

Abstract
Objective
The regeneration of the hematopoietic system in bone marrow after chemotherapy depends on a balance between the quiescence and proliferation of lineage-specific progenitor cells. Even though the vascular network in bone is damaged by cytoablation, the transcriptional control of quiescence in endothelial cells is not well known. In this study, we investigated the role of the transcription factor ELF4 in the proliferation of endothelial cells in bone marrow.
Methods and Results
Loss-of-function models were used to study the role of ELF4 in human and murine endothelial cells. ELF4 promotes cell-cycle entry by activating CDK4 in human umbilical vein endothelial cells. Elf4-null mice exhibited enhanced recovery of bone marrow CD45− CD31+ endothelial cells and sinusoidal blood vessels following administration of 5-fluorouracil.
Conclusion
Loss of ELF4 leads to increased quiescence in bone marrow endothelial cells by the deregulation of CDK4 expression and to enhanced regeneration of sinusoidal blood vessels.
Keywords: endothelial cells, bone marrow ablation, transcription factor, ELF4, quiescence
Introduction
Cellular quiescence protects tissue-specific progenitor cells from cell-cycle dependent cytotoxicity to ensure longevity and regenerative potential. This reversible cell-cycle arrest is actively regulated by cell-intrinsic factors, in addition to proliferation-inducing signals from the milieu1. Chemotherapeutic drugs that target dividing cells cause ablation of hematopoietic and endothelial cells in the bone marrow (BM) 2, 3. The molecular events that protect endothelial cells from apoptosis and disruption of cell-cell junctions following administration of cytotoxic drugs that ultimately lead to the regression of sinusoidal blood vessels in BM are not well-understood 4.
ELF4 is a member of the ETS family of transcription factors defined by a highly conserved DNA-binding domain (ETS domain) 5, 6. ELF4 has a tumor suppressor function in acute myelogenous leukemias by gene repression or chromosomal translocation with the ERG gene 7-10. However, ELF4 can also have oncogenic properties in ovarian cancer 11. In the hematopoietic system, ELF4 activates the cytotoxicity of natural killer cells and negatively regulates quiescence in hematopoietic stem cells 12, 13. We recently reported that ELF4 also inhibits T cell proliferation downstream of TCR and mTOR signaling by activating the tumor suppressor KLF4 14, 15. In this work, we addressed whether ELF4 controls proliferation in a non-hematopoietic cell lineage.
ETS proteins are important transcriptional regulators for the development and function of endothelial cells 16. Furthermore, the enhancers and promoters of many endothelial-specific genes are regulated by ETS proteins 16. For instance, ETS1, ERG, Fli-1, ELF1 and TEL are involved in vascular development and angiogenesis 17. ELF4 is highly homologous to ELF1, ELF2 (a.k.a. NERF2), ELF3 and ELF5 6, a sub-family involved in the regulation of Tie2 gene expression 18, 19. Similarly to ETS1, deletion of the Elf4 gene is not embryonic lethal, suggesting either functional redundancy or function in adult tissues. Despite the lack of developmental defects observed in loss-of-function mouse models generated by homozygous gene deletion, they can still provide information about unique functions in processes of vascular remodeling in adult tissues not compensated by homologous ETS proteins 20-24.
The vasculature in BM is a complex network that includes sinusoidal blood vessels (SBV) that traverse the bone cavity from the endosteum to the central sinus. In contrast to other blood vessels, SBVs are formed by a discontinuous single layer of endothelial cells to allow maximal permeability 25. Thus, hematopoietic cells and metastatic tumor cells (i.e. breast and prostate cancer) can exit or home in BM via SBVs. An additional function of BM endothelial cells is to provide HSCs with a microenvironment that signals proliferation, differentiation, and mobilization 2, 26, 27. Chemically induced cytoablation leads to a collapse and regression of SBVs due to a reduction of BM cellularity that normally supports the vascular network in BM 2, 3, 28. The regulation of quiescence in endothelial cells during homeostasis has not been previously addressed. We postulated that quiescent endothelial cells in BM would be spared from chemical ablation, as happens with HSCs, resulting in heightened regeneration of blood vessels in BM.
In this work, we report that ELF4 promotes cell-cycle entry in human umbilical-cord endothelial cells (HUVEC). Thus, silencing of ELF4 gene expression led to increased quiescence in HUVEC cells due to the downregulation of CDK4 expression. In agreement with this finding, Elf4−/− mice revealed enhanced recovery of BM CD45− CD31+ endothelial cells and regeneration of sinusoidal blood vessels after cytoablation, due to a lower proliferation rate at steady state. These data support the hypothesis that ELF4 negatively regulates quiescence in endothelial cells similarly to hematopoietic stem cells13.
Methods
Cells
Human umbilical vein endothelial cells (HUVEC) were obtained from Lonza and cultured following the manufacturer’s instructions. NIH3T3 and COS-7 cells were obtained from ATCC.
Mice
Elf4−/− mice were obtained from S. Nimer and described previously 12. C57BL/6 mice were purchased from Jackson Laboratories. For cytoablation, a single dose of 5-fluorouracil (5-FU) was administered intraperitoneally (150 mg/kg). All mice were bred and maintained in specific pathogen-free conditions at the Baylor College of Medicine. All experiments were approved by the Institutional Animal Care and Usage Committee of Baylor College of Medicine.
