

Abstract
This study investigated the effect of a knockout of the caspase 2 gene on the sensitivity of murine nigral dopaminergic neurons to 1-methyl-4-1,2,3,6-tetrahydropyridine (MPTP)-induced toxicity. Female wild type (WT), heterozygous caspase 2 NL (HET) and homozygous caspase 2 null (NL) mice were treated with cumulative dosages of 0, 10, 15 or 20 mg/kg MPTP free base. Without MPTP treatment, one week later dopamine (DA) levels were not significantly different in HET or NL versus WT mice. Twenty mg/kg MPTP reduced striatal DA in WT and HET (p<0.01) but not NL mice. This same MPTP dosage regimen also induced a significantly greater decrease in tyrosine hydroxylase immunopositive (TH+) protein in striata of WT compared to NL mice (p<0.001).
Subsequently, WT and NL mice were treated daily with 20 mg/kg MPTP for 3 days and 25 mg/kg MPTP for 2 additional days, and TH+ neurons in the substantia nigra (SN) were estimated using unbiased stereology. When compared to untreated WT, the numbers of TH+ neurons were significantly lower in the SN of untreated NL mice (p<0.05). Treatment with the MPTP regimen significantly reduced TH+ neurons in WT mice but not NL mice.
In primary mesencephalic cultures both the cell bodies and the neuronal processes of TH immunopositive (TH+) neurons from NL embryos were significantly (p<0.001) more resistant to 10µM MPP+ compared to WT. Following MPP+ treatment, features of apoptotic cell death were also significantly (p<0.001) more prevalent in nuclei of TH+ neurons in cultures prepared from WT versus NL mouse pups.
These results suggest that caspase 2 may play a role in modulating the MPTP-induced damage to the nigrostriatal dopaminergic system.
Keywords: dopamine, MPTP, Parkinson’s disease, caspase 2, cell death, neurodegeneration, tyrosine hydroxylase
Introduction
Although it was the second to be discovered (Kumar et al.,1994), caspase 2 is among the least understood of the known caspases (Troy and Shelanski,2003). This enzyme is highly conserved (Sato et al.,1997), and its involvement in apoptosis is well supported. Early studies showed that over expression of this protease induced programmed cell death in some cultured cells (Kumar et al.,1994) (Wang et al.,1994) and treatment with antisense RNA for caspase 2 suppressed apoptosis in murine hematopoietic cells (Kumar,1995), PC12 cells (Haviv et al.,1998;Troy et al.,1997), and cultured rat sympathetic neurons (Troy et al.,1997). The transient elevation of caspase 2 in the brain and other tissues during embryogenesis suggested that this protease might also play a role in apoptosis during development (Kumar et al.,1994).
On the other hand, the absence of a clearly abnormal phenotype in caspase 2 null mice (Bergeron et al.,1998), suggested for a time that caspase 2 was a relatively unimportant component of one or more redundant or parallel apoptotic pathways (Troy and Shelanski,2003). However, although appearing normal, caspase 2 null mice do show a reduced life span associated with an age-related reduction in bone density, increased bone remodeling, reduced hair growth and an increased level of irreversibly oxidized protein (Zhang et al.,2007).
Further, more recent studies suggest that in some forms of genotoxic stress-induced apoptotic cell death caspase 2 may act as an initiator protease. In some cases the activation of caspase 2 involves an interaction with two additional proteins, i.e., RAIDD (Guo et al.,2002;Tu et al.,2006) and PIDD (Tinel and Tschopp,2004), which is prerequisite for the release of cytochrome c in the mitochondrial apoptotic pathway. At least in part, the initiator role of caspase 2 appears to involve a Bid-mediated activation of Bcl-2 family members, e.g., Bax and/or Bak (Gao et al.,2005).
Caspase 2 may also play a role in the cell death associated with certain neurotoxins and in some models of neurodegenerative disease. Exposure to 1-methyl-4-phenylpyridinium (MPP+), the active metabolite of 1-methyl-4-1,2,3,6-tetrahydropyridine (MPTP), increased caspase 2 in SH-SY5Y cells (Bando et al.,2005), and treatment with MPTP also elevated caspase 2 in mice in vivo (Yang et al.,1998). Further, the proteolytic cleavage of caspase 2 was observed in a mouse mesencephalic dopaminergic hybrid cell line treated with MPP+ (Chee et al.,2005). Cultured sympathetic neurons derived from caspase 2 null mice were also resistant to the toxicity of β-amyloid (Troy et al.,2000). In addition, caspase 2 may play a key role in apoptotic cell death of medium spiny neurons in a mouse model of Huntington's disease (Hermel et al.,2004). A recent investigation suggests a role for caspase 2 in ischemia-induced apoptotic cell death in rat hippocampal CA1 neurons (Niizuma et al.,2008). Further, our laboratory has recently shown that caspase 2 acts as an initiator caspase in rotenone-induced apoptotic cell death in cultured young adult cortical neurons (Tiwari et al.,2011).
Here we show that caspase 2 may also play an important role in the mediation of a neurodegenerative response, i.e., that tyrosine hydroxylase immunopositive (TH+) neurons in primary cultures of embryonic mesencephalic cells as well as the nigrostriatal dopaminergic pathway in mice null for caspase 2 expression have reduced sensitivity to MPTP-induced toxicity.
Materials and Methods
Materials
Glutamine, B27 supplement, fibroblast growth factor-2, penicillin/streptomycin and Neurobasal-A media were purchased from Life Technologies (Gibco BRL, Grand Island, NY, USA). Hibernate-A was from BrainBits (LLC). Laminin, OptiPrep 1.32, papain, Poly-D-Lysine, 1-methyl-4-phenylpyridinium (MPP+), 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine hydrochloride (MPTP) and trypan blue were purchased from Sigma (St. Louis, MO). O-phosphoric acid, perchloric acid and disodium ethylenediamine tetraethylacetate (EDTA) were purchased from Fisher Scientific. Acetonitrile was obtained from Burdick-Jackson. Alexa Fluor 488 IgG secondary antibody and Hoechst-33258 were purchased from Molecular Probes (Inc., Eugene, OR). Vectashield mounting medium was from Vector Laboratories, Inc (Burlingame, CA). The BCA protein assay kit and Chemiluminescence system were from Pierce (Rockford, IL). Beta-actin antibody, RIPA buffer, and HRP-labeled anti-mouse IgG secondary antibodies were from Santa Cruz Biotechnology. Antibodies against TH were from Pel-Freeze (Biologicals Division, Rogers, Arkansas) for immunocytochemistry or from Chemicon (Temecula, CA) for western blotting. If not mentioned, the chemicals were purchased from Sigma. All the cell culture plastic and glassware were from Nunc (Nalgene Nunc International, Rochester NY).
Animals
The caspase-2 knockout mice used in these studies were originally generated by Dr. Junying Yuan (Bergeron et al.,1998) and kindly provided to us by Dr. Carol Troy of Columbia University following Dr. Yuan's approval. Littermate wild type (WT), heterozygous caspase 2 null (HET) and caspase 2 null (NL) mice were derived from a breeding colony maintained at the Audie Murphy Veterans Administration Hospital or the University of Texas Health Science Center, San Antonio, Texas. The animals used were females which had been backcrossed for more than 10 generations into a C57BL6 background. The animals were genotyped by PCR amplification of DNA collected from each animal. After weaning, the mice used in these experiments were housed in groups of 4 in micro-isolator topped cages in an AAALAC-accredited facility maintaining strict adherence to the NIH Guidelines for the Care and Use of Laboratory Animals. The animals were 2–3 months old at the time of experimentation, were maintained specific pathogen free and were exposed to a 12:12 light-dark lighting regimen with the lights on from 0600 to 1800. All animals were fed a standard laboratory chow (Irradiated Harlan Teklad LM-485 Mouse/Rat Sterilizable Diet) ad libitum. All protocols involving the use of animals were reviewed and approved by the Institutional Animal Care and Use Committees located at the Audie Murphy Veterans Administration Hospital or the University of Texas Health Science Center.
MPTP treatment in vivo
MPTP hydrochloride was dissolved in physiological saline and was administered subcutaneously in a volume of 0.2 mL to randomly selected animals. The MPTP dosage was 5 mg/kg (free base) per administration, and mice were given 0, 2, 3 or 4 administrations of the drug for total doses of 0, 10, 15 or 20 mg/kg. The administrations of the drug were given at 2-hour intervals.
In one study WT and NL mice were treated daily with 20 mg/kg MPTP free base for 3 days and 25 mg/kg MPTP for 2 additional days. This dosage regimen was similar to that previously reported to promote apoptotic (Tatton and Kish,1997) rather than necrotic cell death (Jackson-Lewis et al.,1995) in nigral dopaminergic neurons. In that earlier study a daily dosage of 30 mg/kg MPTP was administered (Tatton and Kish,1997). However, in our hands the acute toxic effects of administering the 30 mg/kg dosage resulted in an unacceptably high death rate.
Unless stated otherwise, the animals were sacrificed one week following the last dosage of MPTP.
Tissue collection for biochemical studies
Tissues were collected between 0900 and 1200 one week following MPTP administration. Each striatum was dissected using clearly defined landmarks as previously described (Morgan et al.,1975), placed in a separate 1.5 mL microcentrifuge tube and immediately frozen on dry ice. The tissues were stored at −80°C until analysis. In one study the ventral midbrain (VMB) was dissected by making a coronal cut with a razor blade immediately in front of the superior colliculus and a second coronal cut just behind the inferior colliculus. This second cut was directed to pass ventrally through the intersection of the pons and midbrain. The VMB was then separated from the dorsal midbrain by a transverse cut made just dorsally to the visible cerebral peduncles.
Dopamine assay
Each striatum was homogenized in cold (4°C) 0.1 N perchloric acid (HCLO4) containing 1 mM sodium metabisulfite (Na2S2O5) and 100 nM dihydroxybenzylamine (DHBA). The latter compound was used as an internal standard. The homogenate was centrifuged for 4 minutes at maximum speed in a Heraeus Biofuge 13, and dopamine (DA) in the supernatant was extracted onto alumina (Proll et al.,1982) and quantified as previously described (Morgan and Nelson, 2001). All DA samples were analyzed in duplicate using a Waters Model 464 electrochemical (EC) detector. The potential on the EC electrode was set at +0.7 volts, and the mobile phase was a 75 mM phosphate buffer (pH 2.5) containing 25 micromolar (µM) EDTA, 2.3 mM octane sulfonate and 3 percent acetonitrile.
Protein concentration was determined in an aliquot of each homogenate (Bradford,1976), and the concentration of DA in each sample was expressed as picomoles per mg of protein.
Western blot analysis of tyrosine hydroxylase
The striata or the VMB of each animal was dissected, and homogenates were prepared in RIPA buffer. Protein concentration was determined using BCA reagent. Proteins (40µg) were resolved using a 4–12% gradient SDS-PAGE and transferred to PVDF membranes. The membranes were blocked in 5% skim milk for 1 hr, and probed overnight at 4°C with a primary antibody (Chemichon; 1:5000) to mouse TH in 1% skim milk or BSA. The membranes were washed and incubated for 1 hr with a HRP-conjugated secondary anti-mouse antibody (Molecular Probes; 1:5000). The blots were washed and developed by using an enhanced chemiluminescence system according to manufacturer's instructions and exposed to Hyperfilm-ECL (Amersham). The blots of striatal proteins were reprobed with beta tubulin and then with GADPH antibodies as loading controls. The blots of VMB proteins, which did not include tissue collected from MPTP-treated animals, were reprobed with an antibody to beta actin. Densitometry analysis band intensity was performed using Image J software.
Immunohistochemistry and stereology
Mice were anesthetized with a lethal dosage of a drug cocktail (5mg/mL Ketazet/ 1mg/mL xylazine) and exsanguinated by intracardiac perfusion with phosphate-buffered saline (pH 7.3; PBS) containing 5 percent sucrose followed by perfusion for 10 minutes with 4 percent paraformaldehyde in the same PBS solution. All perfusions were performed using a myNeuroLab Perfusion One pump. Each brain was subsequently stored overnight in the same fixative at 4°C and then cryoprotected in 30 percent sucrose for an additional 24 hours or until the tissue sank to the bottom of the sucrose solution.
A cryostat was used to cut serial 30 micron coronal sections through the midbrain of each animal. In our hands, sections thicker than 30 microns did not show complete penetration of the TH antibody. The floating sections were immunoblocked with 4 percent normal goat serum and then incubated with 1:1000 primary antibody to rabbit TH (Pel-Freeze) for 48 hours. The antigen-antibody complex was visualized using a Vectastain Peroxidase Kit and a diaminobenzidine substrate.
Stereology was performed at 960X using a Axioplan 2 Imaging microscope (Carl Zeiss Inc.; Göttingen, Germany), fitted with a DEI-750 CE video camera (Optronics; Goleta, California) and a LEP MAC5000 motorized stage controller (Ludl Electronic Products; Hawthorne, New York). The software package was Stereo Investigator (MBF Bioscience, Williston, Vermont). From a random start the numbers of TH+ neurons were counted in every fourth section through the SN of each brain. The Gundersen Coefficient of Error for the individual counts were routinely 0.05. Data were expressed as TH+ neurons/striatum (Malagelada et al.,2010).
Mesencephalic cell culture
Cultures of dopaminergic neurons were established from postnatal mice (8–10 days old) following the protocol published by Brewer with modifications (Brewer and Torricelli,2007). Briefly, mice were euthanatized and brains were extracted. The mesencephalon of each embryo was dissected and transferred to Hibernate-A with B27 (4°C). The tissue was minced and incubated with media containing papain (12 mg/6 ml in Hibernate A) at 30°C in a shaker for 20 minutes. The tissue was then transferred to room temperature (RT), triturated 10–15 times using a siliconized Pasteur pipet and then centrifuged at 800×g. The resulting pellets were resuspended in 3 ml of conditional medium (Neurobasal A/B27, 0.5mM glutamine, 10 ng/ml fibroblast growth factor-2 and penicillin/ streptomycin) and viability was determined by trypan blue exclusion. The cells were plated on glass cover-slips, previously coated with 100 microgram (µg) poly-D-lysine and laminin and placed in 24-multi-well plates. Approximately, 1.25 × 104 cells were plated per coverslip and were allowed to attach and attain morphology.
After 7 days in-vitro (DIV), depending on the experiment, cells were treated for 48 or 96 hours with 10 micromolar (µM) 1-methyl-4-phenylpyridinium (MPP+) in B27 without antioxidants or with the medium alone.
For neurite length measurements, a Olympus Fluoview Ver. 2.0a Viewer was used to quantify the length of the longest neurite for each of 100 TH immunoreactive neurons in six wells per treatment group. To determine neurite length, a sample tool was used to draw manually along the length of the longest visible neurite. Rudimentary processes were defined as being 10 µm or less in length at 20× magnification. Results were expressed as a percentage of cells with only rudimentary processes.
In a separate experiment features of apoptotic cell death, i.e., nuclear condensation and clumping, were investigated and compared in TH+ neurons in mesencephalic cultures prepared from WT and NL mice treated for 48 hours with MPP+. This was a time interval following MPP+ treatment when TH+ neurons were continuing to disappear following MPP+ exposure.
