

Abstract
Mammary gland growth and differentiation during pregnancy is a developmental process that is sensitive to the toxic effects of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD). TCDD is a widespread environmental contaminant and a potent ligand for the aryl hydrocarbon receptor (AhR). We demonstrate reduced β-casein protein induction in mouse mammary glands and in cultured SCp2 mammary epithelial cells following exposure to TCDD. SCp2 cells exposed to TCDD also show reduced cell clustering and less alveolar-like structure formation. SCp2 cells express transcriptionally active AhR, and exposure to TCDD induces expression of the AhR target gene CYP1B1. Exposure to TCDD during pregnancy reduced expression of the cell adhesion molecule E-cadherin in the mammary gland and decreased phosphorylation of STAT5, a known regulator of β-casein gene expression. These data provide morphological and molecular evidence that TCDD-mediated AhR activation disrupts structural and functional differentiation of the mammary gland, and present an in vitro model for studying the effects of TCDD on mammary epithelial cell function.
Keywords: TCDD, Aryl hydrocarbon receptor, Mammary gland, Mouse, Pregnancy, SCp2 cell, E-cadherin, STAT5 phosphorylation
1. Introduction
The aryl hydrocarbon receptor (AhR) is a ligand-activated transcription factor with widespread expression throughout many tissues and cell types. The mechanism of action of AhR in regulating the transcription of its target genes is well-characterized [1-3]. Prior to ligand binding, AhR is primarily cytosolic and in complex with molecular chaperones such as Hsp90, XAP2, and p23. Once activated through ligand binding, AhR translocates to the cell nucleus and dimerizes with the aryl hydrocarbon nuclear translocator (ARNT) [4]. The AhR/ARNT complex further associates with coregulator proteins that modulate interaction of the complex with aryl hydrocarbon receptor response elements (AhREs) found in the upstream promoter regions of various target genes [5-9]. The normal physiological functions of AhR are beginning to be explored; however, much of what is known about the AhR has resulted from studies using xenobiotic ligands to activate receptor function [3, 10]. Inappropriate activation of AhR by exogenous ligands, such as 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD), has been shown to cause disruption of many cellular and developmental processes, including cell proliferation, fate determination, and differentiation [2]. AhR, through AhRE binding, is known to regulate genes involved in drug metabolism, cell cycle, proliferation, differentiation, and other cellular processes [3, 10], implicating direct gene regulation as a target for disruption by xenobiotic chemicals.
The capacity of AhR agonists to cause endocrine disruption has been demonstrated in both the male and female reproductive systems, with effects observed in wildlife, experimental animals, and humans [11-15]. Hormone signaling pathways may be sensitive to extremely low dose exposure to various chemicals [16], and TCDD has been strongly implicated in disruption of estrogen-mediated signaling [17, 18]. There are various mechanisms by which AhR has been shown to affect estrogen receptor or other nuclear receptor signaling pathways, and these include modulation of hormone synthesis and/or metabolism, direct transcriptional regulation of nuclear receptors, stimulation of nuclear receptor degradation, as well as competition for coregulators necessary for transcriptional regulation by nuclear receptors [18]. Additionally, as a lipophilic, persistent organic pollutant, TCDD exposure may increase over time via bioaccumulation [19] [20].
The mammary gland serves as both a likely target for bioaccumulation of xenobiotic AhR ligands, as well as a model system to study how AhR regulates many aspects of development. The mammary gland develops rudimentary ductal structures during fetal and prepubertal stages, and these structures develop further at puberty in the female. However, it is not until pregnancy that the full developmental process proceeds with extensive cell proliferation and differentiation, resulting in further ductal branching and development of the milk-producing alveolar structures. Upon the weaning of offspring, the mammary gland undergoes involution, which is characterized by widespread apoptosis [21]. Our laboratory discovered that mice exposed to TCDD during pregnancy display impaired mammary gland differentiation and an inability to nutritionally support their offspring. Expression of the milk protein WAP gene (whey acidic protein) was also reduced in TCDD-exposed dams [22]. In mice, the coordinated induction of milk protein genes occurs around the 9th day of pregnancy [23], and we have found that the coordinated induction of these genes is impaired by TCDD (our unpublished data). These changes occurred in the absence of alterations in circulating estradiol, progesterone and prolactin levels [22], suggesting that AhR modulates other pathways important for mammary gland differentiation. Further evidence that AhR ligands disrupt mammary gland differentiation during pregnancy was provided in a study using mammary gland explants from estrogen and progesterone-primed mice that were then maintained under hormonal stimulation in culture. The explants were also exposed to the AhR agonist ligand 2,3,7,8-tetrachlorodibenzofuran, which acted as a negative regulator of mammary gland growth and differentiation by suppressing development of lobuloalveolar structures [24]. In order to determine the molecular mechanisms responsible for AhR-mediated disruption of mammary gland development and function, we have continued to study the effects of TCDD exposure in mice during pregnancy, and we have extended our studies using SCp2 mammary epithelial cells in culture.
Mammary gland development during pregnancy involves the differentiation of epithelial cells through formation of alveolar structures, cell polarization, and the production, secretion, and luminal sequestration of milk proteins [25, 26]. Primary mouse mammary epithelial cells and derived cell lines, cultured on extracellular matrix (ECM) in the presence of lactogenic hormones, have been shown to undergo morphological and functional changes that mirror those that occur in the mammary gland of the pregnant mouse [27-30]. The SCp2 mammary epithelial cell line is a clonal derivation of cells isolated from BALB/c mice at mid-pregnancy [29]. The original COMMA-1D cell line was heterogeneous, with only a subset of the cells expressing β-casein after the addition of ECM and lactogenic hormones [31]. COMMA-1D cells were then enriched for ECM-responsive, β-casein producing cells that were designated CID-9 [32]. Finally, the homogeneous SCp2 cell line was generated through expansion of a single cell-derived clone isolated from the CID-9 cell line [29]. SCp2 cells have a distinctly epithelial morphology, express cytokeratins, and lack the fibroblast-type filament protein vimentin. These characteristics, along with the capacity for β-casein expression, suggest that the SCp2 cell clone originated from mouse mammary epithelial cells.