Promoter assays
ELF4 cDNA was cloned into the CMV5 expression vector. cMYC cDNA was obtained from Addgene and then cloned into the CMV5 plasmid. The human CDK4 promoter obtained from Addgene was cloned into the pGL3-basic vector (Promega). COS-7 and NIH3T3 cells were transfected with a mixture of plasmids (CMV5-ELF4 or CMV5-cMYC), pGL3-CDK4 and renilla luciferase plasmids using Lipofectamine 2000 (Invitrogen). Luciferase activity was measured using the dual-luciferase reporter assay system (Promega) and a luminometer (Sirius, Berthold).
Detection of ELF4 expression
HUVEC cells were grown on slides and fixed with 1% paraformaldehyde in PBS for 10 minutes at room temperature. Cells were permeabilized with 0.1% Triton X-100 in PBS for 15 minutes and then incubated with 5% bovine serum albumin in PBS for 30 minutes. HUVEC cells were incubated with rabbit anti-human ELF4 (Orbigen) followed by anti-rabbit IgG conjugated with Alexa 488 (Invitrogen). Phalloidin-Alexa 555 (Invitrogen) was used to detect F-actin. Murine endothelial cells were purified from BM using anti-CD31 and BD-IMag (BD Biosciences) and spun onto glass slides. Following fixation and permeabilization as described above, cells were incubated with anti-CD45-FITC or anti-CD31- FITC, and anti-ELF412. Slides were mounted with ProLong Gold antifade reagent (Invitrogen), and images were taken using a NIKON 90i microscope. ELF4 expression was also determined in paraffin fixed sections of mouse embryos (14.5 d.p.c.) using an affinity purified polyclonal antibody against ELF4 as previously described13.
Cell cycle analysis
DNA content was determined using propidium iodine as described previously13 and analyzed using a FACScanto cytometer (BD Bioscience) and ModFit LT software (Verity). For the nuclear detection of Ki67, cells were fixed and permeabilized using reagents from the apo-BrdU kit (BD Bioscience) and stained with anti-Ki67-FITC (Vector Laboratories) and 7-AAD. Murine endothelial cells were identified by flow cytometry using anti-CD31 and anti-CD45 antibodies. Samples were analyzed using FACScanto cytometer and FlowJo software (Tree Star).
Levels of cell-cycle regulators in HUVEC cells treated with control shRNA or ELF4shRNA were evaluated by immunoblots using polyclonal anti-p21 (eBioscience) and rabbit polyclonal anti-p27, mouse anti-cyclin D1, mouse anti-cyclin D3, mouse anti-cdk4, and rabbit anti-phospho-Rb (Cell Signaling Technology). ELF4 was detected using rabbit polyclonal anti-ELF4. β-actin was used to normalize sample loading.
Detection of blood vessels
Femurs were fixed and decalcified (Fisher Diagnostic) and embedded in paraffin. To identify bone vasculature, sections were stained with anti-MECA32 (Becton Dickinson). Morphometric analysis was performed to enumerate capillaries and sinusoidal blood vessels in different sections using NEI-Elements software (Nikon). Capillaries and sinusoidal blood vessels were identified based on morphology (hematoxylin and eosin staining) and MECA32 expression 2, 3. Tissue sections were reviewed by a pathologist at Texas Children’s Hospital. BM cells were cultured on fibronectin-coated plates for 14 days in F12K media (ATCC) supplemented with 10% FBS, 0.1 mg/ml heparin and 0.03 mg/ml of endothelial cell growth supplement (ECGS). Adherent cells were incubated with Alexa Fluor 488 Dil-ac-LDL (5 ug/ml) for 3-4 hours at room temperature and positive colonies were counted under fluorescence microscope. Percentage of Dil-ac-LDL positive endothelial cells was confirmed by flow cytometry using F4/80 antibody to rule out macrophages.
Immunofluorescence in yolk sac
Mouse embryos (9.5 d.p.c.) were fixed with 4% paraformaldehyde, permeabilized with 0.5% Triton X-100 in PBS for 10 minutes, and blocked with MOM solution (Vector Laboratories). Yolk sac was stained overnight with mouse anti-smooth-muscle actin (DAKO), rat anti-CD31 (Beckton Dickinson) and affinity purified rabbit anti-ELF4 at 4 °C. Tissues were then incubated with anti-mouse Alexa 488, anti-rat Alexa 633 and anti-rabbit Alexa 568 (Invitrogen). Images were taken using a Zeiss Axioplan 2 microscope.
Gene silencing in HUVEC cells
HUVEC cells were transfected with 30 pmol ELF4 Stealth RNAi or Stealth RNAi negative control (medium GC content) using lipofectamine RNAiMax (Invitrogen) for 6 hours at 37 °C. Cells were cultured for an additional 48 hours in EGM-2 medium. Expression of ELF4 was measured by real-time PCR using the following primers: 5’-TCCGAAATGC TTCCAGACTC-3’ and 5’-GGGTCAGTGACAGGTGAGGT-3’ and for βactin: 5’-CAAACA TGATCTGGGTCATCTTCTC-3’ and 5’-GCTCGTCGTCGA C AACGGCTC-3’
Results
Loss of ELF4 leads to enhanced regeneration of sinusoidal blood vessels in bone marrow
We previously described ELF4 as a negative regulator of quiescence in HSC during homeostasis but not during regenerative hematopoiesis 13. The increased hematological recovery post cytoablative treatment observed in Elf4−/− mice was due to a larger pool of more quiescent HSCs 13. Based on this finding, we tested whether ELF4 also controls neovascularization of BM by regulating quiescence of endothelial progenitor cells. We first confirmed that ELF4 is expressed in the nuclei of HUVEC cells, endothelium of blood vessels and endocardium of 14.5 d.p.c. mouse embryos (Figure 1A, B). To correlate ELF4 expression with blood vessels, we performed whole-mount immunofluorescence of the yolk sac of 9.5 d.p.c. embryos using CD31 as marker of endothelial cells (Figure 1C). Finally, we purified CD31+ murine BM cells and observed that ELF4 is expressed in both hematopoietic (CD45 positive) and non-hematopoietic (CD45 negative) cells (Figure 1D). ELF4 expression showed a nuclear punctuate pattern in CD45− CD31+ BM cells, which was reminiscent of the expression pattern in HSC 13, suggesting that ELF4 is not associated with any well-defined nuclear domain. Collectively, these findings confirm that ELF4 is expressed in endothelial cells.
