

Abstract
Inhaled corticosteroids (ICS) are a mainstay anti-inflammatory therapy for the management of asthma. ICS are synthetic glucocorticoids that are structurally similar to the natural active human glucocorticoid cortisol. Steroid transforming enzymes of the aldo-keto reductase (AKR) family, namely AKR1D1 (5β-steroid reductase) and AKR1C1-4 (ketosteroid reductases) are implicated in the systemic metabolism of cortisol in liver. In this study, the activities of these AKR1 enzymes on cortisol and two ICS compounds budesonide (BUD) and flunisolide (FLU) were investigated. It was found that the catalytic efficiency of AKR1D1 for the reduction of the double bond in cortisol was 4 and 10 fold higher than the catalytic efficiencies of AKR1D1 with FLU and BUD, respectively. This suggests that compared to cortisol, for which the 5β-reduction is a major metabolic pathway, a lower degree of systemic (hepatic) metabolism of BUD and FLU via AKR1D1 takes place. In addition, BUD potently inhibited AKR1D1 and AKR1C4, the key steroid metabolizing enzymes in liver, which may disrupt endogenous steroid hormone metabolism and thus contribute to BUD-induced systemic effects. Activities of AKR1C1-AKR1C3 on cortisol and the two ICS compounds (targeting the 20-keto group) suggest these enzymes may be involved in the local (lung) metabolism of these glucocorticoids.
Keywords: asthma, steroid metabolism, insensitivity, systemic effects
1. Introduction
Asthma is a chronic inflammatory disease of lung with increasing prevalence. Asthma affects about 300 million people worldwide, including up to one in four urban children [1, 2]. Inhaled corticosteroids (ICS) are the main anti-inflammatory agents used to effectively treat persistent asthma. ICS are synthetic glucocorticoids developed based on cortisol that contain chemical modifications to optimize anti-inflammatory effects and minimize mineralocorticoid and other systemic effects [3, 4].
Cortisol is the natural human glucocorticoid and functions by binding to the glucocorticoid receptor (GR), which is found in almost all tissues and regulates many important metabolic, cardiovascular, immunologic, and homeostatic functions [5]. As synthetic glucocorticoids, ICS activate GR and regulate the transcription of various gene products involved in inflammation [6]. The level of cortisol is tightly regulated to control activity of GR in different tissues. Cortisol synthesis and clearance are intricately linked by a compensatory mechanism. With increasing metabolic clearance of cortisol there is an associated increase in adrenocorticotropic hormone (ACTH) secretion and cortisol production in the adrenal gland. Thus, the level of circulating active glucocorticoid (cortisol or synthetic glucocorticoids) plays a critical role in determining the activity of the hypothalamic-pituitary-adrenal axis for ACTH secretion.
Cortisol is metabolized predominantly in liver to inactive metabolites for eventual clearance [7-9]. The major metabolic pathways involve the reduction of the Δ4-3-oxo functionality of cortisol first by human steroid 5α- or 5β-steroid reductases to form 5α- or 5β-dihydrocortisol, and subsequently by ketosteroid reductases to form 3α,5α/β-tetrahydrocortisols. Steroid transforming enzymes of the aldo-keto reductase (AKR) 1 family, namely AKR1D1 (5β-reductase) and AKR1C1-4 (ketosteroid reductases) are implicated. Cortisol can also be inactivated at the 11-position to cortisone by 11β-hydroxysteroid dehydrogenase (HSD). However, the 11β-HSD enzyme in liver functions exclusively as a high-affinity reductase to generate cortisol from cortisone [10]. Reduction at the 20-position and hydroxylation at 6-, 7-, or 16-positions by cytochrome P450 enzymes also occur, but are minor metabolic pathways of cortisol in liver.
Cortisol can also be metabolized in peripheral tissues such as lung, however contributions from different pathways are less clear. Significant 11β-HSD type 2 activity was found in human bronchial epithelial cells [11], which can inactivate cortisol to cortisone. While expression of AKR1D1 or 5α-Rs in lung are unknown, AKR1C1-3 are known to be expressed [12], suggesting a possible contribution to cortisol metabolism by 20-keto reduction.
There are two major concerns in ICS therapy for asthma: (i) systemic side-effects, especially for long term or high dosage users [13] and (ii) drug insensitivity/resistance in up to one third of patients [14, 15]. Metabolic properties of these reagents may affect their pharmacological actions. For example, efficient metabolism in liver is desired to minimize the amount of active reagent getting into systemic circulation causing adverse effects, whereas metabolism in lung is undesired so that the inactivation of the drug does not occur. However, limited information is available regarding the metabolic properties of ICS [4,5].
Due to their structural similarity to cortisol, ICS have the potential to undergo similar metabolic reactions at 3-, 5-, and 20- positions, suggesting that AKR enzymes may be involved. In this study, the activity of AKR1 enzymes on cortisol and two ICS drugs budesonide (BUD) and flunisolide (FLU) were examined and compared. Results suggest that the AKR enzymes affect the pharmacological effects of ICS.
2. Materials and Methods
2.1 Materials
All steroids were obtained from Steraloids (Wilton, NH, U.S.A.). Pyridine nucleotides were purchased from Roche Applied Science (Indianapolis, IN, USA). All other reagents were purchased from Sigma-Aldrich (St. Louis, MO) and were of ACS (American Chemical Society) grade or better. Recombinant AKR1 enzymes were over-expressed and purified to homogeneity as previously described [16, 17]. Under standard assay conditions, the specific activities of the purified AKR enzymes were 1.8 μmol/min/mg for AKR1C1, 2.3 μmol/min/mg for AKR1C2, and 1.3 μmol/min/mg for AKR1C3 for 0.2 mM 1-acenaphthenol oxidation, 0.3 μmol/min/mg for AKR1C4 for 75 μM androsterone oxidation, and 85 nmol/min/mg for AKR1D1 for 10 μM testosterone reduction [18, 19].
2.2 Kinetic Assays
Initial rates of the NADPH dependent reduction of steroids catalyzed by AKR1 enzymes were measured with a Hitachi F-2500 fluorescence spectrophotometer (Hitachi America, Ltd.; New York, NY) by monitoring the change in fluorescence emission of NADPH. Excitation and emission wavelengths were set at 340 nm and 450 nm, respectively. Changes in fluorescence units were converted to nanomoles of cofactor by using standard curves of fluorescence emission versus known NADPH concentrations. Typical reaction samples contained 100 mM potassium phosphate buffer, pH 7.0, enzyme (2.1 μg AKR1D1, 2.9 μg AKR1C1, 2.4 μg AKR1C2, 4.3 μg AKR1C3, or 1.7 μg AKR1C4), NADPH at fixed concentration (saturating concentration 8 μM or 25 μM), steroid at varied concentrations (0.1 μM – 50 μM), and 4% methanol in a total volume of 1 mL. Reactions were carried out at 37 °C. Analysis of kinetic data was carried out as previously described to determine the maximum velocity Vmax (nmol/min) and the apparent Michaelis-Menten constant Km (μM) [18]. For slow reactions such as the reduction of glucocorticoids by AKR1C1-4, complete substrate concentration profile was not possible, the initial velocities at 2 μM of steroid substrate were determined (reported as activity nmol/min/mg).
To evaluate BUD and FLU as inhibitor of AKR1D1 and AKR1C4 enzymes, NADPH-dependent reduction of cortisol catalyzed by AKR1D1 and NAD+-dependent oxidation of 1-tetralol catalyzed by AKR1C4 were monitored [20, 21]. Initial rates were measured in the presence of the inhibitor at varying concentrations while holding substrate concentration at Km. Percent activity was plotted against inhibitor concentration to determine IC50 values. The 100% value was the initial velocity in the absence of the inhibitor.
2.4 TLC Analysis of Cortisol Metabolites
To characterize products, reactions were conducted in 0.4 ml systems containing 100 mM potassium phosphate (pH 7.0), 2.3 mM NADPH and 25 μM steroid in 4% methanol. Reactions were initiated by the addition of 5 μg enzyme and incubated at 37 °C for 90 min. Reactions were quenched and exacted twice by 0.8 ml of water-saturated ethyl acetate. The combined extracts were dried, re-dissolved in 100 μl methanol and applied to Partisil LK6D Silica TLC (thin layer chromatography) plates (Whatman International Ltd.). Chromatograms were developed in cyclohexane/ethyl acetate (1:4, v/v). Products were identified by co-migration with authentic standards applied to the same plate following visualization by spraying with acetic acid/sulfuric acid/anisaldehyde (100:2:1, v/v/v) and heat.
3. Results and Discussion
3.1 Reduction of Cortisol, BUD and FLU by AKR1D1 and AKR1C4 - Implications for ICS Systemic Metabolism
Cortisol and ICS all contain a Δ4-3-oxo moiety in the A ring of the steroid (Figure 1), which can potentially undergo sequential 4,5-double bond reduction and 3-ketoreduction. Liver is known to be the primary site of systemic cortisol metabolism and forms the A-ring reduced metabolites. In vitro metabolism of cortisol in human liver cytosolic fractions gave 3α,5α/β-tetrahydrocortisols as the major metabolites [22], implicating AKR1D1 and AKR1C enzymes, where AKR1D1 is the first enzyme for the 5β-pathway and AKR1C enzymes follow both 5α-reductase and AKR1D1.
Fig.1.

(A) Chemical structures of cortisol, BUD and FLU and (B) metabolic reactions catalyzed by AKRs.
Cortisol, BUD and FLU were used as substrates for AKR1D1 and the steady state kinetic parameters of the reaction were determined (Table 1). When compared to cortisol, ICS compounds BUD and FLU displayed reduced Vmax values and increased Km values. As a result, the catalytic efficiency of AKR1D1 for cortisol reduction is 10 and 4 fold higher than those for BUD and FLU reduction, respectively. The identities of the products formed from the in vitro reactions were analyzed by TLC. By reference to authentic standards, it was confirmed that the reduction of cortisol by AKR1D1 formed the 5β-reduced cortisol. TLC analysis also showed significant disappearance of BUD and FLU and appearance of unknown product bands in samples containing AKR1D1 compared with the no enzyme control (data not shown).
Table 1.
Kinetic Parameters for the Reactions Catalyzed by AKR1D1 and AKR1C4.
| Steroid | AKR1D1 (double bond reduction) |
AKR1C4 (20-keto reduction) |
||
|---|---|---|---|---|
|
Vmax (nmol min−1) |
Km (μM) |
Vmax/Km (nmol min−1μM−1) |
Activity (nmol min−1mg−1) |
|
| cortisol | 58 ± 4 | 13 ± 2 | 4.4 | 13.2 ± 0.6 |
| BUD | 9 ± 1 | 22 ± 6 | 0.4 | 0.8 ± 0.2 |
| FLU | 30 ± 3 | 31 ± 7 | 1.0 | 4.9 ± 0.5 |
AKR1C4 is believed to be the principal AKR1C isoform responsible for the formation of tetrahydro metabolites, since it displays the highest catalytic efficiencies among four AKR1C enzymes for the 3-keto reduction with most steroid substrates that have been studied [23, 24]. When 5β-dihydrocortisol was used as substrate for AKR1C4, the Vmax value was estimated to be 400 nmol/min and Km value was estimated to be 0.5 μM. Compared to the reaction of AKR1D1 for the formation of 5β-dihydrocortisol, the reaction of AKR1C4 for the 3-keto reduction of 5β-dihydrocortisol is about 200 fold more catalytically efficient. Authentic 5β-reduced FLU was not available to accurately determine the kinetic parameters for the formation of 5β-tetrahydro FLU. Instead, the reduction of 5β-reduced FLU catalyzed by AKR1C4 was assessed in AKR1D1/AKR1C4 coupled assays. In the coupled assay, FLU was incubated with AKR1D1 and excess NADPH and the fluorescence signal of NADPH was monitored. When the reaction of AKR1D1 was complete signaled by the leveled NADPH fluorescence, AKR1C4 was added to the system. The addition of AKR1C4 caused rapid disappearance of NADPH fluorescence, consistent with rapid 3-keto reduction of the product of the AKR1D1 reaction. This also suggested that AKR1D1 was rate-determining for the 5β-metabolic pathway of FLU.
AKR1C4 can also act on cortisol, BUD, and FLU directly by reducing the 20-keto position. However, this was found to be a weak activity of this enzyme, with reaction rates significantly lower than those of AKR1D1 (Table 1).
Liver is the primary site of systemic metabolism for not only the natural glucocorticoid cortisol but also for ICS, since significant portions of ICS will be absorbed into the bloodstream through inhalation or swallowed and go through first-pass metabolism. To minimize the amount of active ICS getting into systemic circulation and causing adverse effects, fast metabolism in liver is desired. Currently available ICS all have very low bioavailability suggesting fast first-pass metabolism [3, 4]. However, AKR1D1 displayed decreased activities on BUD and FLU compared to cortisol. This indicates that the double bond at 1,2-position reduces the propensity for A-ring metabolism and that for the systemic metabolism of ICS other enzymes such as the CYP450 enzymes may play a more significant role.
3.2 Inhibition of AKR1D1 and AKR1C4 - Implications for Systemic Side-Effects of BUD
The observation that BUD is a poor substrate for AKR1D1 and AKR1C4 raised the question whether BUD would inhibit the reaction of other steroids catalyzed by the two enzymes. The reaction systems monitored were NADPH-dependent reduction of cortisol for AKR1D1 and NAD+-dependent oxidation of 1-tetralol for AKR1C4 [20, 21]. IC50 values of BUD for these reactions were determined to be 1.1 and 1.4 μM, respectively (Figure 2). In contrast to BUD, IC50 values of FLU were 7.8 and 96 μM for AKR1D1 and AKR1C4 catalyzed reactions, respectively. This suggests IC50 values against the same enzyme can vary significantly between ICS.
Fig.2.
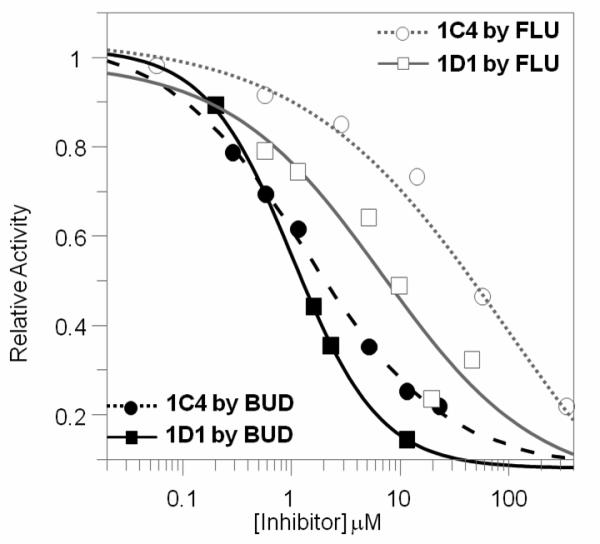
Inhibition of AKR1D1 and AKR1C4 reactions by BUD and FLU.
Potent inhibition of AKR1D1 and AKR1C4 by BUD suggests BUD may disrupt endogenous metabolism of cortisol in the liver. This is possible since following inhalation the majority of the dose (up to 90 %) impacts on the oropharyngeal region, is swallowed and gets into the liver. Clinical observations of effects of BUD on cortisol levels also point to disruption of hepatic cortisol metabolism by BUD. One study showed that low dosage BUD treatment of children with mild to moderate asthma increased mean cortisol levels [25]. This may be explained by inhibitory effects of BUD on AKR1D1 and AKR1C4 in liver to block cortisol inactivation, resulting in an immediate elevation of circulating cortisol. In another study where higher dose of BUD and longer term of treatment were used, the plasma and urinary levels of cortisol were shown to be significantly suppressed after doses of ICS compared to placebo [26]. Interestingly, a surge in plasma level of cortisol was seen after the first dose of BUD, which may be explained by the immediate inhibitory effects of BUD on cortisol metabolism. This initial elevated cortisol level and systemic BUD can suppress ACTH and lead to decrease in cortisol production. So the long term effects of BUD on cortisol level manifested itself as suppression.
Inhibitory effects of BUD on hepatic AKR enzymes may not be limited to cortisol metabolism. Previous studies have shown these enzymes play critical role in the biosynthesis and/or metabolism of steroid hormones including sex hormones and neurosteroids, as well as bile acids [18, 23, 24, 27-33]. Because these agents are important for normal functions of many systems, for example, sex hormones for bone absorption and growth, reproductive, and cardiovascular functions, neurosteroids for central nervous function, bile acids for gastrointestinal functions, disruption of AKR activity may lead to serious detrimental consequences in the above systems. It is well known that while ICS exert anti-inflammatory effects in lung, ICS can cause many undesired side-effects, such as adrenal suppression, reduction of bone mineral density, retardation of vertical growth in children, and ocular toxicity, etc [13, 34]. Adverse effects in other systems such as central nervous system, gastrointestinal system and cardiovascular have also been noted. ICS-induced adrenal suppression is most widely studied and is believed to be caused by active drug or metabolite that escapes metabolism and enters systemic circulation. However, the mechanisms for most adverse effects are not entirely understood. The observation that BUD inhibits key AKR enzymes provides a new mechanism for ICS-induced side effects. Interestingly, BUD and FLU behaved quite differently as inhibitors of AKR. As a result, their pharmacological actions in terms of inducing side effects would differ. This agrees with the observation that all ICS have similar efficacy but vary in their propensity to cause adverse effects.
3.3 Reduction of Cortisol, BUD and FLU by AKR1C1-3 Enzymes-Implications for Local Metabolism of ICS and Drug Insensitivity
Cortisol and ICS all have a 20-keto group in their structure, which can be potentially reduced by AKR1C enzymes. The ability of AKR1C1-3 to metabolize BUD and FLU were characterized in reconstituted systems using purified enzymes. In the fluorescence assays where the consumption of NADPH was monitored, cortisol, BUD, and FLU were found to support the turnovers of AKR1C enzymes. Specific activities for these reactions were determined (Table 2).
Table 2.
Specific activities of AKR1C Enzymes for the Reductions of Cortisol and ICS.
| Activity (nmol min−1mg−1) | |||
|---|---|---|---|
| Steroid | AKR1C1 | AKR1C2 | AKR1C3 |
| cortisol | 10.9 | 11.4 | 9.8 |
| BUD | 8.2 | 2.5 | 4.3 |
| FLU | 8.7 | 4.9 | 6.0 |
AKR1C1-3 are known to be abundantly expressed in lung and the expression levels are significantly increased in the lungs of smokers [35]. The activity of AKR1C1-3 on BUD and FLU may have important implications for one of the major concerns of ICS therapy for asthma, ICS insensitivity. Reportedly, up to 30% of asthma patients experience glucocorticoid insensitivity or resistance [14, 15]. Cigarette smoking worsens asthma and is associated with reduced response to corticosteroid therapy [36]. The mechanism of ICS insensitivity is complex [37]. This raises the possibility of local metabolism of ICS by AKR1C enzymes contributing to drug insensitivity. As AKR1C enzymes are known to be over-expressed in smokers [35], it is speculated that increased metabolism of ICS by AKR1C may lead to enhanced inactivation of ICS and render smokers with asthma less responsive to the drug.
3.4 Conclusion
ICS have the same Δ4-3-oxo moiety as cortisol, thus potentially can be A-ring reduced in the liver. Herein, it is shown that AKR1D1, the first and rate-determining enzyme for the 5β-pathway, displayed reduced activity with BUD and FLU relative to cortisol. In particular, BUD is a very poor substrate for AKR1D1 and instead potently inhibits the reactions catalyzed by AKR1D1 and AKR1C4. Since AKR1D1 and AKR1C4 play critical roles in the biosynthesis and/or metabolism of steroid hormones including sex hormones, neurosteroids, and bile acids, inhibition by BUD could lead to serious detrimental consequences in many systems where ICS adverse effects have been observed. In addition, it is shown the 20-keto group in cortisol and ICS can be reduced by the AKR1C enzymes. AKR1C1-3 are known to be expressed in lung cells, suggesting local metabolism by AKR1C enzymes may occur. Differential expression levels of metabolizing enzymes may lead to different levels of local metabolism/inactivation resulting in differences in drug response.
Acknowledgements
The author thanks Mr. Gagan Sarawgi for technical assistance and Drs. Trevor M. Penning and Michael C. Byrns for insightful discussions and critical reading of the manuscript.
This research has been made possible through a pilot-project fund awarded to Y.J. from NIH grant P30 ES015857. Y.J. is also supported by a FOCUS-Junior Faculty Investigator Award for Research in Woman's Health funded by the Edna G. Kynett Memorial Foundation and by NIH grant R01-DK47015 awarded to T.M. Penning.
Abbreviations used
- ACTH
adrenocorticotropic hormone
- AKR
aldo-keto reductase (also visit www.med.upenn.edu/akr)
- AKR1D1
5β-reductase
- AKR1C1-4
ketosteroid reductases
- BUD
budesonide
- FLU
flunisolide
- GR
glucocorticoid receptor
- HSD
hydroxysteroid dehydrogenase
- ICS
inhaled corticosteroid(s)
- 5α-R
5α-reductase
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Masoli M, Fabian D, Holt S, Beasley R. The global burden of asthma: executive summary of the GINA Dissemination Committee report. Allergy. 2004;59(5):469–478. doi: 10.1111/j.1398-9995.2004.00526.x. [DOI] [PubMed] [Google Scholar]
- 2.Ricciardolo FL. The treatment of asthma in children: inhaled corticosteroids. Pulm Pharmacol Ther. 2007;20(5):473–482. doi: 10.1016/j.pupt.2005.11.007. [DOI] [PubMed] [Google Scholar]
- 3.Barnes NC. The properties of inhaled corticosteroids: similarities and differences. Prim Care Respir J. 2007;16(3):149–154. doi: 10.3132/pcrj.2007.00038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cerasoli F., Jr. Developing the ideal inhaled corticosteroid. Chest. 2006;130(1 Suppl):54S–64S. doi: 10.1378/chest.130.1_suppl.54S. [DOI] [PubMed] [Google Scholar]
- 5.Munck A, Naray-Fejes-Toth A. The ups and downs of glucocorticoid physiology. Permissive and suppressive effects revisited. Mol Cell Endocrinol. 1992;90(1):C1–4. doi: 10.1016/0303-7207(92)90091-j. [DOI] [PubMed] [Google Scholar]
- 6.van der Velden VH. Glucocorticoids: mechanisms of action and anti-inflammatory potential in asthma. Mediators Inflamm. 1998;7(4):229–237. doi: 10.1080/09629359890910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Roberts S, Szego CM. Biochemistry of the steroid hormones. Annu Rev Biochem. 1955;24:543–596. doi: 10.1146/annurev.bi.24.070155.002551. [DOI] [PubMed] [Google Scholar]
- 8.Tomkins GM. Enzymatic mechanisms of hormone metabolism. I. Oxidation-reduction of the steroid nucleus. Recent Prog Horm Res. 1956;12:125–133. [PubMed] [Google Scholar]
- 9.Andrew R. Clinical measurement of steroid metabolism. Best Pract Res Clin Endocrinol Metab. 2001;15(1):1–16. doi: 10.1053/beem.2001.0116. [DOI] [PubMed] [Google Scholar]
- 10.Jamieson PM, Chapman KE, Edwards CR, Seckl JR. 11β-hydroxysteroid dehydrogenase is an exclusive 11β- reductase in primary cultures of rat hepatocytes: effect of physicochemical and hormonal manipulations. Endocrinology. 1995;136(11):4754–4761. doi: 10.1210/endo.136.11.7588203. [DOI] [PubMed] [Google Scholar]
- 11.Feinstein MB, Schleimer RP. Regulation of the action of hydrocortisone in airway epithelial cells by 11β-hydroxysteroid dehydrogenase. Am J Respir Cell Mol Biol. 1999;21(3):403–408. doi: 10.1165/ajrcmb.21.3.3560. [DOI] [PubMed] [Google Scholar]
- 12.Penning TM, Burczynski ME, Jez JM, Hung CF, Lin HK, Ma H, Moore M, Palackal N, Ratnam K. Human 3α-hydroxysteroid dehydrogenase isoforms (AKR1C1-AKR1C4) of the aldo-keto reductase superfamily: functional plasticity and tissue distribution reveals roles in the inactivation and formation of male and female sex hormones. Biochem J. 2000;351(Pt 1):67–77. doi: 10.1042/0264-6021:3510067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lipworth BJ. Systemic adverse effects of inhaled corticosteroid therapy: A systematic review and meta-analysis. Arch Intern Med. 1999;159(9):941–955. doi: 10.1001/archinte.159.9.941. [DOI] [PubMed] [Google Scholar]
- 14.Leung DY, Bloom JW. Update on glucocorticoid action and resistance. J Allergy Clin Immunol. 2003;111(1):3–22. doi: 10.1067/mai.2003.97. quiz 23. [DOI] [PubMed] [Google Scholar]
- 15.Barnes PJ, Adcock IM. Glucocorticoid resistance in inflammatory diseases. Lancet. 2009;373(9678):1905–1917. doi: 10.1016/S0140-6736(09)60326-3. [DOI] [PubMed] [Google Scholar]
- 16.Burczynski ME, Harvey RG, Penning TM. Expression and characterization of four recombinant human dihydrodiol dehydrogenase isoforms: oxidation of trans-7, 8-dihydroxy-7,8-dihydrobenzo. Biochemistry. 1999;38(32):10626. doi: 10.1021/bi995085z. [DOI] [PubMed] [Google Scholar]
- 17.Ratnam K, Ma H, Penning TM. The arginine 276 anchor for NADP(H) dictates fluorescence kinetic transients in 3α-hydroxysteroid dehydrogenase, a representative aldoketo reductase. Biochemistry. 1999;38(24):7856–7864. doi: 10.1021/bi982838t. [DOI] [PubMed] [Google Scholar]
- 18.Jin Y, Duan L, Lee SH, Kloosterboer HJ, Blair IA, Penning TM. Human cytosolic hydroxysteroid dehydrogenases of the aldo-ketoreductase superfamily catalyze reduction of conjugated steroids: implications for phase I and phase II steroid hormone metabolism. J Biol Chem. 2009;284(15):10013–10022. doi: 10.1074/jbc.M809465200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Drury JE, Di Costanzo L, Penning TM, Christianson DW. Inhibition of human steroid 5β-reductase (AKR1D1) by finasteride and structure of the enzyme-inhibitor complex. J Biol Chem. 2009;284(30):19786–19790. doi: 10.1074/jbc.C109.016931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Matsuura K, Shiraishi H, Hara A, Sato K, Deyashiki Y, Ninomiya M, Sakai S. Identification of a principal mRNA species for human 3α-hydroxysteroid dehydrogenase isoform (AKR1C3) that exhibits high prostaglandin D2 11-ketoreductase activity. J Biochem. 1998;124(5):940–946. doi: 10.1093/oxfordjournals.jbchem.a022211. [DOI] [PubMed] [Google Scholar]
- 21.Usami N, Yamamoto T, Shintani S, Ishikura S, Higaki Y, Katagiri Y, Hara A. Substrate specificity of human 3(20)α-hydroxysteroid dehydrogenase for neurosteroids and its inhibition by benzodiazepines. Biol Pharm Bull. 2002;25(4):441–445. doi: 10.1248/bpb.25.441. [DOI] [PubMed] [Google Scholar]
- 22.Abel SM, Maggs JL, Back DJ, Park BK. Cortisol metabolism by human liver in vitro--I. Metabolite identification and inter-individual variability. J Steroid Biochem Mol Biol. 1992;43(7):713–719. doi: 10.1016/0960-0760(92)90297-v. [DOI] [PubMed] [Google Scholar]
- 23.Penning TM, Burczynski ME, Jez JM, Hung CF, Lin HK, Ma H, Moore M, Palackal N, Ratnam K. Human 3α-hydroxysteroid dehydrogenase isoforms (AKR1C1-AKR1C4) of the aldo-keto reductase superfamily: functional plasticity and tissue distribution reveals roles in the inactivation and formation of male and female sex hormones. Biochem J. 2000;351:67–77. doi: 10.1042/0264-6021:3510067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Steckelbroeck S, Jin Y, Gopishetty S, Oyesanmi B, Penning TM. Human cytosolic 3α-hydroxysteroid dehydrogenases of the aldo-keto reductase superfamily display significant 3β-hydroxysteroid dehydrogenase activity: Implications for steroid hormone metabolism and action. J Biol Chem. 2003;279:10784–10795. doi: 10.1074/jbc.M313308200. [DOI] [PubMed] [Google Scholar]
- 25.Hoekx JC, Hedlin G, Pedersen W, Sorva R, Hollingworth K, Efthimiou J. Fluticasone propionate compared with budesonide: a double-blind trial in asthmatic children using powder devices at a dosage of 400 μg × day(−1) Eur Respir J. 1996;9(11):2263–2272. doi: 10.1183/09031936.96.09112263. [DOI] [PubMed] [Google Scholar]
- 26.Martin RJ, Szefler SJ, Chinchilli VM, Kraft M, Dolovich M, Boushey HA, Cherniack RM, Craig TJ, Drazen JM, Fagan JK, Fahy JV, Fish JE, Ford JG, Israel E, Kunselman SJ, Lazarus SC, Lemanske RF, Jr., Peters SP, Sorkness CA. Systemic effect comparisons of six inhaled corticosteroid preparations. Am J Respir Crit Care Med. 2002;165(10):1377–1383. doi: 10.1164/rccm.2105013. [DOI] [PubMed] [Google Scholar]
- 27.Bauman DR, Steckelbroeck S, Penning TM. The roles of aldo-keto reductases in steroid hormone action. Drug News Perspect. 2004;17(9):563–578. doi: 10.1358/dnp.2004.17.9.872570. [DOI] [PubMed] [Google Scholar]
- 28.Berseus O, Danielsson H, Kallner A. Synthesis and metabolism of cholest-4-ene-7α,12α-diol-3-one and 5β-cholestane-7α,12α-diol-3-one. J Biol Chem. 1965;240:2396–2401. [PubMed] [Google Scholar]
- 29.Berseus O. Conversion of cholesterol to bile acids in rat: Purification and properties of delta-4-3-ketosteroid 5β-reductase and a 3α-hydroxysteroid dehydrogenase. Eur J Biochem. 1967;2:493–502. doi: 10.1111/j.1432-1033.1967.tb00163.x. [DOI] [PubMed] [Google Scholar]
- 30.Clayton PT, Mills KA, Johnson AW, Barabino A, Marazzi MG. Δ4-3-oxosteroid 5β-reductase deficiency: failure of ursodeoxycholic acid treatment and response to chenodeoxycholic acid plus cholic acid. Gut. 1996;38:623–628. doi: 10.1136/gut.38.4.623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Di Costanzo L, Drury JE, Penning TM, Christianson DW. Crystal structure of human liver Δ4-3-oxosteroid 5β-reductase (AKR1D1) and implications for substrate binding and catalysis. J Biol Chem. 2008;283(24):16830–16839. doi: 10.1074/jbc.M801778200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Steckelbroeck S, Jin Y, Oyesanmi B, Lee SH, Kloosterboer HJ, Penning TM. Tibolone is metabolized by the 3α/3β-hydroxysteroid dehydrogenase activities of the four human isozymes of the aldo-keto reductase 1C subfamily: Inversion of stereospecificity with a Δ5(10)-3-ketosteroid. Mol Pharmacol. 2004;66:1702–1711. doi: 10.1124/mol.104.004515. [DOI] [PubMed] [Google Scholar]
- 33.Steckelbroeck S, Oyesanmi B, Jin Y, Lee SH, Kloosterboer HJ, Penning TM. Tibolone metabolism in human liver is catalyzed by 3α/3β-hydroxysteroid dehydrogenase activities of the four isoforms of the aldo-keto reductase (AKR)1C subfamily. J Pharmacol & Exp Therap. 2006;316:1300–1309. doi: 10.1124/jpet.105.091587. [DOI] [PubMed] [Google Scholar]
- 34.Lipworth BJ, Jackson CM. Effects of oral and inhaled corticosteroids on the hypothalamic-pituitary-adrenal axis. J Allergy Clin Immunol. 1999;104(3 Pt 1):713–714. doi: 10.1016/s0091-6749(99)70352-0. [DOI] [PubMed] [Google Scholar]
- 35.Zhang L, Lee JJ, Tang H, Fan YH, Xiao L, Ren H, Kurie J, Morice RC, Hong WK, Mao L. Impact of smoking cessation on global gene expression in the bronchial epithelium of chronic smokers. Cancer Prev Res (Phila Pa) 2008;1(2):112–118. doi: 10.1158/1940-6207.CAPR-07-0017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Thomson NC, Chaudhuri R, Livingston E. Asthma and cigarette smoking. Eur Respir J. 2004;24(5):822–833. doi: 10.1183/09031936.04.00039004. [DOI] [PubMed] [Google Scholar]
- 37.Adcock IM, Barnes PJ. Molecular mechanisms of corticosteroid resistance. Chest. 2008;134(2):394–401. doi: 10.1378/chest.08-0440. [DOI] [PubMed] [Google Scholar]