-remitting multiple sclerosis. Proc Assoc Am Physicians. 1999;111:35–44. doi: 10.1046/j.1525-1381.1999.09115.x. [DOI] [PubMed] [Google Scholar]
- 123.Giovannoni G, Comi G, Cook S, CLARITY Study Group A placebo-controlled trial of oral cladribine for relapsing multiple sclerosis. N Engl J Med. 2010;362:416–426. doi: 10.1056/NEJMoa0902533. [DOI] [PubMed] [Google Scholar]
- 124.Rieckmann P, Giovannoni G, Cook SD. Cladribine tablets in relapsing-remitting multiple sclerosis: study design of the 2-year, Phase IIIb CLARITY (CLAdRibine tablets Treating multiple sclerosis orallY) extension study. Mult Scler. 2008;14:S161–S162. [Google Scholar]
- 125.Montalban X, Cohen BA, Jeffery DR, et al. ONWARD Study Group Oral cladribine added to interferon beta-1a for active multiple sclerosis: a 96-week, double- blind, placebo-controlled phase IIb study. Mult Scler. 2007;13(Suppl 2):S245. [Google Scholar]
- 126.Merck Serono Merck Serono Announces Initiation of the ORACLE MS Trial to Evaluate Cladribine Tablets in Patients at Risk of Developing Multiple Sclerosis. Press release 18-09-2008. http://www.merckserono.net/corp.merckserono/en/images/20080918_en_tcm112_21562.pdf. Accessed October 21, 2010.
- 127.Dasanu CA, Alexandrescu DT. Risk of additional cancers in untreated and treated hairy cell leukemia patients. Expert Opin Pharmacother. 2010;11:41–50. doi: 10.1517/14656560903405647. [DOI] [PubMed] [Google Scholar]
- 128.US Food and Drugs Administration FDA approves first oral drug to reduce MS relapses. 2010. Available at: http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm226755.htm. Accessed December 27, 2010.
- 129.Fujino M, Funeshima N, Kitazawa Y, et al. Amelioration of experimental autoimmune encephalomyelitis in Lewis rats by FTY720 treatment. J Pharmacol Exp Ther. 2003;305:70–77. doi: 10.1124/jpet.102.045658. [DOI] [PubMed] [Google Scholar]
- 130.Webb M, Tham C-S, Lin F-F, et al. Sphingosine 1-phosphate receptor agonists attenuate relapsing-remitting experimental autoimmune encephalitis in SJL mice. J Neuroimmunol. 2004;153:108–121. doi: 10.1016/j.jneuroim.2004.04.015. [DOI] [PubMed] [Google Scholar]
- 131.Kappos L, Antel J, Comi G, et al. Oral fingolimod (FTY720) for relapsing multiple sclerosis. N Engl J Med. 2006;355:1124–1140. doi: 10.1056/NEJMoa052643. [DOI] [PubMed] [Google Scholar]
- 132.O’Connor P, Comi G, Montalban X, et al. Oral fingolimod (FTY720) in multiple sclerosis: two-year results of a phase II extension study. Neurology. 2009;72:73–79. doi: 10.1212/01.wnl.0000338569.32367.3d. [DOI] [PubMed] [Google Scholar]
- 133.Montalban X, O’Connor P, Antel J, et al. Oral fingolimod (FTY720) shows sustained low rates of clinical and MRI disease activity in patients with relapsing multiple sclerosis: four-year results from a phase II extension. Neurology. 2009;72(Suppl 3):A313. Abstract. [Google Scholar]
- 134.Kappos L, Radue EW, O’Connor P, et al. FREEDOMS Study Group A placebo-controlled trial of oral fingolimod in relapsing multiple sclerosis. N Engl J Med. 2010;362:387–401. doi: 10.1056/NEJMoa0909494. [DOI] [PubMed] [Google Scholar]
