

Abstract
BACKGROUND
Several microtubule targeting agents are capable of inducing CYP3A4 via activation of the pregnane X receptor (PXR; NR1I2).
OBJECTIVE
To evaluate the CYP3A4 induction potential of vinblastine both clinically and in vitro and determine the involvement of the nuclear receptors NR1I2 and the constitutive androstane receptor (NR1I3).
METHODS
Midazolam pharmacokinetics were evaluated in 6 patients who were enrolled in a Phase 1/2 study of infusional vinblastine given in combination with the ABCB1 (P-glycoprotein) antagonist valspodar (PSC 833) and received the CYP3A4 phenotyping probe midazolam on more than 1 occasion. Genotyping was conducted in CYP3A4, CYP3A5, and ABCB1 to rule out potential pharmacogenetic influences. Clinical data were followed-up by Western blotting and reporter assays in HepG2 and NIH3T3 cells treated with vinblastine over a dose range of 150-4800 ng/mL for 48 hours.
RESULTS
In 6 patients with cancer, vinblastine increased the median (95% CI) clearance of the CYP3A4 phenotyping probe midazolam from 21.7 L/h (12.6 to 28.1) to 32.3 L/h (17.3 to 53.9) (p = 0.0156, Wilcoxon signed-rank test). No obvious effect of polymorphisms in CYP3A4, CYP3A5, and ABCB1 on midazolam clearance was observed. In vitro, vinblastine induced CYP3A4 protein. Furthermore, cell-based reporter gene assays using transiently transfected HepG2 and NIH3T3 cells indicated that vinblastine (150-4800 ng/mL) weakly activated human and mouse full-length NR1I2, but had no influence on NR1I3.
CONCLUSIONS
Collectively, these findings suggest that vinblastine is able to induce CYP3A4, at least in part, via an NR1I2-dependent mechanism, and thus has the potential to facilitate its own elimination and cause interactions with other CYP3A4 substrates.
Keywords: CYP3A4, induction, NR1I2 (PXR), vinblastine
Vinblastine is a naturally occurring vinca alkaloid that is found in the Madagascar periwinkle plant Catharanthus roseus (L.) G. Don (formerly known as Vinca rosea L.). This antimicrotubule agent has been used widely in cancer chemotherapy for several decades as a single agent and in combination with other antineoplastic drugs. It is approved for the treatment of testicular cancer, Hodgkin’s disease, malignant lymphoma, Kaposi’s sarcoma, breast cancer, choriocarcinoma, mycosis fungoides, Letterer-Siwe disease, and malignant histiocytosis.1
Large intra- and interindividual variability in the pharmacokinetics of vinblastine has been reported.2,3 This agent undergoes extensive hepatic metabolism and is mainly excreted in feces. Several studies have demonstrated that members of the cytochrome P450 (CYP) 3A sub-family are involved in the metabolism of vinblastine and other vinca alkaloids.4-10 CYP3A4-mediated metabolism of vinblastine was first demonstrated using human liver microsomes. Microsomal metabolism of vinblastine was highly variable and correlated with CYP3A4 protein and mRNA expression.10 In addition to being a substrate of CYP3A4, vinblastine has been shown to inhibit the metabolism of vindesine11 and dihydropyridine denitronifedipine,4 both of which are metabolized by CYP3A4.
Other microtubule-targeting agents, including paclitaxel, discodermolide, and the epothilone B analogue ixabepilone, are capable of inducing CYP3A4 expression in vitro.12-14 For each of these agents, induction of CYP3A4 is mediated via activation of the pregnane X receptor (PXR, SXR, NR1I2),13,14 a member of the nuclear receptor family of ligand-activated transcription factors that have a central role in regulating the transcription of genes involved in xenobiotic metabolism and transport.15 The objective of this study was to evaluate whether vinblastine also acts as an inducer of CYP3A4, both in vivo and in vitro, and, if so, to determine the role of NR1I2 in this process.
Methods
PATIENTS
A total of 6 patients (Table 1), who were enrolled in a Phase 1/2 study of infusional vinblastine given in combination with the ABCB1 (P-glycoprotein) antagonist valspodar (PSC 833)16 and received the CYP3A4 phenotyping probe midazolam on more than 1 occasion, were included in this study. All patients were white and had a diagnosis of renal cell carcinoma. Inclusion and exclusion criteria have been described previously and all patients had adequate hepatic function (aspartate aminotransferase <2.5 times the upper limit of normal; total bilirubin <1.5 times the upper limit of normal).16 Based on the knowledge that valspodar is metabolized by CYP3A4,17 the same enzyme that is involved in cyclosporine metabolism, the use of strong inducers of cyclosporine metabolism or CYP3A4 (including nafcillin, rifampin, carbamazepine, phenobarbital, and phenytoin) or the use of inhibitors (including diltiazem, nicardipine, verapamil, fluconazole, clarithromycin, erythromycin, bromocriptine, and danazol) was avoided. The study was approved by the institutional review board and all patients gave informed consent.