Figure 1. The transcription factor ELF4 is expressed in endothelial cells.
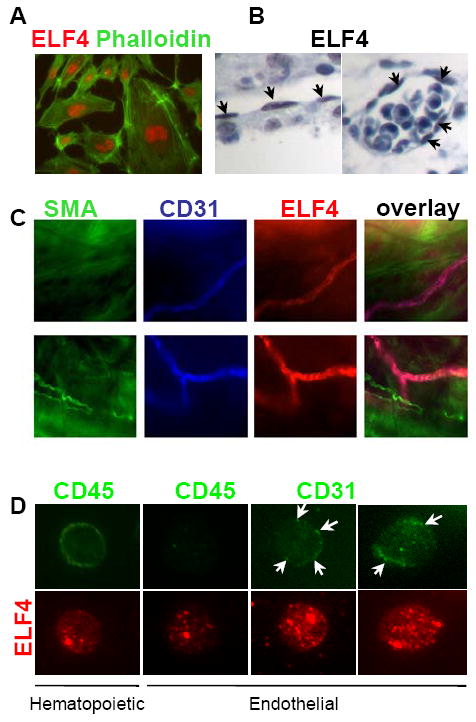
(A) Nuclear expression of ELF4 was determined by immunofluorescence in HUVEC cells using anti-ELF4 (red) and phalloidin (green). Magnification: 40x. (B) Immunohistochemistry in E14.5 embryos using an anti-ELF4 antibody. Arrows indicate positive endothelial cells in endocardium (left) and a blood vessel (right). Magnification: 20x. (C) Immunofluorescence of yolk sac (E9.4) using smooth muscle actin (SMA, green), anti-CD31 (blue), and anti-ELF4 (red). (D) Immunofluorescence of murine hematopoietic (CD45+) and non-hematopoietic endothelial cells (CD45− CD31+). Arrows indicate CD31 surface expression. Magnification: 40x.
Both hematopoietic and vasculature systems are disrupted in the BM after administration of 5-FU 3. Because ELF4 promotes cell-cycle entry of HSCs at steady state 13, we monitored the expansion of BM endothelial cells and regeneration of sinusoidal blood vessels in ablated Elf4-null mice. We enumerated CD45− CD31+ BM cells in femurs of 5-FU-treated mice by flow cytometry (Figure 2). Increased expansion of BM CD45− CD31+ endothelial cells was observed in Elf4−/− mice between 9 and 12 days. Elf4−/− femurs also exhibited increased numbers of endothelial cells 3 days after 5-FU administration, indicating that more endothelial cells survived this treatment compared to wild type mice. We confirmed this acute effect by culturing BM cells on fibronectin-coated plates and incubating them with Dil-Ac-LDL to count colonies of endothelial cells representative of the initial number in BM isolate. Three days after 5-FU administration, the number of Dil-Ac-LDL-positive colonies recovered from BM was significantly higher in Elf4−/− femurs compared to wild type femurs (Figure 3A).
Figure 2. Enhanced recovery of endothelial cells in BM of Elf4−/− mice.
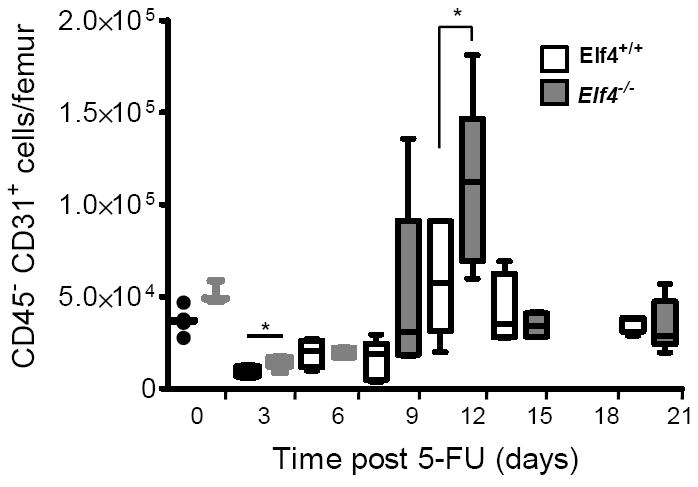
The number of CD45− CD31+ cells after administration with a single dose of 5-FU was analyzed by flow cytometry in wild type and Elf4−/− femurs and represented in box plots (5 mice per time point). The solid horizontal lines represent median values (Two-tailed Student’s t-test, * P< 0.05).
Figure 3. Loss of ELF4 leads to increased numbers of sinusoidal blood vessels in BM.