The differentiation potential of SCp2 cells has been well-characterized, beginning with morphological and functional response to ECM and lactogenic hormones. When cultured with lactogenic hormones, but without ECM, less than 0.1% of cells express β-casein. With the addition of ECM, more than 90% of SCp2 cells form cell aggregates and express β-casein [29], demonstrating dependence on the addition of exogenous ECM for differentiation in culture. In contrast, the fibroblastic cell line SCg6, also derived from a CID-9 clone, did not form cell aggregates or express β-casein under any of the inductive culture conditions, indicating that a mixture of cell types was present in the parental cell line [29]. Further analysis showed that even in permissive cultures, SCp2 cells that remained at the periphery of cell clusters did not express β-casein, underscoring the importance of cell aggregation for functional differentiation [29].
As a functional endpoint for our studies, we have focused on impaired expression of the milk protein β-casein. At the molecular level, we have also found changes in expression of the cell adhesion molecule E-cadherin along with defects in activation of STAT5, a transcription factor essential for mammary gland development and lactogenesis [33]. Our results demonstrate an in vitro model system used to assess the effects of TCDD on mammary epithelial cell differentiation and identify TCDD-mediated defects in molecular pathways that are important for normal mammary gland function.
2. Materials and methods
2.1 In vivo TCDD exposure
TCDD (Cambridge Isotopes, Cambridge, MA) was dissolved in anisole and diluted in peanut oil. Vehicle (VEH) control consisted of peanut oil containing an equivalent concentration of anisole. Female C57BL/6 mice were treated by gavage with 5 μg/kg TCDD on days of pregnancy (DP) 0 and 7, or on DP0, 7 and 14, as described previously [22]. RNA was isolated from DP9 mammary glands using Trizol reagent (Invitrogen, Carlsbad, CA). Purity, integrity and concentration of the RNA in each sample were determined using a spectrophotometer and gel electrophoresis. Individual samples from each mouse were hybridized separately to Affymetrix Murine Genome chips (Set 430A), using an Affymetrix instrument system (scanner, fluidics station, and hybridization oven). Raw data were examined using Microarray Suite software (Affymetrix, Santa Clara, CA) and the overall quality of each chip was visually examined and found to be within normal parameters. Quality control using spiked genes and housekeeping genes was conducted to evaluate each chip. Mammary glands collected on DP17 and homogenized for preparation of protein extracts for immunoblot analyses. Protein concentrations were determined by the BCA assay (Pierce, Rockford, IL) according to the manufacturer’s instructions.
2.2 SCp2 cell culture
The SCp2 mouse mammary epithelial cell line [29] was maintained in DMEM-F12 medium (Gibco, Grand Island, NY) containing 5 μg/mL insulin (Sigma Chemical Co., St. Louis, MO), 50 μg/mL Gentamycin (Gibco, Grand Island, NY), and 5% fetal bovine serum (HyClone, Logan, UT). To induce differentiation, cells were plated on growth factor-reduced Matrigel (BD Biosciences, San Jose, CA) in serum-free DMEM-F12 medium and treated with 3-6 μg/mL prolactin (Sigma Chemical Co., St. Louis, MO) and 1 μg/mL hydrocortisone (Pharmacia & Upjohn, Bridgewater, NJ). Cells were exposed to either vehicle (0.1% DMSO, Sigma Chemical Co., St. Louis, MO) or 1 nM TCDD at the time of plating, or after a delay of 24 or 48 hours relative to the time of plating. The culture medium containing prolactin, hydrocortisone, and either vehicle or TCDD was replaced daily for 7-8 days. SCp2 cell morphology and differentiation- induced cluster formation was monitored using an Olympus CKX41 inverted microscope. Cell cultures were photographed using the Spot Insight Firewire 4 digital camera.
2.3 Preparation of SCp2 cell extracts
SCp2 cells were harvested using BD Cell Recovery Solution (BD Biosciences, San Jose, CA) according to the manufacturer’s instructions. Protein extracts were prepared for immunoblot analyses. Briefly, extraction buffer (25 mM HEPES pH 7.4, 5 mM MgCl2, 1 mM EDTA, 1mM dithiothreitol, 0.1% Triton X-100, and protease inhibitors) was added to harvested cells and samples were incubated on ice for 20 min. Samples were then centrifuged at 14K RPM, at 4°C, for 20 min. Sample supernatants were removed for analysis and/or storage.
2.4 Immunoblotting
Proteins were fractionated on either 8% or 10% polyacrylamide gels and transferred to PVDF membrane (PerkinElmer, Waltham, MA). Membranes were probed with antibodies specific for ß-casein (1:1000 or 1:500; M. Bissell or Santa Cruz Biotechnology, Santa Cruz, CA, respectively), AhR (1:2500; BioMol, Plymouth Meeting, PA), CYP1B1 (1:500; Santa Cruz Biotechnology, Santa Cruz, CA), E-cadherin (1:2500; BD Biosciences, San Jose, CA), phosphorylated STAT5A/B (pSTAT5A/B, 1:500; Millipore, Billerica, MA), STAT5 (1:500; Santa Cruz Biotechnology, Santa Cruz, CA), or actin (1:5000; Sigma Chemical Co., St. Louis, MO) overnight at 4°C. Each membrane was then probed with the appropriate horseradish peroxidase (HRP)-conjugated secondary antibody, and proteins were visualized using Western Lightning chemiluminescent substrate (Perkin Elmer, Waltham, MA) and film exposure. Immunoblot films were scanned and blot band density was analyzed using ImageJ v.1.38x.
2.5 Statistics
Student’s t-tests or 1-way ANOVA were used to evaluate immunoblot densitometry data (GraphPad Prism 4.0). Results were considered statistically significant at p<0.05. Results are expressed as mean ± standard error of the mean (SEM). Sample sizes are indicated in the figure legends.