MPP+ Quantitation
Wild type and caspase 2 NL female mice were injected subcutaneously with a single dose of 20 mg/kg MPTP free base, and groups of 3 mice of each genotype were sacrificed as 1, 2, 4 and 6 hours post drug treatment. The striata of each mouse was dissected as described above, frozen in liquid nitrogen and stored at −80°C. Subsequently, one striatum from each animal was homogenized in 0.34 N perchloric acid, and the homogenate was centrifuged. The resulting supernatant was mixed with an equal volume of mobile phase (70 percent 20 mM potassium monobasic phosphate, 30 percent acetonitrile), and 100 microliters of the resulting mixture was injected onto a Grace C18 (5 micron, 4.6mm ID × 150 mm) reverse phase column. The HPLC system consisted of a Waters Model 515 pump, and MPP+ was detected with a Waters Model 2487 UV Detector at a fixed wavelength of 295 nm. The flow rate of the mobile phase was 0.5 mL/minute. Quantitation of MPP+ was determined with reference to a linear standard curve prepared by running blank brain homogenate calibrator samples spiked with known concentrations MPP+. Final MPP+ concentrations were expressed as ng MPP+/mg tissue weight.
Statistical analysis
Where appropriate, the data were analyzed by either a two-way or one-way analysis of variance (ANOVA). If significant effects were found, statistically significant differences between individual treatment groups were further resolved by Student-Newman-Keuls (SNK) tests or, if so stated, by Bonferroni correction tests.
Results
In the first experiment the effects of MPTP toxicity on striatal DA were investigated in WT, HET and NL mice (Figure 1). Statistical evaluation of these data with a 2 way ANOVA showed (1) that overall, treatment with MPTP produced a highly statistically significant reduction in striatal DA (F = 16.4; v1 = 3, v2 = 114, p<0.001) and (2) that the effect of MPTP on striatal DA was markedly different among the 3 genotypes of mice (F = 8.53; v1 = 2, v2 = 114; p<0.001). Further statistical resolution of these observations showed that in the absence of MPTP treatment striatal DA levels were not different in WT versus HET or NL mice. Treatment with a cumulative dose of 20 mg/kg MPTP significantly reduced striatal DA in WT and HET mice (p<0.01) but did not significantly reduce this neurotransmitter in NL mice.
Fig. 1.
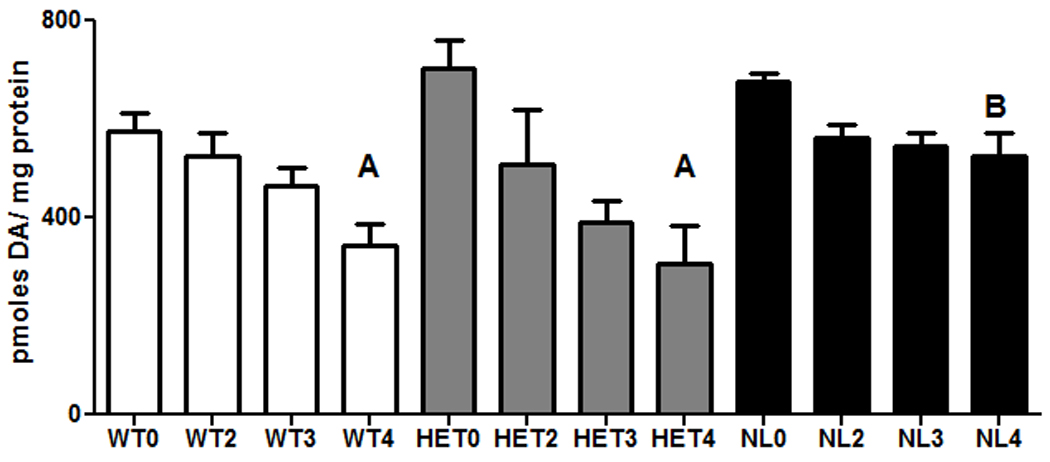
Dopamine levels (picomoles / mg protein) in the striata of wild type (WT; clear bars) compared to mice heterozygous (HET; gray bars) or homozygous for a knockout of the caspase 2 gene (NL; dark bars) following treatment with MPTP. The dose of MPTP (free base) was 5 mg/kg. The numbers on the abscissa indicate the number of times that this dose was administered subcutaneously. MPTP was administered at 2 hour intervals. The cumulative dosages of MPTP were 0, 10, 15 or 20 mg/kg, respectively. Each bar represents the mean ± the standard error of the mean (SEM) of 5 (HET), 10 (WT) or 16–17 (NL) animals per group. The data presented in the figure represent the pooled results of two separate experiments. Statistically significant differences were determined by a two way analysis of variance followed by Student-Newman-Keuls tests. A - p<0.01 from WT0, HET0, respectively; B shows significant protection from WT4 and HET4 (p<0.01).
The levels of TH immunoreactive protein were also examined in the striata of both NL and WT mice. In the absence of MPTP treatment, TH levels were comparable in these two groups of animals. However, treatment with MPTP, significantly reduced the level of striatal TH immunoreactive protein in WT (p<0.001) but not NL mice (Figure 2).
Fig. 2.
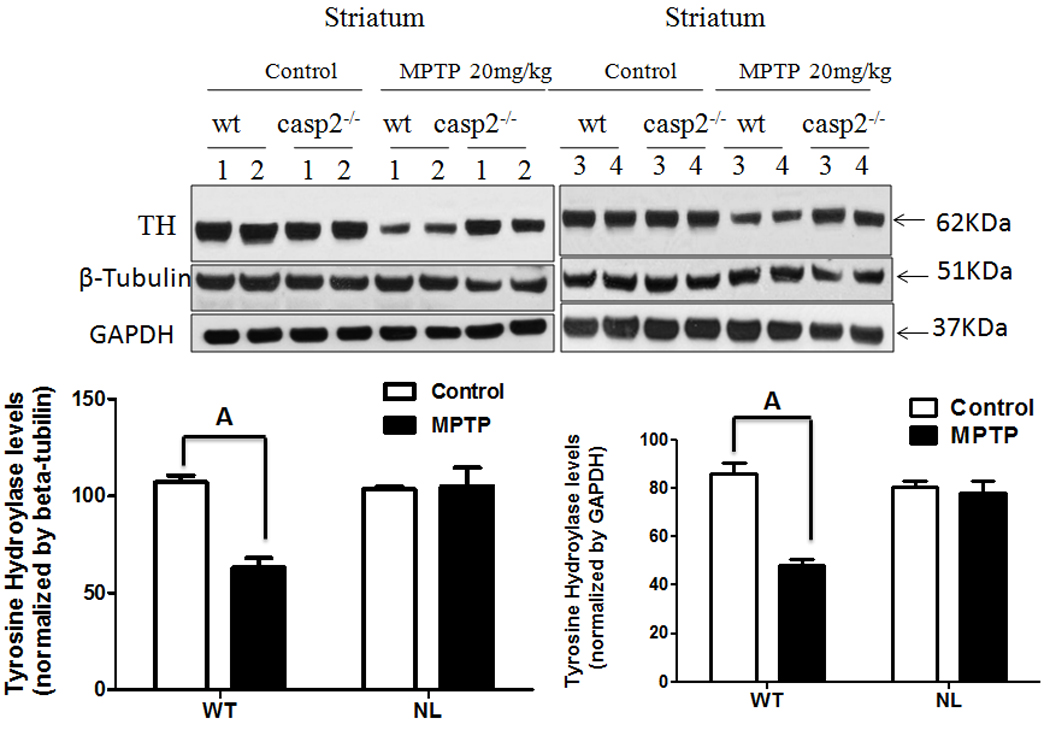
Comparison of TH immunoreactive (TH+) protein in the striata of WT versus NL mice. Mice were treated with 4 dosages of 5 mg/kg MPTP free base given at 2 hour intervals (cumulative dose - 20 mg/kg) or were not treated with this drug. One week later the striata were collected from each animal, the tissue was homogenized, and proteins were resolved by SDS-PAGE and transferred to PVDF membranes. Subsequently, western blots were performed by first probing the membranes with a primary antibody to mouse TH and later with antibodies to beta-tubulin and GADPH, as a loading controls. Densitometry analysis of band intensity was performed using Image J software. Statistically significant differences were determined by a one way analysis of variance followed by Bonferroni multiple range tests. A – p<0.001 from untreated WT, untreated NL and NL MPTP-treated
The comparatively low dosages of MPTP used in the above studies were unlikely to induce a significant loss of nigral dopaminergic cell bodies in the SN (Jackson-Lewis et al.,1995). Therefore, a higher dosage and a different dosing regimen were used to test the potential effects of a knockout of caspase 2 on the MPTP-induced loss of nigral dopaminergic cell bodies. Interestingly, in the absence of MPTP treatment, significantly fewer (p<0.05) TH+ neurons were observed in the SN of NL versus WT mice (Figure 3). Further, treatment with MPTP significantly reduced the numbers of TH+ neurons (p<0.05) in the WT mice compared to WT untreated. By contrast, MPTP did not significantly reduce TH+ in the NL mice versus NL untreated.