Table 1.
Patient Demographics and Genotypes
| Pts. |
ECOG PS |
% ↑ Midazolam Cl |
CYP3A4*1B | CYP3A4*17 | CYP3A4*18A | CYP3A5*3C | CYP3A5*6 | ABCB13435C>T | |
|---|---|---|---|---|---|---|---|---|---|
| Sex | Age, y | ||||||||
| Male | 55 | 1 | 51.7 | *1A/*1A | *1A/*1A | *1A/*1A | *3C/*3C | *1A/*1A | CT |
| Male | 60 | 0 | 35.9 | *1A/*1B | *1A/*1A | *1A/*1A | *1A/*3C | *1A/*1A | TT |
| Male | 58 | 1 | 216.7 | *1A/*1A | *1A/*1A | *1A/*1A | *3C/*3C | *1A/*1A | CT |
| Male | 40 | 1 | 2.5 | *1A/*1A | *1A/*1A | *1A/*1A | *3C/*3C | *1A/*1A | CT |
| Male | 50 | 1 | 46.9 | *1A/*1A | *1A/*1A | *1A/*1A | *3C/*3C | *1A/*1A | CC |
| Female | 66 | 1 | 6.9 | *1A/*1A | *1A/*1A | *1A/*1A | *3C/*3C | *1A/*1A | CT |
Cl = clearance; ECOG PS = Eastern Cooperative Oncology Group Performance Status.
DRUG ADMINISTRATION
Valspodar, vinblastine, and midazolam were obtained from commercial sources by the National Institutes of Health Clinical Center Pharmacy Department, Bethesda, MD. Briefly, valspodar was prepared for a 24-hour infusion (50 mg/mL in polyoxyethylated castor oil [Cremophor EL] diluted in 250 mL or 500 mL of 5% dextrose or 0.9% sodium chloride) and was administered on the first 2 days of therapy in cycle 1 followed 4 days later by a 6-day infusion of valspodar. Vials containing 10 mg of lyophilized vinblastine were reconstituted and the drug was then further diluted in 250 mL of sterile 0.9% sodium chloride injection. Vinblastine was given as a 3-day continuous intravenous infusion, which began 24 hours after the initiation of valspodar (1-2.6 mg/m2/day) every 28 days (days 9-11 of cycle 1 and days 2-4 during subsequent cycles). Midazolam was administered as an intravenous bolus injection at a dose of 0.0145 mg/kg on day 1 of cycles 1 and 4 to assess phenotypic CYP3A4 activity.
MIDAZOLAM PHARMACOKINETICS
Blood samples were drawn into heparinized tubes at 0.083, 0.25, 0.5, 1, 2, 3, 4, and 5 hours following midazolam administration. Samples were centrifuged immediately to obtain plasma, which was stored at −80 °C until analysis. Plasma concentrations of midazolam were determined by high-performance liquid chromatography with mass spectrometric detection, as described previously.18 The lower limit of quantitation of this assay is 1 ng/mL, with values for precision and accuracy of ≤15% and <12% relative error, respectively. Pharmacokinetic parameters were determined by noncompartmental analysis using WinNonlin version 4.0 (Pharsight Corporation, Mountain View, CA). The area under the plasma concentration versus time curve (AUC) was calculated using the linear trapezoidal method from time zero to the time of the final quantifiable concentration. The AUC was then extrapolated to infinity by dividing the last measured concentration by the rate constant of the terminal phase (k), which was determined by linear-regression analysis of the final 3 time points of the log-linear concentration-time plot. All reported AUC values were dose normalized. The systemic clearance of midazolam was calculated by dividing the administered dose in milligrams by the observed AUC.
STATISTICAL ANALYSIS
Based on the standard deviation of the changes expected in phenotypic CYP3A4 activity (sd), a power (1-β) of 0.8 (80%), a clinically relevant change in the clearance of midazolam of 30% (δ; standardized difference, 2δ/sd), and a 2-sided significance level (α) of 0.05 (5%), a patient sample size of 6 was required in a paired, 2-sided analysis. A nonparametric 2-sided Wilcoxon signed-rank test was used to assess the differences in the median AUC and clearance of midazolam between day 1 during cycles 1 and 4. All pharmacokinetic data are reported as median values with 95% confidence interval. This calculation was performed with the software package NCSS version 2004 (Number Cruncher Statistical Systems; J. L. Hintze, Kaysville, UT). The cutoff value for statistical significance was set at p < 0.05.