- 135.Cohen JA, Barkhof F, Comi G, TRANSFORMS Study Group Oral fingolimod or intramuscularinterferon for relapsing multiple sclerosis. N Engl J Med. 2010;362:402–415. doi: 10.1056/NEJMoa0907839. [DOI] [PubMed] [Google Scholar]
- 136.Kappos L, Miller DH, MacManus DG, et al. BG00012, a novel oral fumarate, is effective in patients with relapsing-remitting multiple sclerosis. Mult Scler. 2006;12(Suppl 1):S85. [Google Scholar]
- 137.Hoefnagel JJ, Thio HB, Willemze R, et al. Long-term safety aspects of systemic therapy with fumaric acid esters in severe psoriasis. Br J Dermatol. 2003;149:363–369. doi: 10.1046/j.1365-2133.2003.05433.x. [DOI] [PubMed] [Google Scholar]
- 138.Ruffini F, Bergamaschi A, Marinaro C, et al. Laquinimod prevents the inflammation-induced derangement of neurogenic niches in experimental autoimmune encephalomyelitis mice. Mult Scler. 2010;16:S41–S196. doi: 10.1177/1352458512469698. Abstract P885. [DOI] [PubMed] [Google Scholar]
- 139.Thöne J, Lee D, Seubert S, et al. Laquinimod ameliorates experimental autoimmune encephalomyelitis via BDNF-dependent mechanisms. Mult Scler. 2010;16:S41–S196. Abstract P881. [Google Scholar]
- 140.Polman C, Barkhof F, Sandberg-Wollheim M, et al. Laquinimod in Relapsing MS Study Group Treatment with laquinimod reduces development of active MRI lesions in relapsing MS. Neurology. 2005;64:987–991. doi: 10.1212/01.WNL.0000154520.48391.69. [DOI] [PubMed] [Google Scholar]
- 141.Comi G, Pulizzi A, Rovaris M, et al. LAQ/5062 Study Group Effect of laquinimod on MRI-monitored disease activity in patients with relapsing-remitting multiple sclerosis: a multicentre, randomised, double-blind, placebo-controlled phase IIb study. Lancet. 2008;371:2085–2092. doi: 10.1016/S0140-6736(08)60918-6. [DOI] [PubMed] [Google Scholar]
- 142.TEVA press release Teva announces successful results of phase III study with oral Laquinomod for multiple sclerosis. 2010. Available at: http://www.tevapharm.com/pr/2010/pr_976.asp. Accessed December 24, 2010.
- 143.Merrill JE, Hanak S, Pu SF, et al. Teriflunomide reduces behavioral, electrophysiological, and histopathological deficits in the Dark Agouti rat model of experimental autoimmune encephalomyelitis. J Neurol. 2009;256:89–103. doi: 10.1007/s00415-009-0075-3. [DOI] [PubMed] [Google Scholar]
- 144.Confavreux C, O’Connor PW, Freedman MS, et al. Safety of teriflunomide in the treatment of relapsing multiple sclerosis: results over an 8-year extension. Mult Scler. 2010;16:S41–S196. Abstract P833. [Google Scholar]
- 145.Li DK, O’Connor PW, Confavreux C, et al. Long-term brain MRI and clinical assessments of teriflunomide for the treatment of multiple sclerosis: extension of a phase II study. Mult Scler. 2010;16:S41–S196. Abstract P431. [Google Scholar]
- 146.O’Connor P, Wolinsky J, Confavreux C, et al. A placebo-controlled phase III trial (TEMSO) of oral teriflunomide in relapsing multiple sclerosis: clinical efficacy and safety outcomes. Mult Scler. 2010;16:S41–S196. Abstract P79. [Google Scholar]
- 147.Wolinsky J, O’Connor P, Confavreux C, et al. A placebo-controlled phase III trial (TEMSO) of oral teriflunomide in relapsing multiple sclerosis: magnetic resonance imaging (MRI) outcomes. Mult Scler. 2010;16:S41–S196. Abstract P982. [Google Scholar]