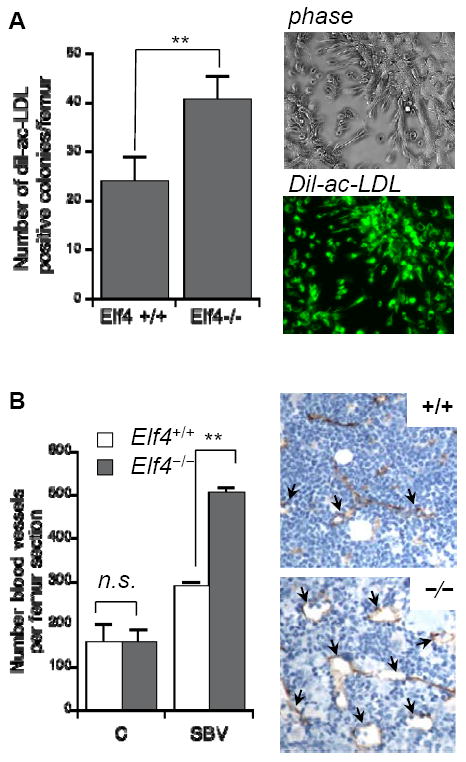
(A) BM cells from wild type and Elf4−/− mice were collected after three days of 5-FU administration, cultured for two weeks on fibronectin-coated plates and incubated with Alexa Fluor 488 Dil-Ac-LDL. The number of Dil-ac-LDL-positive colonies is shown on the left, and a representative colony is shown on the right. (B) The pan-endothelial marker MECA32 was used to identify capillaries (C) and sinusoidal blood vessels (SBV) in femurs of mice treated with 5-FU 14 days earlier. Arrows indicate representative SBV. Two-tailed Student’s t-test, n = 3, ** P< 0.01.
To investigate whether this increase in number of BM endothelial cells leads to heightened regeneration of sinusoidal blood vessels (SBVs), we examined the expression of MECA-32, a pan-endothelial marker, in femurs of wild type and Elf4−/− mice after 5-FU treatment (Supplemental Figure 1). Even though the number of capillaries in Elf4−/− femurs was similar to wild type femurs, the number of SBVs was significantly higher in Elf4−/− femurs, correlating with a peak of expansion in CD45− CD31+ BM cells (Figure 3B). This finding suggested increased quiescence of cells at steady state and/or enhanced regenerative proliferation of Elf4−/− CD45− CD31+ BM cells. Even though the enumeration of BM endothelial cells by flow cytometry has limitations, Elf4−/− mice showed increased numbers of CD45− CD31+ cells at the nadir (Figure 2, 9,498±2,497 in +/+ versus 15,076±3,362 in −/−, n = 5) consistent with a higher number of Dil-ac-LDL-positive colonies (Figure 3A). Therefore, a paucity of proliferation in Elf4−/− BM endothelial cells could account for the improved regeneration of BM vasculature.
ELF4 regulates proliferation in bone marrow endothelial cells
We previously reported that ELF4 promotes cell-cycle entry in HSCs and induces cell cycle arrest in naïve CD8+ T cells 13, 14. These findings suggest that ELF4 activates the expression of genes involved in cell cycle regulation in a cell context-dependent manner. Therefore, we investigated whether ELF4 also controls the proliferation of BM endothelial cells during homeostasis. Analysis of DNA content revealed a lower percentage of Elf4−/− CD45− CD31+ BM cells in S-phase during homeostasis (Figure 4A, B). Next, a combination of Ki67 expression in nuclei and DNA content was used to distinguish cells in G0 from G1. Elf4−/− CD45− CD31+ BM cells showed a significant increase in G0 phase (Ki67−/lo 2n DNA) of the cell cycle (Figure 4C,D). This finding suggests that a relatively low proliferative capacity of Elf4−/− BM endothelial cells, defined as CD45− CD31+, leads to accelerated survival and regeneration of SBVs, coinciding with the hematological recovery previously reported 13. Notably, we did not detect differences in the proliferative capacity of BM endothelial cells after 5-FU administration (not shown).
Figure 4. ELF4 inhibits quiescence in bone marrow endothelial cells.
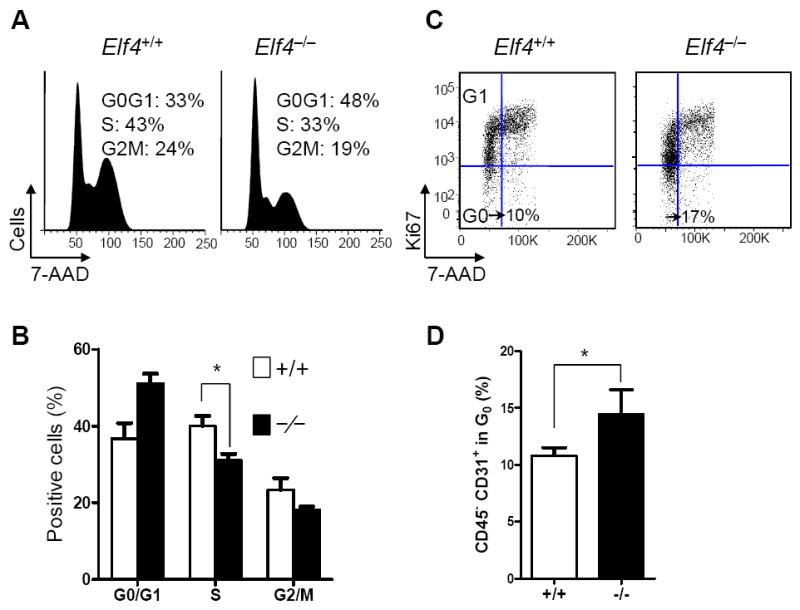
(A) DNA content was measured with 7-AAD in CD45− CD31+ BM cells at steady state. A representative profile is shown. (B) Statistical analysis of DNA content (Two-tailed Student’s t-test, n = 3, * P< 0.05). (C) Dual detection of Ki67 and 7-AAD in CD45− CD31+ BM cells isolated from untreated wild type and Elf4−/− mice. A representative profile is shown. (D) Statistical analysis of cells in G0 phase of the cell cycle (Two-tailed Student’s t-test, n = 3, * P < 0.05).