GENOTYPING
DNA was isolated from plasma and individual CYP3A4*1B (−392A>G; 5′-regulatory region; dbSNP rs ID number, rs2740574; see http://www.ncbi.nlm.nih.gov/), CYP3A4*17 (15615T>C; F189S; rs4987161), CYP3A4*18A (20070T>C; L293P; no rs ID number), CYP3A5*3C (6986A>G; splicing defect; rs776746), CYP3A5*6 (14690G>A; splicing defect; rs10264272), and ABCB1 (MDR1) (3435C>T; I1145I; rs1045652) genotypes were determined as described previously.19
WESTERN BLOTTING
HepG2 cells, maintained in RPMI 1640 supplemented with 10% fetal bovine serum and 1% penicillin-streptomycin, were treated with vinblastine (150-4800 ng/mL) for 48 hours. CYP3A4 protein expression was determined by Western blot analysis as described previously.13 For microsome preparation, cells were centrifuged at 750 × g for 5 minutes and washed immediately in a storage buffer composed of 50 mM potassium phosphate, 1 mM EDTA, and 20% glycerol (pH 7.4). This was followed by homogenization using a glass-Teflon homogenizer and a buffer containing 1.15 M potassium chloride, 10 mM EDTA, 100 mM potassium phosphate, and 0.2 mM phenylmethylsulfonyl fluoride (pH 7.5). Following differential centrifugation (12,000 × g for 12 minutes; 100,000 × g for 90 minutes), the pellet was resuspended in storage buffer to a concentration of 4 mg/mL. Fifty micrograms of microsomal protein were loaded per lane and transferred to a nitrocellulose membrane following separation by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). Human CYP3A4 + reductase BD Supersomes (BD Biosciences, Bedford, MS) were used as a positive control. The WB-MAB-3A mouse monoclonal antibody and an HRP-conjugated rabbit anti-mouse IgG were purchased from BD Biosciences and diluted as per manufacturer’s instructions. Bands on film were optically scanned (Epson Expression 1600, San Diego, CA).
TRANSIENT TRANSFECTION EXPERIMENTS
The reporter plasmids pSG5-hPXR (human NR1I2) and pSG5-mPXR (mouse NR1I2) were provided by Dr. Steven Kliewer (University of Texas Southwestern Medical Center, Dallas, TX).20 The reporter plasmids pDR-hCAR (human NR1I3) and pcDNA3-mCAR (mouse NR1I3) were provided by Drs. David Moore (Baylor College of Medicine, Houston, TX) and Masahiko Negishi (National Institute of Environmental Health Sciences, Research Triangle Park, NC), respectively. The CYP3A4-PXRE-LUC reporter plasmid containing the proximal promoter (−362/+53) and distal XREM (−7836/−7208) was prepared as previously described.21 Vinblastine (Sigma-Aldrich, St. Louis, MO) and valspodar (Novartis Pharmaceutical Corp., East Hanover, NJ; distributed by the Cancer Therapy Evaluation Program, National Cancer Institute, Bethesda, MD) were dissolved in dimethyl sulfoxide (DMSO) and stored at −20 °C. HepG2 and NIH3T3 cells were maintained in minimum Eagle’s medium supplemented with 10% fetal bovine serum, 1% penicillin-streptomycin, and 1% l-glutamine. Experiments were performed as described previously.22 Briefly, approximately 3 × 105 cells were seeded into 24-well plates, 24 hours before co-transfection by calcium phosphate precipitation with 0.5 μg of the reporter plasmid and 0.1 μg of the expression plasmid or empty vector plasmid. The SV40-β-galactosidase control vector (0.1 μg) was used as an internal control for transfection efficiency. Twelve hours later, cells were washed and then incubated with drug prepared in medium containing 10% charcoal dextran-treated fetal bovine serum (Hyclone Laboratories, Logan, UT) for 24 hours. Cells were harvested, lysed, and centrifuged at 1500 xg for 4 min, and an aliquot of supernatant was used to determine luciferase (Luciferase Assay System, Promega, Madison, WI) and β-galactosidase (β-Galactosidase Enzyme Assay System, Promega, Madison, WI) activities according to the manufacturers’ instructions. Luciferase activity was normalized to β-galactosidase activity. All experiments were performed twice in triplicate. As positive controls, rifampin and pregnenolone-16α-carbonitrile (PCN) were used for human and mouse NR1I2, and CITCO (6-(4-chlorophenyl)imidazo[2,1-b][1,3]thiazole-5-carbaldehyde O-(3,4-dichlorobenzyl)oxime) and TCPOBOP (1,4-bis[2-(3,5-dichloropyridyloxy)]benzene) for human and mouse NR1I3, respectively. The final concentration of DMSO was ≤0.2% in all experiments.