- 148.Lily O, McFadden E, Hensor E, et al. Disease-specific quality of life in multiple sclerosis: the effect of disease modifying treatment. Mult Scler. 2006;12:808–813. doi: 10.1177/1352458506070946. [DOI] [PubMed] [Google Scholar]
- 149.Jongen PJ, Lehnick D, Sanders E, et al. FOCUS study group Health-related quality of life in relapsing remitting multiple sclerosis patients during treatment with glatiramer acetate: a prospective, observational, international, multi-centre study. Health Qual Life Outcomes. 2010;15:133. doi: 10.1186/1477-7525-8-133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Daugherty KK, Butler JS, Mattingly M, et al. Factors leading patients to discontinue multiple sclerosis therapies. J Amer Pharm Assoc. 2005;45:371–375. doi: 10.1331/1544345054003804. [DOI] [PubMed] [Google Scholar]
- 151.Devonshire V, Lapierre Y, Macdonell R, et al. GAP Study Group The Global Adherence Project (GAP): a multicenter observational study on adherence to disease-modifying therapies in patients with relapsing-remitting multiple sclerosis. Eur J Neurol. 2011;18:69–77. doi: 10.1111/j.1468-1331.2010.03110.x. [DOI] [PubMed] [Google Scholar]
- 152.Bermel RA, Weinstock-Guttman B, Bourdette D, et al. Intramuscular interferon beta-1a therapy in patients with relapsing-remitting multiple sclerosis: a 15-year follow-up study. Mult Scler. 2010;16:588–596. doi: 10.1177/1352458509360549. [DOI] [PubMed] [Google Scholar]
- 153.Piehl F, Holmén C, Hillert J, et al. Swedish natalizumab (Tysabri) multiple sclerosis surveillance study. Neurol Sci. 2011;31(Suppl 3):289–293. doi: 10.1007/s10072-010-0345-y. [DOI] [PubMed] [Google Scholar]
- 154.Rudick RA, Miller D, Hass S, et al. AFFIRM and SENTINEL Investigators Health-related quality of life in multiple sclerosis: effects of natalizumab. Ann Neurol. 2007;62:335–346. doi: 10.1002/ana.21163. [DOI] [PubMed] [Google Scholar]
- 155.O’Connor P, Filippi M, Arnason B, et al. BEYOND Study Group 250 microg or 500 microg interferon beta-1b versus 20 mg glatiramer acetate in relapsing-remitting multiple sclerosis: a prospective, randomised, multicentre study. Lancet Neurol. 2009;8:889–897. doi: 10.1016/S1474-4422(09)70226-1. [DOI] [PubMed] [Google Scholar]
- 156.Borner M, Scheithauer W, Twelves C, et al. Answering patients’ needs: oral alternatives to intravenous therapy. Oncologist. 2001;6(Suppl 4):12–16. doi: 10.1634/theoncologist.6-suppl_4-12. [DOI] [PubMed] [Google Scholar]
- 157.Meca-Lallana JE, de Mingo-Casado P, Amorin-Díaz M, et al. Effects of glatiramer acetate on spasticity in previously interferon-beta-treated and treatment-naive patients with relapsing-remitting multiple sclerosis: a prospective, nonrandomized, open-label, uncontrolled, observational pilot study. Clin Ther. 2010;32:1061–1066. doi: 10.1016/j.clinthera.2010.06.005. [DOI] [PubMed] [Google Scholar]
- 158.Goeb JL, Even C, Nicolas G, et al. Psychiatric side effects of interferon-beta in multiple sclerosis. Eur Psychiatry. 2006;21:186–193. doi: 10.1016/j.eurpsy.2005.09.013. [DOI] [PubMed] [Google Scholar]
- 159.Buljevac D, Flach HZ, Hop WC, et al. Prospective study on the relationship between infections and multiple sclerosis exacerbations. Brain. 2002;125:952–960. doi: 10.1093/brain/awf098. [DOI] [PubMed] [Google Scholar]