Fig. 3.
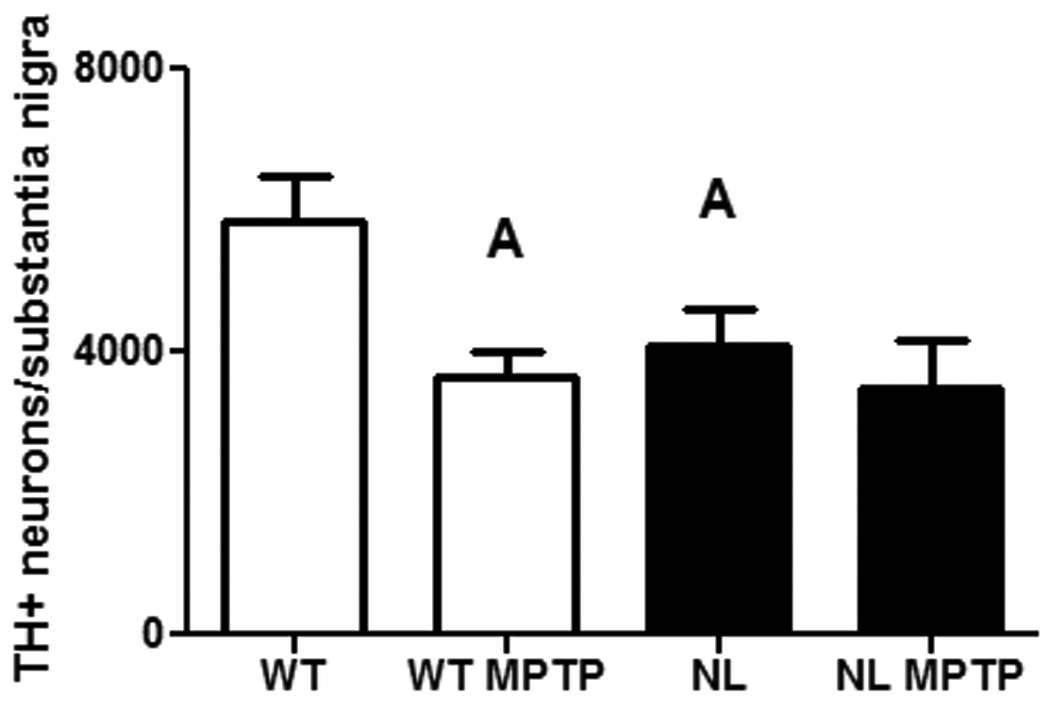
The number of TH+ neurons per substantia nigra in untreated and MPTP-treated wild type mice (WT; clear bars) and homozygous caspase 2 null mice (NL; dark bars). WT and NL mice were treated with 20 mg/kg MPTP free base for 3 days and 25 mg/kg MPTP for 2 additional days. The cumulative dosage of MPTP (free base) was 110 mg/kg. Each bar represents the mean ± SEM. The sample sizes were 6 for WT and NL and 7 for WT MPTP and NL MPTP. Statistically significant differences were determined by a one-way analysis of variance followed by Student-Newman-Keuls tests. A – p<0.05 from WT0
To determine if the reduced number of TH+ neurons observed in the SN of NL mice could be in part the result of reduced levels of TH in this brain region, a subsequent experiment compared the levels of TH immunoreactive protein in the VMB of WT versus NL. However, the results show that TH immunoreactive protein is actually significantly increased (p<0.02) in this brain region of the NL as compared to the WT mice (Fig.4).
Fig. 4.
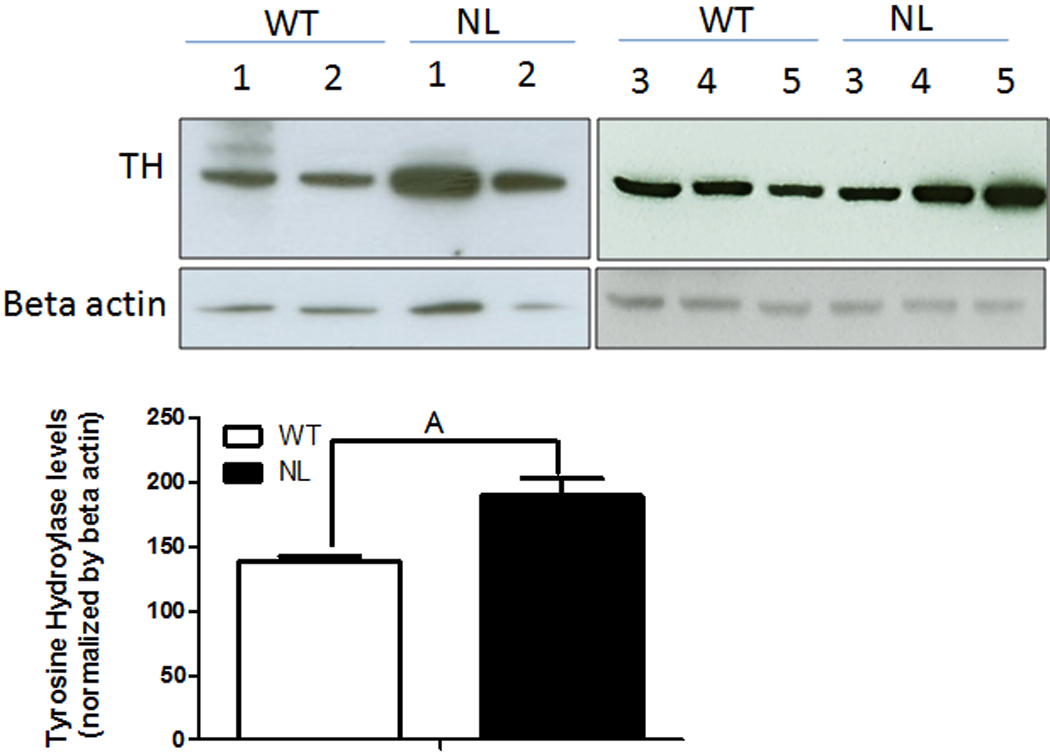
Comparison of TH+ immunoreactive protein levels in the ventral midbrain (VMB) of WT versus NL mice. The VMB was dissected by performing 2 coronal cuts, one immediately anterior to the superior colliculus and a second immediately posterior to the inferior colliculus which was directed to then pass ventrally through the margin between the midbrain and the pons. A transverse cut just superior to the visible cerebral peduncles was used to separate the VMB from the dorsal midbrain. The tissues were homogenized and western blots were subsequently performed as outlined in the caption to Figure 2 except that the TH+ immunoreactive protein was normalized to beta actin. A - p<0.02 from WT
In order to assess whether the reduced sensitivity of the nigrostriatal dopaminergic pathway to MPTP-induced toxicity in NL versus WT mice could be explained by a difference in the clearance of MPP+, the active metabolite of MPTP, this parameter was measured and compared in mice of these two genotypes. The results show that following treatment with 20 mg/kg MPTP free base there was a highly statistically significant time-dependent decrease in MPP+ (p<0.001) in the striata of both the WT and NL animals (Figure 5). However, the levels of MPP+ were comparable in WT and caspase 2 NL mice at 1, 2, 4 and 6 hours following the subcutaneous administration of MPTP.