Results
IN VIVO STUDY
The CYP3A4 phenotyping probe midazolam (0.0145 mg/kg) was administered by intravenous bolus to 6 patients with renal cell carcinoma undergoing treatment with vinblastine (72-hour intravenous infusion given every 28 days). None of the patients received any prescription drug other than vinblastine that is known or suspected to alter CYP3A4 activity that could have confounded the results, albeit that valspodar is a very weak CYP3A4 inhibitor.23 Midazolam clearance and AUC were determined on day 1 and during cycle 4 (days 92-119; Figure 1). On day 1 of cycle 1, the clearance of midazolam ranged from 16.2 to 28.1 L/h (median 21.7; 95% CI 12.6 to 28.1). All 6 patients exhibited increased midazolam clearance between cycle 1 and cycle 4, indicative of increased CYP3A4 activity following repeat administration of vinblastine (Figure 1). The observed increase in midazolam clearance ranged from 2% to 217%, with a median value of 32.3 L/h (95% CI 17.3 to 53.9; p = 0.0156). The corresponding median AUC values decreased from 46.4 ng·h/mL per mg (95% CI 35.6 to 61.7) to 31.0 ng·h/mL per mg (95% CI 18.6 to 57.7; p = 0.0313). The half-life of midazolam ranged from 2.19 to 3.52 hours (median 2.54; 95% CI 2.19 to 3.52) on day 1 of cycle 1, and from 2.52 to 4.81 hours (median 3.08; 95% CI 2.52 to 4.81) on day 1 of cycle 4 (p = 0.0793). One patient received midazolam on 5 occasions over a period of 1.3 years, at the end of which midazolam clearance was 3.6-fold above the clearance value prior to treatment with vinblastine. The interindividual variability in the extent of CYP3A4 induction by vinblastine was not obviously associated with any of the studied CYP3A4, CYP3A5, and ABCB1 genotypes (Table 1).
Figure 1.

Effect of vinblastine on midazolam clearance (Cl; L/h) and area under the curve (AUC) (ng·h/mL per mg). Data were obtained from 6 patients treated with vinblastine as a 72-hour intravenous infusion every 28 days. Midazolam (0.0145 mg/kg, intravenous bolus) was used as a CYP3A4 phenotyping probe on day 1 of cycle 1 (period 1) and on day 1 of cycle 4 (period 2). A nonparametric 2-sided Wilcoxon signed-rank test was used to assess the differences in medians.
IN VITRO CYP3A4 PROTEIN EXPRESSION
Expression of CYP3A4 protein was determined following exposure of HepG2 cells to vinblastine for 48 hours at concentrations ranging between 150 and 4800 ng/mL. Vinblastine, at concentrations of 300 ng/mL and above, substantially increased CYP3A4 protein expression compared to that seen in DMSO -treated control cells (Figure 2).
Figure 2.
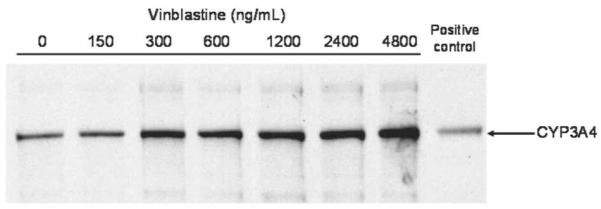
Effect of vinblastine on the expression of CYP3A4 protein in HepG2 cells. Cells were treated with vinblastine for 48 hours as indicated and CYP3A4 protein expression was determined by Western blot analysis. Human CYP3A4 plus reductase BD Supersomes were used as a positive control (far right lane). The range used corresponds to vinblastine concentrations of 0.165 μM (150 ng/mL) to 5.28 μM (4800 ng/mL)
REPORTER GENE ASSAYS
Cell-based luciferase reporter gene assays for the determination of NR1I2 transcriptional activity were performed to establish whether vinblastine is an activating ligand of NR1I2. HepG2 cells were transiently transfected with the expression plasmid for full-length human NR1I2 (pSG5-hPXR) or mouse NR1I2 (pSG5-mPXR) and the respective luciferase reporter plasmids. Treatment with vinblastine resulted in a concentration-dependent increase in transcriptional activity of both human and mouse NR1I2 (Figure 3). The extent of activation by vinblastine (2-fold to 3-fold), however, was relatively weak compared to that of the positive controls rifampin and PCN, respectively, which resulted in a 5-fold to 8-fold increase in reporter activity. Vinblastine had no effect on the transcriptional activity of human and mouse full-length NR1I3 in transiently transfected NIH3T3 cells (Figure 4). Increased activity of the reporter was not observed after treatment with valspodar, suggesting that this compound is not an activating ligand of NR1I2 or NR1I3 (Figure 3 and Figure 4).