- 160.Giannantoni A, Scivoletto G, Di Stasi SM, Grasso MG, Vespasiani G, Castellano V. Urological dysfunctions and upper urinary tract involvement in multiple sclerosis patients. Neurourol Urodyn. 1998;17:89–98. doi: 10.1002/(sici)1520-6777(1998)17:2<89::aid-nau2>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- 161.Stuve O, Gold R, Chan A, et al. Alpha4-Integrin antagonism with natalizumab: effects and adverse effects. J Neurol. 2008;255(Suppl 6):58–65. doi: 10.1007/s00415-008-6011-0. [DOI] [PubMed] [Google Scholar]
- 162.Clifford DB, De Luca A, Simpson DM, et al. Natalizumab-associated progressive multifocal leukoencephalopathy in patients with multiple sclerosis: lessons from 28 cases. Lancet Neurol. 2010;9:438–446. doi: 10.1016/S1474-4422(10)70028-4. [DOI] [PubMed] [Google Scholar]
- 163.Gorelik L, Lerner M, Bixler S, et al. Anti-JC virus antibodies: implications for PML risk stratification. Ann Neurol. 2010;68:295–303. doi: 10.1002/ana.22128. [DOI] [PubMed] [Google Scholar]
- 164.Panzara MA, Bozic C, Sandrock AW. More on melanoma with transdifferentiation. N Engl J Med. 2008 Jul 3;359:99. 99–100. doi: 10.1056/NEJMc086089. author reply. [DOI] [PubMed] [Google Scholar]
- 165.Amato MP, Portaccio E, Ghezzi A, MS Study Group of the Italian Neurological Society Pregnancy and fetal outcomes after interferon-β exposure in multiple sclerosis. Neurology. 2010;75:1794–1802. doi: 10.1212/WNL.0b013e3181fd62bb. [DOI] [PubMed] [Google Scholar]
- 166.Biogen . Avonex (interferon-1a): product monograph. Biogen; 1998. [Google Scholar]
- 167.Francis G, Collins W, Burtin P. Fingolimod (NDA 22-527), Briefing document. 2010. Available at: http://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/PeripheralandCentralNervousSystemDrugsAdvisoryCommittee/UCM214675.pdf. Accessed January 1, 2011.
- 168.Muraro PA, Uccelli A. Immuno-therapeutic potential of haematopoietic and mesenchymal stem cell transplantation in MS. Results Probl Cell Differ. 2010;51:237–257. doi: 10.1007/400_2008_14. [DOI] [PubMed] [Google Scholar]
- 169.Chataway J. When confronted by a patient with the radiologically isolated syndrome. Pract Neurol. 2010;10:271–277. doi: 10.1136/jnnp.2010.224931. [DOI] [PubMed] [Google Scholar]
- 170.Korteweg T, Tintore M, Uitdehaag B, et al. MRI criteria for dissemination in space in patients with clinically isolated syndromes: a multicentre follow-up study. Lancet Neurol. 2006;5:221–227. doi: 10.1016/S1474-4422(06)70353-2. [DOI] [PubMed] [Google Scholar]
- 171.Barkhof F, Rocca M, Francis G, et al. Early Treatment of Multiple Sclerosis Study Group Validation of diagnostic magnetic resonance imaging criteria for multiple sclerosis and response to interferon beta1a. Ann Neurol. 2003;53:718–724. doi: 10.1002/ana.10551. [DOI] [PubMed] [Google Scholar]
- 172.Hutchinson M, Kappos L, Calabresi PA, et al. AFFIRM and SENTINEL Investigators The efficacy of natalizumab in patients with relapsing multiple sclerosis: subgroup analyses of AFFIRM and SENTINEL. J Neurol. 2009;256:405–415. doi: 10.1007/s00415-009-0093-1. [DOI] [PubMed] [Google Scholar]