3. Results
3.1 Activation of AhR by TCDD reduces the expression of β-casein both in vivo and in vitro
Mouse whole mammary tissue harvested at day 17 of pregnancy (Fig. 1A) and differentiated SCp2 cells collected after 8d in culture (Fig. 1C) were subjected to immunoblot analyses, revealing reduced levels of β-casein protein following exposure to TCDD. Densitometric analysis of immunoblots indicated significantly reduced β-casein expression after TCDD exposure in both mammary tissue (Fig. 1B, p<0.05) and SCp2 cells (Fig. 1D, p<0.05). When the addition of TCDD to SCp2 cell cultures was delayed by 24h or 48h relative to the time of plating, the effects of TCDD on the suppression of β-casein induction were lessened (Fig 1E and F). This indicates that very early exposure to TCDD during differentiation of cultured mammary epithelial cells is required for the most profound suppression of β-casein production.
Fig. 1.
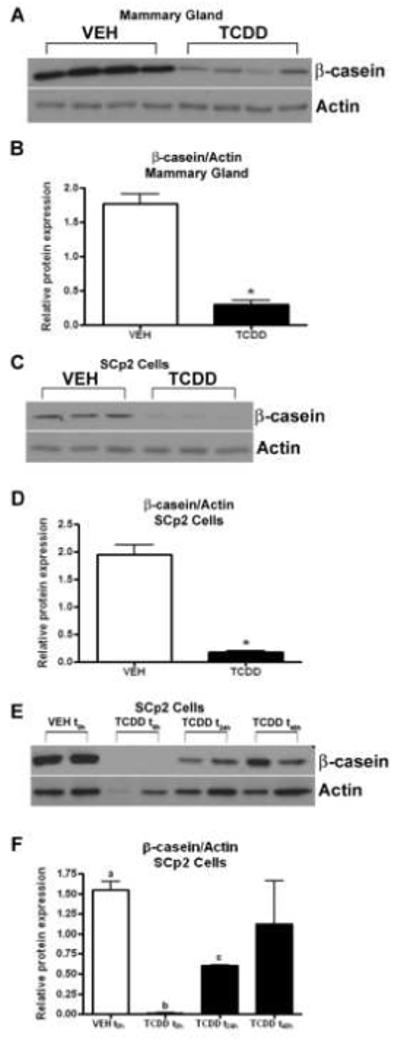
β-casein protein expression following TCDD exposure both in vivo and in vitro. Induction of β-casein is reduced in the mammary gland at day 17 of pregnancy (A) and in differentiated SCp2 cells (C) after exposure to TCDD. The bar graphs (B and D) depict results of densitometric analysis. Data represent mean ± SEM (n=3-4; *p<0.05 compared to vehicle control). Mammary gland: VEH (peanut oil), TCDD (5 μg/kg). SCp2 Cells: VEH (0.1% DMSO), TCDD (1 nM). Delayed addition of TCDD to SCp2 cell cultures reduced effects of TCDD on the suppression of β-casein induction (E and F). Data represent mean ± SEM (n=2; p<0.01, a vs b and a vs c; p<0.05, b vs c). h, hours; t0h, t24h, t48hindicate the time of first exposure to TCDD. Results are representative of at least two independent experiments.
3.2 Differentiating SCp2 cells exposed to TCDD show reduced alveolar-like structure formation
SCp2 cells induced to differentiate with lactogenic hormones were exposed to vehicle (0.1% DMSO) or TCDD (1 nM) for 8d. Cells were photographed early following plating and exposure to TCDD (24h), and just prior to harvest (8d) (Fig. 2). At both early and late time points, cells exposed to TCDD appeared to undergo less cell clustering, and formed fewer and smaller alveolar-like structures compared to vehicle-exposed control cells.
Fig. 2.
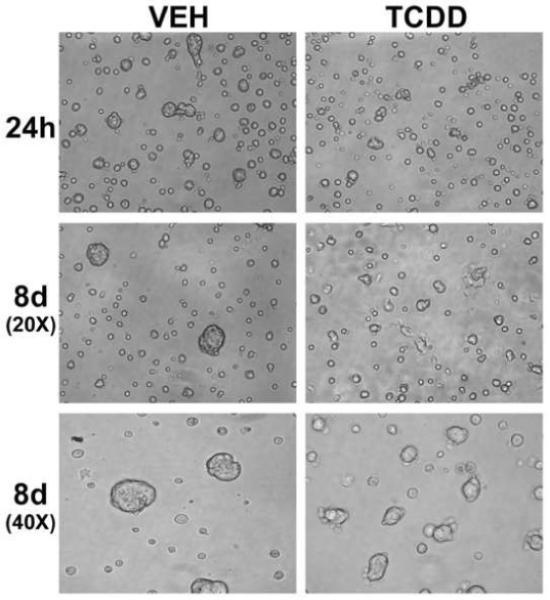
Morphology of differentiating SCp2 cells exposed to TCDD. Photomicrographs of SCp2 cells, induced to differentiate and exposed to vehicle (0.1% DMSO) or TCDD (1 nM), were taken at 24h and 8d after plating. Reduced alveolar-like structure formation is observed after TCDD exposure. Images of the 8d cultures were obtained using both low (20X) and high (40X) magnification objective lenses. h, hours; d, days.
3.3 AhR is expressed and transcriptionally active in the mouse mammary gland and SCp2 cells in culture
Immunoblot analyses of mouse mammary tissue collected at day 17 of pregnancy (Fig. 3A) and of differentiated SCp2 cells harvested after 8d in culture (Fig. 3B) demonstrate strong expression of AhR protein. AhR protein levels decline following exposure to TCDD in both model systems. Furthermore, undifferentiated SCp2 cells harvested after 24h of exposure to TCDD (Fig. 3C) show increased expression of the AhR target gene CYP1B1. TCDD treatment also induces cyp1a1 and cyp1b1 gene expression in mammary tissue of virgin and pregnant mice (Fig. 3D), further supporting functional activation of AhR in this tissue, and parallel effects of AhR activation on cultured mammary epithelial cells and whole tissue.
Fig. 3.
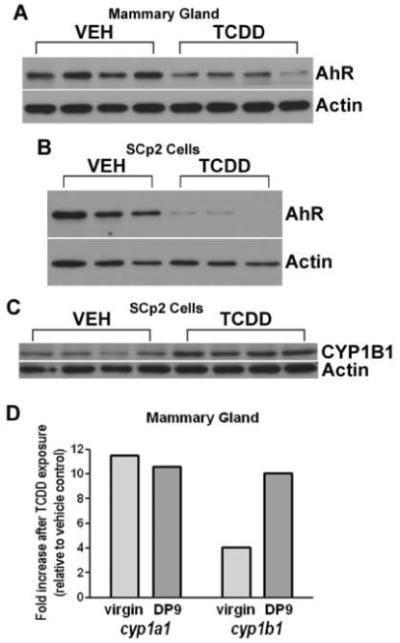
AhR and CYP1B1 expression in mammary tissue and SCp2 cells following TCDD exposure. AhR protein expression is evident in the day of pregnancy (DP) 17 mammary gland (A) and in differentiated SCp2 cells (B), with reduced expression following exposure to TCDD. SCp2 cells analyzed 24h after exposure to TCDD show increased expression CYP1B1 protein (C). Cyp1a1 and cyp1b1 gene expression is induced in mammary glands from both virgin and pregnant mice exposed to TCDD (D). Age-matched virgin mice (n=2/group) were treated with two doses of vehicle or TCDD administered 9 and 2 days prior to sacrifice. Impregnated mice were treated with vehicle or TCDD (n=3/group) on DP0 and DP7, and glands were collected for analysis on DP9. The graph depicts the average fold-change in gene expression in TCDD-exposed mammary glands relative to mammary glands from vehicle-treated control virgins or DP9 dams (expression in TCDD sample/expression in VEH sample). Statistical comparisons were made using original expression data, and mean gene expression was significantly different between vehicle-treated and TCDD-treated mammary glands in both virgin and DP9 groups (p<0.05). Mammary gland: VEH (peanut oil), TCDD (5 μg/kg). SCp2 Cells: VEH (0.1% DMSO), TCDD (1 nM).
3.4 Exposure to TCDD reduces the expression of E-cadherin both in vivo and in vitro
Mouse whole mammary tissue harvested at day 17 of pregnancy (Fig. 4A) and differentiated SCp2 cells collected after 8d in culture (Fig. 4C) were subjected to immunoblot analyses, showing reduced levels of E-cadherin protein following exposure to TCDD. Densitometric analysis of immunoblots (Fig. 4B and D) indicate significantly reduced E-cadherin expression after TCDD exposure in whole mammary tissue (Fig. 4B, p<0.05).
Fig. 4.

E-cadherin levels following TCDD exposure both in vivo and in vitro. Expression of E-cadherin protein is reduced in the mammary gland at day 17 of pregnancy (A) and in differentiated SCp2 cells (C) after exposure to TCDD. The bar graphs (B and D) depict results of densitometric analysis. Data represent mean ± SEM (n=3-4); p-values indicate comparison to vehicle control. Mammary gland: VEH (peanut oil), TCDD (5 μg/kg). SCp2 Cells: VEH (0.1% DMSO), TCDD (1 nM). Results are representative of at least two independent experiments.
3.5 Exposure to TCDD reduces the level of STAT5 phosphorylation in mammary glands during pregnancy
Immunoblot analyses of mouse whole mammary tissue harvested at day 17 of pregnancy show reduced levels of phosphorylated STAT5 (pSTAT5) protein following exposure to TCDD (Fig. 5A). The levels of total STAT5 protein were not altered by TCDD exposure. Densitometric analysis of immunoblots indicates a significant reduction in STAT5 phosphorylation after TCDD exposure (Fig. 5B, p<0.05) with no change in total STAT5 expression (Fig. 5C).
Fig. 5.
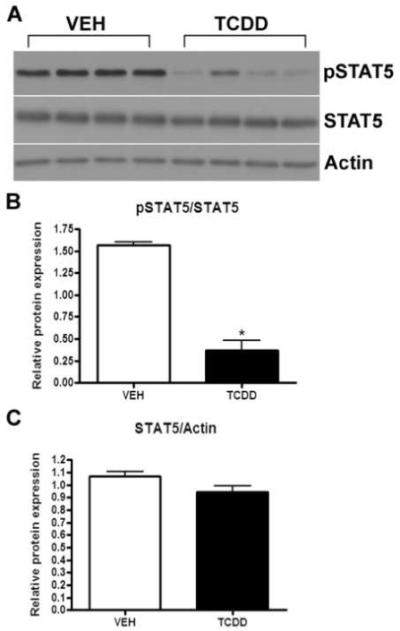
Levels of STAT5 phosphorylation in TCDD-exposed mammary glands during pregnancy. Expression of pSTAT5 is reduced in the mammary gland at day 17 of pregnancy, while expression of total STAT5 is unchanged, after exposure to TCDD (A). The bar graphs (B and C) depict results of densitometric analysis. Data represent mean ± SEM (n=3-4; *p<0.05 compared to vehicle control). Mammary gland: VEH (peanut oil), TCDD (5 μg/kg). Results are representative of two independent experiments.
4. Discussion
Our results describe a new in vitro model to examine the effects of TCDD on mammary epithelial cell differentiation and reveal defects in molecular pathways critical for normal mammary gland function. To demonstrate a functional mammary epithelial cell culture model, we first confirmed that milk protein expression, represented by β-casein protein levels, is reduced in SCp2 cells following TCDD exposure. Importantly, this reduction in β-casein expression in mammary epithelial cells parallels that observed in the mammary glands of mice exposed to the same toxicant during pregnancy. AhR is expressed in the mouse mammary gland and both cyp1a1 and cyp1b1 are induced by TCDD in this tissue. Through experiments to determine whether the disruptive effects of TCDD on mammary epithelial cell function in vitro could be the result of AhR activation, we confirmed expression of both AhR and the TCDD-induced AhR target gene CYP1B1 [34, 35] at the protein level in SCp2 cells. An additional indication of AhR activation by TCDD is the reduction in AhR protein levels in both whole mammary gland and SCp2 cells in culture following TCDD exposure. Ligand-induced activation of ubiquitin-dependent proteosomal degradation of AhR has been characterized in other model systems and is a hallmark downstream effect of transcriptional activation of AhR by TCDD [36, 37]. Finally, the changes in cultured mammary epithelial cell morphology following TCDD exposure is another indication of disrupted development of alveolar-like structures and mirrors the defects in late-stage alveolar development observed in mouse mammary glands following in vivo exposure to TCDD [22].
