

Abstract
Multidrug regimens and corresponding drug interactions cause many adverse reactions and treatment failures. Drug efflux transporters: P-gp, MRP, BCRP in conjunction with metabolizing enzymes (CYPs) are major factors in such interactions. Most effective combination antiretrovirals (ARV) therapy includes a PI or a NNRTI or two NRTI. Coadministration of such ARV may induce efflux transporters and/or CYP3A4 resulting in sub-therapeutic blood levels and therapeutic failure due to reduced absorption and/or increased metabolism. A similar prognosis is true for ARV-compounds and drugs of abuse combinations. Morphine and nicotine enhance CYP3A4 and MDR1 expression in vitro. A 2.5 fold rise of cortisol metabolite was evident in smokers relative to nonsmokers. Altered functions of efflux transporters and CYPs in response to ARV and drugs of abuse may result in altered drug absorption and metabolism. Appropriate in vitro models can be employed to predict such interactions. Influence of genetic polymorphism, SNP and inter-individual variation in drug response has been discussed. Complexity underlying the relationship between efflux transporters and CYP makes it difficult to predict the outcome of HAART as such, particularly when HIV patients taking drugs of abuse do not adhere to HAART regimens. HIV+ pregnant women on HAART medications, indulging in drugs of abuse, may develop higher viral load due to such interactions and lead to increase in mother to child transmission of HIV. A multidisciplinary approach with clear understanding of mechanism of interactions may allow proper selection of regimens so that desired therapeutic outcome of HAART can be reached without any side effects.
Keywords: P-glycoprotein, Multidrug resistance-associated protein, Breast cancer resistance protein, Cytochrome P-450, Antiretrovirals agents, Protease inhibitors and drugs of abuse
Introduction
Multidrug therapy is routinely indicated for the treatment of AIDS. Drug–drug interactions can alter pharmacological or toxicological response resulting from concomitant intake of anti-HIV agents along with drugs of abuse. Absorption of orally administered drugs may be limited by efflux and metabolism by enterocytes and hepatocytes respectively. The role of efflux transporters in drug disposition is now widely accepted. Multidrug resistance (MDR) proteins – P-glycoprotein (P-gp), multidrug resistance-associated protein (MRP), and breast cancer resistant protein (BCRP) along with metabolizing enzyme cytochrome P450 (CYP) are the main factors responsible for low oral bioavailability. Two key factors i.e. drug efflux proteins (P-gp, MRP and BCRP) or efflux pump (EP) and CYP3A4 regulate all pharmacokinetic and pharmacodynamic interactions during the process of drug absorption and metabolism. In general, drug interactions occur when one drug may change the disposition of another resulting in altered therapeutic outcomes. Pharmacodynamic interactions may produce additive, synergistic, or antagonistic effects. Overexpression of these proteins in response to antiretrovirals (ARV) and/or drugs of abuse can compromise therapeutic outcome.
General principles of drug–drug interactions
When two or more drugs are co-administered, such interactions can cause additive, synergistic or antagonistic effects. In Kaletra, both lopinavir and ritonavir act synergistically to produce higher antiretroviral activity. Ritonavir produces higher area under the curve (AUC) of lopinavir possibly through inhibition of both P-gp and CYP3A4. For treating opportunistic infections, concomitant treatment of saquinavir with ketoconazole can elevate AUC of saquinavir due to inhibition of both P-gp and CYP3A4. Therefore, any combination therapy with HIV protease inhibitor (PI) requires dosage adjustment.
In general, administration of nucleoside/nucleotide reverse transcriptase inhibitors (NRTIs) leads to pharmacodynamic interactions due to additive/synergistic effects. These compounds are not substrates for CYP3A4 and are eliminated by kidney. As a result, these compounds do not produce any significant drug–drug interactions with PIs and nonnucleoside reverse transcriptase inhibitors (NNRTI). In contrast, all NNRTI are either inducers or inhibitors of CYP3A4. Nevirapine is a CYP3A4 inducer, whereas delavirdine serves as an inhibitor and efavirenz causes both induction and inhibition. Therefore, selection of NNRTI in HAART regimen requires more attention since some combinations can lead to low bioavailability and others may exacerbate PI toxicity. PI dosages need to be considered carefully, since these drugs can induce/inhibit both P-gp and CYP3A4. Therefore, these compounds can cause a number of drug–drug interactions, particularly if the drug combinations are also substrates for P-gp and/or CYP3A4. Another complicated factor in this equation is dosage adjustment, because many drugs can be substrates, inducers or inhibitors of P-gp and/or CYP depending on dose. Appropriate dose adjustment can exert therapeutic effects without causing toxicity. In Kaletra, ritonavir (100 mg) and lopinavir (400 mg) combination produces maximal antiretroviral activity despite the fact that ritonavir can act as an inhibitor, or inducer of P-gp and CYP3A4. Selection of appropriate drug combination can result in an improved therapeutic outcome.
Drugs of abuse which include alcohol, morphine, heroin, codeine, hydrocodone, oxycodone, and buprenorphine, amphetamine, 3,4-methylenedioxymethamphetamine (MDMA), benzodiazepines (midazolam, alprazolam, lorazepam, diazepam), cocaine, marijuana, opiates, narcotic analgesics [phenylpiperidines, meperidines, fentanyl, pseudopiperidines (methadone) and propoxyphen] and nicotine can significantly interact with ARV causing treatment failures or toxicity. Such interactions may result from induction or inhibition of EP and/or metabolizing enzymes. Induction of these proteins may lead to a significant loss in therapeutic activity whereas inhibition can result in toxicity. Also, lopinavir plus ritonavir (Kaletra), ritonavir, and efavirenz were found to cause opiate withdrawal when given in combination with methadone (Iribarne et al., 1996; Beauverie et al., 1998; Pinzani et al., 2000; Bart et al., 2001; Baker et al., 2006). Conversely, inhibition(s) of EP and/or CYP3A4 can cause enhanced plasma concentration and therefore, drug toxicity. Overlapping substrate specificities to these proteins make it difficult to understand perplexing pharmacokinetic interactions with multidrug regimens. Inter-patient variability of drug response can occur due to change in genetic profiles, intake of food, herbal supplement, and recreational drugs.
The mechanisms behind opiate withdrawal may involve drug–drug interaction (Wynn et al., 2005). Induction of P-gp and CYP3A4 by antiretrovirals results in enhanced efflux and metabolism of metha-done. Nicotine induces several drug-metabolizing enzymes i.e. CYP3A4, CYP1A2, CYP2D6, CYP2B6, CYP2E1 and UGT. However information on therapeutic interactions and their mechanisms with drugs of abuse following chronic exposure are extremely limited. Brain MDR1 expression in morphine tolerant rats has been known to rise (unpublished data from our laboratory). Such over expression of MDR1 can reduce brain levels of therapeutic agents which are substrates for this efflux pump. In this section we have outlined several important drug–drug interactions which occur in clinical management of AIDS patients involving several antiretroviral agents i.e. PI: ritonavir, saquinavir, lopinavir and tipranavir; NRTI: didanosine and zidovudine and NNRTI: efavirenz and nevirapine. In the following sections, clinical relevance of drug–drug interactions involving both efflux proteins (P-gp, MRP and BCRP) and metabolizing enzymes, CYP450 have been described.
MDR products
P-glycoprotein
P-gp is the most extensively studied efflux transporter which functions as a biological barrier by extruding toxins and xenobiotics into extracellular fluid. Several studies have demonstrated that it plays a significant role in drug absorption and disposition. It is a 170 kDa transmembrane protein, which is a product of multidrug resistance (MDR1) gene. This protein is one of well-characterized drug efflux pump which plays an important role in protecting cells against cytotoxic drugs by extruding xenobiotics (Borst et al., 2000). Two variants of MDR products, MDR1 and MDR3 were reported in human whereas three members of this family i.e. mdr1a, mdr1b and mdr2 were detected in mice (Gottesman and Pastan, 1993; Schinkel et al., 1997). P-gp encoded by human MDR1 and mouse mdr1a/1b genes is responsible for drug efflux whereas, P-gp transcribed by human MDR3 and mouse mdr2 generally serve as phospholipid transporter (Ruetz and Gros, 1994; van Helvoort et al., 1996). This protein is characterized as the ATP binding cassette (ABC), and is responsible for efflux of chemotherapeutic agents from resistant cancer cells (Leslie et al., 2001). Several HIV protease inhibitors are either inducers or inhibitors of P-gp (Leslie et al., 2001). Similarly, drugs of abuse can also act as inhibitors or inducers. Recent reviews by Troutman et al. (2008) and from our laboratory (Katragadda et al., 2005; Pal and Mitra, 2006a; Pal and Mitra, 2006b) have elaborated on the role of P-gp in oral drug delivery. Approximately 60–65% homology with other species is present in mammalian P-gp, indicating that the protein structure is highly conserved throughout evolution because of their importance in xenobiotics trafficking.
Multidrug resistance-associated proteins
MRP efflux pumps belong to the same ABC super family. These are 190 kD proteins which are responsible for the transport of drugs across lipid membranes (Krishnamachary and Center, 1993; Zaman et al., 1994). Unlike P-gp, these proteins primarily transport conjugated organic anions including cysteinyl leukotrienes (LTC) (Larkin et al., 2004; Xiao et al., 2005). So far eight different MRP has been reported for ABC proteins. Human MRP-1, MRP-2 and MRP-3 are known to be involved in efflux of anti-HIV drugs and their conjugated metabolites (Bakos et al., 2000; Huisman et al., 2002; Naruhashi et al., 2002; Hulot et al., 2005; Letourneau et al., 2005). A number of NRTI, NNRTI and nucleotide reverse transcriptase inhibitors (NtRTIs), particularly tenofovir appear to inhibit MRP1, MRP2 and MRP3 in a concentration dependent manner (Ketabi-Kiyanvash et al., 2003).
Breast cancer resistance protein
BCRP/ABCG2 is considered as ABC half transporter, having only six transmembrane domains. A recent report suggests significant inhibition of BCRP by several anti-HIV compounds. Based on in vitro IC50values, the rank order for BCRP inhibition was lopinavir>nelfinavir>delavirudine>efavirenz>saquinavir>atazanavir> amprenavir>abacavir (Weiss et al., 2007a,b). Conversely, several NRTI, NtRTI (tenofovir) and NNRT have been characterized as P-gp inducer (Rumiantsev Iu, 1991). Substrate specificity and tissue localization of MDR protein differs from MRP.
While the expression of P-gp is more restricted to tissues involved in absorption and secretion, human P-gp was initially discovered in cancer cells, and later found to be constitutively present in a number of normal tissues such as intestine, brain, liver, kidney, pancreas and adrenal gland. In the intestine, it is localized almost exclusively within the brush border region of the apical surface of mature enterocytes and thus significantly restricts oral absorption of xenobiotics. Endothelial cells at blood–brain barrier (BBB) and the choroid plexus express high levels of P-gp (Gottesman and Pastan, 1993; Ambudkar et al., 1999; Yu, 1999). All multidrug transporters are localized predominantly in the plasma membrane. In polarized cells, P-gp is localized in the apical (luminal) membrane surface such as in the epithelial cells of the intestine, proximal tubules of kidney, and in the biliary canalicular membrane of hepatocytes. MRP1 is ubiquitously expressed. Its expression in polarized cells is restricted to the basolateral membrane. Expression of MRP2, MDR3, and bile salt export pumps (BSEP/ABCB11) predominantly appear in the canalicular membrane of hepatocytes, while MRP3 and MRP5 are expressed in the basolateral membranes (Stieger et al., 2007). MRP2 is also abundant in the apical membranes of intestine and proximal tubules of kidney. BCRP is highly expressed in the placenta, liver, and most interestingly, in various stem cells (Bates et al., 2001; Zhou et al., 2001). In polarized cells BCRP expression was found to be mostly apical (Thiebaut et al., 1987; van Helvoort et al., 1996; Jonker et al., 2000; Kipp and Arias, 2000; Zhou et al., 2001). The localizations of these transporters in the intestine are depicted in the Fig. 1. While P-gp transports mostly large hydrophobic, either neutral or positively charged compounds, the MRP family primarily exports hydrophobic anionic conjugates and neutral molecules. Neutral drug transport by MRP1 is quite an enigma, and is probably linked to allosteric effect of intra-cellular transport of free reduced gluthatione (Borst et al., 2000). The exact spectrum of BCRP transported substrates has not yet been established. These efflux transporters interact directly with non-polar substrates within the membrane lipid bilayer, and may act as a drug filppase, transferring drugs from the inner to the outer leaflet. However, a clear mechanism of such translocation has not been established. As a consequence of such efflux, drug absorption is reduced and bioavailability of xenobiotics diminishes at the target sites (Sharom, 1997; Wacher et al., 1995, 1998).
Fig. 1.

Localization of efflux transporters on intestinal epithelium. ‘A’ represents ATP binding sites.
Metabolizing enzymes: Cytochrome P450
Basic hypothesis of drug–drug interactions is based on altered oral bioavailability of one drug due to altered absorption and/or metabolism in the presence of another drug. Despite the fact that liver is the primary site for metabolism, small intestine contributes significantly to break down of orally absorbed drugs (Krishna and Klotz, 1994). CYP, the principal enzyme system with several isozymes regulates metabolism of a large number of drugs. Human genome analysis has revealed the presence of about 57 CYP genes. However, about a dozen genes appear to play significant role in drug clearance (Evans and Relling, 1999; Nelson, 2002). At least six isozymes of CYP (CYP1A2, CYP2C9, CYP2C19, CYP2D6, CYP2E1 and CYP3A4/5) are responsible for oxidation of drugs in human (Slaughter and Edwards, 1995). CYP enzymes associated with drug metabolism are present in tissues such as liver, small intestine, kidney, lung, brain and placenta (Watkins, 1997; Ding and Kaminsky, 2003; Zhao and Imig, 2003; Liu et al., 2004). The major congener of CYP family is CYP3A4, which is highly expressed in intestine and liver. It is the principal enzyme responsible for metabolism of more than 70% of all drugs in human (Krishna and Klotz, 1994; Watkins, 1997). A significant amount of CYP3A4 is expressed in the brush border epithelium of small intestine (Kolars et al., 1992; Shen et al., 1997). Also, a moderate amount of CYP3A5 is polymorphically expressed in the enterocytes (Lown et al., 1994). Such localization of CYP3A4/5 in enterocytes has significant impact on drug absorption because it is capable of metabolizing xenobiotics during transport across intestinal epithelium (Shen et al., 1997).
Clinical significance of P-gp and CYP3A4 mediated interactions
A combination of metabolic enzymes and efflux proteins play central role in determining the rate and extent of xenobiotics absorption from intestinal lumen into systemic circulation (Katoh et al., 2001). Interestingly, most compounds, which are substrates for P-gp, are also substrates for CYP3A4. It is also remarkable that both CYP3A4 and P-gp are expressed in the enterocytes, liver and kidney. Because of coexpression and overlapping substrate specificities, it is plausible that both P-gp and CYP act synergistically (Chiou et al., 2000) in limiting drug absorption and metabolism and consequently bioavailability (Fig. 2). Therefore, significant impact of these two proteins on drug–drug interactions is undeniable. However, the magnitude of such interactions and their effect on individual protein depend on their spatial relationship and whether the drug is a substrate, inducer or inhibitor of CYP3A4 and /or P-gp. Interdependence of CYP3A4 and P-gp has been experimentally shown. Intestinal first-pass metabolism of indinavir augmented significantly from 6% in the control to 34% in dexamethasone treated rats (Osterberg and Norinder, 2000). Dexamethasone pretreatment caused 2.5-fold rise in both CYP3A4 and P-gp expressions in rat intestine. However, a 6-fold increase in indinavir first-pass metabolism in dexamethasone pretreated rats cannot be explained by a 2.5-fold increase in CYP3A4. In another in vitro study, metabolism of indinavir was evaluated in Caco-2 cell monolayers. Metabolism rate was 6-fold higher only when the drug solution is added on the apical side which indicates that P-gp-mediated efflux enhances indinavir metabolism (Chiba et al., 1996). Therefore, these studies indicate that enhanced expressions of P-gp and CYP3A4 and higher P-gp-mediated efflux in dexamethasone treated rats caused higher intestinal indinavir metabolism. Such coordinated function of active efflux and metabolism in the small intestine can result in poor absorption of xenobiotics (Watkins et al., 1987; Wacher et al., 1998). Moreover, it is apparent that in multidrug regimens, one agent can change the bioavailability of another through altered functions of P-gp and CYP3A4 (Fig. 2).
Fig. 2.
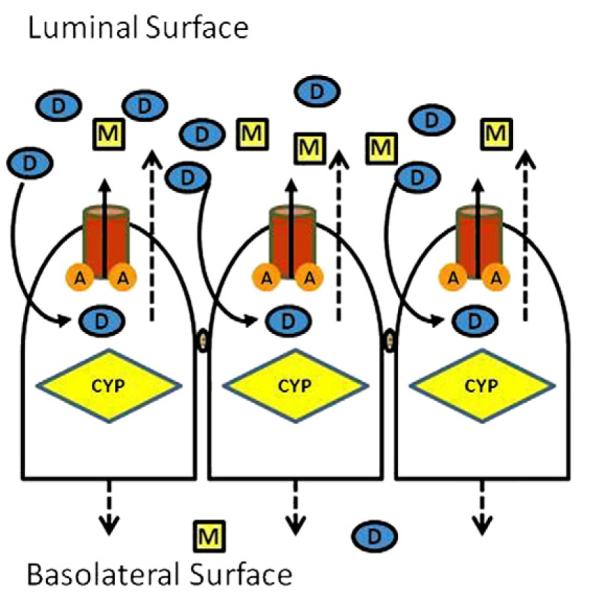
Drug (D) diffusing into the cells will be pumped out by P-gp/MRP/BCRP and have another chance to diffuse in: more metabolite (M) formed and less parent drug will cross the membrane to the blood. ‘A’ represents ATP binding sites.
For certain therapeutic agents, MDR, particularly appears to be a key determinant in drug disposition as well as elimination. Recently it has been demonstrated that changes in MDR-mediated efflux can have profound effect on the extent of drug metabolism. Also co-administration of two or more ARV agents can affect these processes due to altered functions of EP and CYP3A4 resulting in poor clinical response and therapeutic outcome. Currently preferred anti-HIV regimen utilizes two NRTI in combination with either an NNRTI, or a ritonavir boosted PI. Antiretroviral compounds can be classified into three broad groups: (A) NRTI, neither a substrate of P-gp nor CYP3A4; (B) NNRTI- metabolized by CYP3A4; and (C) PI-substrates for both EP and CYP3A4. Coadministration of any such anti-HIV compounds may induce the activity of EP and /or CYP3A4 resulting in sub-therapeutic blood levels and therapeutic failure due to reduced absorption and/or increased metabolism. A similar condition may occur when anti-retroviral compounds are combined with drugs of abuse. As a consequence, a withdrawal symptom may appear due to drug–drug/drugs of abuse interactions. Here we have outlined several important P-gp and CYP-mediated drug–drug interactions.
Antiretroviral agents
Interaction with NRTI may occur due to competition for nucleoside transporter during influx and competitive inhibition of efflux by MRP (Patel et al., 2004; Weiss et al., 2007b). As we mentioned earlier, inclusions of appropriate modulating agents which are substrates for either CYP3A4 and/or P-gp, can lead to higher oral bioavailability initially. However, interactions between two protease inhibitors in chronic therapy are not always productive. After co-administration, agents may induce the activity of efflux pump and/or CYP3A4 resulting in sub-therapeutic concentrations. Currently, more than 20 antiretroviral medications are approved for combination therapy by FDA (Table 1). Most effective combination antiretroviral therapy regimens include a PI or a NNRTI along with a backbone of 2 NRTI. Usually preferred regimens utilize two NRTI in combination with either an NNRTI or a ritonavir-boosted PI. Currently, Department of Health and Human Services (DHHS) of United States prefer antiretroviral drug regimens to include PI [atazanavir (Reyataz), darunavir (Prezista), fosamprenavir (Lexiva), and coformulated lopinavir/ritonavir (Kaletra)], NNRTI [efavirenz (Sustiva), and nevirapine (Viramune)], and NRTI [didanosine/lamivudine, lamivudine and emtricitabine, coformulated zidovudine/lamivudine (Combivir) and abacavir/lamivudine (Epzicom).
Table 1.
FDA approved medications for highly active antiretroviral therapy (HAART).
| Abbreviation | Generic name |
Brand name | Mechanism of interaction |
|---|---|---|---|
| Nucleoside/nucleotide reverse transcriptase inhibitors (NRTIs) | |||
| 3TC | Lamivudine | Epivir | P-gp substrate |
| ABC | Abacavir | Ziagen | P-gp, BCRP substrate |
| AZT or ZDV | Zidovudine | Retrovir | MRP-4,5, BCRP substrate |
| d4T | Stavudine | Zerit | P-gp substrate |
| ddI | Didanosine | Videx EC | MRP-4,5 substrate |
| FTC | Emtricitabine | Emtriva | MRP-1,5 substrate and MRP1 inhibitor |
| TDF | Tenofovir | Viread | P-gp, MRP-4 substrate |
| Non-nucleoside reverse transcriptase inhibitors (NNRTIs) | |||
| DLV | Delavirdine | Rescriptor | P-gp and BCRP inhibitor |
| EFV | Efavirenz | Sustiva (US)/ Stocrin (UK) |
P-gp and BCRP inhibitor, MRP- 1,6 inducer |
| ETR | Etravirine | Intelence | – |
| NVP | Nevirapine | Viramune | P-gp and BCRP inhibitor, P-gp inducer |
| Multi-combinations of NRTIs and NNRTIs | ||
|---|---|---|
| Abbreviation | Generic name | Brand name |
| EFV+TDF+FTC | – | |
| d4T+3TC+NVP | – | |
| AZT+3TC+NVP | – | Atripla |
| Protease inhibitors (PIs) | |||
|---|---|---|---|
| Abbreviation | Generic name | Brand name | Mechanism of interaction |
| APV | Amprenavir | Agenerase | P-gp substrate |
| FOS-APV | Fosamprenavir | Lexiva (US)/Telzir (UK) |
– |
| ATV | Atazanavir | Reyataz | P-gp substrate |
| DRV | Darunavir | Prezista | Pgp and MRP2 substrate |
| IDV | Indinavir | Crixivan | P-gp and MRP2 substrate |
| LPV/RTV | Lopinavir +ritonavir |
Kaletra/aluvia (developing world) |
– |
| NFV | Nelfinavir | Viracept | P-gp substrate |
| RTV | Ritonavir | Norvir | P-gp Substrate, MRP-1 and BCRP inhibitor |
| SQV | Saquinavir | Invirase | P-gp substrate |
| TPV | Tipranavir | Aptivus | P-gp substrate |
Interactions of drugs of abuse with ARV
Drug–drug interactions in HAART are very complex because of high prevalence of renal, hepatic, neurological and pancreatic diseases in HIV-infected population. Treatment of HIV is further complicated by substance abuse. PI and NNRTI are either substrates, inducers or, inhibitors of CYP3A4 and/or P-gp. Any compound interacting with efflux proteins and metabolizing enzymes will precipitate drug–drug interactions, which may result in altered bioavailability leading to synergism or antagonism. Interactions between recreational drugs such as benzodiazepines, cocaine, lysergic acid dithylamide (LSD), amphetamine (Meth), 3,4-methylenedioxymethamphetamine (MDMA), and opiates with anti-HIV medications may cause drug toxicity.
Interaction of benzodiazepines with ritonavir
In a recent review, Wynn et al. (2005) have described the interactions of antiretrovirals with drugs of abuse. Since many benzodiazepines (midazolam, trizolam and alprazolam) are substrates and inhibitors for P-gp and CYP3A4, simultaneous administration of ritonavir with alprazolam reduced the clearance of alprazolam initially leading to elevated alprazolam-toxicity (Greenblatt et al., 2000a). However, it was reversed following prolonged coadministration (12 days) of these two drugs. Inhibition of CYP3A4 and/or P-gp by ritonavir may lead to rise in alprazolam, midazolam and trizolam levels, and therefore, may cause toxicity. Induction of CYP3A4 and/or P-gp leads to withdrawal symptoms and therapeutic failure. In this case, AUC of alprazolam was significantly reduced (Venkatakrishnan et al., 1998). Such discrepancy may be due to ritonavir’s initial inhibitory effect on CYP3A4 followed by induction of CYP3A4 upon chronic dosing. Such effects may also result from both short term inhibition and long term induction of P-gp by ritonavir. Clinically relevant pharmacokinetic interactions between ARV and benzodiazepines are presented in Table 2. Flunitrazepam is primarily metabolized by CYP3A4 and CYP2C19. Ritonavir coadministration may result in flunitrazepam toxicity including hypotension, confusion and aggressive behavior (Smith et al., 2002). Though other benzodiazepines i.e., lorazepam, oxazepam, temazepam, and diazepam are metabolized by non CYP3A4 enzymes such as uridine 5′-diphosphate glucuronosyltransferase (UGT), UGT1A1, UGT1A3, and UGT2B7, but have the possibility to undergo drug interactions with antiretrovirals.
Table 2.
Pharmacokinetic interactions between ARV and benzodiazepines.
| ARV | Benzodiazepines | Interaction | Mechanism | Reference |
|---|---|---|---|---|
| RIT | Midazolam | Midazolam AUC ↑ by a factor of 28.4, oral clearance ↓by 4.2 | CYP3A4 inhibition by ritonavir |
(Greenblatt et al., 2009) |
| Saquinavir/ RIT |
Midazolam | Two weeks treatment ↑AUC and Cmax by 12.4 and 4.3 fold respectively Post oral midazolam AUC and Cmax ↑ by 5 and 2.3 fold respectively. i.v. ↓clearance and ↑ t1/2 of midazolam |
Inhibition of CYP3A4 activity |
(Palkama et al., 1999; Schmitt et al., 2009) |
| RIT | Ttriazolam | RIT↓triazolam clearance, ↑elimination t1/2: sedation and performance impairment | Inhibition of CYP3A4 |
(Greenblatt et al., 2000b) |
| RIT | Alprazolam | RIT↓ triazolam clearance to 41% of control, ↑elimination t1/2: sedation and performance impairment | Inhibition of CYP3A4A, |
(Greenblatt et al., 2000a) |
Interaction of MDMA and methamphetamine with ritonavir
Metabolism of both MDMA and methamphetamine was reduced significantly due to inhibition of CYP3A4 by ritonavir (Table 3). Conversely, ritonavir 500 mg twice daily lowered meperidine AUC by 67% and Cmax from 126 ng/ml to 51 ng/ml but AUC of nor-meperidine elevated by 47% indicating induction of CYP-mediated metabolism (Piscitelli et al., 2000). Both amphetamine and MDMA can cause potential interactions with ritonavir and delaviridine (Henry and Hill, 1998). MDMA is demethylated to 3, 4-dihydroxymethamphetamine primarily by CYP2D6 with minor contributions from CYP1A2, CYP2B6 and CYP3A4 (Maurer et al., 2000; Armstrong and Cozza, 2003a,b). Besides CYP3A4, ritonavir can inhibit CYP2D6, CYP2C9 and CYP2C19 (Harrington et al., 1999). Fetal drug–drug interactions has been reported in HIV patients taking MDMA with zidovudine, lamivudine and ritonavir in daily regimens (Henry and Hill, 1998). Inhibition of CYP2D6 by ritonavir is likely the cause of enhanced MDMA toxicity. In a clinical study of 14 patients with CYP2D6 genotype, a nonlinear MDMA pharmacokinetic profile was noted (de la Torre et al., 2000; Totah et al., 2007). Metabolism of amphetamine and MDMA are not completely understood. However, genetic variability of CYP2D6 and CYP2B6, and possibly P-gp-effect may cause adverse toxicity from inhibition by ritonavir, bupropion, fluoxetine and quinidine (Ketabi-Kiyanvash et al, 2003; Weiss et al., 2007a,b).
Table 3.
Clinically relevant pharmacokinetic interactions between ARV and natural/semi-synthetic opium derivatives (illicit drugs).
| ARV | Substance of abuse | Interaction | Mechanism | Reference |
|---|---|---|---|---|
| HAART | Crack-cocaine | Viral load significantly higher in cocaine users | Lower adherence to medication | (Baum et al., 2009) |
| RIT | MDMA | Cmax of MDMA ↑ by 10 fold after ingestion of RIT leading to death | Inhibition of CYP2D6 mediated metabolism of MDMA by RIT |
(Henry and Hill, 1998) |
| RIT | Methamphetamine | Fatal ↑plasma concentrations of methamphetamine upon concurrent use of RIT |
Inhibition of CYP2D6 mediated metabolism of methamphetamine by RIT |
(Hales et al., 2000) |
| RIT | Meperidine and Nor-meperidine |
Meperidine AUC ↓ by 67%, Cmax ↓ from 126 ng/ml to 51 ng/ml. Nor-meperidine AUC ↑ by 47% |
Induction of CYP-mediated hepatic metabolism of meperidine |
(Piscitelli et al., 2000) |
Interaction of cocaine with ARV
Cocaine is metabolized to nor-cocaine primarily by CYP3A4 (LeDuc et al., 1993; Ladona et al., 2000). Therefore, concomitant administration of cocaine with any PI, particularly ritonavir will result in overdose-effect due to inhibition (acute response) or withdrawal symptom due to induction of CYP3A4. Simultaneous exposure of cocaine and ritonavir (CYP3A4 inhibitor) will result in cocaine toxicity. Such action can be reversed when nevirapine and efavirenz are added to the regimens due to induction of CYP3A4. It may also shift the metabolic pathway from hydroxylation to N-demethylation producing higher levels of toxic metabolites (Pellinen et al., 1994; Bornheim, 1998). Many HIV patients use gamma-hydroxybutyrate (GHB) whose metabolism has not been clearly delineated in human. GHB toxicity in HIV-positive individuals undergoing ritonavir and saquinavir therapy has been reported by Harrington et al. (1999). The authors concluded that PI mediated inhibition of CYP3A4 was the likely cause of toxicity. The effect of ritonavir (induction or inhibition) on CYP may differ, depending on dosage and duration of therapy.
Opiate interactions with ARV
Mean trough plasma concentrations of lopinavir and atazanavir were substantially reduced in drug abuser HIV subjects relative to non abuser group (Higgins et al., 2007). A detailed review of opiate interactions with other agents has been published by Armstrong and Cozza (2003a,b). Narcotic analgesics i.e. phenylpiperidines, meperidines, fentanyl and pseudopiperidines (methadone) and propoxyphen along with alkaloids i.e. natural: heroin, morphine and codeine; semisynthetics: hydromorphone, oxymorphone, hydrocodone, oxycodone, dihydrooxycodeine and buprenorphine are widely prescribed for AIDS patients. Many of these opiates are metabolized by CYP3A4 and are also substrates/inhibitors/inducers of P-gp (Pal and Mitra, 2006a,b). In a two week clinical trial, plasma methadone concentration was substantially diminished due to ritonavir treatment and the effect was attributed to CYP3A and P-gp induction (Kharasch et al., 2008). In a similar study, these authors indicated 40–50% reduction in plasma methadone by nelfinavir (Kharasch et al., 2009). These two studies concluded that methadone metabolism is mostly regulated by CYP2B6 and CYP2C29 (Totah et al., 2007, 2008). These agents pose a serious risk of causing drug–drug interactions with multidrug regimens in HAART. Clinical pharmacokinetic interactions between ARV and methadone are presented in the Table 4. Methadone, a long-acting opiate is widely administered as therapeutic alternative to heroin dependence. Methadone administration augmented AUC of AZT by approximately 40%. However, AZT did not affect plasma methadone levels (Schwartz et al., 1992; McCance-Katz et al., 1998). Another clinical study indicates that neither ddI nor d4T affected methadone plasma concentration. However, methadone caused lowering of d4T- and ddI-AUC by 18% and 60% respectively (Staszewski et al., 1998). Administration of nevirapine resulted in narcotic withdrawal symptoms in patients stabilized on methadone. In these patients, on an average 45% increase in methadone dose was required to prevent withdrawal symptoms (Staszewski et al., 1998; Altice et al., 1999). Also, lopinavir with ritonavir (Kaletra), ritonavir, and efavirenz were found to cause opiate withdrawal when given in combination with methadone (Beauverie et al., 1998; Pinzani et al., 2000; Bart et al., 2001; McCance-Katz et al., 2000). Methadone is primarily metabolized by CYP3A4 with minor contributions from CYP2D6, CYP2C9 and CYP2E1 (Wu et al., 1993; Iribarne et al., 1996). The mechanisms behind this opiate withdrawal may involve drug–drug interaction due to induction of P-gp and CYP3A4 by antiretrovirals resulting in enhanced efflux and metabolism of methadone. In a recent review, Gourevitch and Friedland (1999a,b) have described in detail methadone interaction with anti-HIV medications.
Table 4.
Clinical pharmacokinetic interactions between ARV and methadone.
| ARV | Interaction | Mechanism | Reference |
|---|---|---|---|
| Efavirenz | Plasma conc. of R-methadone and S-methadone ↓ from 168 to 90 ng/ml, and 100 to 28 ng/ml respectively. An 80% ↑ in the dose was needed to overcome withdrawal symptoms. |
Induction of CYP3A4 | (Marzolini et al., 2000) |
| Efavirenz | The peak plasma concentration of methadone ↓from 689 to 358 ng/ml and the AUC ↓ by 67%. A 22% ↑ of methadone daily dose was required. |
Induction of CYP3A4 | (Clarke et al., 2001b) |
| Efavirenz | A 70% ↓ in methadone plasma levels after 6 days on efavirenz therapy. Stable mean plasma concentrations reached after 133% ↑ of methadone dose. |
Induction of CYP3A4 | (Boffito et al., 2002) |
| Nevirapine | One week after starting stavudine, nevirapine, nelfinavir and saquinavir therapy, patient started showing withdrawal symptoms. Methadone dose had to be adjusted from 80 mg/day to 130 mg/day. |
Induction of CYP3A4 | (Heelon and Meade, 1999) |
| Nevirapine/ efavirenz |
Inclusion of nevirapine caused symptoms similar to opiate withdrawal. Symptoms discontinued after removal of nevirapine, and recurred after nevirapine re-challenged. |
Induction of CYP3A4 | (Pinzani et al., 2000) |
| Nevirapine | Methadone dose had to be ↑ 3 patients on nevirapine therapy, peak plasma concentrations were sub-therapeutic level. |
Induction of CYP3A4 | (Altice et al., 1999) |
| Nevirapine | Development of acute opiate withdrawal symptoms after inclusion of nevirapine in the treatment regimen, 2 patients discontinued nevirapine. |
Increased metabolism due to Induction of CYP3A4 |
(Otero et al., 1999) |
| Nevirapine | Both AUC (51%) and Cmax (36%) of plasma methadone concentrations decreased when nevirapine was administered. An average of 16% ↑ in methadone dose was required. |
Induction of CYP3A4 | (Clarke et al., 2001a) |
| Nelfinavir | Subtherapeutic levels of methadone attained within 6 weeks after inclusion of nelfinavir and methadone dose was adjusted to 285 mg/day. |
Nelfinavir induced CYP3A4 mediated methadone metabolism |
(McCance-Katz et al., 2000) |
| Nelfinavir | Methadone plasma concentration ↓ by 40–50% | Nelfinavir induced renal and hepatic clearance of methadone. Induction of CYP3A4/5 by nelfinavir |
(Kharasch et al., 2009) |
| Abacavir/ amprenavir |
Plasma concentrations of methadone decreased by 35% of the control and patients reported withdrawal symptoms. |
Induction of CYP3A4 by amprenavir | (Bart et al., 2001) |
| Ritonavir | Patient experienced methadone withdrawal symptoms, dose needed to be increased from 90 mg/kg to 130 mg/kg. |
Induction of CYP3A4 by ritonavir | (Geletko and Erickson, 2000) |
| Lopinavir/ ritonavir |
A 26%↓in AUC, oral methadone clearance ↑, Cmax and Cmin each ↓ by 28% and significant ↑ in the opiate withdrawal symptoms. |
Induction of CYP3A4 mediated methadone metabolism by lopinavir |
(McCance-Katz et al., 2003) |
| Lopinavir/ ritonavir |
Removing lopinavir/ritonavir combination induced increased blood levels of methadone leading to Torsade de pointes |
Lack of methadone metabolism | (Luthi et al., 2007) |
Metabolism of heroin and morphine occur independently of CYP 450 system. These compounds are first metabolized by liver and plasma esterases and then by glucuronidation with UGT2B7 and UGT1A3. Also, several sedatives i.e. hydromorphone, oxymorphone, and dehydrocodeine undergo conjugation mainly by UGT2B7 and UGT1A3 with a minor contribution from CYP2D6. These compounds are unlikely to interact with ARV. Pharmacokinetic interactions between ARV and buprenorphine are summarized in Table 5. In contrast, semi-synthetic opiates i.e. hydrocodone, oxycodone, and buprenorphine are mainly metabolized by CYP3A4 and CYP2D6. Therefore, inhibition or induction of CYP2D6, CYP2B6 and CYP3A4 by ARV is likely to produce variable analgesic effects.
Table 5.
Clinical pharmacokinetic interactions between ARV and buprenorphine.
| ARV | Interaction | Mechanism | Reference |
|---|---|---|---|
| Atazanavir (ATZ) and atazanavir/ritonavir(RIT) |
Both ATZ alone and ATZ/RIT combination after five days of exposure caused significant ↑ in AUC and ↓ clearance of buprenorphine and its glucoronide metabolites |
Via inhibition of CYP3A4 or inhibition of secretion of glucoronides into the bile |
(McCance-Katz et al., 2007) |
| Efavirenz, delaverdine, nelfinavir, RIT, and lopinavir/RIT |
Altered metabolism M1 ↓and ↑M3 by efavirenz and ↑M1 and ↓ M3 by delavirdine. Both nelfinavir and ritonavir caused M1 and M3↓. |
While efavirenz induced CYP3A, delaverdine, nelfinavir and ritonavir inhibited CYP3A |
(Moody et al., 2009) |
| Delaverdine and RIT | Prolongation of QT intervals in opiod dependent subjects. | Inhibition of CYP3A4 | (Baker et al., 2006) |
| Tiprinavir/RIT | AUC and Cmax of tiprinavir ↓ by 19% and 25% respectively | Mechanism unclear | (Bruce et al., 2009) |
| Didanosine, atazanavir, tenofovir and RIT |
Three patients received the ARV combination therapy and resulted in ↑ levels and clinical symptoms of opiate excess |
Inhibition of CYP3A metabolism of buprenorphine by atazanavir/ritonavir |
(Bruce and Altice, 2006) |
M1, M3=Known metabolites of buprenorphine.
Opiate interactions with P-gp and BCRP
Due to the overlapping substrate specificity of P-gp and CYP3A4, opiate analgesics have been studied for their potential to interact with P-gp. Indeed, Stormer et al. (2001) have shown that methadone inhibits P-gp mediated rhodamine transport across Caco-2 cell monolayers. Similar in vitro results were reported where methadone increased cellular accumulation of P-gp substrates (Callaghan and Riordan, 1993; Nanovskaya et al., 2005). One recent report suggests the interaction of buprenorphine and its major metabolite norbuprenorphine with BCRP (Tournier et al., 2009). Several studies using P-gp knockout mice models have revealed that methadone and buprenorphine are transported by P-gp, implicating its role in modulating drug absorption and their potential in predicting clinically relevant for drug–drug interactions (Thompson et al., 2000; Hamabe et al., 2006; Suzuki et al., 2007). These studies were further corroborated by a clinical study delineating the effect of quinidine, a specific P-gp inhibitor, on oral and intravenously administered methadone (Kharasch et al., 2008). The authors concluded that co-administration of quinidine significantly elevated plasma methadone levels and consequently resulted in an increased pharmacodynamic response, elucidating the role of P-gp in methadone disposition. Since protease inhibitors are known dual substrates, inducers and inhibitors of P-gp and CYP3A4, clinicians should be made aware of potential drug interactions when prescribing ARV agents to the AIDS patients with co-morbid opiate addiction.
Genetic polymorphism in drug response
Growing evidence suggests that genetic factors influence inter-individual variation in drug response. Polymorphism has been indicated to vary with ethnicity. Mutations in genes are common which may lead to genetic polymorphism. Several reports suggest inter-individual variability in drug response is linked to single nucleotide polymorphisms (SNP). Polymorphism in a number of genes encoding for drug-metabolizing enzymes and transporters has been reported. Hoffmeyer et al. (2000) first demonstrated SNP on human P-gp through systemic screening. So far 28 SNPs have been detected at 27 positions on MDR1 gene. Mutations at exon 26 (C3435T) and 21(G2677T/A) of MDR are responsible for duodenal expression of P-gp. A significantly reduced duodenal P-gp expression in homozygous (3435TT) individuals has been linked to higher plasma digoxin level (Huisman et al., 2002). Also C3435T has been reported to be a risk factor for HIV infection (Fellay et al., 2002). Chowbay et al. (2005) have described ethnic variability among Chinese, Malays, and Indians. Pharmacogenetics of drug-metabolizing enzymes (CYP1A1, CYP3A4, CYP3A5 and UGT1A1) and MDR1 in Asian populations are different from those in Caucasian and African populations. The CYP3A4*1B allele which has an A-290G substitution in the promoter region of CYP3A4 is not present in all three Asian population studied in Singapore but present in more than 54% of Africans and 5% of Caucasians. MDR1 is a well conserved gene. However, current evidence indicates that its polymorphism affects substrate specificity. Three SNP frequently arise at positions 1236C>T, 2677G>T and 3435C>T. In a recent review Fung and Gottesman (2009) have indicated that the frequency of synonymous 3435C>T polymorphism appears to vary significantly with ethnicity. ABCB1 3435C>T genotype was also found to alter serum levels of cortisol and aldosterone during postmenstrual phase of the normal menstrual cycle (Nakamura et al., 2009). Common haplotype plays a significant role in drug response and efficacy. Even variability in CYP3A4 alleles cannot alone explain variation in CYP3A4 expression. An evidence for higher CYP3A4 expression in MDR1 2677T carriers was found in human intestine (Lamba et al., 2006). The influence of MDR1 genotype on CYP3A4 expression adds additional complexity in drug–drug interaction.
Importance of in vitro models for assessment of potential interactions between HAART medications and substance of abuse
Multidrug regimens and corresponding drug–drug interactions cause many adverse drug reactions and treatment failures. Mechanisms by which such drug interactions occur has been brieflydiscussed in some of the examples. Most of the clinical studies have inferred CYP as a principle factor for such anti-HIV medications/drugs of abuse interactions (Table 6). Involvement of P-gp and other efflux transporters in these interactions were not evaluated. In fact no systematic mechanism based study has so far been undertaken to determine these changes in drug efflux and metabolism. It is not practical to conduct clinical pharmacokinetic studies with various permutations and combinations of antiretroviral agents and drugs of abuse. Since combination therapy effectively raises the efficacy of viral reduction, it concurrently increases the probability of drug–drug interactions. Each of these anti-HIV drugs acts differently, such as substrate, inducer or inhibitor. Therefore, corresponding drug–drug interactions are very complicated. Delineation of such drug–drug interactions through clinical pharmacokinetic studies involves substantial cost, time and exposing human volunteers to a high degree of risk. The complexity of the treatment regimens employed in HIV-infected patients undergoing opiate dependence therapy and the ethical issues involved do not allow pharmaceutical companies to carry out clinical trials for potential drug interactions at a large scale. If all the required clinical studies are planned and carried out as per the traditional protocols, it is inevitable that the drug development process will be prolonged, expensive and many of the studies will be inconclusive statistically. Also, some real and potentially important interactions will be missed simply because it is not possible to do each and every needed study due to time constraints and cost. Even though, clinical studies are the ultimate means of studying drug interactions, various models have been developed to eliminate ethical and economical hurdles involved in human studies. Animal experimentation is one of the most popular non human means to study these interactions. Such studies to assess drug interactions in animals are also costly and time consuming. Moreover, involvements of efflux transporters in addition to CYP metabolizing enzymes further complicate drug disposition and metabolism. Therefore, current drug development process may need to devote substantial efforts on the generation of credible data on drug–drug interactions involving in vitro cell culture models for each new drug entity. Clearly such data may alert clinicians of potential complications as well as benefits of various drug combinations.
Table 6.
Preclinical and clinical data on the effects of alcohol and nicotine on CYP3A activity.
| Substance | Study | Interaction | Reference(s) |
|---|---|---|---|
| Alcohol | Human | Moderate alcohol consumption caused increased intestinal CYP3A activity and subsequently reduced the oral bioavailability of midazolam |
(Liangpunsakul et al., 2005) |
| Ethanol/ isopentanol |
Rats | Combined treatment resulted in a greater increase in hepatic CYP3A | (Louis et al., 1994) |
| Smoking | Human | Irinotecan AUC was significantly lowered in smokers as compared to nonsmokers. Modulation of CYP3A. | (van der Bol et al., 2007) |
| Nicotine | Human | Nicotine activated PXR and induced CYP3A4 transcription | (Lamba et al., 2004) |
PXR=Pregnane xenobiotic receptor.
Literature suggests single or two PI can cause induction/inhibition of P-gp, MRP, BCRP and CYP3A4. But our in vitro works suggest that appropriate combination of two PI can result in reduction of P-gp expression (Fig. 3). Several NNRTI and NRTI/NtRTI can cause P-gp induction and inhibition of MRP and BCRP (Weiss et al., 2007a,b). Hence, there is a great need to develop appropriate in vitro models suitable for the assessment of long term efficacy of anti-HIV treatments which can mimic the in vivo conditions. These results must enable clinicians to predict the safest and the most efficacious dosage regimen for HAART. Such pharmacokinetic studies involving several permutations and combinations of various drugs and substance of abuse are, however, possible employing in vitro cell culture models. In vitro screening techniques may play a major role in identifying the possible drug interactions and thus create an ideal stage for pivotal clinical studies to emerge. Currently MDCK cells are replacing Caco-2 for its rapid growth and expression of very tight junctions. MDCK cells transfected with MDR1/MRP2/BCRP expressing individual efflux protein lacking metabolizing enzyme CYP3A4 are not suitable model for drug interaction studies following oral dosing. MDCK cells which express specific combinations of efflux proteins (MDR1/MRP2/BCRP) and metabolizing enzymes (CYP3A4) have been developed in our laboratory to identify contribution of these proteins to drug interactions. These cell lines can be designated as MDCK-EM (efflux and metabolism) and may be suitable for in vivo predictable results employing dual chamber model (Fig. 4). MDCK-MDR1 cell line has shown induction of MDR1 mRNA in response to two PI combinations.
Fig. 3.

Effect of PI, their binary and ternary combinations on the expression of MDR1 mRNA expression in MDCK-MDR1 cells after 72 h treatment. 0.5% DMSO was used as the control. R: Ritonavir, S: Saquinavir, I: Indinavir, N: Nelfinavir. (*indicates significant difference compared to control; p<0.05, n=8±S.D.).
Fig. 4.

MDCK cell culture models for drug interaction studies. E: efflux transporter (P-gp or MRP2 or BCRP) and M: metabolizing enzyme (CYP3A4). This figure is a schematic representation of an integrated cell culture system. The upper reservoir containing MDCK-EM monolayer resembles the mucosal surface of intestine can be exposed to the given drug or combination of drugs. The lower reservoir resembling the serosal compartment contains the HepG2-transfected with CYP3A4 and/CYP2D6 mimicking liver enzymes or hepatocytes. The transported drug molecules will be metabolized in the basal chamber.
Unpublished preliminary results show that the proposed cell lines express significantly higher levels of the transfected genes. Transfected MDCK cells indeed express functionally active protein of the transfected genes (Fig. 5). Both P-gp and CYP3A4 may be functionally linked because (a) two proteins are co-localized within enterocytes, (b) share common substrates and (c) are co-inducible in response to xenobiotics (Watkins, 1997). Highest cortisol metabolite formation is observed in the MDCK-MDR1-CYP3A4 cells due to the combined activity of CYP3A5, CYP3A4 and MDR1 (Fig. 6). To further confirm these results, cellular transport of 50 μM methadone (another P-gp and CYP3A4 substrate) was carried out in transfected MDCK-MDR1 with CYP3A4 in apical to basolateral direction. Results indicate intact methadone transport in transfected cell line was significantly lower than non CYP3A4 transfected cell line (Fig. 7). This decrease in flux can be attributed to functionally active CYP3A4 in MDCK-MDR1 cells. More importantly, the results from these in vitro models correlate well with in vivo data obtained from animal studies. Subsequently in vivo oral pharmacokinetic studies for a drug combination (lopinavir/ketoconazole) were carried out in rats and compared with the results from the in vitro models for the same combination (Fig. 8 unpublished data). A 2 fold increase in permeability was observed with the combination as compared to control. Hence a clear correlation is evident between in vitro and in vivo results.
Fig. 5.

Immunoblot showing 56.5 kD band for CYP3A4 protein: lane 1) molecular marker, lane 2) MDCK-WT, lane 3) MDCK-WT-CYP3A4, lane 4) MDCK-MDR1, lane 5) MDCK-MDR1-CYP3A4, lane 6) blank, lanes 7 through 9) human intestinal microsomes. Lanes 2–5 were loaded with 20 μg microsomal protein where as lanes 7 to 9 were loaded with 10, 5 and 2.5 μg microsomal proteins respectively.
Fig. 6.

Transport of 500 μM cortisol in MDCK-WT and MDCK-MDR1 cells transfected with CYP3A4. Time dependent formation of CYP3A4-mediated metabolite of cortisol (6β-hydroxy cortisol) is shown in this figure. p<0.05, n=6±S.D.
Fig. 7.

Transport of 50 μM methadone in MDCK-MDR1 cells transfected with CYP3A4. p<0.05, n=6±S.D.
Fig. 8.
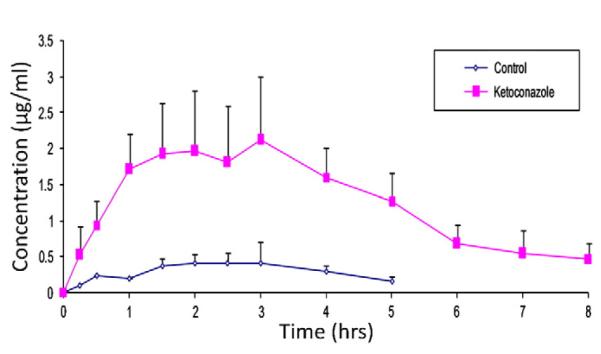
Oral pharmacokinetics of lopinavir in Sprague-Dawley rats. p<0.05, n=3±S.D.
Induction of MDR1 mRNA in MDCK-MDR1 cell line
Four protease inhibitors (ritonavir, saquinavir, nelfinavir and indinavir) showed marked increase in the expression of MDR1 mRNA (Fig. 3). However, their combination effects differed in the expression of MDR1 mRNA. Ritonavir and saquinavir combinations have completely silenced the expression MDR1 mRNA relative to control. Clinically, ritonavir causes boosting effect of lopinavir against HIV viral load in combination treatment. Results from our laboratory clearly indicate similar trend in ritonavir–saquinavir combination. The combinations of ritonavir and nelfinavir however resulted in increased expression of MDR1. These in vitro results suggest, in certain combination, such efficacy can be reversed due to induction of MDR1. While ritonavir–indonavir caused maximum induction, ritonavir–saquinavir was completely ineffective on causing induction of MDR1 mRNA. In fact this combination was comparable to ritonavir alone with 50% reduction relative to control. It is important to realize that induction or inhibition of gene expressions is dose dependent. In a clinical study, plasma methadone concentration was substantially reduced due to ritonavir treatment and the effect was attributed to CYP3A and P-gp induction (Kharasch et al., 2009), but boosting effect of ritonavir in Kaletra is reflection of its inhibitory effects on both P-gp and CYP. In former case, 300 mg of ritonavir twice daily was administered while in Kaletra only 100 mg of ritonavir with 400 mg of lopinavir are generally recommended once/twice daily. Our study suggests that judicial selection of appropriate drug combination can result in beneficial effect. In contrast inappropriate selection of drug combination may cause treatment failure with unexpected toxicity.
Induction of MDR and CYP3A4 in Caco2 and HepG2 cells in response to drugs of abuse
We hypothesize that chronic exposure to methadone, morphine and nicotine may alter expression of genes encoding for efflux transporters (e.g., P-gp, MRP, and BCRP). Since intestine is the primary barrier for oral absorption, we first examined our hypothesis in Caco-2 cells, an intestinal derived human colon-carcinoma cell line. Once confluent, cells are exposed to morphine (3.0 μM) or, nicotine (2.5 μM) for fifteen days. A significant induction in MDR1 mRNA levels was observed following chronic morphine and nicotine exposure in Caco-2 cells. Morphine and nicotine exhibited a five and four fold induction of MDR1 mRNA respectively (Fig. 9). In contrast, only nicotine exposure resulted in significant elevation of CYP3A4 mRNA in HepG2 cells (Fig. 10). Nicotine treatment raised CYP3A4 mRNA levels by six fold in HepG2 cells. In fact, apart from nicotine, both alcohol and morphine treatment for five days resulted in induction of CYP3A4 mRNA levels (Fig. 11). Dose dependent induction of BCRP by nicotine also suggests heavy smokers will generate higher expression of efflux transporter (BCRP) than non-smoker or moderate smoker (Fig. 12). Cortisol metabolism in human lung microsomes showed four-fold increase in 6-hydroxycortisol in smokers’ lung than nonsmokers (Fig. 13). Similar data have been obtained from rat study where nicotine treatment for five days resulted in enhanced cortisol metabolism in rat lung (Fig. 14). This study suggests that drugs of abuse such as alcohol, morphine and nicotine are capable of inducing P-gp and/or CYP3A4 in intestine/liver which can cause reduced drug absorption of many orally administered agents particularly those are substrates for P-gp and/or CYP3A4 (unpublished data from our laboratory). These data suggest that suitable in vitro model may be employed to generate data for drug–drug/drugs of abuse interactions. An understanding of the mechanisms of interactions between drugs of abuse and antiretroviral agents may aid in improving the treatment of AIDS.
Fig. 9.

Expression of MDR1 mRNA in Caco-2 in presence of 3 μM morphine and 2.5 μM nicotine (* indicates significant difference compared to control; p<0.05, n=3±S.D).
Fig. 10.

Quantitative changes in CYP3A4 mRNA levels following induction with 3 μM morphine and 2.5 μM nicotine in HepG2. Results are expressed as relative fold difference (* indicates significant difference compared to control; p<0.05, n=4±S.D.).
Fig. 11.

Enhanced activity of CYP3A4 in HepG2 in the presence of morphine and nicotine. HepG2 cells were treated with DMSO (0.1% v/v), morphine (3 μM), nicotine (2.5 μM), omeprazole (100 μm) and rifampicin (50 μm). (* indicates significant difference relative to control; p<0.05, n=8±S.D).
Fig. 12.
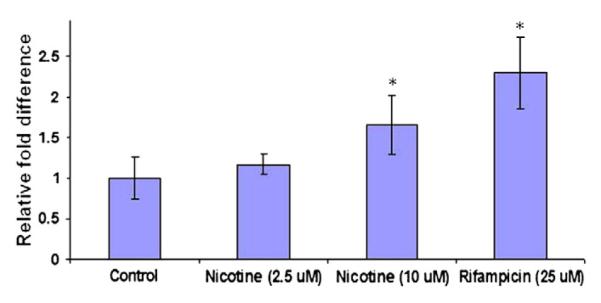
Dose dependent induction of BCRP in Calu-3 cells by nicotine suggests heavy smokers will have more expression of efflux transporter (BCRP) than nonsmoker or moderate smoker. (* indicates significant difference compared to control; p value <0.05%, n=3±S.D).
Fig. 13.

Cortisol metabolism in human lung microsomes (commercially available). (* indicates significant difference compared to control; p value <0.05%).
Fig. 14.
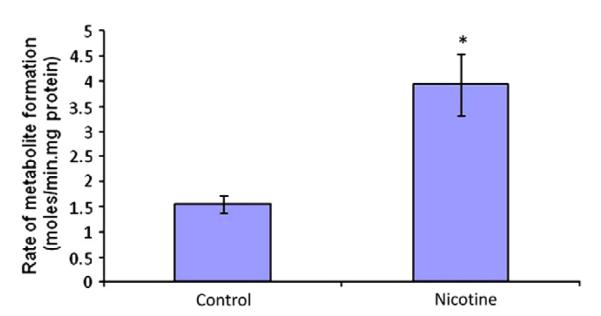
Cortisol metabolism in lung microsomes from control and nicotine (3.0 mg /kg body weight, twice daily for five days) treated rats. (* indicates significant difference compared to control; p value <0.05%).
In combination therapy, one drug can also potentially change clinical outcomes of the other drug. Such interactions include induction or inhibition of metabolizing enzymes and/or drug efflux proteins. Therefore, mechanism based studies involving gene and functional expression of various efflux proteins (MDR1, MRP and BCRP), and metabolizing enzymes (CYP) following chronic exposure to selected anti-HIV medications along with drugs of abuse need to be carried out in vitro and drug–drug interaction database must be created. Preliminary results from our laboratory also indicate an up-regulation of MDR1 mRNA and protein expression in Caco-2 cells following chronic exposure to nicotine and morphine. Similarly MDR1 and BCRP mRNA expressions are also elevated in rat brain. Moreover, morphine and nicotine caused induction of CYP3A4 mRNA expression and functional activity in HepG2 cells. A properly validated in vitro model can quantitatively estimate such drug–drug interactions of clinical significance utilizing both metabolic and efflux pathways. Such in vitro models are now currently available in our laboratory. Such database will allow a priori dose adjustment to avoid toxicity, treatment failure and maximize the efficacy of HAART medications particularly in AIDS patients indulging in drugs of abuse. It remains unclear what final outcome would be when two PIs along with NNRTI and/or NRTI are given to HIV patients indulging drugs of abuse. What impact such combinations make on these efflux transporters and/or CYP? Will such combinations induce more efflux or reduce metabolism? Can such combinations along with substance of abuse result in treatment failure for HIV infection? What would be the outcome if HIV patients have SNP? All these questions are yet to be answered.
Conclusions
P-gp, originally found as an adaptive response of cancer cells towards chemotherapeutic agents, is now well recognized, broadly distributed and constitutively expressed protein present in many tissues and organs. We are just beginning to realize that P-gp along with other efflux transporters (MRP and BCRP) having overlapping substrate specificity plays critical role in drug disposition across many biological barriers. Many drug–drug interactions can be explained by altered drug absorption and metabolism due to enhanced or reduced functions of efflux transporters (MDR gene products) along with metabolizing enzymes (CYPs) in response to drug combinations. Many exhaustive reviews have emerged focusing on various aspects of these efflux transporter and metabolizing enzyme based interactions (Gourevitch and Friedland, 1999a; Pal and Mitra, 2006a; Troutman et al., 2008). Apart from efflux transporters and metabolizing enzymes, multiple organic anion transporter (MOAT) involved in drug elimination process in the kidney can also play a critical role in this drug interaction process. P-gpco-localizing with CYP in the intestine, acts synergistically for ARV absorption with enhanced metabolism (Fig. 2). P-gp not only limits drug absorption but participate in excretion of xenobiotics and their metabolites, minimizing ARV systemic as well as tissue/organ exposure. Also the presence of P-gp in many sensitive organs (brain, testis, placenta), provide a driving force in limiting ARV exposure to HIV sanctuary sites. Therefore, induction or inhibition of such efflux transporters and metabolizing enzymes may change both therapeutic and toxicity profiles. Such knowledge may alert healthcare professional about possible drug interactions and allow dosage adjustment. Appropriate selection of ARV and their dosages may result in desired therapeutic outcomes. Moreover, genetic polymorphisms of drug transporter and metabolizing enzyme may be responsible for many adverse drug interactions. It is very important that potential ARV/drugs of abuse interactions should be identified either by in vitro and/or in vivo testing at an early stage of drug development process, so that a priori informed decision could be made to avoid adverse drug interactions. Many factors can influence drug–drug interactions such as food, herbal supplement, substance abuse, disease and genetic polymorphisms (Pal and Mitra, 2006b). Elucidation of these factors and their relationship with P-gp will definitely advance our ability in predicting anti-HIV medication drugs of abuse interaction. Interaction between HAART and drugs of abuse may lower the efficacy of HAART which may result in higher plasma viral load in HIV patients. Particularly, HIV-infected pregnant women, even with HAART medications, indulging in cocaine, may also develop higher viral load due to drug–drug interactions and consequently lead to increase in mother to child transmission of HIV. Complexity underlying the relationship between P-gp and CYP makes it difficult to predict the outcome of HAART as such, particularly when HIV patients indulging in drugs of abuse do not adhere to treatment regimens. However, a multidisciplinary approach along with a clear understanding of the mechanism of drug–drug interactions may allow proper selection of drug regimens so that desired therapeutic outcome of HAART can be reached generating no unwanted side effects.
Acknowledgement
Support by NIH grants GM 64320-03 and U19 AT003264.
Footnotes
Mother-to-child transmission of HIV and drugs of abuse: Post-HARRT era.
Conflict of interest statement The authors declare that there are no conflicts of interest.
References
- Altice FL, Friedland GH, Cooney EL. Nevirapine induced opiate withdrawal among injection drug users with HIV infection receiving methadone. AIDS. 1999;13(8):957–62. doi: 10.1097/00002030-199905280-00012. [DOI] [PubMed] [Google Scholar]
- Ambudkar SV, Dey S, Hrycyna CA, Ramachandra M, Pastan I, Gottesman MM. Biochemical, cellular, and pharmacological aspects of the multidrug transporter. Annu Rev Pharmacol Toxicol. 1999;39:361–98. doi: 10.1146/annurev.pharmtox.39.1.361. [DOI] [PubMed] [Google Scholar]
- Armstrong SC, Cozza KL. Pharmacokinetic drug interactions of morphine, codeine, and their derivatives: theory and clinical reality, part I. Psychosomatics. 2003a;44(2):167–71. doi: 10.1176/appi.psy.44.2.167. [DOI] [PubMed] [Google Scholar]
- Armstrong SC, Cozza KL. Pharmacokinetic drug interactions of morphine, codeine, and their derivatives: theory and clinical reality, Part II. Psychosomatics. 2003b;44(6):515–20. doi: 10.1176/appi.psy.44.6.515. [DOI] [PubMed] [Google Scholar]
- Baker JR, Best AM, Pade PA, McCance-Katz EF. Effect of buprenorphine and antiretroviral agents on the QT interval in opioid-dependent patients. Ann Pharmacother. 2006;40(3):392–6. doi: 10.1345/aph.1G524. [DOI] [PubMed] [Google Scholar]
- Bakos E, Evers R, Sinko E, Varadi A, Borst P, Sarkadi B. Interactions of the human multidrug resistance proteins MRP1 and MRP2 with organic anions. Mol Pharmacol. 2000;57(4):760–8. doi: 10.1124/mol.57.4.760. [DOI] [PubMed] [Google Scholar]
- Bart PA, Rizzardi PG, Gallant S, Golay KP, Baumann P, Pantaleo G, et al. Methadone blood concentrations are decreased by the administration of abacavir plus amprenavir. Ther Drug Monit. 2001;23(5):553–5. doi: 10.1097/00007691-200110000-00010. [DOI] [PubMed] [Google Scholar]
- Bates SE, Robey R, Miyake K, Rao K, Ross DD, Litman T. The role of half-transporters in multidrug resistance. J Bioenerg Biomembr. 2001;33(6):503–11. doi: 10.1023/a:1012879205914. [DOI] [PubMed] [Google Scholar]
- Baum MK, Rafie C, Lai S, Sales S, Page B, Campa A. Crack-cocaine use accelerates HIV disease progression in a cohort of HIV-positive drug users. J Acquir Immune Defic Syndr. 2009;50(1):93–9. doi: 10.1097/QAI.0b013e3181900129. [DOI] [PubMed] [Google Scholar]
- Beauverie P, Taburet AM, Dessalles MC, Furlan V, Touzeau D. Therapeutic drug monitoring of methadone in HIV-infected patients receiving protease inhibitors. AIDS. 1998;12(18):2510–1. [PubMed] [Google Scholar]
- Boffito M, Rossati A, Reynolds HE, Hoggard PG, Back DJ, DiPerri G. Undefined duration of opiate withdrawal induced by efavirenz in drug users with HIV infection and undergoing chronic methadone treatment. AIDS Res Hum Retroviruses. 2002;18(5):341–2. doi: 10.1089/088922202753519115. [DOI] [PubMed] [Google Scholar]
- Bornheim LM. Effect of cytochrome P450 inducers on cocaine-mediated hepatotoxicity. Toxicol Appl Pharmacol. 1998;150(1):158–65. doi: 10.1006/taap.1998.8403. [DOI] [PubMed] [Google Scholar]
- Borst P, Evers R, Kool M, Wijnholds J. A family of drug transporters: the multidrug resistance-associated proteins. J Natl Cancer Inst. 2000;92(16):1295–302. doi: 10.1093/jnci/92.16.1295. [DOI] [PubMed] [Google Scholar]
- Bruce RD, Altice FL. Three case reports of a clinical pharmacokinetic interaction with buprenorphine and atazanavir plus ritonavir. AIDS. 2006;20(5):783–4. doi: 10.1097/01.aids.0000216384.22432.9a. [DOI] [PubMed] [Google Scholar]
- Bruce RD, Altice FL, Moody DE, Lin SN, Fang WB, Sabo JP, et al. Pharmacokinetic interactions between buprenorphine/naloxone and tipranavir/ritonavir in HIV-negative subjects chronically receiving buprenorphine/naloxone. Drug Alcohol Depend. 2009;105(3):234–9. doi: 10.1016/j.drugalcdep.2009.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callaghan R, Riordan JR. Synthetic and natural opiates interact with P-glycoprotein in multidrug-resistant cells. J Biol Chem. 1993;268(21):16059–64. [PubMed] [Google Scholar]
- Chiba P, Ecker G, Schmid D, Drach J, Tell B, Goldenberg S, et al. Structural requirements for activity of propafenone-type modulators in P-glycoprotein-mediated multidrug resistance. Mol Pharmacol. 1996;49(6):1122–30. [PubMed] [Google Scholar]
- Chiou WL, Chung SM, Wu TC. Potential role of P-glycoprotein in affecting hepatic metabolism of drugs. Pharm Res. 2000;17(8):903–5. doi: 10.1023/a:1007570517183. [DOI] [PubMed] [Google Scholar]
- Chowbay B, Zhou S, Lee EJ. An interethnic comparison of polymorphisms of the genes encoding drug-metabolizing enzymes and drug transporters: experience in Singapore. Drug Metab Rev. 2005;37(2):327–78. doi: 10.1081/dmr-28805. [DOI] [PubMed] [Google Scholar]
- Clarke SM, Mulcahy FM, Tjia J, Reynolds HE, Gibbons SE, Barry MG, et al. Pharmacokinetic interactions of nevirapine and methadone and guidelines for use of nevirapine to treat injection drug users. Clinical infectious diseases : an official publication of the Infectious Diseases Society of America. 2001a;33(9):1595–7. doi: 10.1086/322519. [DOI] [PubMed] [Google Scholar]
- Clarke SM, Mulcahy FM, Tjia J, Reynolds HE, Gibbons SE, Barry MG, et al. The pharmacokinetics of methadone in HIV-positive patients receiving the non-nucleoside reverse transcriptase inhibitor efavirenz. Br J Clin Pharmacol. 2001b;51(3):213–7. doi: 10.1046/j.1365-2125.2001.00342.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Torre R, Farre M, Ortuno J, Mas M, Brenneisen R, Roset PN, et al. Non-linear pharmacokinetics of MDMA (‘ecstasy’) in humans. Br J Clin Pharmacol. 2000;49(2):104–9. doi: 10.1046/j.1365-2125.2000.00121.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding X, Kaminsky LS. Human extrahepatic cytochromes P450: function in xenobiotic metabolism and tissue-selective chemical toxicity in the respiratory and gastrointestinal tracts. Annu Rev Pharmacol Toxicol. 2003;43:149–73. doi: 10.1146/annurev.pharmtox.43.100901.140251. [DOI] [PubMed] [Google Scholar]
- Evans WE, Relling MV. Pharmacogenomics: translating functional genomics into rational therapeutics. Science. 1999;286(5439):487–91. doi: 10.1126/science.286.5439.487. [DOI] [PubMed] [Google Scholar]
- Fellay J, Marzolini C, Meaden ER, Back DJ, Buclin T, Chave JP, et al. Response to antiretroviral treatment in HIV-1-infected individuals with allelic variants of the multidrug resistance transporter 1: a pharmacogenetics study. Lancet. 2002;359(9300):30–6. doi: 10.1016/S0140-6736(02)07276-8. [DOI] [PubMed] [Google Scholar]
- Fung KL, Gottesman MM. A synonymous polymorphism in a common MDR1 (ABCB1) haplotype shapes protein function. Biochim Biophys Acta. 2009;1794(5):860–71. doi: 10.1016/j.bbapap.2009.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geletko SM, Erickson AD. Decreased methadone effect after ritonavir initiation. Pharmacotherapy. 2000;20(1):93–4. doi: 10.1592/phco.20.1.93.34654. [DOI] [PubMed] [Google Scholar]
- Gottesman MM, Pastan I. Biochemistry of multidrug resistance mediated by the multidrug transporter. Annu Rev Biochem. 1993;62:385–427. doi: 10.1146/annurev.bi.62.070193.002125. [DOI] [PubMed] [Google Scholar]
- Gourevitch MN, Friedland GH. Methadone and antiretroviral medications, part I. AIDS Clin Care. 1999a;11(4):30–31. contd. [PubMed] [Google Scholar]
- Gourevitch MN, Friedland GH. Methadone and antiretroviral medications, part II. AIDS Clin Care. 1999b;11(5):37, 43, 45–36. [PubMed] [Google Scholar]
- Greenblatt DJ, von Moltke LL, Harmatz JS, Durol AL, Daily JP, Graf JA, et al. Alprazolamritonavir interaction: implications for product labeling. Clin Pharmacol Ther. 2000a;67(4):335–41. doi: 10.1067/mcp.2000.105757. [DOI] [PubMed] [Google Scholar]
- Greenblatt DJ, von Moltke LL, Harmatz JS, Durol AL, Daily JP, Graf JA, et al. Differential impairment of triazolam and zolpidem clearance by ritonavir. J Acquir Immune Defic Syndr. 2000b;24(2):129–36. doi: 10.1097/00126334-200006010-00007. [DOI] [PubMed] [Google Scholar]
- Greenblatt DJ, Peters DE, Oleson LE, Harmatz JS, MacNab MW, Berkowitz N, et al. Inhibition of oral midazolam clearance by boosting doses of ritonavir, and by 4, 4-dimethyl-benziso-(2H)-selenazine (ALT-2074), an experimental catalytic mimic of glutathione oxidase. Br J Clin Pharmacol. 2009;68(6):920–7. doi: 10.1111/j.1365-2125.2009.03545.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hales G, Roth N, Smith D. Possible fatal interaction between protease inhibitors and methamphetamine. Antivir Ther. 2000;5(1):19. [PubMed] [Google Scholar]
- Hamabe W, Maeda T, Fukazawa Y, Kumamoto K, Shang LQ, Yamamoto A, et al. P-glycoprotein ATPase activating effect of opioid analgesics and their P-glycoprotein-dependent antinociception in mice. Pharmacol Biochem Behav. 2006;85(3):629–36. doi: 10.1016/j.pbb.2006.10.018. [DOI] [PubMed] [Google Scholar]
- Harrington RD, Woodward JA, Hooton TM, Horn JR. Life-threatening interactions between HIV-1 protease inhibitors and the illicit drugs MDMA and gamma-hydroxybutyrate. Arch Intern Med. 1999;159(18):2221–4. doi: 10.1001/archinte.159.18.2221. [DOI] [PubMed] [Google Scholar]
- Heelon MW, Meade LB. Methadone withdrawal when starting an antiretroviral regimen including nevirapine. Pharmacotherapy. 1999;19(4):471–2. doi: 10.1592/phco.19.6.471.31046. [DOI] [PubMed] [Google Scholar]
- Henry JA, Hill IR. Fatal interaction between ritonavir and MDMA. Lancet. 1998;352(9142):1751–2. doi: 10.1016/s0140-6736(05)79824-x. [DOI] [PubMed] [Google Scholar]
- Higgins N, Zingman BS, Slish J, Reichman RC, Fischl MA, Gripshover B, et al. Factors associated with altered pharmacokinetics in substance users and non-substance users receiving lopinavir and atazanavir. The Am J Addict. 2007;16(6):488–94. doi: 10.1080/10550490701641256. [DOI] [PubMed] [Google Scholar]
- Hoffmeyer S, Burk O, von Richter O, Arnold HP, Brockmoller J, Johne A, et al. Functional polymorphisms of the human multidrug-resistance gene: multiple sequence variations and correlation of one allele with P-glycoprotein expression and activity in vivo. Proc Natl Acad Sci USA. 2000;97(7):3473–8. doi: 10.1073/pnas.050585397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huisman MT, Smit JW, Crommentuyn KM, Zelcer N, Wiltshire HR, Beijnen JH, et al. Multidrug resistance protein 2 (MRP2) transports HIV protease inhibitors, and transport can be enhanced by other drugs. AIDS. 2002;16(17):2295–301. doi: 10.1097/00002030-200211220-00009. [DOI] [PubMed] [Google Scholar]
- Hulot JS, Villard E, Maguy A, Morel V, Mir L, Tostivint I, et al. A mutation in the drug transporter gene ABCC2 associated with impaired methotrexate elimination. Pharmacogenet Genomics. 2005;15(5):277–85. doi: 10.1097/01213011-200505000-00002. [DOI] [PubMed] [Google Scholar]
- Iribarne C, Berthou F, Baird S, Dreano Y, Picart D, Bail JP, et al. Involvement of cytochrome P450 3A4 enzyme in the N-demethylation of methadone in human liver microsomes. Chem Res Toxicol. 1996;9(2):365–73. doi: 10.1021/tx950116m. [DOI] [PubMed] [Google Scholar]
- Jonker JW, Smit JW, Brinkhuis RF, Maliepaard M, Beijnen JH, Schellens JH, et al. Role of breast Cancer researchistance protein in the bioavailability and fetal penetration of topotecan. J Natl Cancer Inst. 2000;92(20):1651–6. doi: 10.1093/jnci/92.20.1651. [DOI] [PubMed] [Google Scholar]
- Katoh M, Nakajima M, Yamazaki H, Yokoi T. Inhibitory effects of CYP3A4 substrates and their metabolites on P-glycoprotein-mediated transport. Eur J Pharm Sci. 2001;12(4):505–13. doi: 10.1016/s0928-0987(00)00215-3. [DOI] [PubMed] [Google Scholar]
- Katragadda S, Budda B, Anand BS, Mitra AK. Role of efflux pumps and metabolising enzymes in drug delivery. Expert Opin Drug Deliv. 2005;2(4):683–705. doi: 10.1517/17425247.2.4.683. [DOI] [PubMed] [Google Scholar]
- Ketabi-Kiyanvash N, Weiss J, Haefeli WE, Mikus G. P-glycoprotein modulation by the designer drugs methylenedioxymethamphetamine, methylenedioxyethylamphetamine and paramethoxyamphetamine. Addict Biol. 2003;8(4):413–8. doi: 10.1080/13556210310001646475. [DOI] [PubMed] [Google Scholar]
- Kharasch ED, Bedynek PS, Walker A, Whittington D, Hoffer C. Mechanism of ritonavir changes in methadone pharmacokinetics and pharmacodynamics: II. Ritonavir effects on CYP3A and P-glycoprotein activities. Clin Pharmacol Ther. 2008;84(4):506–12. doi: 10.1038/clpt.2008.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kharasch ED, Walker A, Whittington D, Hoffer C, Bedynek PS. Methadone metabolism and clearance are induced by nelfinavir despite inhibition of cytochrome P4503A (CYP3A) activity. Drug Alcohol Depend. 2009;101(3):158–68. doi: 10.1016/j.drugalcdep.2008.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kipp H, Arias IM. Intracellular trafficking and regulation of canalicular ATP-binding cassette transporters. Semin Liver Dis. 2000;20(3):339–51. doi: 10.1055/s-2000-9388. [DOI] [PubMed] [Google Scholar]
- Kolars JC, Schmiedlin-Ren P, Schuetz JD, Fang C, Watkins PB. Identification of rifampininducible P450IIIA4 (CYP3A4) in human small bowel enterocytes. J Clin Investig. 1992;90(5):1871–8. doi: 10.1172/JCI116064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishna DR, Klotz U. Extrahepatic metabolism of drugs in humans. Clin Pharmacokinet. 1994;26(2):144–60. doi: 10.2165/00003088-199426020-00007. [DOI] [PubMed] [Google Scholar]
- Krishnamachary N, Center MS. The MRP gene associated with a non-P-glycoprotein multidrug resistance encodes a 190-kDa membrane bound glycoprotein. Cancer Res. 1993;53(16):3658–61. [PubMed] [Google Scholar]
- Ladona MG, Gonzalez ML, Rane A, Peter RM, de la Torre R. Cocaine metabolism in human fetal and adult liver microsomes is related to cytochrome P450 3A expression. Life Sci. 2000;68(4):431–43. doi: 10.1016/s0024-3205(00)00952-8. [DOI] [PubMed] [Google Scholar]
- Lamba V, Yasuda K, Lamba JK, Assem M, Davila J, Strom S, et al. PXR (NR1I2): splice variants in human tissues, including brain, and identification of neurosteroids and nicotine as PXR activators. Toxicol Appl Pharmacol. 2004;199(3):251–65. doi: 10.1016/j.taap.2003.12.027. [DOI] [PubMed] [Google Scholar]
- Lamba J, Strom S, Venkataramanan R, Thummel KE, Lin YS, Liu W, et al. MDR1 genotype is associated with hepatic cytochrome P450 3A4 basal and induction phenotype. Clin Pharmacol Ther. 2006;79(4):325–38. doi: 10.1016/j.clpt.2005.11.013. [DOI] [PubMed] [Google Scholar]
- Larkin A, O’Driscoll L, Kennedy S, Purcell R, Moran E, Crown J, et al. Investigation of MRP-1 protein and MDR-1 P-glycoprotein expression in invasive breast cancer: a prognostic study. Int J Cancer. 2004;112(2):286–94. doi: 10.1002/ijc.20369. [DOI] [PubMed] [Google Scholar]
- LeDuc BW, Sinclair PR, Shuster L, Sinclair JF, Evans JE, Greenblatt DJ. Norcocaine and N-hydroxynorcocaine formation in human liver microsomes: role of cytochrome P-450 3A4. Pharmacology. 1993;46(5):294–300. doi: 10.1159/000139058. [DOI] [PubMed] [Google Scholar]
- Leslie EM, Deeley RG, Cole SP. Toxicological relevance of the multidrug resistance protein 1, MRP1 (ABCC1) and related transporters. Toxicology. 2001;167(1):3–23. doi: 10.1016/s0300-483x(01)00454-1. [DOI] [PubMed] [Google Scholar]
- Letourneau IJ, Bowers RJ, Deeley RG, Cole SP. Limited modulation of the transport activity of the human multidrug resistance proteins MRP1, MRP2 and MRP3 by nicotine glucuronide metabolites. Toxicol Lett. 2005;157(1):9–19. doi: 10.1016/j.toxlet.2004.12.014. [DOI] [PubMed] [Google Scholar]
- Liangpunsakul S, Kolwankar D, Pinto A, Gorski JC, Hall SD, Chalasani N. Activity of CYP2E1 and CYP3A enzymes in adults with moderate alcohol consumption: a comparison with nonalcoholics. Hepatology. 2005;41(5):1144–50. doi: 10.1002/hep.20673. [DOI] [PubMed] [Google Scholar]
- Liu M, Hurn PD, Alkayed NJ. Cytochrome P450 in neurological disease. Curr Drug Metab. 2004;5(3):225–34. doi: 10.2174/1389200043335540. [DOI] [PubMed] [Google Scholar]
- Louis CA, Wood SG, Kostrubsky V, Sinclair PR, Sinclair JF. Synergistic increases in rat hepatic cytochrome P450s by ethanol and isopentanol. J Pharmacol Exp Ther. 1994;269(2):838–45. [PubMed] [Google Scholar]
- Lown KS, Kolars JC, Thummel KE, Barnett JL, Kunze KL, Wrighton SA, et al. Interpatient heterogeneity in expression of CYP3A4 and CYP3A5 in small bowel. Lack of prediction by the erythromycin breath test. Drug Metab Dispos. 1994;22(6):947–55. [PubMed] [Google Scholar]
- Luthi B, Huttner A, Speck RF, Mueller NJ. Methadone-induced Torsade de pointes after stopping lopinavir–ritonavir. Eur J Clin Microbiol Infect Dis. 2007;26(5):367–9. doi: 10.1007/s10096-007-0293-5. [DOI] [PubMed] [Google Scholar]
- Marzolini C, Troillet N, Telenti A, Baumann P, Decosterd LA, Eap CB. Efavirenz decreases methadone blood concentrations. AIDS. 2000;14(9):1291–2. doi: 10.1097/00002030-200006160-00036. [DOI] [PubMed] [Google Scholar]
- Maurer HH, Bickeboeller-Friedrich J, Kraemer T, Peters FT. Toxicokinetics and analytical toxicology of amphetamine-derived designer drugs (‘Ecstasy’) Toxicol Lett. 2000;112–113:133–42. doi: 10.1016/s0378-4274(99)00207-6. [DOI] [PubMed] [Google Scholar]
- McCance-Katz EF, Rainey PM, Jatlow P, Friedland G. Methadone effects on zidovudine disposition (AIDS Clinical Trials Group 262) J Acquir Immune Defic Syndr Hum Retrovirol. 1998;18(5):435–43. doi: 10.1097/00042560-199808150-00004. [DOI] [PubMed] [Google Scholar]
- McCance-Katz EF, Farber S, Selwyn PA, O’Connor A. Decrease in methodone levels with nelfinavir mesylate. Am J Psychiatry. 2000;157(3):481. doi: 10.1176/appi.ajp.157.3.481. [DOI] [PubMed] [Google Scholar]
- McCance-Katz EF, Rainey PM, Friedland G, Jatlow P. The protease inhibitor lopinavir–ritonavir may produce opiate withdrawal in methadone-maintained patients. Clinical infectious diseases : an official publication of the Infectious Diseases Society of America. 2003;37(4):476–82. doi: 10.1086/376907. [DOI] [PubMed] [Google Scholar]
- McCance-Katz EF, Moody DE, Morse GD, Ma Q, DiFrancesco R, Friedland G, et al. Interaction between buprenorphine and atazanavir or atazanavir/ritonavir. Drug Alcohol Depend. 2007;91(2–3):269–78. doi: 10.1016/j.drugalcdep.2007.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moody DE, Chang Y, Huang W, McCance-Katz EF. The in vivo response of novel buprenorphine metabolites, M1 and M3, to antiretroviral inducers and inhibitors of buprenorphine metabolism. Basic Clin Pharmacol Toxicol. 2009;105(3):211–5. doi: 10.1111/j.1742-7843.2009.00432.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura T, Okamura N, Yagi M, Omatsu H, Yamamori M, Kuwahara A, et al. Effects of ABCB1 3435C>T genotype on serum levels of cortisol and aldosterone in women with normal menstrual cycles. Genet Mol Res. 2009;8(2):397–403. doi: 10.4238/vol8-2gmr574. [DOI] [PubMed] [Google Scholar]
- Nanovskaya T, Nekhayeva I, Karunaratne N, Audus K, Hankins GD, Ahmed MS. Role of P-glycoprotein in transplacental transfer of methadone. Biochem Pharmacol. 2005;69(12):1869–78. doi: 10.1016/j.bcp.2005.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naruhashi K, Tamai I, Inoue N, Muraoka H, Sai Y, Suzuki N, et al. Involvement of multidrug resistance-associated protein 2 in intestinal secretion of grepafloxacin in rats. Antimicrob Agents Chemother. 2002;46(2):344–9. doi: 10.1128/AAC.46.2.344-349.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson DR. Introductory remarks on human CYPs. Drug Metab Rev. 2002;34(1–2):1–5. doi: 10.1081/dmr-120001385. [DOI] [PubMed] [Google Scholar]
- Osterberg T, Norinder U. Theoretical calculation and prediction of P-glycoprotein-interacting drugs using MolSurf parametrization and PLS statistics. Eur J Pharm Sci. 2000;10(4):295–303. doi: 10.1016/s0928-0987(00)00077-4. [DOI] [PubMed] [Google Scholar]
- Otero MJ, Fuertes A, Sanchez R, Luna G. Nevirapine-induced withdrawal symptoms in HIV patients on methadone maintenance programme: an alert. AIDS. 1999;13(8):1004–5. doi: 10.1097/00002030-199905280-00025. [DOI] [PubMed] [Google Scholar]
- Pal D, Mitra AK. CYP3A4 and MDR mediated interactions in drug therapy. Clin Res Regul Aff. 2006a;23(3):125–63. [Google Scholar]
- Pal D, Mitra AK. MDR- and CYP3A4-mediated drug-herbal interactions. Life Sci. 2006b;78(18):2131–45. doi: 10.1016/j.lfs.2005.12.010. [DOI] [PubMed] [Google Scholar]
- Palkama VJ, Ahonen J, Neuvonen PJ, Olkkola KT. Effect of saquinavir on the pharmacokinetics and pharmacodynamics of oral and intravenous midazolam. Clin Pharmacol Ther. 1999;66(1):33–9. doi: 10.1016/S0009-9236(99)70051-2. [DOI] [PubMed] [Google Scholar]
- Patel J, Buddha B, Dey S, Pal D, Mitra AK. In vitro interaction of the HIV protease inhibitor ritonavir with herbal constituents: changes in P-gp and CYP3A4 activity. Am J Ther. 2004;11(4):262–77. doi: 10.1097/01.mjt.0000101827.94820.22. [DOI] [PubMed] [Google Scholar]
- Pellinen P, Honkakoski P, Stenback F, Niemitz M, Alhava E, Pelkonen O, et al. Cocaine N-demethylation and the metabolism-related hepatotoxicity can be prevented by cytochrome P450 3A inhibitors. Eur J Pharmacol. 1994;270(1):35–43. doi: 10.1016/0926-6917(94)90078-7. [DOI] [PubMed] [Google Scholar]
- Pinzani V, Faucherre V, Peyriere H, Blayac JP. Methadone withdrawal symptoms with nevirapine and efavirenz. Ann Pharmacother. 2000;34(3):405–7. doi: 10.1345/aph.19156. [DOI] [PubMed] [Google Scholar]
- Piscitelli SC, Kress DR, Bertz RJ, Pau A, Davey R. The effect of ritonavir on the pharmacokinetics of meperidine and normeperidine. Pharmacotherapy. 2000;20(5):549–53. doi: 10.1592/phco.20.6.549.35162. [DOI] [PubMed] [Google Scholar]
- Ruetz S, Gros P. Phosphatidylcholine translocase: a physiological role for the mdr2 gene. Cell. 1994;77(7):1071–81. doi: 10.1016/0092-8674(94)90446-4. [DOI] [PubMed] [Google Scholar]
- Iu V Rumiantsev. Oxygen barotherapy in kidney diseases. Vrach Delo. 1991;10:26–31. [PubMed] [Google Scholar]
- Schinkel AH, Mayer U, Wagenaar E, Mol CA, van Deemter L, Smit JJ, et al. Normal viability and altered pharmacokinetics in mice lacking mdr1-type (drug-transporting) P-glycoproteins. Proc Natl Acad Sci USA. 1997;94(8):4028–33. doi: 10.1073/pnas.94.8.4028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt C, Hofmann C, Riek M, Patel A, Zwanziger E. Effect of saquinavir–ritonavir on cytochrome P450 3A4 activity in healthy volunteers using midazolam as a probe. Pharmacotherapy. 2009;29(10):1175–81. doi: 10.1592/phco.29.10.1175. [DOI] [PubMed] [Google Scholar]
- Schwartz EL, Brechbuhl AB, Kahl P, Miller MA, Selwyn PA, Friedland GH. Pharmaco-kinetic interactions of zidovudine and methadone in intravenous drug-using patients with HIV infection. J Acquir Immune Defic Syndr. 1992;5(6):619–26. [PubMed] [Google Scholar]
- Sharom FJ. The P-glycoprotein efflux pump: how does it transport drugs? J Membr Biol. 1997;160(3):161–75. doi: 10.1007/s002329900305. [DOI] [PubMed] [Google Scholar]
- Shen DD, Kunze KL, Thummel KE. Enzyme-catalyzed processes of first-pass hepatic and intestinal drug extraction. Ed Drug Deliv Rev. 1997;27(2–3):99–127. doi: 10.1016/s0169-409x(97)00039-2. [DOI] [PubMed] [Google Scholar]
- Slaughter RL, Edwards DJ. Recent advances: the cytochrome P450 enzymes. Ann Pharmacother. 1995;29(6):619–24. doi: 10.1177/106002809502900612. [DOI] [PubMed] [Google Scholar]
- Smith KM, Larive LL, Romanelli F. Club drugs: methylenedioxymethamphetamine, flunitrazepam, ketamine hydrochloride, and gamma-hydroxybutyrate. Am J Health-Syst Pharm. 2002;59(11):1067–76. doi: 10.1093/ajhp/59.11.1067. [DOI] [PubMed] [Google Scholar]
- Staszewski S, Haberl A, Gute P, Nisius G, Miller V, Carlebach A. Nevirapine/didanosine/lamivudine once daily in HIV-1-infected intravenous drug users. Antivir Ther. 1998;3(Suppl 4):55–6. [PubMed] [Google Scholar]
- Stieger B, Meier Y, Meier PJ. The bile salt export pump. Pflügers Arch Eur J Physiol. 2007;453(5):611–20. doi: 10.1007/s00424-006-0152-8. [DOI] [PubMed] [Google Scholar]
- Stormer E, Perloff MD, von Moltke LL, Greenblatt DJ. Methadone inhibits rhodamine123 transport in Caco-2 cells. Drug Metab Dispos. 2001;29(7):954–6. [PubMed] [Google Scholar]
- Suzuki T, Zaima C, Moriki Y, Fukami T, Tomono K. P-glycoprotein mediates brain-to-blood efflux transport of buprenorphine across the blood–brain barrier. J Drug Target. 2007;15(1):67–74. doi: 10.1080/10611860601141606. [DOI] [PubMed] [Google Scholar]
- Thiebaut F, Tsuruo T, Hamada H, Gottesman MM, Pastan I, Willingham MC. Cellular localization of the multidrug-resistance gene product P-glycoprotein in normal human tissues. Proc Natl Acad Sci USA. 1987;84(21):7735–8. doi: 10.1073/pnas.84.21.7735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson SJ, Koszdin K, Bernards CM. Opiate-induced analgesia is increased and prolonged in mice lacking P-glycoprotein. Anesthesiology. 2000;92(5):1392–9. doi: 10.1097/00000542-200005000-00030. [DOI] [PubMed] [Google Scholar]
- Totah RA, Allen KE, Sheffels P, Whittington D, Kharasch ED. Enantiomeric metabolic interactions and stereoselective human methadone metabolism. J Pharmacol Exp Ther. 2007;321(1):389–99. doi: 10.1124/jpet.106.117580. [DOI] [PubMed] [Google Scholar]
- Totah RA, Sheffels P, Roberts T, Whittington D, Thummel K, Kharasch ED. Role of CYP2B6 in stereoselective human methadone metabolism. Anesthesiology. 2008;108(3):363–74. doi: 10.1097/ALN.0b013e3181642938. [DOI] [PubMed] [Google Scholar]
- Tournier N, Chevillard L, Megarbane B, Pirnay S, Scherrmann JM, Decleves X. Interaction of drugs of abuse and maintenance treatments with human P-glycoprotein (ABCB1) and breast Cancer researchistance protein (ABCG2) Int J Neuropsychopharmacol. 2009:1–11. doi: 10.1017/S1461145709990848. [DOI] [PubMed] [Google Scholar]
- Troutman M, Luo G, Knight B, Thakker D, Gan L-S. The Role of P-Glycoprotein in Drug Disposition: Significance to Drug Development. In: Rodrigues D, editor. Drug–Drug Interactions. informa healthcare; NY: 2008. [Google Scholar]
- van der Bol JM, Mathijssen RH, Loos WJ, Friberg LE, van Schaik RH, de Jonge MJ, et al. Cigarette smoking and irinotecan treatment: pharmacokinetic interaction and effects on neutropenia. J Clin Oncol. 2007;25(19):2719–26. doi: 10.1200/JCO.2006.09.6115. [DOI] [PubMed] [Google Scholar]
- van Helvoort A, Smith AJ, Sprong H, Fritzsche I, Schinkel AH, Borst P, et al. MDR1 P-glycoprotein is a lipid translocase of broad specificity, while MDR3 P-glycoprotein specifically translocates phosphatidylcholine. Cell. 1996;87(3):507–17. doi: 10.1016/s0092-8674(00)81370-7. [DOI] [PubMed] [Google Scholar]
- Venkatakrishnan K, Greenblatt DJ, von Moltke LL, Shader RI. Alprazolam is another substrate for human cytochrome P450-3A isoforms. J Clin Psychopharmacol. 1998;18(3):256. doi: 10.1097/00004714-199806000-00015. [DOI] [PubMed] [Google Scholar]
- Wacher VJ, Wu CY, Benet LZ. Overlapping substrate specificities and tissue distribution of cytochrome P450 3A and P-glycoprotein: implications for drug delivery and activity in cancer chemotherapy. Mol Carcinog. 1995;13(3):129–34. doi: 10.1002/mc.2940130302. [DOI] [PubMed] [Google Scholar]
- Wacher VJ, Silverman JA, Zhang Y, Benet LZ. Role of P-glycoprotein and cytochrome P450 3A in limiting oral absorption of peptides and peptidomimetics. J Pharm Sci. 1998;87(11):1322–30. doi: 10.1021/js980082d. [DOI] [PubMed] [Google Scholar]
- Watkins PB. The barrier function of CYP3A4 and P-glycoprotein in the small bowel. Ed Drug Deliv Rev. 1997;27(2–3):161–70. doi: 10.1016/s0169-409x(97)00041-0. [DOI] [PubMed] [Google Scholar]
- Watkins PB, Wrighton SA, Schuetz EG, Molowa DT, Guzelian PS. Identification of glucocorticoid-inducible cytochromes P-450 in the intestinal mucosa of rats and man. J Clin Investig. 1987;80(4):1029–36. doi: 10.1172/JCI113156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss J, Rose J, Storch CH, Ketabi-Kiyanvash N, Sauer A, Haefeli WE, et al. Modulation of human BCRP (ABCG2) activity by anti-HIV drugs. J Antimicrob Chemother. 2007a;59(2):238–45. doi: 10.1093/jac/dkl474. [DOI] [PubMed] [Google Scholar]
- Weiss J, Theile D, Ketabi-Kiyanvash N, Lindenmaier H, Haefeli WE. Inhibition of MRP1/ABCC1, MRP2/ABCC2, and MRP3/ABCC3 by nucleoside, nucleotide, and nonnucleoside reverse transcriptase inhibitors. Drug Metab Dispos. 2007b;35(3):340–4. doi: 10.1124/dmd.106.012765. [DOI] [PubMed] [Google Scholar]
- Wu D, Otton SV, Sproule BA, Busto U, Inaba T, Kalow W, et al. Inhibition of human cytochrome P450 2D6 (CYP2D6) by methadone. Br J Clin Pharmacol. 1993;35(1):30–4. doi: 10.1111/j.1365-2125.1993.tb05666.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wynn GH, Cozza KL, Zapor MJ, Wortmann GW, Armstrong SC. Med-psych drug–drug interactions update. Antiretrovirals, part III: antiretrovirals and drugs of abuse. Psychosomatics. 2005;46(1):79–87. doi: 10.1176/appi.psy.46.1.79. [DOI] [PubMed] [Google Scholar]
- Xiao JJ, Foraker AB, Swaan PW, Liu S, Huang Y, Dai Z, et al. Efflux of depsipeptide FK228 ( FR901228, NSC-630176) is mediated by P-glycoprotein and multidrug resistance-associated protein 1. J Pharmacol Exp Ther. 2005;313(1):268–76. doi: 10.1124/jpet.104.072033. [DOI] [PubMed] [Google Scholar]
- Yu DK. The contribution of P-glycoprotein to pharmacokinetic drug–drug interactions. J Clin Pharmacol. 1999;39(12):1203–11. doi: 10.1177/00912709922012006. [DOI] [PubMed] [Google Scholar]
- Zaman GJ, Flens MJ, van Leusden MR, de Haas M, Mulder HS, Lankelma J, et al. The human multidrug resistance-associated protein MRP is a plasma membrane drug-efflux pump. Proc Natl Acad Sci USA. 1994;91(19):8822–6. doi: 10.1073/pnas.91.19.8822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X, Imig JD. Kidney CYP450 enzymes: biological actions beyond drug metabolism. Curr Drug Metab. 2003;4(1):73–84. doi: 10.2174/1389200033336892. [DOI] [PubMed] [Google Scholar]
- Zhou S, Schuetz JD, Bunting KD, Colapietro AM, Sampath J, Morris JJ, et al. The ABC transporter Bcrp1/ABCG2 is expressed in a wide variety of stem cells and is a molecular determinant of the side-population phenotype. Nat Med. 2001;7(9):1028–34. doi: 10.1038/nm0901-1028. [DOI] [PubMed] [Google Scholar]