We next investigated the effects of ectopic expression of ELF4 in the proliferation of HUVEC cells using a fusion protein. Cells were nucleofected with an ELF4-GFP expression plasmid, purified by cell sorting and analyzed by DNA content. ELF4 induced the proliferation of HUVEC cells, as shown by a higher proportion of cells in S-phase (Figure 5A). Conversely, silencing ELF4 in HUVEC cells using gene-specific shRNA led to a significant reduction of the number of cells in S-phase (Figure 5B). To further discriminate cells in G0 and G1 and determine whether quiescence was specifically altered by ELF4, we measured the expression of Ki67 in conjunction with DNA content. As shown for BM cells, quiescent HUVEC cells were defined as cells with 2n DNA content that were negative for Ki67. Loss of ELF4 resulted in a significant increase in the percentage of cells in G0, suggesting that ELF4 promotes cell cycle entry of endothelial cells (Figure 5C, D). In contrast to ELF4, silencing of ELF1 did not significantly affect the percentage of 2n Ki67− cells (Figure 5C, D). The difference in the magnitude of quiescence between primary murine CD45− CD31+ and HUVEC cells is likely due to cellular heterogeneity and the fact that commercial HUVEC cells are pre-selected for proliferation. Nevertheless, it is clear in both systems that loss of ELF4 reduces proliferation of endothelial cells and increases the proportion of cells in G0. In addition to regulating proliferation of HUVEC cells, ELF4 modulates tube formation and wound healing (Supplemental Figure 2).
Figure 5. ELF4 promotes cell-cycle entry in HUVEC cells.
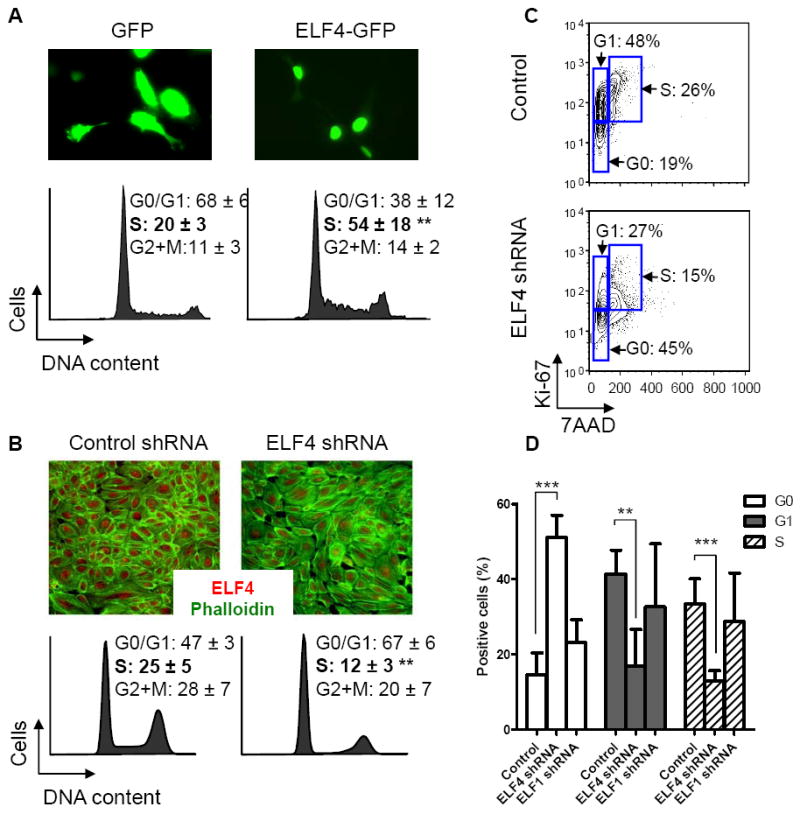
(A) HUVEC cells expressing the fusion protein ELF4-GFP (top) were used to measure DNA content (bottom). After cell sorting of GFP-positive cells, cell cycle was analyzed using ModFit software. The percentage in each phase of the cell cycle is shown (Two-tailed Student’s t-test, ** P < 0.01, n = 4). (B) ELF4 expression was silenced in HUVEC cells using shRNA to study its effect on the cell cycle. ELF4 (red) and phalloidin (green) expression in HUVEC cells was performed by immunofluorescence. The percentage in each phase of the cell cycle is shown on the bottom (Two-tailed Student’s t-test, ** P < 0.01, n = 4). (C) Percentage of quiescent HUVEC cells was analyzed as cells positive for Ki67 with 2n DNA. A representative profile is shown for HUVEC cells transfected with control shRNA or ELF4 shRNA. (D) Statistical analysis of HUVEC cells transfected with control, ELF4 or ELF1 shRNA (Two-tailed Student’s t-test, **P < 0.01, ***P < 0.001 n = 3)
The use of HUVEC cells allowed us to further investigate the mechanism of cell cycle regulation by immunoblot analysis. As expected from the proliferation assays, loss of ELF4 activity resulted in decreased phosphorylation of Rb (Figure 6A). In contrast to normal levels of Cyclin D1, Cyclin D3, and p21, the expression of CDK4 was diminished by 40% (Figure 6A). A rather discrete reduction in CDK4 levels may be due to a combination of transient ELF4 mRNA silencing and the half-life of CDK4 protein. Reduced proliferation of HUVEC cells treated with ELF4 shRNA could be due to lower expression of CDK4. We performed promoter assays to confirm that ELF4 directly regulates expression of the CDK4 gene. We found that ELF4 activates the CDK4 promoter as efficiently as c-MYC, a known activator of CDK4 (Figure 6B). Collectively, our data suggest that ELF4 can promote cell-cycle entry of endothelial cells, at least in part by regulating CDK4 expression.