Figure 3.

Effect of vinblastine and valspodar (PSC 833) on the transcriptional activity of NR1I2 in HepG2 cells co-transfected with the pSG5-hPXR and pSG5-mPXR expression plasmids and CYP3A4-PXRE2-LUC reporter plasmid. Rifampin (10 μM) and pregnenolone-16α-carbonitrile (PCN; 50 μM) were used as positive controls. Experiments were performed twice in triplicate, except for vinblastine 150 ng/mL for pSG5-hPXR (n = 1). Error bars are standard error of the mean. The range used corresponds to vinblastine concentrations of 0.165 μM (150 ng/mL) to 5.28 μM (4800 ng/mL).
Figure 4.

Effect of vinblastine and valspodar (PSC 833) on the transcriptional activity of NR1I3 in NIH3T3 cells co-transfected with the pDR6-hCAR and pcDNA3-mCAR expression plasmids and CYP3A4-PXRE2-LUC reporter plasmid. CITCO (6-(4-chlorophenyl)imidazo[2,1-b][1,3]thiazole-5-carbaldehyde O-(3,4-dichlorobenzyl)oxime; 100 nM) and TCPOBOP (1,4-bis[2-(3,5-dichloropyridyloxy)]benzene; 250 nM) were used as positive controls. Experiments were performed twice in triplicate. Error bars are standard error of the mean. The range used corresponds to vinblastine concentrations of 0.165 μM (150 ng/mL) to 5.28 μM (4800 ng/mL).
Discussion
In this study, the ability of the antimicrotubule agent vinblastine to induce CYP3A4 was evaluated both clinically and in vitro. Repeated exposure to vinblastine resulted in significantly increased clearance of the CYP3A4 phenotyping probe midazolam in all patients, indicative of induction of CYP3A4. Midazolam does not interact with ABCB1.24 In addition to vinblastine, patients received the ABCB1 inhibitor valspodar,16 which might be considered a drawback of this study. However, since valspodar is reported to be a potential competitive inhibitor of CYP3A4,17 it is unlikely that this agent contributed to the observed induction of this drug-metabolizing enzyme; rather, valspodar would be expected to mask the effect of vinblastine on the clearance of midazolam. Moreover, valspodar is only a very weak inhibitor of midazolam metabolism through CYP3A4/5 in human liver microsomes, and selective ABCB1 inhibitors do not affect midazolam clearance mechanisms in humans.23,25
To provide mechanistic evidence that induction of CYP3A4 expression by vinblastine is responsible for the increase in midazolam clearance observed clinically, HepG2 cells were exposed to increasing concentrations of vinblastine for 48 hours. Consistent with the clinical observation, vinblastine substantially induced CYP3A4 protein expression in vitro, even at low concentrations. The majority of compounds that induce CYP3A4 do so via activation of the nuclear receptor NR1I2. Furthermore, this has recently been shown to be the mechanism of CYP3A4 induction by other microtubule-targeting agents, including paclitaxel and epothilones,13,14 and the mechanism ABCB1 induction through vinblastine itself at a higher concentration (10 μM, or 8110 ng/mL).26 Therefore, cell-based luciferase reporter gene assays were performed to determine whether vinblastine is an activating ligand of NR1I2 between 150 ng/mL and 4800 ng/mL. The results from these experiments suggest that vinblastine is a weak activator of both human and mouse NR1I2 and that the observed induction of CYP3A4 expression is likely to occur, at least in part, via an NR1I2-dependent mechanism. Of note, activation of another closely related nuclear receptor that can regulate CYP3A4, namely NR1I3,27 by vinblastine was not observed, even at high concentrations. However, activation of another nuclear receptor that can regulate CYP3A4, such as the vitamin D receptor (NR1I1),28-31 or the glucocorticoid receptor (NR3C1),32 cannot be excluded. Additionally, CYP3A4 induction may occur as a post-translational event as a result of stabilization of mRNA or decreased protein turnover.
The observed induction of CYP3A4 by vinblastine has considerable clinical implications. First, since vinblastine itself is a substrate of this enzyme,10 autoinduction leading to accelerated metabolism may occur in patients receiving vinblastine over prolonged periods. Second, there is the possibility for a previously unrecognized type of pharmacokinetic interaction between vinblastine and concomitantly administered drugs that are CYP3A4 substrates. Both of these events could result in altered efficacy or toxicity of the drugs involved. Indeed, CYP3A4 induction by vinblastine may provide a plausible explanation for the outcomes of 2 earlier clinical studies involving this drug.33,34 For example, it was reported that treatment of patients with testicular cancer using a combination regimen of cisplatin, vinblastine, and bleomycin resulted in a 30% increase in the clearance of antipyrine, which lasted for 6 weeks.34 Although antipyrine is metabolized by several CYP isoenzymes, CYP3A4 is responsible for the formation of a major metabolite, 4-hydroxyantipyrine.35 Additionally, the combination of paclitaxel, vinblastine, and cisplatin was found to be less effective and less toxic than expected in the treatment of advanced transitional cell carcinoma of the urothelium.33 In this combination, vinblastine was administered as a bolus prior to paclitaxel, which is very extensively metabolized by CYP2C8 and CYP3A4.36,37 Thus, induction of the CYP3A4-mediated metabolism of paclitaxel by vinblastine could contribute to the low response rate and the low incidence of peripheral neuropathy reported. However, vinblastine was administered as a bolus injection in the above study, while our data were obtained in patients receiving a continuous infusion, and it remains unclear whether or not the inductive effect on CYP3A4 through vinblastine treatment is schedule dependent in vivo.
Finally, there is substantial evidence for the involvement of CYP3A4 in the metabolism of other vinca alkaloids in addition to vinblastine. Studies with human liver microsomes revealed that CYP3A4 is responsible for the metabolism of vindesine.11 Vincristine and vinorelbine were shown to inhibit vinblastine metabolism,10 and vincristine also inhibited the metabolism of dihydropyridine denitronifedipine.4 The CYP3A4-inducing antiepileptic agents carbamazepine and phenytoin increased the clearance of vincristine in patients with brain tumors.7 Furthermore, nifedipine and itraconazole, which are inhibitors of CYP3A4, decreased the clearance6 and increased the neurotoxicity5 of vincristine, respectively. Since vinblastine acts as an inducer of CYP3A4, in addition to being a substrate and inhibitor of this enzyme, it is possible that similar induction phenotypes are associated with other vinca alkaloids.
In conclusion, this study indicates that vinblastine acts as an inducer of CYP3A4 both clinically and in vitro. Similar to other microtubule-targeted agents known to induce CYP3A4, induction of CYP3A4 by vinblastine appears to occur, at least in part, via an NR1I2-dependent mechanism. This observation has clinical ramifications not only for vinblastine, but also for other vinca alkaloids in clinical use or development.
Acknowledgments
This work was supported in part by the Intramural Research Program of the National Institutes of Health, National Cancer Institute, Center for Cancer Research, and by a grant from the Damon Runyon Cancer Research Foundation (CI: 15-02 to SM).
The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organization imply endorsement by the US Government.
Footnotes
Conflict of Interest: Authors reported none
Contributor Information
Nicola F Smith, Clinical Pharmacology Program, Medical Oncology Branch, National Cancer Institute, Bethesda, MD.
Sridhar Mani, Assistant Professor of Medicine and Molecular Pharmacology, Albert Einstein Cancer Center and Department of Medicine, Albert Einstein College of Medicine, Bronx, NY.
Erin G Schuetz, Department of Pharmaceutical Sciences, St. Jude Children’s Research Hospital, Memphis, TN.
Kazuto Yasuda, Department of Pharmaceutical Sciences, St. Jude Children’s Research Hospital.
Tristan M Sissung, Clinical Pharmacology Program, Medical Oncology Branch, National Cancer Institute.
Susan E Bates, Experimental Therapeutics Section, Medical Oncology Branch, Center for Cancer Research, National Cancer Institute.
William D Figg, Head of the Clinical Pharmacology Program, Medical Oncology Branch, National Cancer Institute.
Alex Sparreboom, Department of Pharmaceutical Sciences, St. Jude Children’s Research Hospital.
References
- 1.Rowinsky EK, Tolcher AW. Cancer: principles and practice of oncology. Lippincott Williams and Wilkins; Philadelphia: 2004. [Google Scholar]
- 2.Rahmani R, Zhou XJ. Pharmacokinetics and metabolism of vinca alkaloids. Cancer Surv. 1993;17:269–81. [PubMed] [Google Scholar]
- 3.Ratain MJ, Vogelzang NJ, Sinkule JA. Interpatient and intrapatient variability in vinblastine pharmacokinetics. Clin Pharmacol Ther. 1987;41:61–7. doi: 10.1038/clpt.1987.9. [DOI] [PubMed] [Google Scholar]
- 4.Baumhakel M, Kasel D, Rao-Schymanski RA, et al. Screening for inhibitory effects of antineoplastic agents on CYP3A4 in human liver microsomes. Int J Clin Pharmacol Ther. 2001;39:517–28. doi: 10.5414/cpp39517. [DOI] [PubMed] [Google Scholar]
- 5.Bohme A, Ganser A, Hoelzer D. Aggravation of vincristine-induced neurotoxicity by itraconazole in the treatment of adult ALL. Ann Hematol. 1995;71:311–2. doi: 10.1007/BF01697985. [DOI] [PubMed] [Google Scholar]
- 6.Fedeli L, Colozza M, Boschetti E, et al. Pharmacokinetics of vincristine in cancer patients treated with nifedipine. Cancer. 1989;64:1805–11. doi: 10.1002/1097-0142(19891101)64:9<1805::aid-cncr2820640908>3.0.co;2-d. [DOI] [PubMed] [Google Scholar]
- 7.Villikka K, Kivisto KT, Maenpaa H, Joensuu H, Neuvonen PJ. Cyochrome P450-inducing antiepileptics increase the clearance of vincristine in patients with brain tumors. Clin Pharmacol Ther. 1999;66:589–93. doi: 10.1053/cp.1999.v66.103403001. [DOI] [PubMed] [Google Scholar]
- 8.Yao D, Ding S, Burchell B, Wolf CR, Friedberg T. Detoxication of vinca alkaloids by human P450 CYP3A4-mediated metabolism: implications for the development of drug resistance. J Pharmacol Exp Ther. 2000;294:387–95. [PubMed] [Google Scholar]
- 9.Zhou XJ, Zhou-Pan XR, Gauthier T, Placidi M, Maurel P, Rahmani R. Human liver microsomal cytochrome P450 3A isozymes mediated vindesine biotransformation. Metabolic drug interactions. Biochem Pharmacol. 1993;45:853–61. doi: 10.1016/0006-2952(93)90169-w. [DOI] [PubMed] [Google Scholar]
- 10.Zhou-Pan XR, Seree E, Zhou XJ, et al. Involvement of human liver cytochrome P450 3A in vinblastine metabolism: drug interactions. Cancer Res. 1993;53:5121–6. [PubMed] [Google Scholar]
- 11.Zhou XJ, Martin M, Placidi M, Cano JP, Rahmani R. In vivo and in vitro pharmacokinetics and metabolism of vincaalkaloids in rat. II. Vinblastine and vincristine. Eur J Drug Metab Pharmacokinet. 1990;15:323–32. doi: 10.1007/BF03190222. [DOI] [PubMed] [Google Scholar]
- 12.Kostrubsky VE, Lewis LD, Strom SC, et al. Induction of cytochrome P4503A by taxol in primary cultures of human hepatocytes. Arch Biochem Biophys. 1998;355:131–6. doi: 10.1006/abbi.1998.0730. [DOI] [PubMed] [Google Scholar]
- 13.Mani S, Huang H, Sundarababu S, et al. Activation of the steroid and xenobiotic receptor (human pregnane X receptor) by nontaxane microtubule-stabilizing agents. Clin Cancer Res. 2005;11:6359–69. doi: 10.1158/1078-0432.CCR-05-0252. [DOI] [PubMed] [Google Scholar]
- 14.Synold TW, Dussault I, Forman BM. The orphan nuclear receptor SXR coordinately regulates drug metabolism and efflux. Nat Med. 2001;7:584–90. doi: 10.1038/87912. [DOI] [PubMed] [Google Scholar]
- 15.Kliewer SA, Goodwin B, Willson TM. The nuclear pregnane X receptor: a key regulator of xenobiotic metabolism. Endocr Rev. 2002;23:687–702. doi: 10.1210/er.2001-0038. [DOI] [PubMed] [Google Scholar]
- 16.Bates SE, Bakke S, Kang M, et al. A phase I/II study of infusional vinblastine with the P-glycoprotein antagonist valspodar (PSC 833) in renal cell carcinoma. Clin Cancer Res. 2004;10:4724–33. doi: 10.1158/1078-0432.CCR-0829-03. [DOI] [PubMed] [Google Scholar]
- 17.Fischer V, Rodriguez-Gascon A, Heitz F, et al. The multidrug resistance modulator valspodar (PSC 833) is metabolized by human cytochrome P450 3A. Implications for drug-drug interactions and pharmacological activity of the main metabolite. Drug Metab Dispos. 1998;26:802–11. [PubMed] [Google Scholar]
- 18.Lepper ER, Hicks JK, Verweij J, Zhai S, Figg WD, Sparreboom A. Determination of midazolam in human plasma by liquid chromatography with mass-spectrometric detection. J Chromatogr B Analyt Technol Biomed Life Sci. 2004;806:305–10. doi: 10.1016/j.jchromb.2004.04.003. [DOI] [PubMed] [Google Scholar]
- 19.Lepper ER, Baker SD, Permenter M, et al. Effect of common CYP3A4 and CYP3A5 variants on the pharmacokinetics of the cytochrome P450 3A phenotyping probe midazolam in cancer patients. Clin Cancer Res. 2005;11:7398–404. doi: 10.1158/1078-0432.CCR-05-0520. [DOI] [PubMed] [Google Scholar]
- 20.Lehmann JM, McKee DD, Watson MA, Willson TM, Moore JT, Kliewer SA. The human orphan nuclear receptor PXR is activated by compounds that regulate CYP3A4 gene expression and cause drug interactions. J Clin Invest. 1998;102:1016–23. doi: 10.1172/JCI3703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang J, Kuehl P, Green ED, et al. The human pregnane X receptor: genomic structure and identification and functional characterization of natral allelic variants. Pharmacogenetics. 2001;11:555–72. doi: 10.1097/00008571-200110000-00003. [DOI] [PubMed] [Google Scholar]
- 22.Lamba V, Yasuda K, Lamba JK, et al. PXR (NR1I2): splice variants in human tissues, including brain, and identification of neurosteroids and nicotine as PXR activators. Toxicol Appl Pharmacol. 2004;199:251–65. doi: 10.1016/j.taap.2003.12.027. [DOI] [PubMed] [Google Scholar]
- 23.Kawahara I, Kato Y, Suzuki H, et al. Selective inhibition of human cytochrome P450 3A4 by N-[2(R)-hydroxy-1(S)-indanyl]-5-[2(S)-(1, 1-dimethylethylaminocarbonyl)-4-[(furo[2, 3-b]pyridin-5-yl)methyl]piperazin-1-yl]-4(S)-hydroxy-2(R)-phenylmethy lpentanamide and P-glycoprotein by valspodar in gene transfectant systems. Drug Metab Dispos. 2000;28:1238–43. [PubMed] [Google Scholar]
- 24.Kim RB, Wandel C, Leake B, et al. Interrelationship between substrates and inhibitors of human CYP3A and P-glycoprotein. Pharm Res. 1999;16:408–14. doi: 10.1023/a:1018877803319. [DOI] [PubMed] [Google Scholar]
- 25.Kurnik D, Wood AJ, Wilkinson GR. The erythromycin breath test reflects P-glycoprotein function independently of cytochrome P450 3A activity. Clin Pharmacol Ther. 2006;80:228–34. doi: 10.1016/j.clpt.2006.06.002. [DOI] [PubMed] [Google Scholar]
- 26.Harmsen S, Meijerman I, Febus CL, Maas-Bakker RF, Beijnen JH, Schellens JH. PXR-mediated induction of P-glycoprotein by anticancer drugs in a human colon adenocarcinoma-derived cell line. Cancer Chemother Pharmacol. 2009;66:765–71. doi: 10.1007/s00280-009-1221-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Goodwin B, Hodgson E, D’Costa DJ, Robertson GR, Liddle C. Transcriptional regulation of the human CYP3A4 gene by the constitutive androstane receptor. Mol Pharmacol. 2002;62:359–65. doi: 10.1124/mol.62.2.359. [DOI] [PubMed] [Google Scholar]
- 28.Drocourt L, Ourlin JC, Pascussi JM, Maurel P, Vilarem MJ. Expression of CYP3A4, CYP2B6, and CYP2C9 is regulated by the vitamin D receptor pathway in primary human hepatocytes. J Biol Chem. 2002;277:25125–32. doi: 10.1074/jbc.M201323200. [DOI] [PubMed] [Google Scholar]
- 29.Makishima M, Lu TT, Xie W, et al. Vitamin D receptor as an intestinal bile acid sensor. Science. 2002;296:1313–6. doi: 10.1126/science.1070477. [DOI] [PubMed] [Google Scholar]
- 30.Schmiedlin-Ren P, Thummel KE, Fisher JM, Paine MF, Lown KS, Watkins PB. Expression of enzymatically active CYP3A4 by Caco-2 cells grown on extracellular matrix-coated permeable supports in the presence of 1alpha,25-dihydroxyvitamin D3. Mol Pharmacol. 1997;51:741–54. doi: 10.1124/mol.51.5.741. [DOI] [PubMed] [Google Scholar]
- 31.Thummel KE, Brimer C, Yasuda K, et al. Transcriptional control of intestinal cytochrome P-4503A by 1alpha,25-dihydroxy vitamin D3. Mol Pharmacol. 2001;60:1399–406. doi: 10.1124/mol.60.6.1399. [DOI] [PubMed] [Google Scholar]
- 32.Pascussi JM, