

Abstract
Voltage-gated K+ channels are key regulators of neuronal excitability, playing major roles in setting resting membrane potential, repolarizing the cell membrane after action potentials and affecting transmitter release. The M-type channel or M-channel is a unique voltage- and ligand-regulated K+ channel. It is composed of the molecular counterparts KCNQ2 and KCNQ3 (also named Kv7.2 and Kv7.3) channels and expressed in the soma and dendrites of neurons. The present investigation examined the hypothesis that KCNQ2/3 channels played a regulatory role in neuronal differentiation and maturation. In cultured mouse embryonic stem (ES) cells undergoing neuronal differentiation and primary embryonic (E15-17) hippocampal cultures, KCNQ2 and KCNQ3 channels and underlying M-currents were identified. Blocking of KCNQ channels in these cells for 5 days using the specific channel blocker XE991 (10 μM) or linopirdine (30 μM) significantly decreased synaptophysin and syntaxin expression without affecting cell viability. Chronic KCNQ2/3 channel block reduced the expression of vesicular GABA transporter (v-GAT), but not vesicular glutamate transporter (v-GluT). Enhanced ERK1/2 phosphorylation was observed in XE991- and linopirdine-treated neural progenitor cells. In electrophysiological recordings, cells undergoing chronic block of KCNQ2/3 channels showed normal amplitude of mPSCs while the frequency of mPSCs was reduced. On the other hand, KCNQ channel opener N-Ethylmaleimide (NEM, 2 μM) increased mPSC frequency. Fluorescent imaging using fluorescent styryl-dye FM4-64 revealed that chronic blockade of KCNQ2/3 channels decreased endocytosis but facilitated exocytosis. These data indicate that KCNQ2/3 channels participate in regulation of neuronal differentiation and show a tonic regulation on pre-synaptic transmitter release and recycling in developing neuronal cells.
Keywords: M-current, synaptogenesis, Neuronal differentiation, Mouse ES cells, Hippocampal neurons, KCNQ channels, ERK1/2
Introduction
Pluripotent stem cells have the potential of differentiating into virtually all types of cells, including excitatory and inhibitory neurons. Transplantation therapy using neural progenitor cells derived from embryonic and adult tissues has emerged as a potential treatment for many neurodegenerative diseases such as ischemic stroke, brain and spinal cord traumatic injuries, Parkinson disease, Alzheimer’s disease, and so on (Burns, et al., 2009, Kim and de Vellis, 2009, Richardson, et al., 2010, Srivastava, et al., 2008, Wei, et al., 2005). Before and after cell transplantation, appropriate cell differentiation is a key maneuver for successful repair of a damaged tissue or neural network. Although many investigations have examined roles of growth/trophic factors and intracellular signals in neuronal differentiation of stem cells, there has been little information about potential roles of ion channels in regulating selective differentiation of neural progenitor cells.
K+ channels are the most diverse class of ion channels. Humans have 70 or more genes that encode subunits of K+ channels. Splice variant and formation of heteromeric channels further suggest great structural and functional diversities (Jentsch, 2000). Opening of these channels generally causes membrane hyperpolarization. Consequently, these channels are responsible for maintaining a negative membrane potential, modulating spike firing rates, and determining the height and width of action potentials. In association with these actions, K+ channels take part in regulating neuronal transmission. Among K+ channels, the delayed rectifier channels, transient A-type channels, slow activating I(Ks) channel, Ca2+- and ATP-gated channels, and S channels have been linked to play foremost roles in affecting transmitter release (Artinian, et al., 2010, Meir, et al., 1999, Ueda and Wu, 2006).
The M-type K+ channel was first identified in the early 1980s in frog and rat sympathetic neurons (Brown and Adams, 1980, Constanti and Brown, 1981). It was later confirmed that the M-channel is a widely expressed voltage-gated, non-inactivating channel in the nervous system and non-neuronal cells. M-channel activity is regulated by multiple factors and signaling pathways including muscarinic receptor activation, intracellular Ca2+, and arachidonic acid metabolites (Brown and Yu, 2000). The M-channel is activated at a voltage near the threshold for action potential initiation. Thus, M-channels function as a ‘brake’ on repetitive action potential firing and regulates neuronal excitability, which may indirectly and acutely affect transmitter release (Delmas and Brown, 2005). Whether the M-channel has broader physiological functions in addition to controlling the membrane excitability has rarely been explored.
Recent progress has identified the KCNQ2 and KCNQ3 channels to be the molecular basis of the M-channel (Brown and Yu, 2000). These channels were later classified as Kv7.2 and Kv7.3 channels according to their molecular structures (Jespersen, et al., 2005). Among five genes of the KCNQ channel family (KCNQ1-5), KCNQ1 is expressed in the heart and smooth muscle cells, the other four KCNQ channels are expressed mainly in the nervous systems. KCNQ2 and KCNQ3 homomultimers or heteromultimers are thought to underlie the M-channel (Shapiro, et al., 2000, Wang, et al., 1998). The recently identified KCNQ5 gene, expressed in the brain and skeletal muscle, can co-assemble with KCNQ3 to play a role in the M-channel heterogeneity (Shah, et al., 2002). KCNQ channels are mostly located on the presynaptic membrane (Lai and Jan, 2006), which puts these channels at a favorable position to regulate transmitter release. Mutations of KCNQ genes have been reported to relate to human diseases. KCNQ/M-channel modulators have been developed for treatments of Alzheimer’s disease, epilepsy, and stroke, mainly based on their effect of causing membrane hyperpolarization and consequently attenuating pathological excitatory activities (Cooper and Jan, 2003).
Neuronal differentiation is a process for which undifferentiated cells are committed to neural progenitors and later become neuronal cells, while neuronal maturation usually referrers to the later phase of differentiation that produces mature neurons. Specifically, maturation includes changes within the branching of the terminals, changes in receptor subunit composition, synapse formation and selective synapse elimination. Synaptogenesis generally refers to the last two processes of the generation of mature synapses (Akins and Biederer, 2006). Since we identified expression of KCNQ2 and/or KCNQ3 channels in mature neurons as well as in undifferentiated stem cells and neural progenitor cells, the present investigation explored the possibility that the activity of KCNQ2/3 channels might play a regulatory role in neuronal differentiation/maturation, synaptogenesis, and transmitter release during neural development.
Methods and Materials
Experimental Materials
Rabbit polyclonal antibodies to v-GluT and v-GAT were obtained from SYSY Synaptic Systems (Goettingen, Germany). The v-GluT and v-GAT antibodies recognize the glutamate transporter and GABA transporter in the membrane of synaptic vesicles, respectively. Rabbit polyclonal antibodies to synaptophysin, GAD67 and GluR1 were purchased from Santa Cruz (Santa Cruz, CA, USA). 10,10-bis(4-pyridinylmethyl)-9(10H)-anthracenone (XE991), linopirdine and N-Ethylmaleimide (NEM) were purchased from Sigma-Aldrich (St. Louis, MO, USA).
Mouse embryonic stem cell culture
ES cell cultures were prepared from stocks of an ES cell line (D3 cells) that was maintained in our laboratory. ES cells of no more than 30 passages were used for experiments. The undifferentiated ES cells were cultured on Gelatin-coated T25 flasks in the presence of 1,000 units/ml of leukemia inhibitory factor from Chemicon (Temecula, CA, USA) and 0.1 mM β-mercaptoethanol in the induction media (ESIM). ESIM consists of Dulbecco’s modified eagle medium with L-glutamine (Gibco, Carlsbad, CA, USA), supplemented with 10% fetal bovine serum, 10% new-born calf serum, 8 μg/ml guanosine, 7.3 μg/ml cytidine, 7.3 μg/ml uridine, and 2.4 μg/ml thymidine.