Figure 6. ELF4 activates CDK4 in HUVEC cells.
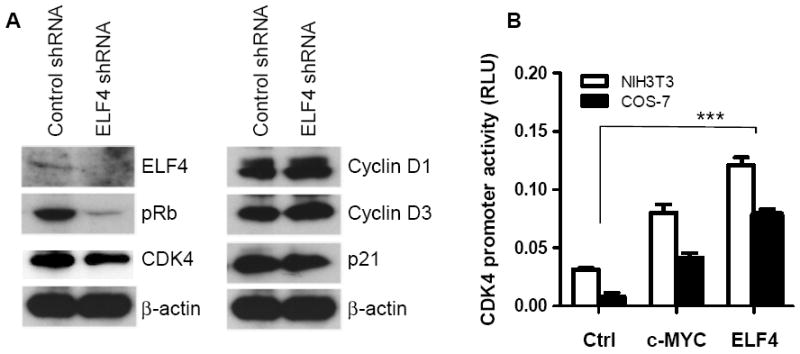
(A) Immunoblot analysis of the expression of ELF4 and cell cycle regulators in HUVEC cells transfected with control or ELF4-specific shRNAs. (B) Capacity of ELF4 to activate the CDK4 gene was determined by promoter reporter assay using COS7 and NIH3T3 cells (Two-tailed Student’s t-test, ***P < 0.001 compared to control, n = 3).
Discussion
Quiescence is a reversible cell cycle arrest that prevents differentiation and ensures longevity of lineage-specific progenitor cells. A better understanding of the regulation of quiescence in endothelial cells is necessary to minimize vascular damage and to accelerate neovascularization of BM in patients undergoing chemo- or radiation therapies. In this study, we demonstrated that the transcription factor ELF4 negatively regulates quiescence in human umbilical cord endothelial cells in vitro and in murine BM endothelial cells in vivo.
The absence of a vascular phenotype in Elf4−/− mice suggests that this transcription factor may have a functional role in vascular remodeling of adult tissues rather than during embryogenesis. Therefore, we tested whether loss of the Elf4 gene leads to alterations in the vascular regeneration post-myelosuppression induced by 5-FU administration 2, 3. Elf4−/− mice showed enhanced recovery of CD45− CD31+ endothelial cells and sinusoidal blood vessels peaking approximately two weeks after cytoablation, consistent with the kinetics of blood vessel recovery previously reported 3. Even though Tie2 signaling is essential for the remodeling and maturation of new vessels and maintenance of a ‘resting state’ in established blood vessels 29, 30, Elf4−/− mice crossed to Tie2-LacZ transgenic mice did not show significant reduction in Tie2 expression in different tissues (not shown). We hypothesized that the enhanced restoration of sinusoidal blood vessels in the BM of Elf4−/− mice was due to cell cycle regulation in BM endothelial cells at steady state.
A limitation in our study is the lack of well-defined cell surface markers to identify BM endothelial progenitor cells by flow cytometry. However, the use of human derived endothelial cells allowed us to study the role of ELF4 in proliferation with greater detail. Interestingly, loss of ELF4 function resulted in increased quiescence of HUVEC cells, reduced phosphorylation of Rb, and lower expression of cyclin-dependent kinase 4 (CDK4). There is emerging evidence that CDK4 activity is involved in cellular quiescence. Inhibition of CDK4/6 activity has been shown to increase resistance to radiation in hematopoietic cells 31. GATA-2 induces HSC quiescence by reducing the levels of CCND3, CDK4 and CDK6 32. In skin stem cells, NFATc1 targets CDK4 to control stem cell quiescence 33. In addition to proliferation, transient silencing of ELF4 in HUVEC cells affected their ability to heal a wound and to form tube-like structures in Matrigel (Supplemental Figure 2). However, ELF4 silencing in HUVEC cells did not alter cell-cell contacts in a monolayer or VE-cadherin levels (not shown). Our findings uncovered a new role of ELF4 in endothelial cells: control of quiescence by activating expression of CDK4.
Our study supports the model that a heightened recovery of sinusoidal blood vessels in Elf4−/− mice is associated with the deregulated proliferation of endothelial cells. The paucity of endothelial cells to proliferate in the absence of ELF4 can explain the increased resistance to cell cycle-dependent toxicity because endothelial cells in G0 phase can escape the deleterious effect of 5-FU. Consequently, more “progenitor” endothelial cells can rapidly regenerate sinusoidal blood vessels in Elf4−/− mice. Collectively, we propose that ELF4 has dual functions in hematological recovery post-BM ablation by regulating cell cycle entry in both hematopoietic stem cells 13 and in BM endothelial cells. As a consequence, ELF4 activity can contribute to the regeneration of a functional vascular niche. Gene modulation of the ELF4 gene could be used in the future to enhance hematological recovery of patients undergoing chemotherapy or radiation by accelerating the regeneration of both the hematopoietic system and the vascular niche. The determination of the genes regulated by ELF4 in BM endothelial cells and the mechanism by which ELF4 is regulated remain to be investigated.
Supplementary Material
Acknowledgments
We thank the Morphology Core Facility at Texas Children’s Hospital for immunohistochemical analysis, Dolores Lopez-Terrada for reviewing MECA-32 staining in the femur, and Natalie Dang for assistance in initial experiments.
Source of Funding This work was supported in part by the National Cancer Institute (KO1 CA099156-01 to H.D.L.) and National Institute of Allergy and Infectious Diseases (1RO1AI077536-01 to H.D.L.) of the US National Institutes of Health, the Curtis Hankamer Basic Research Fund (H.D.L.), and the Dan Duncan Cancer Center at Baylor College of Medicine (H.D.L).
Footnotes
Disclosures None
References
- 1.Tzachanis D, Lafuente EM, Li L, Boussiotis VA. Intrinsic and extrinsic regulation of T lymphocyte quiescence. Leuk Lymphoma. 2004;45:1959–1967. doi: 10.1080/1042819042000219494. [DOI] [PubMed] [Google Scholar]
- 2.Kopp HG, Avecilla ST, Hooper AT, Rafii S. The bone marrow vascular niche: home of HSC differentiation and mobilization. Physiology (Bethesda) 2005;20:349–356. doi: 10.1152/physiol.00025.2005. [DOI] [PubMed] [Google Scholar]
- 3.Kopp HG, Avecilla ST, Hooper AT, Shmelkov SV, Ramos CA, Zhang F, Rafii S. Tie2 activation contributes to hemangiogenic regeneration after myelosuppression. Blood. 2005;106:505–513. doi: 10.1182/blood-2004-11-4269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lapidot T, Dar A, Kollet O. How do stem cells find their way home? Blood. 2005;106:1901–1910. doi: 10.1182/blood-2005-04-1417. [DOI] [PubMed] [Google Scholar]
- 5.Sharrocks AD. The ETS-domain transcription factor family. Nat Rev Mol Cell Biol. 2001;2:827–837. doi: 10.1038/35099076. [DOI] [PubMed] [Google Scholar]
- 6.Lacorazza HD, Nimer SD. The emerging role of the myeloid Elf-1 like transcription factor in hematopoiesis. Blood Cells Mol Dis. 2003;31:342–350. doi: 10.1016/s1079-9796(03)00162-1. [DOI] [PubMed] [Google Scholar]
- 7.Moore SD, Offor O, Ferry JA, Amrein PC, Morton CC, Dal Cin P. ELF4 is fused to ERG in a case of acute myeloid leukemia with a t(X;21)(q25-26;q22) Leuk Res. 2005 doi: 10.1016/j.leukres.2005.10.014. [DOI] [PubMed] [Google Scholar]
- 8.Alcalay M, Meani N, Gelmetti V, Fantozzi A, Fagioli M, Orleth A, Riganelli D, Sebastiani C, Cappelli E, Casciari C, Sciurpi MT, Mariano AR, Minardi SP, Luzi L, Muller H, Di Fiore PP, Frosina G, Pelicci PG. Acute myeloid leukemia fusion proteins deregulate genes involved in stem cell maintenance and DNA repair. J Clin Invest. 2003;112:1751–1761. doi: 10.1172/JCI17595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Park DJ, Vuong PT, de Vos S, Douer D, Koeffler HP. Comparative analysis of genes regulated by PML/RAR alpha and PLZF/RAR alpha in response to retinoic acid using oligonucleotide arrays. Blood. 2003;102:3727–3736. doi: 10.1182/blood-2003-02-0412. [DOI] [PubMed] [Google Scholar]
- 10.Muller-Tidow C, Steffen B, Cauvet T, Tickenbrock L, Ji P, Diederichs S, Sargin B, Kohler G, Stelljes M, Puccetti E, Ruthardt M, deVos S, Hiebert SW, Koeffler HP, Berdel WE, Serve H. Translocation products in acute myeloid leukemia activate the Wnt signaling pathway in hematopoietic cells. Molecular and cellular biology. 2004;24:2890–2904. doi: 10.1128/MCB.24.7.2890-2904.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yao JJ, Liu Y, Lacorazza HD, Soslow RA, Scandura JM, Nimer SD, Hedvat CV. Tumor promoting properties of the ETS protein MEF in ovarian cancer. Oncogene. 2007 doi: 10.1038/sj.onc.1210170. [DOI] [PubMed] [Google Scholar]
- 12.Lacorazza HD, Miyazaki Y, Di Cristofano A, Deblasio A, Hedvat C, Zhang J, Cordon-Cardo C, Mao S, Pandolfi PP, Nimer SD. The ETS protein MEF plays a critical role in perforin gene expression and the development of natural killer and NK-T cells. Immunity. 2002;17:437–449. doi: 10.1016/s1074-7613(02)00422-3. [DOI] [PubMed] [Google Scholar]
- 13.Lacorazza HD, Yamada T, Liu Y, Miyata Y, Sivina M, Nunes J, Nimer SD. The transcription factor MEF/ELF4 regulates the quiescence of primitive hematopoietic cells. Cancer Cell. 2006;9:175–187. doi: 10.1016/j.ccr.2006.02.017. [DOI] [PubMed] [Google Scholar]
- 14.Yamada T, Park CS, Mamonkin M, Lacorazza HD. Transcription factor ELF4 controls the proliferation and homing of CD8+ T cells via the Kruppel-like factors KLF4 and KLF2. Nature immunology. 2009;10:618–626. doi: 10.1038/ni.1730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yamada T, Gierach K, Lee PH, Wang X, Lacorazza HD. Cutting edge: Expression of the transcription factor E74-like factor 4 is regulated by the mammalian target of rapamycin pathway in CD8+ T cells. J Immunol. 2010;185:3824–3828. doi: 10.4049/jimmunol.1000718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.De Val S, Black BL. Transcriptional control of endothelial cell development. Dev Cell. 2009;16:180–195. doi: 10.1016/j.devcel.2009.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dejana E, Taddei A, Randi AM. Foxs and Ets in the transcriptional regulation of endothelial cell differentiation and angiogenesis. Biochim Biophys Acta. 2007;1775:298–312. doi: 10.1016/j.bbcan.2007.05.003. [DOI] [PubMed] [Google Scholar]