The toxic effects of exposure to environmental contaminants are often linked to the timing of the exposure. Timing is particularly important in tissues during periods of development, which typically involve widespread cell proliferation, apoptosis, and tissue reorganization, as well as structural and functional differentiation. Virgin female rodents prenatally exposed to TCDD have persistent mammary gland abnormalities as well as predisposition to mammary cancer as adults [38-41]. In the adult, few tissues undergo the same level of development and differentiation that occurs during fetal development. The exception to this is the mammary gland, which undergoes extensive development at the onset of pregnancy, producing a functional milk-producing structure for the nourishment of offspring. It is the later stage of epithelial cell clustering and differentiation to form the milk protein-producing alveolar structures [26] that is modeled by prolactin and hydrocortisone treatment of SCp2 cells in culture [29]. Few studies have initiated exposure to AhR ligands at the onset of pregnancy, or in conjunction with hormonal treatment to model the effects of pregnancy, for the purpose of analyzing effects on lobuloalveolar development and lactation in the exposed dam [22, 24]. Our model of TCDD exposure using SCp2 cells allowed variation in the timing of exposure while focusing the analysis on effects that occur in epithelial cells. We found that delaying addition of TCDD reduced the detrimental effects of exposure on the functional differentiation of mammary epithelial cells in culture. Our results suggest that a window of time very early in the process of alveolar development and functional differentiation contains critical molecular signaling events that are disrupted upon activation of AhR by an exogenous ligand. An alternative explanation is that decreased β-casein is due to the duration of TCDD treatment, not absence of AhR-mediated signals during early stages of cellular differentiation. However, the critical cellular and morphological changes occur within 3 days of plating on Matrigel and induction with lactogenic hormones [30]. With TCDD-exposure initiated prior to accumulation of significant levels of β-casein protein and continued exposure for at least 5 days, cultures with delayed addition of TCDD are able to produce and accumulate β-casein. This supports the idea of an early window of exposure during which AhR activation is able to interfere with the production and/or accumulation of milk protein in SCp2 cells.
Lactogenic differentiation occurs during mid-pregnancy in the mouse, beginning when epithelial cell clusters, or alveoli, form at the ends of the mammary ducts. In late pregnancy, the alveoli become dilated due to pressure from milk protein secretions produced by the functional alveolar epithelial cells [26]. E-cadherin is a cell adhesion molecule critical for the formation of epithelial cell layers as well as the polarization and function of epithelial cells that make up various tissues, including the mammary gland [42]. Deletion of E-cadherin in a knockout mouse model resulted in embryonic lethality [43]; therefore, alternative methods have been used to study the specific function of this protein in the mammary gland. Transgenic expression of a truncated form of E-cadherin lacking its extracellular domain resulted in a dominant-negative effect on cell-cell adhesion, cell polarity, and cell-matrix interactions that disrupted the structure and function of the fully differentiated mammary epithelium [44]. Additionally, a conditional knockout of the E-cadherin gene in differentiating alveolar epithelial cells of the mammary gland resulted in dramatic reduction in milk protein production with concomitant loss of prolactin-induced STAT5a activation [45]. Activation of STAT5a, a protein essential for pregnancy-induced mammary gland development and lactogenesis [33, 46], is necessary for maximal transcription of multiple milk protein genes [47-49]. Thus, our observation that E-cadherin levels and STAT5 activation are altered by TCDD activation of AhR in the mammary gland and mammary epithelial cells in culture is of interest. E-cadherin protein levels are reduced following TCDD exposure in the MCF-7 breast cancer cell line [50], however this is the first study showing the same effect in untransformed mammary epithelial cells and mammary glands from exposed mice. The lack of cell clustering and fewer, as well as smaller, alveolar-like structures observed in SCp2 mammary epithelial cells in our experiments may then result from the loss of cell-cell adhesion due to the reduction in E-cadherin expression. In addition to fewer clusters forming, the loss of E-cadherin expression may affect cell-cell signaling, limiting cellular polarization and differentiation that ultimately reduces the ability of these cells to produce β-casein and other milk proteins.
In parallel with the reduced levels of E-cadherin, which is linked with reduced activation of STAT5 in vivo [45], our studies show altered post-translational modification of STAT5 in the mammary glands of mice exposed to TCDD throughout pregnancy. STAT5 is functionally activated by phosphorylation [51], and our results show reduced phosphorylation of STAT5 following TCDD exposure, with no alteration in total levels of STAT5 protein. Once activated, STAT5 dimerization and nuclear translocation prepares this transcription factor for binding to promoter regions of target genes [52, 53]. Less phosphorylation, and therefore less activation, of STAT5 may result in reduced induction of transcription of the β-casein gene as well other milk-protein target genes, ultimately resulting in overall suppression of milk-protein expression. The paucity of putative AHREs in the upstream promoter region of the β-casein gene itself makes it an unlikely candidate for direct transcriptional regulation by TCDD-activated AhR. However, the alterations we observed in STAT5 phosphorylation in the mammary gland after exposure to TCDD raise the possibility that AhR regulates expression of kinases responsible for the activation of STAT5. The kinase JAK2 is a component of the well-characterized JAK-STAT signaling pathway leading to β-casein protein expression [51, 54], and is responsible for phosphorylation-mediated activation of STAT5 during mammary gland development and differentiation in pregnancy. JAK2 is also a phospho-protein [55], and it is possible that activation of AhR signaling by TCDD inhibits the phosphorylation of JAK2, alters the total amount of JAK2 protein present in the mammary gland, or impacts molecular targets upstream of the JAK-STAT pathway.
We have reported that reduced β-casein production and alterations in the morphology of differentiating SCp2 mammary epithelial cells exposed to TCDD mirror the disrupted mammary gland structure and function observed in vivo. Thus, we have described a new in vitro model for examining the molecular mechanisms by which AhR ligands disrupt normal mammary epithelial cell differentiation and function. Additionally, very early exposure to TCDD during differentiation of cultured mammary epithelial cells is required for the most profound suppression of β-casein production. This suggests that inappropriate activation of AhR during a critical window of time at the onset of alveolar differentiation disrupts molecular signaling pathways that control β-casein expression. Finally, our data suggest that AhR activation by TCDD disrupts mammary gland differentiation by altering expression and post-translational modification of proteins critical in the initiation and maintenance of the functional capacity of the gland during pregnancy. The xenobiotic AhR ligand TCDD and other dioxin-like compounds are the byproducts of industrial processes, and are found in all humans, with higher levels in those living in industrialized nations [56]. Exposure to these ubiquitous environmental contaminants, combined with cumulative effects from the build-up of these chemicals in fatty tissues, make real the possibility for AhR-mediated disruption of mammary development in women during pregnancy. Continued research is necessary to examine the effects of these toxicants on mammary development in humans and wildlife populations, to monitor environmental exposure levels, and to determine relevant exposures to inform the planning of subsequent experimental analyses. Meanwhile, further elucidation of the mechanisms involved in the toxic effects of TCDD exposure using both in vitro and in vivo experimental models is critical for understanding the effects of dioxin exposure and the cellular signaling pathways that are targets of AhR in the mammary gland.
Acknowledgements
This work was supported by research grants from the National Institutes of Health: R01-ES013958 and K02-ES012409 (B.P.L.). The authors gratefully acknowledge Dr. Mina J. Bissell (Lawrence Berkeley National Laboratory) for generously providing SCp2 cells and anti-β-casein antibodies, Dr. Beth A. Vorderstrasse (Washington State University) for helpful discussion, and Mr. Kevin Kipp for technical assistance.
This work was supported by research grants from the National Institutes of Health: R01-ES013958 and K02-ES012409 (B.P.L.). The authors gratefully acknowledge Dr. Mina J. Bissell (Lawrence Berkeley National Laboratory) for generously providing SCp2 cells and anti-β-casein antibodies, Dr. Beth A. Vorderstrasse (Washington State University) for helpful discussion, and Mr. Kevin Kipp for technical assistance.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest statement
The authors declare that there are no conflicts of interest.
References
- [1].Safe SH. Modulation of gene expression and endocrine response pathways by 2,3,7,8-tetrachlorodibenzo-p-dioxin and related compounds. Pharmacol Ther. 1995;67:247–81. doi: 10.1016/0163-7258(95)00017-b. [DOI] [PubMed] [Google Scholar]
- [2].Barouki R, Coumoul X, Fernandez-Salguero PM. The aryl hydrocarbon receptor, more than a xenobiotic-interacting protein. FEBS Lett. 2007;581:3608–15. doi: 10.1016/j.febslet.2007.03.046. [DOI] [PubMed] [Google Scholar]
- [3].Gasiewicz TA, Henry EC, Collins LL. Expression and activity of aryl hydrocarbon receptors in development and cancer. Crit Rev Eukaryot Gene Expr. 2008;18:279–321. doi: 10.1615/critreveukargeneexpr.v18.i4.10. [DOI] [PubMed] [Google Scholar]
- [4].Hankinson O. The aryl hydrocarbon receptor complex. Annu Rev Pharmacol Toxicol. 1995;35:307–40. doi: 10.1146/annurev.pa.35.040195.001515. [DOI] [PubMed] [Google Scholar]
- [5].Beischlag TV, Wang S, Rose DW, et al. Recruitment of the NCoA/SRC-1/p160 family of transcriptional coactivators by the aryl hydrocarbon receptor/aryl hydrocarbon receptor nuclear translocator complex. Mol Cell Biol. 2002;22:4319–33. doi: 10.1128/MCB.22.12.4319-4333.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Nguyen TA, Hoivik D, Lee JE, Safe S. Interactions of nuclear receptor coactivator/corepressor proteins with the aryl hydrocarbon receptor complex. Arch Biochem Biophys. 1999;367:250–7. doi: 10.1006/abbi.1999.1282. [DOI] [PubMed] [Google Scholar]
- [7].Lai ZW, Pineau T, Esser C. Identification of dioxin-responsive elements (DREs) in the 5′ regions of putative dioxin-inducible genes. Chem Biol Interact. 1996;100:97–112. doi: 10.1016/0009-2797(96)03691-5. [DOI] [PubMed] [Google Scholar]
- [8].Denison MS, Fisher JM, Whitlock JP., Jr. The DNA recognition site for the dioxin-Ah receptor complex. Nucleotide sequence and functional analysis. J Biol Chem. 1988;263:17221–4. [PubMed] [Google Scholar]
- [9].Boutros PC, Moffat ID, Franc MA, et al. Dioxin-responsive AHRE-II gene battery: identification by phylogenetic footprinting. Biochem Biophys Res Commun. 2004;321:707–15. doi: 10.1016/j.bbrc.2004.06.177. [DOI] [PubMed] [Google Scholar]
- [10].Nguyen LP, Bradfield CA. The search for endogenous activators of the aryl hydrocarbon receptor. Chem Res Toxicol. 2008;21:102–16. doi: 10.1021/tx7001965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Peterson RE, Theobald HM, Kimmel GL. Developmental and reproductive toxicity of dioxins and related compounds: cross-species comparisons. Crit Rev Toxicol. 1993;23:283–335. doi: 10.3109/10408449309105013. [DOI] [PubMed] [Google Scholar]
- [12].Colborn T. The wildlife/human connection: modernizing risk decisions. Environ Health Perspect. 1994;102(Suppl 12):55–9. doi: 10.1289/ehp.94102s1255a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Carney SA, Prasch AL, Heideman W, Peterson RE. Understanding dioxin developmental toxicity using the zebrafish model. Birth Defects Res Part A Clin Mol Teratol. 2006;76:7–18. doi: 10.1002/bdra.20216. [DOI] [PubMed] [Google Scholar]
- [14].Hotchkiss AK, Rider CV, Blystone CR, et al. Fifteen years after “Wingspread”--environmental endocrine disrupters and human and wildlife health: where we are today and where we need to go. Toxicol Sci. 2008;105:235–59. doi: 10.1093/toxsci/kfn030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Heiden TC, Struble CA, Rise ML, Hessner MJ, Hutz RJ, Carvan MJ., 3rd Molecular targets of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) within the zebrafish ovary: insights into TCDD-induced endocrine disruption and reproductive toxicity. Reprod Toxicol. 2008;25:47–57. doi: 10.1016/j.reprotox.2007.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Brucker-Davis F, Thayer K, Colborn T. Significant effects of mild endogenous hormonal changes in humans: considerations for low-dose testing. Environ Health Perspect. 2001;109(Suppl 1):21–6. doi: 10.1289/ehp.01109s121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Safe S, Wang F, Porter W, Duan R, McDougal A. Ah receptor agonists as endocrine disruptors: antiestrogenic activity and mechanisms. Toxicol Lett. 1998;102-103:343–7. doi: 10.1016/s0378-4274(98)00331-2. [DOI] [PubMed] [Google Scholar]
- [18].Matthews J, Gustafsson JA. Estrogen receptor and aryl hydrocarbon receptor signaling pathways. Nucl Recept Signal. 2006;4:e016. doi: 10.1621/nrs.04016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Long M, Deutch B, Bonefeld-Jorgensen EC. AhR transcriptional activity in serum of Inuits across Greenlandic districts. Environ Health. 2007;6:32. doi: 10.1186/1476-069X-6-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Regoli F, Nigro M, Benedetti M, et al. Interactions between metabolism of trace metals and xenobiotic agonists of the aryl hydrocarbon receptor in the antarctic fish Trematomus bernacchii: environmental perspectives. Environ Toxicol Chem. 2005;24:1475–82. doi: 10.1897/04-514r.1. [DOI] [PubMed] [Google Scholar]
- [21].Hennighausen L, Robinson GW. Signaling pathways in mammary gland development. Dev Cell. 2001;1:467–75. doi: 10.1016/s1534-5807(01)00064-8. [DOI] [PubMed] [Google Scholar]
- [22].Vorderstrasse BA, Fenton SE, Bohn AA, Cundiff JA, Lawrence BP. A novel effect of dioxin: exposure during pregnancy severely impairs mammary gland differentiation. Toxicol Sci. 2004;78:248–57. doi: 10.1093/toxsci/kfh062. [DOI] [PubMed] [Google Scholar]
- [23].Rudolph MC, McManaman JL, Hunter L, Phang T, Neville MC. Functional development of the mammary gland: use of expression profiling and trajectory clustering to reveal changes in gene expression during pregnancy, lactation, and involution. J Mammary Gland Biol Neoplasia. 2003;8:287–307. doi: 10.1023/b:jomg.0000010030.73983.57. [DOI] [PubMed] [Google Scholar]
- [24].Hushka LJ, Williams JS, Greenlee WF. Characterization of 2,3,7,8-tetrachlorodibenzofuran-dependent suppression and AH receptor pathway gene expression in the developing mouse mammary gland. Toxicol Appl Pharmacol. 1998;152:200–10. doi: 10.1006/taap.1998.8508. [DOI] [PubMed] [Google Scholar]
- [25].Daniel CW, Silberstein GB. Postnatal development of the rodent mammary gland. In: Neville M, Daniel C, editors. The Mammary Gland: Development, Regulation and Function. Plenum Press Publishing Corporation; New York: 1987. pp. 3–36. [Google Scholar]
- [26].Brisken C, Rajaram RD. Alveolar and lactogenic differentiation. J Mammary Gland Biol Neoplasia. 2006;11:239–48. doi: 10.1007/s10911-006-9026-0. [DOI] [PubMed] [Google Scholar]
- [27].Li ML, Aggeler J, Farson DA, Hatier C, Hassell J, Bissell MJ. Influence of a reconstituted basement membrane and its components on casein gene expression and secretion in mouse mammary epithelial cells. Proc Natl Acad Sci U S A. 1987;84:136–40. doi: 10.1073/pnas.84.1.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Barcellos-Hoff MH, Aggeler J, Ram TG, Bissell MJ. Functional differentiation and alveolar morphogenesis of primary mammary cultures on reconstituted basement membrane. Development. 1989;105:223–35. doi: 10.1242/dev.105.2.223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Desprez PY, Roskelley C, Judith C, Bissell MJ. Isolation of functional cell lines from a mouse mammary epithelial cell strain: the importance of basement membrane and cell–cell interaction. Molecular and Cellular Differentiation. 1993;1:99–110. [Google Scholar]
- [30].Roskelley CD, Desprez PY, Bissell MJ. Extracellular matrix-dependent tissue-specific gene expression in mammary epithelial cells requires both physical and biochemical signal transduction. Proc Natl Acad Sci U S A. 1994;91:12378–82. doi: 10.1073/pnas.91.26.12378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Danielson KG, Oborn CJ, Durban EM, Butel JS, Medina D. Epithelial mouse mammary cell line exhibiting normal morphogenesis in vivo and functional differentiation in vitro. Proc Natl Acad Sci U S A. 1984;81:3756–60. doi: 10.1073/pnas.81.12.3756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Schmidhauser C, Bissell MJ, Myers CA, Casperson GF. Extracellular matrix and hormones transcriptionally regulate bovine beta-casein 5′ sequences in stably transfected mouse mammary cells. Proc Natl Acad Sci U S A. 1990;87:9118–22. doi: 10.1073/pnas.87.23.9118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Liu X, Robinson GW, Wagner KU, Garrett L, Wynshaw-Boris A, Hennighausen L. Stat5a is mandatory for adult mammary gland development and lactogenesis. Genes Dev. 1997;11:179–86. doi: 10.1101/gad.11.2.179. [DOI] [PubMed] [Google Scholar]
- [34].Zhang L, Savas U, Alexander DL, Jefcoate CR. Characterization of the mouse Cyp1B1 gene. Identification of an enhancer region that directs aryl hydrocarbon receptor-mediated constitutive and induced expression. J Biol Chem. 1998;273:5174–83. doi: 10.1074/jbc.273.9.5174. [DOI] [PubMed] [Google Scholar]
- [35].Zhang L, Zheng W, Jefcoate CR. Ah receptor regulation of mouse Cyp1B1 is additionally modulated by a second novel complex that forms at two AhR response elements. Toxicol Appl Pharmacol. 2003;192:174–90. doi: 10.1016/s0041-008x(03)00276-x. [DOI] [PubMed] [Google Scholar]
- [36].Ma Q, Baldwin KT. 2,3,7,8-tetrachlorodibenzo-p-dioxin-induced degradation of aryl hydrocarbon receptor (AhR) by the ubiquitin-proteasome pathway. Role of the transcription activaton and DNA binding of AhR. J Biol Chem. 2000;275:8432–8. doi: 10.1074/jbc.275.12.8432. [DOI] [PubMed] [Google Scholar]
- [37].Pollenz RS. The mechanism of AH receptor protein down-regulation (degradation) and its impact on AH receptor-mediated gene regulation. Chem Biol Interact. 2002;141:41–61. doi: 10.1016/s0009-2797(02)00065-0. [DOI] [PubMed] [Google Scholar]
- [38].Fenton SE, Hamm JT, Birnbaum LS, Youngblood GL. Persistent abnormalities in the rat mammary gland following gestational and lactational exposure to 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) Toxicol Sci. 2002;67:63–74. doi: 10.1093/toxsci/67.1.63. [DOI] [PubMed] [Google Scholar]
- [39].Jenkins S, Rowell C, Wang J, Lamartiniere CA. Prenatal TCDD exposure predisposes for mammary cancer in rats. Reprod Toxicol. 2007;23:391–6. doi: 10.1016/j.reprotox.2006.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Miller KP, Borgeest C, Greenfeld C, Tomic D, Flaws JA. In utero effects of chemicals on reproductive tissues in females. Toxicol Appl Pharmacol. 2004;198:111–31. doi: 10.1016/j.taap.2003.07.016. [DOI] [PubMed] [Google Scholar]
- [41].Lamartiniere CA. Timing of exposure and mammary cancer risk. J Mammary Gland Biol Neoplasia. 2002;7:67–76. doi: 10.1023/a:1015722507237. [DOI] [PubMed] [Google Scholar]
- [42].Knudsen KA, Wheelock MJ. Cadherins and the mammary gland. J Cell Biochem. 2005;95:488–96. doi: 10.1002/jcb.20419. [DOI] [PubMed] [Google Scholar]
- [43].Larue L, Ohsugi M, Hirchenhain J, Kemler R. E-cadherin null mutant embryos fail to form a trophectoderm epithelium. Proc Natl Acad Sci U S A. 1994;91:8263–7. doi: 10.1073/pnas.91.17.8263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Delmas V, Pla P, Feracci H, Thiery JP, Kemler R, Larue L. Expression of the cytoplasmic domain of E-cadherin induces precocious mammary epithelial alveolar formation and affects cell polarity and cell-matrix integrity. Dev Biol. 1999;216:491–506. doi: 10.1006/dbio.1999.9517. [DOI] [PubMed] [Google Scholar]
- [45].Boussadia O, Kutsch S, Hierholzer A, Delmas V, Kemler R. E-cadherin is a survival factor for the lactating mouse mammary gland. Mech Dev. 2002;115:53–62. doi: 10.1016/s0925-4773(02)00090-4. [DOI] [PubMed] [Google Scholar]
- [46].Miyoshi K, Shillingford JM, Smith GH, et al. Signal transducer and activator of transcription (Stat) 5 controls the proliferation and differentiation of mammary alveolar epithelium. J Cell Biol. 2001;155:531–42. doi: 10.1083/jcb.200107065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Burdon TG, Maitland KA, Clark AJ, Wallace R, Watson CJ. Regulation of the sheep beta-lactoglobulin gene by lactogenic hormones is mediated by a transcription factor that binds an interferon-gamma activation site-related element. Mol Endocrinol. 1994;8:1528–36. doi: 10.1210/mend.8.11.7877621. [DOI] [PubMed] [Google Scholar]
- [48].Li S, Rosen JM. Nuclear factor I and mammary gland factor (STAT5) play a critical role in regulating rat whey acidic protein gene expression in transgenic mice. Mol Cell Biol. 1995;15:2063–70. doi: 10.1128/mcb.15.4.2063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Liu X, Robinson GW, Gouilleux F, Groner B, Hennighausen L. Cloning and expression of Stat5 and an additional homologue (Stat5b) involved in prolactin signal transduction in mouse mammary tissue. Proc Natl Acad Sci U S A. 1995;92:8831–5. doi: 10.1073/pnas.92.19.8831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Diry M, Tomkiewicz C, Koehle C, et al. Activation of the dioxin/aryl hydrocarbon receptor (AhR) modulates cell plasticity through a JNK-dependent mechanism. Oncogene. 2006;25:5570–4. doi: 10.1038/sj.onc.1209553. [DOI] [PubMed] [Google Scholar]
- [51].Liu X, Robinson GW, Hennighausen L. Activation of Stat5a and Stat5b by tyrosine phosphorylation is tightly linked to mammary gland differentiation. Mol Endocrinol. 1996;10:1496–506. doi: 10.1210/mend.10.12.8961260. [DOI] [PubMed] [Google Scholar]
- [52].Darnell JE., Jr. STATs and gene regulation. Science. 1997;277:1630–5. doi: 10.1126/science.277.5332.1630. [DOI] [PubMed] [Google Scholar]
- [53].Barash I. Stat5 in the mammary gland: controlling normal development and cancer. J Cell Physiol. 2006;209:305–13. doi: 10.1002/jcp.20771. [DOI] [PubMed] [Google Scholar]
- [54].Wagner KU, Rui H. Jak2/Stat5 signaling in mammogenesis, breast cancer initiation and progression. J Mammary Gland Biol Neoplasia. 2008;13:93–103. doi: 10.1007/s10911-008-9062-z. [DOI] [PubMed] [Google Scholar]
- [55].Rui H, Kirken RA, Farrar WL. Activation of receptor-associated tyrosine kinase JAK2 by prolactin. J Biol Chem. 1994;269:5364–8. [PubMed] [Google Scholar]
- [56].Schecter A, Birnbaum L, Ryan JJ, et al. Dioxins: an overview. Environ Res. 2006;101:419–28. doi: 10.1016/j.envres.2005.12.003. [DOI] [PubMed] [Google Scholar]