Neural lineage induction of embryoid bodies (EB) were completed by withdrawal of LIF for 4 days followed by exposure to retinoic acid (RA) for 4 days, also known as 4−/4+ RA protocol (Bain, et al., 1995, Bain, et al., 1996, Fischer, et al., 2000). ES cells were harvested and placed into a standard 100-mm bacterial Petri dish in ESIM without adding LIF or β-mercaptoethanol. Four days later, the medium was replaced with fresh ESIM containing 5 × 10−7 M all-trans RA (Sigma-Aldrich, St. Louis, MO, USA) and incubated for 4 more days.
Following the 4−/4+ RA induction, embryoid bodies were dissociated with 0.25% trypsin-EDTA for 10 min at 37 °C. ESIM was added and the cells were spun at 1000g for 5 min. The medium was removed and cells were resuspended in modified Sato medium (Fischer, et al., 2000, McDonald, et al., 1999). Cells were then plated on poly-D-lysine and laminin (PDL) coated 35-mm dishes for imaging studies, Western blotting analysis, and whole cell recording.
Primary embryonic neuronal cultures
Primary hippocampal neurons were obtained from E15-E17 mice as described previously (Shen, et al., 2007). Embryos were harvested acutely from pregnant female mice sacrificed by isoflurane anesthesia and cervical dislocation. Embryos were decapitated, and the hippocampus was dissected. Dissociated hippocampal neurons using trypsin were plated on 35 mm poly-D-lysine and laminin coated dishes (VWR, West Chester, PA, USA) at the density of 3×105 cells/ml in Neurobasal media (Invitrogen, Carlsbad, CA, USA) supplemented with 2% B-27 (Invitrogen) and L-glutamine (0.5 mM). After 3 days in vitro (DIV), 5 μM β-cytosine arabinoside (Sigma-Aldrich) was added to the medium to inhibit cell division. After 7 DIV, half of the medium was changed with fresh medium then patch clamp recordings and other tests were performed 12 to 16 DIV.
Electrophysiological recordings of M-currents and mPSC
Mouse embryonic stem cell-derived neurons or hippocampal neurons were placed on the stage of an inverted microscope. Membrane currents were recorded by whole-cell configuration using an EPC-9 amplifier (List-Electronic, Germany). Recording electrodes of 8–10 MΩ (fire-polished) were pulled from Coming Kovar Sealing #7052 glass pipettes (PG52151-4, WPI, Sarasota, FL, USA), by a Flaming-Brown micropipette puller (P-80/PC, Sutter Instrument Co,. Movat, CA, USA). A high resistant gigaseal (1–3 GΩ) was formed between the cell membrane and the electrode tip. Whole-cell configuration was then established by breaking through the cell membrane. Series resistance compensation was routinely applied during recording. Current and voltage signals were displayed on a computer monitor and collected by a data acquisition/analysis program PULSE (HEKA, Lambrect, Germany). Currents were digitally sampled at 50 μs (20 kHz) and were filtered at 3 kHz by a 3-pole Bessel filter. Pipettes were filled with an internal solution containing the following (in mM) KCl 120, MgCl2 1.5, Na2-ATP 2, CaCl2 1.0, BAPTA 1.0, and HEPES 10. The external solution contains (in mM): NaCl 115, KCl 2.5, MnCl2 2.0, HEPES 10, BAPTA 0.1, Glucose 10, and 0.1 μM tetrodotoxin (TTX). The membrane potential was held at −30 mV for M-current activation. Hyperpolarizing steps to −70 mV were initiated by a digital pulse to deactivate M-current. The membrane potential was held at −70 mV, and mPSCs were recorded for 2 min as one episode. All experiments were performed at room temperature (22–25 °C) and pH 7.4.
Western blotting and immunoprecipitation analysis
Cell lysates were prepared by extracting proteins with lysis buffer (HEPEs 50 mM, 1% sodium deoxycholate, 1% Triton X-100, 0.1% SDS, 150 mM NaCl, 1 mM EDTA, 5 mM NaF, and 2.5 mM NaVO3) supplemented with protease inhibitors. Lysates were centrifuged at 14,000g for 20 min at 4°C and the supernatants were collected. Protein concentration in the supernatants was determined using bicinchoninic acid (BCA) protein assay and adjusted to 1 mg/ml with lysis buffer. Samples containing 500 μg of proteins were incubated with 2 μg of synaptophysin overnight at 4°C. Immunoprecipitates were collected by the addition of 30 μl protein G and incubation for 2 hr at 4°C. Complexes were collected by centrifugation and washed with lysis buffer. Finally the pelleted beads were resuspended in 2× SDS sample buffer (50 mM Tris-HCl, 2% SDS, 10% glycerol, and 5% β-mercaptoethanol). Proteins were separated by SDS-PAGE and transferred to a nitrocellulose membrane. The membrane was blocked using 5% BSA in Tris-buffered saline, and then incubated with primary antibodies for 24hrs at 4 °C. Blots were developed using alkaline peroxidase-conjugated secondary (Cell Signaling Technology), and proteins were visualized by addition of BCIP/NBT solution (Sigma-Aldrich). The membranes were scanned and analyzed by densitometry using Adobe Photoshop (Adobe Systems Inc., San Jose, CA, USA) and Image J.
Immunocytochemical staining
Cells were fixed with 4% paraformaldyhyde for 5 min. A blocking solution of 1% fish gel in PBS was applied for 1 hr, followed by overnight incubation in PBS containing primary antibody v-GluT (1:1000), v-GAT (1:1000) or synaptophysin (1:1000). Following several washes with 1× PBS, TRITC- or FITC-coupled secondary antibodies (1:600/1:200; Invitrogen) were applied for 1 hr at room temperature. Cultures were washed and mounted with Vectashield Mounting Medium (Vector Laboratories, Burlingame, CA, USA) and were visualized under an Olympus IX61 fluorescent microscope. After determination of background levels and calibration for uneven lighting, tissues were analyzed with regard to the specifically stained area using the in-built function of the Image J software (NIH).
TUNEL staining
DeadEnd™ Fluorometric TUNEL System (Promega corporation, Madison, WI, USA) was used for TUNEL staining in primary hippocampal or cortical neurons to detect cell death. After incubation in control media or testing drugs for specified length of time, cells were fixed, permeabilized, incubated in TUNEL reaction solution and counted to calculate the mean percentage of TUNEL-positive cells of randomly selected 6 fields..
FM4-64 dye staining, imaging and analysis
To visualize recycling synaptic vesicles, DIV 7 or 14 hippocampal neurons or mouse embryonic stem cell-derived neurons after 5 or 12 days harvesting were treated for 2 min with medium containing the fluorescent dye N-(3-triethylammonium-propyl)-4-(6-(4-diethylamino)phenyl)-hexatrienyl)pyridi nium dibromide (FM4-64; 10 μM; Invitrogen). Since membrane-associated dye is much more fluorescent than the dye in solution, quantification of the intensity of FM4-64 fluorescence could be used as a measurement of dye-uptake, as a marker of endocytosis. For dye up-take experiments, after the membrane was depolarized with high K+ (75 mM) external solution containing FM4-64 for 2 min, the cells were changed back to culture medium with the dye for 15 min, and non-endocytosed dye was washed off with medium and incubated for another 15 min to image the fluorescence for endocytosis. For dye release experiments, neurons were stimulated with high K+ solution for 2 min. Cells were washed three times with medium to remove non-endocytosed FM dye bound to the plasma membrane and incubated with culture medium for 30 min to image the fluorescence for exocytosis. Stem cell-derived neurons were examined by fluorescence microscopy during FM4-64 experiments with excitation at 568 nm, objective: 40X. Fluorescent intensities of neurons were analyzed using Image J (NIH). Background fluorescence was subtracted from the cell fluorescence values.
Statistics analysis
One-way ANOVA test followed by Tukey test was used for multiple comparisons; Student’s two-tailed t-test was performed for comparison of two experimental groups. Changes were identified as significant if P value was less than 0.05. Data was reported as mean ± SEM (standard error of mean). The statistical analysis was performed using SigmaPlot (Systat Software Inc, Chicago, IL, USA) or GraphPad (GraphPad Software Inc, La Jolla, CA, USA).
Results
Identification of KCNQ2/3 channel expression in mouse embryonic stem cell-derived neural progenitors and hippocampal neurons
Mouse ES cells were subjected to the neural induction 4−/4+ protocol (4 days without retinoic acid (RA) followed by 4 days with RA) (Bain, et al., 1995, Bain, et al., 1996, Fischer, et al., 2000). Primary cultured hippocampal neurons were obtained from E15-E17 prenatal mice. The examination on two different neuronal cells helped to determine the expression and functional role of KCNQ2/3 channels at different stages of neuronal differentiation and in different types of cells. Western blot analysis determined that KCNQ2 protein was highly expressed in undifferentiated mouse ES cells (Fig. 1A). The KCNQ2 expression decreased during the 4−/4+ process but was still easily detectable; at day 5 into the neural induction, KCNQ3 expression began in these neural progenitor cells (Fig. 1A). Both KCNQ2 and KCNQ3 were expressed in primary cultured hippocampal neurons (14 days in vitro or DIV)(Fig. 1A).
Figure 1. KCNQ2/3 channel expression and M-currents in mouse ES cell-derived neurons and hippocampal neurons.

Expression and functional activity of KCNQ2 and KCNQ3 channels were tested in mouse stem-derived neurons and hippocampal neurons (12–15 DIV). A. Western blotting demonstrated that KCNQ2 and KCNQ3 are expressed during differentiation in mouse stem cell-derived neurons or hippocampal neurons. Representative of N ≥ 3 assays. B. Whole cell recording showed M-currents are present in neurally differentiating cells 3 and 5 days after neuronal induction from stem cells or in hippocampal neurons of 14 DIV. Representative of ≥ 20 cells in each recording. C. Bath application (10 min) of 10 μM XE991, a selective KCNQ channel inhibitor, blocked the M-currents recorded in hippocampal neurons. N ≥ 5 cells in each group.
Whole-cell parch clamp recordings revealed that functional M-currents were present in ES cell-derived neural progenitors and hippocampal neurons (14 DIV) (Fig. 1B). The selective KCNQ channel blocker XE991 at 10 μM essentially blocked the whole-cell M-current (Fig. 1C) (Rennie, et al., 2001).
Association of KCNQ2/3 channels with synaptic vesicle proteins
Immunoprecipitation showed that KCNQ2 and KCNQ3 were co-precipitated with the presynaptic vesicle protein, synaptophysin (Fig. 2A). In immunostaining assays, KCNQ2 staining was overlapping with synaptophysin staining in stem cell-derived neuronal cells (Fig. 2B) and hippocampal neurons (Fig. 2C). KCNQ3 was also co-localized with synaptophysin in these two types of cells (data not shown). The similar results in two different neuronal cells suggest that KCNQ2/3 channels are associated with vesicle proteins, which is consistent with previous observations that KCNQ channel subunits are expressed in presynaptic locations (Lai and Jan, 2006).
Figure 2. Association of KCNQ2 and KCNQ3 proteins with synaptophysin.

Immunoprecipitation, Western blotting and immunostaining were applied to examine possible association of KCNQ2/3 channels with the presynaptic protein synaptophysin. A. Proteins were extracted from the adult mouse brain and used in immunoprecipitation assays against synaptophysin. Western blotting was then performed to detect KCNQ2, KCNQ3 or synaptophysin expression as an indication of the formation of protein complex between KCNQ2/KCNQ3 with synaptophysin. Both KCNQ2 and KCNQ3 were presented in the protein complex with synaptophysin. N = 3 assays. B and C. Immunostaining showed that KCNQ2 was also co-localized with synaptophysin in mouse stem cell-derived neurons (B) and hippocampal neurons (12–15 DIV) (C).
KCNQ channel activity and synaptogenesis
To determine whether KCNQ channel activity played an important role in neuronal differentiation, we tested expression of neurofilament and synaptic vesicle proteins using Western blotting and immunocytochemical staining. Neurofilament was tested as a specific marker of neurite outgrowth during neuronal differentiation/maturation. In mouse ES cell-derived neural progenitors and hippocampal neurons, the KCNQ2/3 channel activity was blocked by the channel blocker XE991 (10 μM) or linopirdine (30 μM) added into the culture media for a range of different time points. The exposure to a KCNQ channel blocker did not affect cell viability, verified by unchanged trypan blue and TUNEL staining (Fig. 3). However, XE991 and linopirdine attenuated synaptophysin and syntaxin expression in immunocytochemical assays without affecting neurofilament distribution in differentiating ES cells (Fig. 4A and 4B) and hippocampal neurons of 7 – 14 DIV (Fig. 5A – 5C). Consistently, Western blotting confirmed that 5-day exposure to XE991 or linopirdine attenuated synaptophysin and syntaxin expression levels, while neurofilament expression was unaffected (Fig. 4C and 4D). The unchanged neurofilament supported the view that reduced synaptic protein expression was not due to cell death or deterioration.
Figure 3. Chronic blocking KCNQ2/3 channels did not affect cell viability.
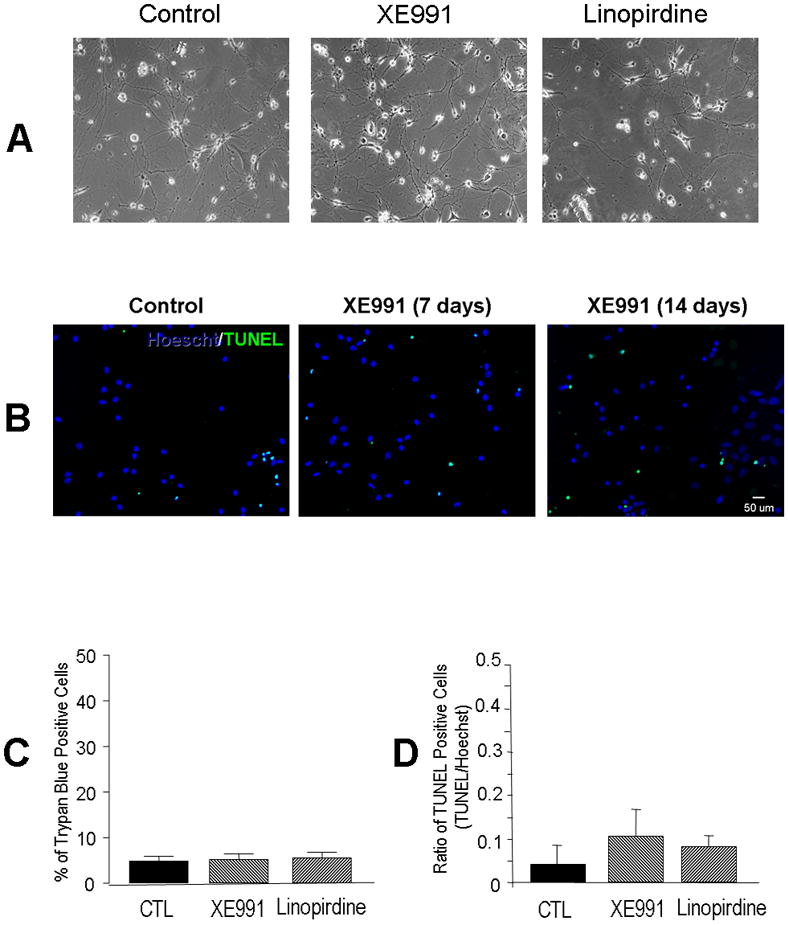
As a control experiment, we evaluated cell viability of hippocampal neurons under our experimental culture conditions. A. Phase contract images of hippocampal cultures 7 days after wash control, XE991 (10 μM) and linopirdine (50 μM). B. Fluorescent images of Hoechst 33342 and TUNEL staining of hippocampal neurons 7–14 days in XE991 (10 μM) treatment. C and D. Quantification of data from the experiments in B. Chronic exposure to XE991 and linopirdine did not cause cell death in hippocampal cultures. N ≥ 3 assays.
Figure 4. Inhibition of KCNQ2/3 channel reduced synaptophysin expression in ES cell-derived neurons.

Expressions of synaptophysin and neurofilament were determined by immunocytochemistry and Western blotting 5 days after XE991 or linopirdine treatment. A and B. Immunostaining images show that chronic exposure to XE991 (10 μM) or linopirdine (30 μM) for 5 days reduced the synaptophysin expression in mouse ES cell-derived neuronal cells, while the exposure showed no effect on the neurofilament staining. Scale bar = 50 μm. C. Western blotting showed that synaptophysin and syntaxin were expressed in differentiating cells 4 – 5 days after the neural induction of ES cells. D and E. Inhibition of KCNQ channel activity reduced both synaptophysin and syntaxin expression revealed by Western blotting. Figure E summarized the results from experiment D. Mean ± S.E. M. N ≥ 3 independent experiments. * P < 0.05 vs. vehicle controls.
Figure 5. Blocking KCNQ2/3 channels reduced synaptophysin expression in hippocampal neurons.

The effects of KCNQ channel inhibitors on synaptophysin expression in hippocampal neurons. A. Hippocampal cultures were exposed for either 7 or 14 days with KCNQ channel inhibitors, XE991 (10 μM) or linopirdine (30 μM). The inhibitor was added into the culture media at 1 DIV. Immunostaining showed that both inhibitors decreased synaptophysin expression. B and C. Quantification of synaptophysin fluorescent intensities in control cells and cells treated with XE991 or linopirdine for 7 (B) and 14 (C) days. Mean ± S.E. M. N ≥ 3 independent experiments. * P < 0.05 vs. vehicle controls.
Since the MEK/ERK pathway and Src family kinases have been reported to be involved in the regulation of neuronal differentiation (Johnson, 2008, Tinel, et al., 1998), we tested these signaling pathways which might be affected by KCNQ2/3 channel activities. In ES cell-derived neural progenitor cells, ERK1/2 phsophorylation was significantly increased in cells subjected to 5-day exposure to XE991 or linopirdine (Fig. 6A). Meanwhile, Src phosphorylation was not significantly changed (Fig. 6B). More specifically, the expression of Src family members Fyn and Lyn kinasis was not affected (data not shown).
Figure 6. Blocking KCNQ2/3 selectively increased ERK1/2 phsophorylation.

Western blot analysis was performed to inspect the total and phosphorylated protein levels of several intracellular signaling molecules in ES cell-derived neural progenitor cells. Both XE991 and linopirdine (5 day exposure) enhanced the levels of phsophorylated ERK1/2 (pERK). The bar graph shows that, due to the increased pERK1/2 and/or decreased total ERK (tERK), the ratio of pERK versus tERK was significantly increased. Meanwhile, Src phosphorylation was not significantly changed. N = 5; *. P < 0.05 vs. controls.
Activation of KCNQ2/3 channels increased synaptophysin expression
Although XE991 and linopirdine are specific KCNQ channel inhibitors, they are not selective blockers for specific KCNQ subtypes. To provide additional evidence for the KCNQ subtype involved in our experiments, we tested the KCNQ2 and KCNQ2/3 channel selective opener N-Ethylmaleimide (NEM) (Roche, et al., 2002). In contrast to the reduced synaptogenesis via inhibition of the KCNQ channel activity, immunostaining showed that activation of KCNQ2/3 channels by NEM (2 μM) in ES cell-derived neuronal cultures for 5 days significantly increased the expression of synaptophysin. The enhanced synaptophysin expression was attenuated by co-applied KCNQ channel inhibitor XE991 (10 μM) (Fig. 7).
Figure 7. KCNQ2/3 channel opener enhanced synaptophysin expression.

Mouse stem cell-derived neurons were treated with the KCNQ2/3 channel opener NEM at different concentrations for 5 days. A. Immunostaining showed NEM (2 μM) dramatically increased synaptophysin expression, the effect was completely prevented by co-applied KCNQ channel blocker, XE991 (10 μM). B. The bar graph summarized quantified the synaptophysin fluorescent intensity in experiments in A. NEM showed a dose-response effect on synaptophysin expression and its blockade by XE991. Mean ± S.E. M. N ≥ 3 independent experiments. * P < 0.05 vs. vehicle controls. # P < 0.05 vs. NEM application.
Selective effect of KCNQ2/3 channel activity on GABAergic synaptic transmission
To investigate whether KCNQ2/3 channel activity was specifically related to formation of a specific type of synapse, XE991 (10 μM) or linopirdine (30 μM) was applied into culture media of ES cell derived-neurons for 5 days and hippocampal neurons for 7 days. XE991 and linopirdine did not show significant effect on expression of vesicular glutamate transporter (v-Glut) but they each attenuated expression of vesicular GABA transporter (v-GAT) in ES cell-derived neurons (Fig. 8A) and hippocampal neurons (Fig. 8B). This suggested that deficient KCNQ channel basal activity selectively interfered with GABAergic synapse composition without affecting the formation of glutamatergic synapses.
Figure 8. Blockage of KCNQ2/3 channel affected synapse formation.

Bar graphs summarized fluorescent intensity of v-GluT and v-GAT immunostaining in ES cell-derived neuronal cells and hippocampal neurons. The expressions of v-GluT and v-GAT were used as markers for excitatory and inhibitory synapses, respectively. A. Quantification of intensities of v-GluT and v-GAT staining in mouse stem cell-derived neurons with and without XE-991 or linopirdine for 5 days after neuronal induction. B. Quantified data from experiments with hippocampal neurons of 14-DIV. XE991 and linopirdine significantly reduced v-GAT expression without affecting v-GluT expression. Mean ± S.E. M. N ≥ 3 independent experiments. * P < 0.05 vs. vehicle controls.
To further identify and more specifically confirm the above observations, we inspected protein levels of the AMPA receptor subunit GluR1 and the 67-kDa isoform (GAD67) of glutamic acid decarboxylase (GAD), markers for glutamatergic and GABAergic neurons, respectively. There was no change in the expression of either GluR1 or GAD67 after 5-day treatment in stem cell-derived neurons (Fig. 9A) or 7-day treatment in hippocampal neurons (Fig. 9B and 9C). However, hippocampal neurons treated with KCNQ channel inhibitors for 14 days showed significant decreases in GAD67 expression, consistent with an inhibitory effect on GABAergic synapse formation (Fig. 9C).
Figure 9. Effects of blocking KCNQ2/3 channels on neuronal differentiation/maturation of ES cell-derived neurons and hippocampal neurons.

Bar graphs summarized Western blotting analyses of GluR1 and GAD67 expression in cells treated with KCNQ2/3 channel inhibitors. A. Mouse ES cell-derived neurons were treated with a KCNQ channel blocker for 5 days, the treatment caused no alteration in the expression of GluR1 and GAD67 in these cells. B and C. Hippocampal neurons were subjected to 7 or 14 days exposure to XE991 (10 μM) or linopirdine (30 μM), respectively. Although the 7 day treatment did not show significant effect, 14 day treatment with XE991 or linopirdine selectively reduced GAD67 expression compare with control cells. Mean ± S.E. M. N ≥ 3 independent experiments. * P < 0.05 vs. vehicle controls.
The role of KCNQ2/3 channels in tonic regulation of neurotransmitter release
Hippocampal neurons were treated from 1 DIV with XE991 (10 μM) or linopirdine (30 μM) for 12 – 14 days, and miniature postsynaptic currents (mPSCs) were recorded using whole-cell recording to examine whether chronically silencing KCNQ2/3 channels might alter spontaneous neurotransmitter release. The KCNQ2/3 channel inhibition did not significantly affect the amplitude of mPSCs. Linoperdine, on the other hand, reduced the frequency of mPSC, although the effect of XE991 on mPSC frequency was not significant (Fig. 10A and 10B). To confirm that the activity of KCNQ2/3 channels played a role in spontaneous neurotransmitter release, we applied the selective KCNQ2/3 channel opener NEM (2 μM) to hippocampal neuronal cultures for 5 days. This maneuver, as expected, enhanced the frequency of mPSCs without affecting the amplitude of miniature currents (Fig. 10C). Thus, the frequency changes indicated that normal KCNQ2/3 activities could play a tonic regulatory role on the pre-synaptic transmission events.
Figure 10. Effects of Inhibiting KCNQ2/3 channels on spontaneous transmitter release in hippocampal neurons.

The effects of manipulating the KCNQ channel activity on mPSCs were studied using whole-cell recordings in hippocampal neurons. A. Hippocampal neurons were subjected to 12–14 day exposure to XE991 (10 μM) or linopirdine (30 μM). Whole cell recording of mPSCs in these neurons were performed between 12 – 16 DIV. B. Bar graph of summarized mPSC recordings in A. Blocking KCNQ2/3 channels did not alter the average amplitude of mPSCs. On the other hand, linopirdine reduced mPSC frequency in these cells. C. The KCNQ2/3 channel opener NEM (2 μM) was applied for 5 days to increase the channel activity. This maneuver showed no effect on the amplitude of mPSCs. However, opposing to channel blockers, the channel opener significantly increased the frequency of mPSCs. N = 10. *. P < 0.05 compared to controls.
The role of KCNQ2/3 channels in regulating neurotransmitter recycling
To further verify whether chronic inhibition of the KCNQ2/3 channel activity had a pre-synaptic effect and to better understand the mechanism of altered transmitter release, we examined the neurotransmitter recycle activity, endocytosis and exocytosis, using fluorescent styryl-dye FM4-64. Mouse ES cell-derived cells were exposed to XE991 (10 μM) or linopirdine (30 μM) for 5 or 12 days after neuronal induction. The schematic diagram in Figure 11 shows the protocol of FM4-64 staining for examining endocytosis and exocytosis (Fig. 11A). The intensity of FM4-64 representing endocytosis decreased in cells exposed to a KCNQ channel blocker (Fig. 11B). When the pre-synaptic membrane was depolarized with a high K+ extracellular solution in the absence of FM4-64, exocytosis can be studied by measuring the residue FM4-64 fluorescence in the cells after returning to the normal solution (Nishiki and Augustine, 2004). The intensities of FM4-64 representing exocytosis were higher in XE991 and linopirdine groups compared to the control group (Fig. 11C). Similarly, decreased endocytosis and facilitated exocytosis was observed in hippocampal neurons subjected to 14-day inhibition of KCNQ2/3 channels. (Fig. 12).
Figure 11. Different effects of KCNQ2/3 channel activities on endocytosis and exocytosis in mouse stem cell-derived neurons.

Fluorescent styryl-dye FM4-64 was used to monitor endocytosis and exocytosis in ES cell-derived neurons after 5 or 12 days differentiation. A. Schematic diagram showed the protocol of FM4-64 staining. B to D. Fluorescent microscopy images and quantification of the intensities of FM4-64 after treatment in XE991 or linopirdine for 5 (C) or 12 (D) days. Both XE991 and linopirdine reduced endocytosis at the two time points, while they equivalently upregulated exocytosis. Scale bar=50 μm. Mean ± S.E. M. N ≥ 3 independent experiments. * P < 0.05 vs. vehicle controls.
Figure 12. The effects of KCNQ2/3 channel activity on endocytosis and exocytosis in hippocampal neurons.

Fluorescent styryl-dye FM4-64 was used to monitor endocytosis and exocytosis in primary cultured hippocampal neurons underwent 7 or 14 day treatment with the KCNQ channel blocker XE991 or linopirdine. The blocker was added into the media at 1 DIV. A. After treated with XE991 or linopirdine for 7 days, there was no significant difference in the intensities of FM4-64 in each group. B. After 14-day treatment, the intensity of FM4-64 for endocytosis was much less in XE991- or linopirdine-treated groups compared to control groups. On the other hand, the FM4-64 intensity for exocytosis was higher in inhibitor-treated groups. Mean ± S.E. M. N ≥ 3 independent experiments. * P < 0.05 vs. vehicle controls.
Discussion
The present investigation reveals a previously unreported tonic regulation of KCNQ2/3 channel activities on neuronal differentiation and synaptogenesis in mouse ES cell-derived neural progenitors and primary cultured embryonic hippocampal neurons. We show that selective block of KCNQ2/3 channels significantly decreases expression of synaptic and vesicular proteins in neuronally differentiating/maturating cells. On the other hand, opening of KCNQ2/3 channels augmented expression of these proteins. The effect appears to be presynaptic and specific to GABAergic neurons but not glutamatergic neurons. The ERK1/2 signaling pathway may participate in the regulation via protein phosphorylation. As a control, we show that chronic blockage of KCNQ channel activity does not affect cell viability.
XE991 and linopiridine are widely used as specific pharmacological tools to block KCNQ channels (Lamas, et al., 1997, Wang, et al., 1998). XE991 is a more potent derivative of linopiridine (Zaczek, et al., 1998). Linopirdine and likely XE991, block the KCNQ2/3 channel by a direct interaction with the channel protein rather than through a second messenger-mediated pathway (Costa and Brown, 1997, Lamas, et al., 1997). The IC50 for XE991 and linopirdine on KCNQ2/3 channel is 0.98 μM and 3.4–7 μM, respectively (Costa and Brown, 1997, Lamas, et al., 1997, Wang, et al., 1998). XE991 and linopirdine may also inhibit eag-related K+ channel ERG1 (Elmedyb, et al., 2007, Wang, et al., 1998). The IC50 values of XE991 and linopirdine on ERG1 are 107 μM and around 59 μM, respectively, which are 10 folds higher than the concentration of XE991 and 2 folds higher than the concentration of linopirdine used in the current investigation. At 10 μM concentrations tested in our experiments, XE991 was unlikely to show a significant effect on ERG1 channels. In addition, the opposite effects of the selective KCNQ2/3 channel opener NEM on neuronal differentiation and synaptic events suggested that specific KCNQ channel/s were involved in the regulation. When XE991 and NEM were co-applied their own effect was cancelled, further supporting the idea that KCNQ2/3 channels were involved in the observed changes. It is worth mentioning that a molecular approach of knocking down KCNQ2/3 channels or an investigation on KCNQ deficient mice may yield valuable evidence on the novel role of these channels on neuronal differentiation and brain development.
Recent work has demonstrated that many ion channels could directly influence biochemical events through their non-ion conducting functions, including regulating the mitogen-activated protein kinase (MAPK) signaling (Kv10.1 channel), tyrosine phosphorylation (Kv1.3 channel), and cell proliferation (Kv11.1, Kv1.3 channels) (Kaczmarek, 2006). A novel role of Kv2.1 as a facilitator of vesicle secretion through a pore independent function has been described (Singer-Lahat, et al., 2008, Singer-Lahat, et al., 2007). Possible models were proposed to account for the Kv2.1-mediated enhancement of vesicle exocytosis in response to stimulation: 1). Kv2.1 protein associates with syntaxin to stabilize the t-SNARE (soluble N-ethylamaleimide-sensitive factor attachment protein receptor) protein complex; 2). Relocation of vesicles to the presynaptic membrane with a high-density Kv2.1 cluster that favors fusion. Both models are favorable for formation of ternary SNARE complexes and lead to an increased neurotransmitter release. Another surprising discovery is the observation from our group that Kv2.1 channel can form a protein complex with the focal adhesion kinase (FAK) thereafter promoting FAK phosphorylation/activation and cell motility (Wei, et al., 2008). In the present investigation, we observed that KCNQ2/3 channels co-localize with the synaptic vesicle protein synaptophysin, suggesting that KCNQ2/3 channels may regulate synapse formation by association with SNARE protein through the non-ion conducting function similar to the Kv2.1 channel. It is noted that KCNQ2/3 channels were studied in cultured cells in this investigation, whether similar regulations take place in the brain in vivo is interesting but currently unknown. A further investigation using pharmacological and molecular biological methods will be necessary to validate and delineate the KCNQ2/3 channel-formed complex and its regulatory roles during CNS development in vivo.
Blocking KCNQ channels reduced the GABA vesicular transporter vGAT expression while GABA synthetic enzyme GAD67 was initially unaffected. Prolonged exposure to a KCNQ channel blocker (14 days) did cause significant and selective reduction in the GAD67 expression. Meanwhile, none of the treatments caused alterations in glutamatergic synapse formation; at least the major AMPA receptor subunit Glut1 and the vascular glutamate transporter v-Glut were not affected. These results indicate a selective consequence of interfering with GABAergic synapse construction/function after a prolonged suppression of KCNQ channels. Synaptogenesis or the formation of synapses is particularly important during a “critical period” when neuronal pruning occurs due to competition for neural growth factors by neurons and synapses. Processes that are not used, or inhibited during this critical period will fail to develop normally later on during development. Whether blocking KCNQ channels reduces GABAergic synapse formation via this mechanism is an attractive idea and remains to be investigated.
KCNQ channels have five subfamily members, KCNQ 1, 2, 3, 4 and 5. Four KCNQ channel subunits except KCNQ1 are expressed in the central nervous system (Maljevic, et al., 2008). Our current investigation focused on KCNQ2 and KCNQ3 that have been shown to be involved in some pathological conditions. For example, mutations in KCNQ2 and KCNQ3 genes are associated with an inherited epilepsy syndrome (Peters, et al., 2005). In the hippocampus, the KCNQ5 subunit is a known contributor to M-currents (Shah, et al., 2002). Current data from this investigation cannot exclude the possibility that KCNQ5 subunit in hippocampal neurons may participate in the regulation observed in this investigation.
Epilepsy is typically thought of as a condition of excessive excitatory transmission. However, an efficient inhibitory system might also be sufficient to prevent excitatory stimuli from producing an excessive level of pervasive activity. Therefore, epilepsy may not only be a product of excessively high excitatory transmission, but also come as a result of abnormally low inhibitory activity (Jobe, 2002). The present investigation provides the potential mechanism underlying a developmental abnormality of reduced inhibitory GAGAergic synaptogenesis or a selective deterioration of inhibitory synaptic regulation in the developing or adult brain.
Some voltage-gated K+ channels are present at presynaptic terminals (Barnes-Davies, et al., 2004, Robitaille, et al., 1993), raising an intriguing question regarding the role of these presynaptic K+ channels in regulation of synaptic transmission. KCNQ channels have been shown to localize at the axon initial segment (AIS) to determine the threshold for firing action potential, thus regulating action potential generation (Hernandez, et al., 2008) and neurotransmitter release (Martire, et al., 2004, Peretz, et al., 2007). Presynaptic KCNQ2/3 channels have been reported to affect neurotransmitter release of loaded [3H] noradrenaline, [3H] GABA and D-[3H] aspartate from hippocampal synaptosomes (Martire, et al., 2004). A novel KCNQ2/3 channel opener NH6 blunted spontaneous spiking behavior and reduced the frequency of spontaneous EPSCs in cultured neurons, whereas the KCNQ channel blocker linopirdine had an opposite effect. The authors explained that M-channel activation caused presynaptic inhibitory effects due to hyperpolarization of the presynaptic terminals, and thereby indirectly depressing vesicular transmitter release (Peretz, et al., 2007). These results from acute experiments are opposite to the data in our study of chronic inhibition of KCNQ2/3 channels. The major differences between these two studies are the timing and duration of the channel inhibition. In the earlier study, KCNQ channel inhibitors were applied transiently during patch clamp recordings that undoubtedly hyperpolarized the membrane potential, which is an established instant mechanism of diminishing neurotransmitter release. In our experiments, we treated cells with a KCNQ2/3 channel inhibitor for several days to study the chronic effect of silencing these channels on neuronal differentiation. The results shown here could not be explained simply by a change in membrane excitability, but may be due to more profound actions of these channels that associate with intracellular regulatory signaling pathways.
Recent studies showed that different differentiation protocols and inducers selectively activated distinct signaling pathways that initiate cell lineage-specific genetic programs to guide the stem cells differentiating into various differentiation populations. Among the signaling pathways, the MAPK pathway can regulate the embryonic stem cell commitment to mature differentiated cells (Binetruy, et al., 2007). As the prototypic of MAPKs, ERK pathway inhibition prevents the self-renewal of ES cells (Binetruy, et al., 2007). Upon differentiation, ERK signaling plays a more dominant role (Chazaud, et al., 2006)(Yang, et al., 2006). However, activation of the ERK pathway by the oncogene Ras interferes with endoderm differentiation from ES cells (Yoshida-Koide, et al., 2004). We observed that chronic inhibition of KCNQ channels suppressed neuronal differentiation while ERK1/2 expression was upregulated. Demonstration of a causal relationship between the upregulation of ERK1/2 and the decreased differentiation requires more systemic investigations. It is clear, however, that a prolonged silencing of KCNQ channels can influence specific intracellular signaling pathways such as ERK1/2 that are important for neuronal differentiation.
Neurotransmitter release from nerve terminals mediates synaptic communication in nervous systems. In exocytosis, termed as ‘full-collapse fusion’, vesicles empty their neurotransmitter fully into synaptic cleft by fusion with the presynaptic membrane. However, repetitive synaptic activity will cause depression if the vesicle pool is depleted at a rate faster than the pool-replenishment process, so endocytosis of fused vesicle membrane is a key step to refill the vesicle pool and facilitate continuous release. To test whether normal KCNQ channel activity is important for functional synaptic function, the present study used FM4-64 to test exocytosis and endocytosis. FM4-64 is a red fluorescent amphiphilic styryl dye that enters into the membranes of synaptic vesicles when endocytosis is stimulated. The dye that is concentrated within the vesicles is expelled when exocytosis is induced by another round of stimulation. The rate of vesicle release is measured from the resulting decrease in fluorescence. Since FM4-64 dye could be applied externally and transiently, it is a useful tool to determine the rates of exocytosis and endocytosis in neuronal cultures. Our data illustrated that the blockage of KCNQ2/3 channels decreased endocytosis and augmented exocytosis. It can be expected that a persistent change like this will eventually lead to failure of transmitter release, and, as a presynaptic event, it could selectively reduced the frequency of mPSCs as shown in the present investigation.
Acknowledgments
This work was supported by NIH grants NS 045810 (LW), NS 058710 (LW), NS057255 (SPY) and the American Heart Association Established Investigator Award (LW). This work was also supported by the NIH grant C06 RR015455 from the Extramural Research Facilities Program of the National Center for Research Resources at MUSC and the NIH grant NS055077 to the ENNCF (Emory Neurology-NINDS Core Facility).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Akins MR, Biederer T. Cell-cell interactions in synaptogenesis. Curr Opin Neurobiol. 2006;16:83–89. doi: 10.1016/j.conb.2006.01.009. [DOI] [PubMed] [Google Scholar]
- 2.Artinian L, Tornieri K, Zhong L, Baro D, Rehder V. Nitric oxide acts as a volume transmitter to modulate electrical properties of spontaneously firing neurons via apamin-sensitive potassium channels. J Neurosci. 2010;30:1699–1711. doi: 10.1523/JNEUROSCI.4511-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bain G, Kitchens D, Yao M, Huettner JE, Gottlieb DI. Embryonic stem cells express neuronal properties in vitro. Dev Biol. 1995;168:342–357. doi: 10.1006/dbio.1995.1085. [DOI] [PubMed] [Google Scholar]
- 4.Bain G, Ray WJ, Yao M, Gottlieb DI. Retinoic acid promotes neural and represses mesodermal gene expression in mouse embryonic stem cells in culture. Biochem Biophys Res Commun. 1996;223:691–694. doi: 10.1006/bbrc.1996.0957. [DOI] [PubMed] [Google Scholar]
- 5.Barnes-Davies M, Barker MC, Osmani F, Forsythe ID. Kv1 currents mediate a gradient of principal neuron excitability across the tonotopic axis in the rat lateral superior olive. Eur J Neurosci. 2004;19:325–333. doi: 10.1111/j.0953-816x.2003.03133.x. [DOI] [PubMed] [Google Scholar]
- 6.Binetruy B, Heasley L, Bost F, Caron L, Aouadi M. Concise review: regulation of embryonic stem cell lineage commitment by mitogen-activated protein kinases. Stem Cells. 2007;25:1090–1095. doi: 10.1634/stemcells.2006-0612. [DOI] [PubMed] [Google Scholar]
- 7.Brown BS, Yu SP. Modulation and genetic identification of the M channel. Prog Biophys Mol Biol. 2000;73:135–166. doi: 10.1016/s0079-6107(00)00004-3. [DOI] [PubMed] [Google Scholar]
- 8.Brown DA, Adams PR. Muscarinic suppression of a novel voltage-sensitive K+ current in a vertebrate neurone. Nature. 1980;283:673–676. doi: 10.1038/283673a0. [DOI] [PubMed] [Google Scholar]
- 9.Burns TC, Verfaillie CM, Low WC. Stem cells for ischemic brain injury: a critical review. J Comp Neurol. 2009;515:125–144. doi: 10.1002/cne.22038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chazaud C, Yamanaka Y, Pawson T, Rossant J. Early lineage segregation between epiblast and primitive endoderm in mouse blastocysts through the Grb2-MAPK pathway. Dev Cell. 2006;10:615–624. doi: 10.1016/j.devcel.2006.02.020. [DOI] [PubMed] [Google Scholar]
- 11.Constanti A, Brown DA. M-Currents in voltage-clamped mammalian sympathetic neurones. Neurosci Lett. 1981;24:289–294. doi: 10.1016/0304-3940(81)90173-7. [DOI] [PubMed] [Google Scholar]
- 12.Cooper EC, Jan LY. M-channels: neurological diseases, neuromodulation, and drug development. Arch Neurol. 2003;60:496–500. doi: 10.1001/archneur.60.4.496. [DOI] [PubMed] [Google Scholar]
- 13.Costa AM, Brown BS. Inhibition of M-current in cultured rat superior cervical ganglia by linopirdine: mechanism of action studies. Neuropharmacology. 1997;36:1747–1753. doi: 10.1016/s0028-3908(97)00155-x. [DOI] [PubMed] [Google Scholar]
- 14.Delmas P, Brown DA. Pathways modulating neural KCNQ/M (Kv7) potassium channels. Nat Rev Neurosci. 2005;6:850–862. doi: 10.1038/nrn1785. [DOI] [PubMed] [Google Scholar]
- 15.Elmedyb P, Calloe K, Schmitt N, Hansen RS, Grunnet M, Olesen SP. Modulation of ERG channels by XE991. Basic Clin Pharmacol Toxicol. 2007;100:316–322. doi: 10.1111/j.1742-7843.2007.00048.x. [DOI] [PubMed] [Google Scholar]
- 16.Fischer S, Cassivi SD, Xavier AM, Cardella JA, Cutz E, Edwards V, Liu M, Keshavjee S. Cell death in human lung transplantation: apoptosis induction in human lungs during ischemia and after transplantation. Ann Surg. 2000;231:424–431. doi: 10.1097/00000658-200003000-00016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hernandez CC, Zaika O, Tolstykh GP, Shapiro MS. Regulation of neural KCNQ channels: signalling pathways, structural motifs and functional implications. J Physiol. 2008;586:1811–1821. doi: 10.1113/jphysiol.2007.148304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jentsch TJ. Neuronal KCNQ potassium channels: physiology and role in disease. Nat Rev Neurosci. 2000;1:21–30. doi: 10.1038/35036198. [DOI] [PubMed] [Google Scholar]
- 19.Jespersen T, Grunnet M, Olesen SP. The KCNQ1 potassium channel: from gene to physiological function. Physiology (Bethesda) 2005;20:408–416. doi: 10.1152/physiol.00031.2005. [DOI] [PubMed] [Google Scholar]
- 20.Jobe PC. Are there specific anatomical and/or transmitter systems (cortical or subcortical) that should be targeted? Int Rev Neurobiol. 2002;49:221–252. doi: 10.1016/s0074-7742(02)49015-0. [DOI] [PubMed] [Google Scholar]
- 21.Johnson DE. Src family kinases and the MEK/ERK pathway in the regulation of myeloid differentiation and myeloid leukemogenesis. Adv Enzyme Regul. 2008;48:98–112. doi: 10.1016/j.advenzreg.2007.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kaczmarek LK. Non-conducting functions of voltage-gated ion channels. Nat Rev Neurosci. 2006;7:761–771. doi: 10.1038/nrn1988. [DOI] [PubMed] [Google Scholar]
- 23.Kim SU, de Vellis J. Stem cell-based cell therapy in neurological diseases: a review. J Neurosci Res. 2009;87:2183–2200. doi: 10.1002/jnr.22054. [DOI] [PubMed] [Google Scholar]
- 24.Lai HC, Jan LY. The distribution and targeting of neuronal voltage-gated ion channels. Nat Rev Neurosci. 2006;7:548–562. doi: 10.1038/nrn1938. [DOI] [PubMed] [Google Scholar]
- 25.Lamas JA, Selyanko AA, Brown DA. Effects of a cognition-enhancer, linopirdine (DuP 996), on M-type potassium currents (IK(M)) and some other voltage- and ligand-gated membrane currents in rat sympathetic neurons. Eur J Neurosci. 1997;9:605–616. doi: 10.1111/j.1460-9568.1997.tb01637.x. [DOI] [PubMed] [Google Scholar]
- 26.Maljevic S, Wuttke TV, Lerche H. Nervous system KV7 disorders: breakdown of a subthreshold brake. J Physiol. 2008;586:1791–1801. doi: 10.1113/jphysiol.2008.150656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Martire M, Castaldo P, D’Amico M, Preziosi P, Annunziato L, Taglialatela M. M channels containing KCNQ2 subunits modulate norepinephrine, aspartate, and GABA release from hippocampal nerve terminals. J Neurosci. 2004;24:592–597. doi: 10.1523/JNEUROSCI.3143-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McDonald JW, Liu XZ, Qu Y, Liu S, Mickey SK, Turetsky D, Gottlieb DI, Choi DW. Transplanted embryonic stem cells survive, differentiate and promote recovery in injured rat spinal cord. Nat Med. 1999;5:1410–1412. doi: 10.1038/70986. [DOI] [PubMed] [Google Scholar]
- 29.Meir A, Ginsburg S, Butkevich A, Kachalsky SG, Kaiserman I, Ahdut R, Demirgoren S, Rahamimoff R. Ion channels in presynaptic nerve terminals and control of transmitter release. Physiol Rev. 1999;79:1019–1088. doi: 10.1152/physrev.1999.79.3.1019. [DOI] [PubMed] [Google Scholar]
- 30.Nishiki T, Augustine GJ. Synaptotagmin I synchronizes transmitter release in mouse hippocampal neurons. J Neurosci. 2004;24:6127–6132. doi: 10.1523/JNEUROSCI.1563-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Peretz A, Sheinin A, Yue C, Degani-Katzav N, Gibor G, Nachman R, Gopin A, Tam E, Shabat D, Yaari Y, Attali B. Pre- and postsynaptic activation of M-channels by a novel opener dampens neuronal firing and transmitter release. J Neurophysiol. 2007;97:283–295. doi: 10.1152/jn.00634.2006. [DOI] [PubMed] [Google Scholar]
- 32.Peters HC, Hu H, Pongs O, Storm JF, Isbrandt D. Conditional transgenic suppression of M channels in mouse brain reveals functions in neuronal excitability, resonance and behavior. Nat Neurosci. 2005;8:51–60. doi: 10.1038/nn1375. [DOI] [PubMed] [Google Scholar]
- 33.Rennie KJ, Weng T, Correia MJ. Effects of KCNQ channel blockers on K+ currents in vestibular hair cells. Am J Physiol Cell Physiol. 2001;280:C473–480. doi: 10.1152/ajpcell.2001.280.3.C473. [DOI] [PubMed] [Google Scholar]
- 34.Richardson RM, Singh A, Sun D, Fillmore HL, Dietrich DW, 3rd, Bullock MR. Stem cell biology in traumatic brain injury: effects of injury and strategies for repair. J Neurosurg. 2010;112:1125–1138. doi: 10.3171/2009.4.JNS081087. [DOI] [PubMed] [Google Scholar]
- 35.Robitaille R, Adler EM, Charlton MP. Calcium channels and calcium-gated potassium channels at the frog neuromuscular junction. J Physiol Paris. 1993;87:15–24. doi: 10.1016/0928-4257(93)90020-t. [DOI] [PubMed] [Google Scholar]
- 36.Roche JP, Westenbroek R, Sorom AJ, Hille B, Mackie K, Shapiro MS. Antibodies and a cysteine-modifying reagent show correspondence of M current in neurons to KCNQ2 and KCNQ3 K+ channels. Br J Pharmacol. 2002;137:1173–1186. doi: 10.1038/sj.bjp.0704989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shah MM, Mistry M, Marsh SJ, Brown DA, Delmas P. Molecular correlates of the M-current in cultured rat hippocampal neurons. J Physiol. 2002;544:29–37. doi: 10.1113/jphysiol.2002.028571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shapiro MS, Roche JP, Kaftan EJ, Cruzblanca H, Mackie K, Hille B. Reconstitution of muscarinic modulation of the KCNQ2/KCNQ3 K+ channels that underlie the neuronal M current. J Neurosci. 2000;20:1710–1721. doi: 10.1523/JNEUROSCI.20-05-01710.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shen H, Korutla L, Champtiaux N, Toda S, LaLumiere R, Vallone J, Klugmann M, Blendy JA, Mackler SA, Kalivas PW. NAC1 regulates the recruitment of the proteasome complex into dendritic spines. J Neurosci. 2007;27:8903–8913. doi: 10.1523/JNEUROSCI.1571-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Singer-Lahat D, Chikvashvili D, Lotan I. Direct interaction of endogenous Kv channels with syntaxin enhances exocytosis by neuroendocrine cells. PLoS One. 2008;3:e1381. doi: 10.1371/journal.pone.0001381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Singer-Lahat D, Sheinin A, Chikvashvili D, Tsuk S, Greitzer D, Friedrich R, Feinshreiber L, Ashery U, Benveniste M, Levitan ES, Lotan I. K+ channel facilitation of exocytosis by dynamic interaction with syntaxin. J Neurosci. 2007;27:1651–1658. doi: 10.1523/JNEUROSCI.4006-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Srivastava AS, Malhotra R, Sharp J, Berggren T. Potentials of ES cell therapy in neurodegenerative diseases. Curr Pharm Des. 2008;14:3873–3879. doi: 10.2174/138161208786898617. [DOI] [PubMed] [Google Scholar]
- 43.Tinel N, Lauritzen I, Chouabe C, Lazdunski M, Borsotto M. The KCNQ2 potassium channel: splice variants, functional and developmental expression. Brain localization and comparison with KCNQ3. FEBS Lett. 1998;438:171–176. doi: 10.1016/s0014-5793(98)01296-4. [DOI] [PubMed] [Google Scholar]
- 44.Ueda A, Wu CF. Distinct frequency-dependent regulation of nerve terminal excitability and synaptic transmission by IA and IK potassium channels revealed by Drosophila Shaker and Shab mutations. J Neurosci. 2006;26:6238–6248. doi: 10.1523/JNEUROSCI.0862-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang HS, Pan Z, Shi W, Brown BS, Wymore RS, Cohen IS, Dixon JE, McKinnon D. KCNQ2 and KCNQ3 potassium channel subunits: molecular correlates of the M-channel. Science. 1998;282:1890–1893. doi: 10.1126/science.282.5395.1890. [DOI] [PubMed] [Google Scholar]
- 46.Wei JF, Wei L, Zhou X, Lu ZY, Francis K, Hu XY, Liu Y, Xiong WC, Zhang X, Banik NL, Zheng SS, Yu SP. Formation of Kv2.1-FAK complex as a mechanism of FAK activation, cell polarization and enhanced motility. J Cell Physiol. 2008;217:544–557. doi: 10.1002/jcp.21530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wei L, Keogh CL, Whitaker VR, Theus MH, Yu SP. Angiogenesis and stem cell transplantation as potential treatments of cerebral ischemic stroke. Pathophysiology. 2005;12:47–62. doi: 10.1016/j.pathophys.2004.11.002. [DOI] [PubMed] [Google Scholar]
- 48.Yang W, Klaman LD, Chen B, Araki T, Harada H, Thomas SM, George EL, Neel BG. An Shp2/SFK/Ras/Erk signaling pathway controls trophoblast stem cell survival. Dev Cell. 2006;10:317–327. doi: 10.1016/j.devcel.2006.01.002. [DOI] [PubMed] [Google Scholar]
- 49.Yoshida-Koide U, Matsuda T, Saikawa K, Nakanuma Y, Yokota T, Asashima M, Koide H. Involvement of Ras in extraembryonic endoderm differentiation of embryonic stem cells. Biochem Biophys Res Commun. 2004;313:475–481. doi: 10.1016/j.bbrc.2003.11.138. [DOI] [PubMed] [Google Scholar]
- 50.Zaczek R, Chorvat RJ, Saye JA, Pierdomenico ME, Maciag CM, Logue AR, Fisher BN, Rominger DH, Earl RA. Two new potent neurotransmitter release enhancers, 10,10-bis(4-pyridinylmethyl)-9(10H)-anthracenone and 10,10-bis(2-fluoro-4-pyridinylmethyl)-9(10H)-anthracenone: comparison to linopirdine. J Pharmacol Exp Ther. 1998;285:724–730. [PubMed] [Google Scholar]