- 18.Dube A, Thai S, Gaspar J, Rudders S, Libermann TA, Iruela-Arispe L, Oettgen P. Elf-1 is a transcriptional regulator of the Tie2 gene during vascular development. Circ Res. 2001;88:237–244. doi: 10.1161/01.res.88.2.237. [DOI] [PubMed] [Google Scholar]
- 19.Gaspar J, Thai S, Voland C, Dube A, Libermann TA, Iruela-Arispe ML, Oettgen P. Opposing functions of the Ets factors NERF and ELF-1 during chicken blood vessel development. Arterioscler Thromb Vasc Biol. 2002;22:1106–1112. doi: 10.1161/01.atv.0000023427.92642.cd. [DOI] [PubMed] [Google Scholar]
- 20.Barton K, Muthusamy N, Fischer C, Ting CN, Walunas TL, Lanier LL, Leiden JM. The Ets-1 transcription factor is required for the development of natural killer cells in mice. Immunity. 1998;9:555–563. doi: 10.1016/s1074-7613(00)80638-x. [DOI] [PubMed] [Google Scholar]
- 21.Hart A, Melet F, Grossfeld P, Chien K, Jones C, Tunnacliffe A, Favier R, Bernstein A. Fli-1 is required for murine vascular and megakaryocytic development and is hemizygously deleted in patients with thrombocytopenia. Immunity. 2000;13:167–177. doi: 10.1016/s1074-7613(00)00017-0. [DOI] [PubMed] [Google Scholar]
- 22.Pham VN, Lawson ND, Mugford JW, Dye L, Castranova D, Lo B, Weinstein BM. Combinatorial function of ETS transcription factors in the developing vasculature. Dev Biol. 2007;303:772–783. doi: 10.1016/j.ydbio.2006.10.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Spyropoulos DD, Pharr PN, Lavenburg KR, Jackers P, Papas TS, Ogawa M, Watson DK. Hemorrhage, impaired hematopoiesis, and lethality in mouse embryos carrying a targeted disruption of the Fli1 transcription factor. Molecular and cellular biology. 2000;20:5643–5652. doi: 10.1128/mcb.20.15.5643-5652.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang LC, Kuo F, Fujiwara Y, Gilliland DG, Golub TR, Orkin SH. Yolk sac angiogenic defect and intra-embryonic apoptosis in mice lacking the Ets-related factor TEL. EMBO J. 1997;16:4374–4383. doi: 10.1093/emboj/16.14.4374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tavassoli M. Structure and function of sinusoidal endothelium of bone marrow. Prog Clin Biol Res. 1981;59B:249–256. [PubMed] [Google Scholar]
- 26.Kiel MJ, Morrison SJ. Uncertainty in the niches that maintain haematopoietic stem cells. Nat Rev Immunol. 2008;8:290–301. doi: 10.1038/nri2279. [DOI] [PubMed] [Google Scholar]
- 27.Yin T, Li L. The stem cell niches in bone. J Clin Invest. 2006;116:1195–1201. doi: 10.1172/JCI28568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Narayan K, Juneja S, Garcia C. Effects of 5-fluorouracil or total-body irradiation on murine bone marrow microvasculature. Exp Hematol. 1994;22:142–148. [PubMed] [Google Scholar]
- 29.Sato TN, Tozawa Y, Deutsch U, Wolburg-Buchholz K, Fujiwara Y, Gendron-Maguire M, Gridley T, Wolburg H, Risau W, Qin Y. Distinct roles of the receptor tyrosine kinases Tie-1 and Tie-2 in blood vessel formation. Nature. 1995;376:70–74. doi: 10.1038/376070a0. [DOI] [PubMed] [Google Scholar]
- 30.Wong AL, Haroon ZA, Werner S, Dewhirst MW, Greenberg CS, Peters KG. Tie2 expression and phosphorylation in angiogenic and quiescent adult tissues. Circ Res. 1997;81:567–574. doi: 10.1161/01.res.81.4.567. [DOI] [PubMed] [Google Scholar]
- 31.Johnson SM, Torrice CD, Bell JF, Monahan KB, Jiang Q, Wang Y, Ramsey MR, Jin J, Wong KK, Su L, Zhou D, Sharpless NE. Mitigation of hematologic radiation toxicity in mice through pharmacological quiescence induced by CDK4/6 inhibition. J Clin Invest. 2010;120:2528–2536. doi: 10.1172/JCI41402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tipping AJ, Pina C, Castor A, Hong D, Rodrigues NP, Lazzari L, May GE, Jacobsen SE, Enver T. High GATA-2 expression inhibits human hematopoietic stem and progenitor cell function by effects on cell cycle. Blood. 2009;113:2661–2672. doi: 10.1182/blood-2008-06-161117. [DOI] [PubMed] [Google Scholar]
- 33.Horsley V, Aliprantis AO, Polak L, Glimcher LH, Fuchs E. NFATc1 balances quiescence and proliferation of skin stem cells. Cell. 2008;132:299–310. doi: 10.1016/j.cell.2007.11.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.