Fig. 5.
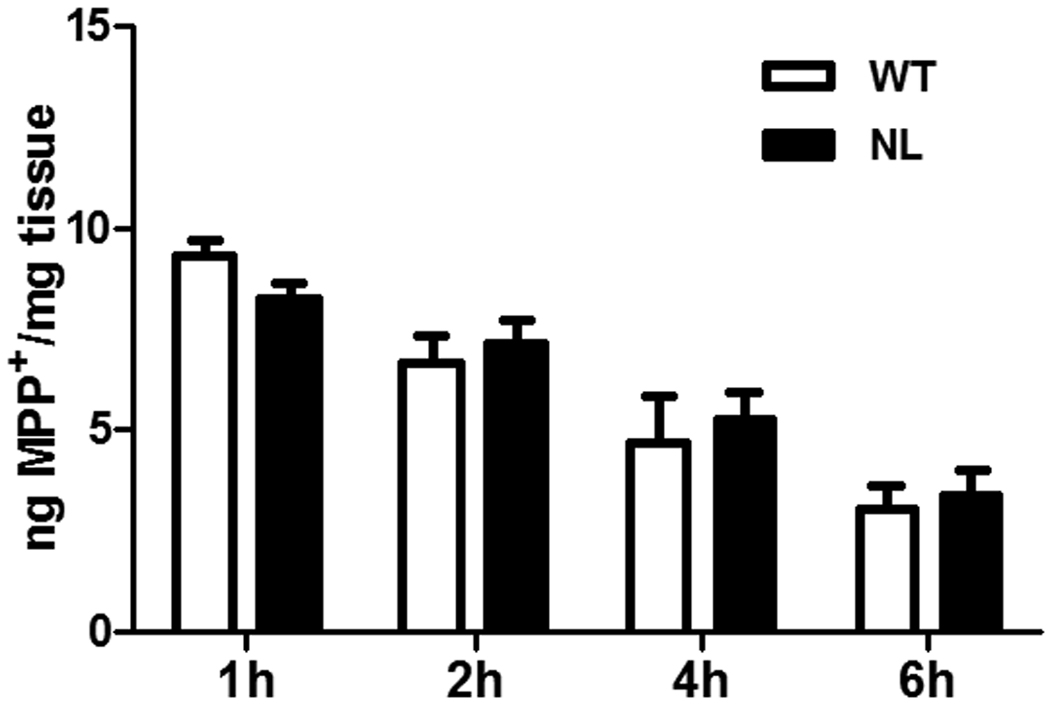
Comparison of the disappearance of MPP+ from striatal tissue of wild type (WT; clear bars) and homozygous caspase 2 null mice (NL; dark bars). WT and NL mice were treated with 20 mg/kg MPTP free base and striata were collected at 1, 2, 4 and 6 hours after treatment. MPP+ was measured by HPLC. Sample sizes were 3 animals per genotype at each time interval. A time-dependent statistically significant decline in MPP+ was observed in both WT and NL mice (p<0.001). However, there was no difference in the time-related decline in this parameter in the WT versus the NL mice.
The neurotoxicity of MPP+ was also compared in primary cultures of mesencephalic neurons prepared from 8–10 day-old WT versus NL mouse pups. After 7 total days in culture the morphology and the numbers of TH+ neurons were comparable in untreated primary cultures prepared from WT and NL mice (Figure 6A and 6B). On the other hand, when these same parameters were compared in primary cultures treated 96 hours previously with 10µM MPP+, a treatment that kills 80 percent of WT TH+ neurons (data not shown), a significantly greater number of TH + neuronal cell bodies (p<0.001) were evident in the primary cultures prepared from NL as opposed to WT mice (Figure 6A, 6B and 6C). Further, following MPP+, the surviving TH+ neurons in the cultures prepared from WT as opposed to NL pups showed a significantly greater (p<0.001) occurrence of either absent or rudimentary TH+ neurites (Figure 7A).
Fig. 6.
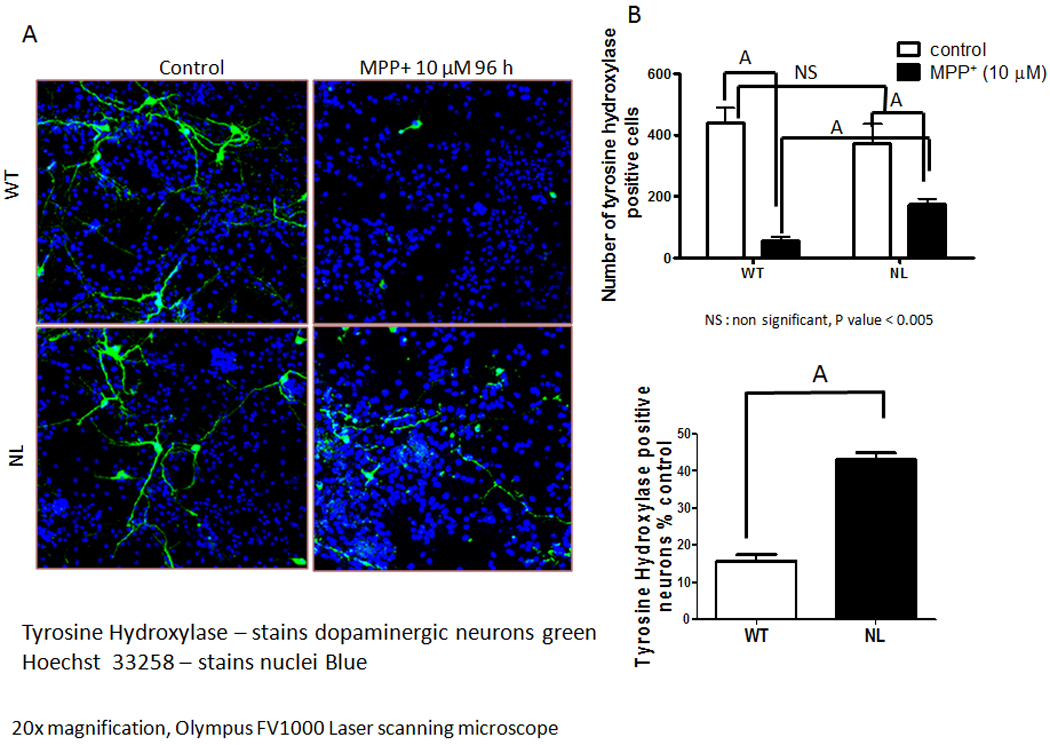
Comparison of the effect of MPP+ toxicity on TH immunopositive (TH+) neurons in primary cultures of mesencephalic cells prepared from 8 – 10 day-old WT versus NL mouse pups. Mesencephalic neurons were cultured from WT and NL midbrains in Neurobasal-TM with B27 supplement and FGF-2. On DIV-7, they were treated with 10 µM MPP+ for 96 hours. (A) Cells were immunostained with an antibody to tyrosine hydroxylase, secondary antibody Alexa Fluor 488 (green) and nuclei (blue) were stained with Hoechest-33258. (B) The total number of TH+ cells were counted in six different cover slips per treatment. (C) The number of TH+ cells remaining following MPP+ treatment of WT versus NL primary cultures are expressed as percentage of those observed in the corresponding control cultures not treated with MPP+. Data shown in each case are the mean ± SEM of three independent experiments. A - p<0.001
Fig. 7.
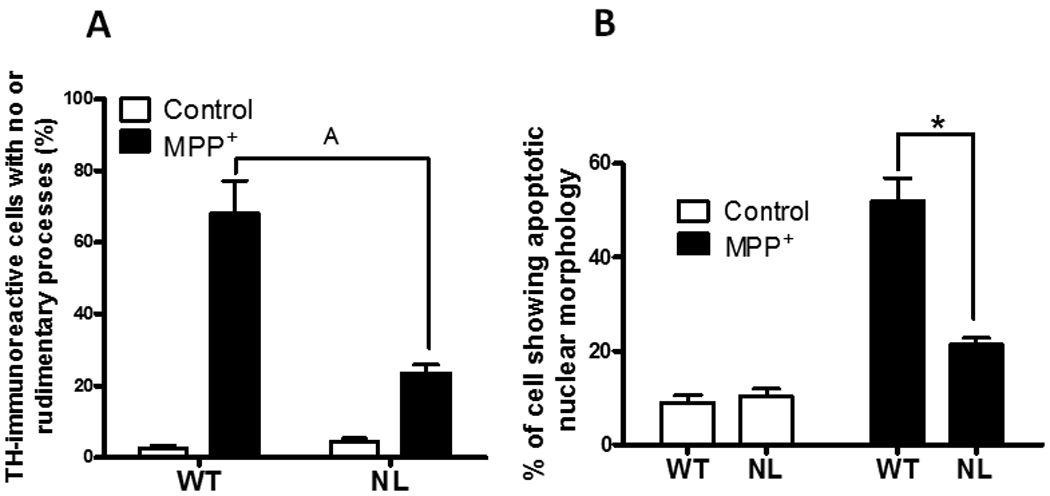
(A) Comparison of the percentage of TH+ cells in mesencephalic cultures prepared from WT or NL mouse pups that show no, or only rudimentary, neurites following treatment with 10µM MPP+. Rudimentary neurites were defined as having the longest process <10 µm in length. These data come from the primary cultures described in Fig. 6. Data shown are the mean ± SEM of three independent experiments. A p<0.001 from MPP+-treated WT. (B) Loss of caspase 2 protects TH+ neurons from MPP+-induced apoptotic cell death. Mesencephalic cultures were prepared from 8–10 day old WT and NL pups and were either untreated or treated with 10µM MPP+ as described in the caption to Figure 6. The cells were exposed to MPP+ for 48 hours. The histogram shows the quantification, i.e., the % of TH+ neurons showing nuclear features of apoptotic cell death, i.e., nuclear condensation and fragmentation, in cells observed in six difference cover slips per treatment. The data (mean ± SEM, n=150–200) represent the average of three independent experiments. A p<0.001 from MPP+-treated WT.
A subsequent study showed that features of apoptotic cell death, i.e., nuclear condensation and clumping, were significantly more prevalent (p<0.001) following 48 hours of exposure to 10 µM MPP+ in mesencephalic cultures prepared from WT compared to NL mouse pups (Figure 7B).
Discussion
The results outlined in this manuscript show that the sensitivity of DA levels in the striatum to the toxicity induced by a low dose regimen of MPTP is significantly reduced in NL mice compared to either WT or HET mice. These data also indicate that one functional caspase 2 gene is sufficient to incur normal sensitivity to MPTP.
Following treatment with MPTP, the levels of TH immunoreactive protein were also significantly less affected in the striata of NL versus WT mice. These data, which represent an independent index of the integrity of the nigrostriatal pathway, provide collaborative evidence that the nigrostriatal dopaminergic pathway of NL mice is significantly less sensitive to the selective neurotoxic effects of MPTP.
In order to increase the probability of detecting potentially small protective effects, the total dosages of MPTP administered to the mice in the initial experiments were kept intentionally low, i.e., roughly one-fourth of that administered in a frequently employed dosing regimen (Mayer et al.,1986). Others have shown that the loss of dopaminergic neurons in the SN following treatment with MPTP is dose-related with a cumulative dosage of 40 mg/kg resulting in roughly an 11 percent decrease (Jackson-Lewis et al.,1995). In our initial in vivo studies the time course of our treatment regimen was the same but the cumulative dosage of 20 mg/kg MPTP was half that given by these other investigators. Therefore, it seems likely that the reduction of striatal DA observed in our initial studies is more the result of toxic effects at the level of the nerve terminals rather than the death of cells bodies. However, it is yet unclear whether the toxic effects observed in these particular experiments reflect damage to the nerve terminal itself or to the enzymatic machinery involved in the synthesis of DA in the terminals (Przedborski and Jackson-Lewis,1998).
The apparently reduced sensitivity to MPTP toxicity in caspase 2 NL versus WT mice cannot be explained by an altered metabolism of this drug as our results show that clearance of the active metabolite MPP+ from the striatum is comparable in both WT and caspase 2 NL mice.
When the neurotoxic effects of MPP+, the active metabolite of MPTP, were studied in primary cultures of mesencephalic neurons; cells isolated from 8–10 day-old NL mouse pups were again shown to be more resistant to the toxicity of MPP+ compared to cells prepared for similarly aged WT mice. These data show that MPP+ produced a markedly greater loss of TH+ neurons in cultures prepared from WT compared to NL mice. Interestingly, there was also a significantly greater preservation of TH+ neurites in the NL-derived cultures while these structures were either absent or more rudimentary in cultures prepared from WT embryos. These data suggest that, at least for neonatal neurons, a homozygous knockout of the caspase 2 gene reduces the sensitivity of both dopaminergic nerve processes and cell bodies to MPTP-associated toxicity.
The results of the studies with primary cultures of mesencephalic neurons along with those obtained from in vivo studies of dopaminergic-related parameters in the striatum suggest, although does not prove, the intriguing possibility that caspase 2 may play a heretofore unsuspected role in the loss of functionally active dopaminergic terminals, which is induced by MPTP-induced toxicity. This possibility is consistent with an earlier report that caspase-related mechanisms may be involved in the degeneration of nerve terminals (Mattson et al.,1998).
An examination of nuclear morphology of the TH+ neurons in mesencephalic cultures indicated that death by apoptosis is responsible for at least a significant portion of the disappearance of these cells following exposure to MPP+. This result is consistent with a previous study which also reported apoptotic cell death following treatment with MPP+ in mesencephalic cultures prepared from rat embryos (Furuya et al.,2004). Our results also show that MPP+ induced significantly less apoptosis in TH+ neurons derived from NL as opposed to WT mouse pups. These latter results provide further evidence that (1) a homozygous knockout of caspase 2 reduces the sensitivity of nigrostriatal dopaminergic pathway to MPTP-induced toxicity and (2) at least in mesencephalic TH+ neurons the knockout of this gene protects dopaminergic cell bodies as well as nerve terminals.
Analysis by unbiased stereology showed that the numbers of TH+ neurons in the SN of untreated NL mice were significantly less that those observed in this same brain region of untreated WT mice. When TH immunopositive protein levels were measured in the VMB, the levels of this parameter in NL mice were actually significantly greater than those observed in WT mice. This increase in TH protein in the VMB may reflect a compensatory response to the presence of a fewer number of TH+ neuron in the SN of the NL mice. With the caveat that the dissection of the VMB likely includes at least a portion of the neurons of the nigrotegmental as well as the nigrostriatal pathway, these data taken together suggest that the reduced number of TH+ neurons determined by stereology reflect a true reduction of nigrostriatal neurons rather than a failure to detect nigrostriatal neurons which were deficient in TH immunoreactive protein. The reduced numbers of TH+ neurons in the SN of the NL mice suggest that caspase 2 may play a role in the development or maturation of the normal constituent of dopaminergic neurons in the SN, but the elucidation of the underlying mechanism will require future investigation.
To determine whether a knockout of caspase 2 could significantly affect the sensitivity of nigral dopaminergic cell bodies in vivo to MPTP-induced toxicity, a dosing regimen was employed which had been previously shown by others to result in an apoptotic-like death of TH+ neurons in the SN of C57BL6 mice (Tatton and Kish,1997). Treatment with this MPTP regimen induced a significant decrease in the numbers of TH+ neurons in the SN of WT mice compared to untreated WT mice. Although we did not also count the number of Nissl-stained neurons in the SN, the fact that the MPTP-regimen employed was similar to that previously shown to induce nigral neuron cell death suggests that the MPTP-induced decrease in TH+ neurons observed in the WT animals is likely due, at least in part, to the cell death. By contrast, this same MPTP treatment did not significantly reduce the numbers of TH+ neurons in the SN of NL mice when compared to untreated NL mice. This latter observation is consistent with our other data which demonstrated (1) a reduced sensitivity of the in vivo nigral dopaminergic pathway to MPTP-induced neurotoxicity and (2) that primary cultures of mesencephalic TH+ neurons prepared from early postnatal caspase 2 NL mice were less sensitive to MPP+.
Interestingly, the stereological data indicate that the numbers of TH+ neurons observed in both the untreated NL and the MPTP-treated NL mice were not significantly different from those observed in the MPTP-treated WT mice. One possible explanation for these data is that normally there are at least two populations of TH+ neurons in the SN, one more sensitive and a second less sensitive to MPTP-induced toxicity. If this were true and if one were to speculate further that caspase 2 plays a preferential role in the development of the population of these neurons which are more sensitive to MPTP, then the population less sensitive to MPTP might predominant in NL mice. This could explain why (1) the number of TH+ neurons are reduced in the SN of untreated NL mice and (2) these neurons in NL mice show reduced sensitivity to MPTP-induced toxicity.
Increased activation of caspase 2 has been observed in association with cell death in cell lines exposed to MPP+ (Bando et al.,2005;Chee et al.,2005), and in one case the cell death was partially prevented by pretreatment with a non-selective caspase 2 antagonist (Chee et al.,2005). However, to our knowledge, the results summarized in this manuscript provide the first evidence for a functional role for caspase 2 in an intact animal model of MPTP-induced neurotoxicity.
Other observations also suggest that caspase 2 may play an important role in at least some neurodegenerative diseases. For example, caspase 2 may play an important role in the neuronal cell loss associated with Huntington’s Chorea (Hermel et al.,2004). Further, primary cultures of sympathetic neurons isolated from homozygous caspase 2 null mice were resistant to the neurotoxic effects of β-amyloid (Troy et al.,2000). Caspase 2 is also the initiator caspase in the apoptotic cell death induced by treatment of primary cultures of neonatal cerebral cortical neurons with rotenone (Tiwari et al.,2011).
In summary, the results of this study suggest that homozygous caspase 2 null mice show resistance to the neurotoxic effects of low dosages of MPTP. Further, the knockout of both copies of the caspase 2 gene is necessary to produce this protective effect. In vivo, this increased resistance is best reflected in parameters associated with nigrostriatal dopaminergic cell processes, i.e., striatal DA and TH immunoreactive protein level. However, the TH+ neurons in the SN of NL mice also appear to be more resistant to MPTP. These in vivo observations are also supported by the in vitro data which show that both TH+ cell processes and cell bodies in caspase 2 NL mice are more resistant to the neurotoxin. Although suggestive, the results of these studies do not conclusively show that caspase 2 is normally part of the MPTP-induced cell death pathway. As noted, some evidence provided by these studies suggest that caspase 2 may play a role in the development of the normal number of TH+ neurons in the SN. It is possible, therefore, that the reduced sensitivity of the dopaminergic nigrostriatal pathway to MPTP-induced toxicity observed in caspase null mice may be more the result of the developmental selection of a population of MPTP-resistant neurons rather than due to the removal of caspase 2 as an important factor in mediating MPTP-induced damage to this neuronal pathway. Studies to resolve these alternative possibilities are underway.
Acknowledgements
This work was supported by grants from the Merit Review Medical Research Program of the Department of Veterans Affairs (WWM) and from the NIH (AG07218, AG19316)(BH). The technical assistance of Elisa Figueroa, Nancy Lopez and Jacqueline Alcala is gratefully acknowledged. Thanks also to Dr. James Roberts (Department of Pharmacology, UTHSCSA and Trinity University, San Antonio) for the use of his microscope and to David Price for his assistance in learning to perform unbiased stereology. The authors also thank Dr. Martin Javors (Department of Psychiatry, UTHSCSA) whose laboratory performed the MPP+ assays. Dr. Carol Troy (Columbia University) kindly provided the caspase 2 null mice.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bando Y, Katayama T, Taniguchi M, Ishibashi T, Matsuo N, Ogawa S, Tohyama M. RA410/Sly1 suppresses MPP+ and 6-hydroxydopamine-induced cell death in SH-SY5Y cells. Neurobiol Dis. 2005;18:143–151. doi: 10.1016/j.nbd.2004.09.008. [DOI] [PubMed] [Google Scholar]
- Bergeron L, Perez GI, MacDonald G, Shi L, Sun Y, Jurisicova A, Varmuza S, Latham KE, Flaws JA, Salter JC, Hara H, Moskowitz MA, Li E, Greenberg A, Tilly JL, Yuan J. Defects in regulation of apoptosis in caspase-2-deficient mice. Genes Dev. 1998;12:1304–1314. doi: 10.1101/gad.12.9.1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- Brewer GJ, Torricelli JR. Isolation and culture of adult neurons and neurospheres. Nat Protoc. 2007;2:1490–1498. doi: 10.1038/nprot.2007.207. [DOI] [PubMed] [Google Scholar]
- Chee JL, Guan XL, Lee JY, Dong B, Leong SM, Ong EH, Liou AK, Lim TM. Compensatory caspase activation in MPP+-induced cell death in dopaminergic neurons. Cell Mol Life Sci. 2005;62:227–238. doi: 10.1007/s00018-004-4413-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furuya T, Hayakawa H, Yamada M, Yoshimi K, Hisahara S, Miura M, Mizuno Y, Mochizuki H. Caspase-11 mediates inflammatory dopaminergic cell death in the 1-methyl-4-phenyl-1, 2, 3, 6-tetrahydropyridine mouse model of Parkinson's disease. J Neurosci. 2004;24:1865–1872. doi: 10.1523/JNEUROSCI.3309-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Z, Shao Y, Jiang X. Essential roles of the Bcl-2 family of proteins in caspase-2-induced apoptosis. J Biol Chem. 2005;280:38271–38275. doi: 10.1074/jbc.M506488200. [DOI] [PubMed] [Google Scholar]
- Guo Y, Srinivasula SM, Druilhe A, Fernandes-Alnemri T, Alnemri ES. Caspase-2 induces apoptosis by releasing proapoptotic proteins from mitochondria. J Biol Chem. 2002;277:13430–13437. doi: 10.1074/jbc.M108029200. [DOI] [PubMed] [Google Scholar]
- Haviv R, Lindenboim L, Yuan J, Stein R. Need for caspase-2 in apoptosis of growth-factor-deprived PC12 cells. J Neurosci Res. 1998;52:491–497. doi: 10.1002/(SICI)1097-4547(19980601)52:5<491::AID-JNR1>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- Hermel E, Gafni J, Propp SS, Leavitt BR, Wellington CL, Young JE, Hackam AS, Logvinova AV, Peel AL, Chen SF, Hook V, Singaraja R, Krajewski S, Goldsmith PC, Ellerby HM, Hayden MR, Bredesen DE, Ellerby LM. Specific caspase interactions and amplification are involved in selective neuronal vulnerability in Huntington's disease. Cell Death Differ. 2004;11:424–438. doi: 10.1038/sj.cdd.4401358. [DOI] [PubMed] [Google Scholar]
- Jackson-Lewis V, Jakowec M, Burke RE, Przedborski S. Time course and morphology of dopaminergic neuronal death caused by the neurotoxin 1-methyl-4-phenyl-1, 2, 3, 6-tetrahydropyridine. Neurodegeneration. 1995;4:257–269. doi: 10.1016/1055-8330(95)90015-2. [DOI] [PubMed] [Google Scholar]
- Kumar S. Inhibition of apoptosis by the expression of antisense Nedd2. FEBS Lett. 1995;368:69–72. doi: 10.1016/0014-5793(95)00602-6. [DOI] [PubMed] [Google Scholar]
- Kumar S, Kinoshita M, Noda M, Copeland NG, Jenkins NA. Induction of apoptosis by the mouse Nedd2 gene which encodes a protein similar to the product of the Caenorhabditis elegans cell death gene ced-3 and the mammalian IL-1 beta-coverting enzyme. Genes Dev. 1994;8:1613–1626. doi: 10.1101/gad.8.14.1613. [DOI] [PubMed] [Google Scholar]
- Malagelada C, Jin ZH, Jackson-Lewis V, Przedborski S, Greene LA. Rapamycin protects against neuron death in in vitro and in vivo models of Parkinson's Disease. J Neurosci. 2010;30:1166–1175. doi: 10.1523/JNEUROSCI.3944-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattson MP, Keller JN, Begley JG. Evidence for synaptic apoptosis. Exp Neurol. 1998;153:35–48. doi: 10.1006/exnr.1998.6863. [DOI] [PubMed] [Google Scholar]
- Mayer RA, Walters AS, Heikkila RE. 1-Methyl-4-phenyl-1, 2, 3, 6-tetrahydropyridine (MPTP) administration to C57-black mice leads to parallel decrements in neostriatal dopamine content and tyrosine hydroxylase activity. Eur J Pharmacol. 1986;120:375–377. doi: 10.1016/0014-2999(86)90482-6. [DOI] [PubMed] [Google Scholar]
- Morgan WW, Nelson JF. Chronic administration of pharmacological levels of melatonin does not ameliorate the MPTP-induced degeneration of the nigrostriatal pathway. Brain Res. 2001;921:115–121. doi: 10.1016/s0006-8993(01)03106-7. [DOI] [PubMed] [Google Scholar]
- Morgan WW, Rudeen PK, Pfeil KA. Effect of immobilization stress on serotonin content and turnover in regions of the rat brain. Life Sci. 1975;17:143–150. doi: 10.1016/0024-3205(75)90250-7. [DOI] [PubMed] [Google Scholar]
- Niizuma K, Endo H, Nito C, Myer DJ, Kim GS, Chan PH. The PIDDosome mediates delayed death of hippocampal CA1 neurons after transient global cerebral ischemia in rats. Proc Natl Acad Sci USA. 2008;105:16368–16373. doi: 10.1073/pnas.0806222105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proll MA, Kamp CW, Morgan WW. Use of liquid chromatography with electrochemistry to measure effects of varying intensities of white light on DOPA accumulation in rat retinas. Life Sci. 1982;30:11–19. doi: 10.1016/0024-3205(82)90630-0. [DOI] [PubMed] [Google Scholar]
- Przedborski S, Jackson-Lewis V. Mechanisms of MPTP toxicity. Mov Disord. 1998;13 Suppl 1:35–38. [PubMed] [Google Scholar]
- Sato N, Milligan CE, Uchiyama Y, Oppenheim RW. Cloning and expression of the cDNA encoding rat caspase-2. Gene. 1997;202:127–132. doi: 10.1016/s0378-1119(97)00463-0. [DOI] [PubMed] [Google Scholar]
- Tatton NA, Kish SJ. In situ detection of apoptotic nuclei in the substantia nigra compact of 1-methyl-4-phenyl-1, 2, 3, 6-tetrahydropyridine-treted mice using terminal deoxynucleotidyl transferase labelling and acridine orange. Neuroscience. 1997;77:1037–1048. doi: 10.1016/s0306-4522(96)00545-3. [DOI] [PubMed] [Google Scholar]
- Tinel A, Tschopp J. The PIDDosome, a protein complex implicated in activation of caspase-2 in response to genotoxic stress. Science. 2004;304:843–846. doi: 10.1126/science.1095432. [DOI] [PubMed] [Google Scholar]
- Tiwari M, Lopez-Cruzan M, Morgan WW, Herman B. Loss of caspase-2 dependent apoptosis induces autophagy following mitochondria oxidative stress in primary cultures of young-adult cortical neurons. J Biol Chem. 2011 doi: 10.1074/jbc.M110.163824. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Troy CM, Rabacchi SA, Friedman WJ, Frappier TF, Brown K, Shelanski ML. Caspase-2 mediates neuronal cell death induced by beta-amyloid. J Neurosci. 2000;20:1386–1392. doi: 10.1523/JNEUROSCI.20-04-01386.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Troy CM, Shelanski ML. Caspase-2 redux. Cell Death Differ. 2003;10:101–107. doi: 10.1038/sj.cdd.4401175. [DOI] [PubMed] [Google Scholar]
- Troy CM, Stefanis L, Greene LA, Shelanski ML. Nedd2 is required for apoptosis after trophic factor withdrawal, but not superoxide dismutase (SOD1) downregulation, in sympathetic neurons and PC12 cells. J Neurosci. 1997;17:1911–1918. doi: 10.1523/JNEUROSCI.17-06-01911.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu S, McStay GP, Boucher LM, Mak T, Beere HM, Green DR. In situ trapping of activated initiator caspases reveals a role for caspase-2 in heat shock-induced apoptosis. Nature Cell Biol. 2006;8:72–77. doi: 10.1038/ncb1340. [DOI] [PubMed] [Google Scholar]
- Wang L, Miura M, Bergeron L, Zhu H, Yuan J. Ich-1, an Ice/ced-3-related gene, encodes both positive and negative regulators of programmed cell death. Cell. 1994;78:739–750. doi: 10.1016/s0092-8674(94)90422-7. [DOI] [PubMed] [Google Scholar]
- Yang L, Matthews RT, Schulz JB, Klockgether T, Liao AW, Martinou JC, Penney JB, Jr., Hyman BT, Beal MF. 1-Methyl-4-phenyl-1, 2, 3, 6-tetrahydropyride neurotoxicity is attenuated in mice overexpressing Bcl-2. J Neurosci. 1998;18:8145–8152. doi: 10.1523/JNEUROSCI.18-20-08145.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Padalecki SS, Chaudhuri AR, De Waal E, Goins BA, Grubbs B, Ikeno Y, Richardson A, Mundy GR, Herman B. Caspase-2 deficiency enhances aging-related traits in mice. Mech Age Dev. 2007;128:213–221. doi: 10.1016/j.mad.2006.11.030. [DOI] [PMC free article] [PubMed] [Google Scholar]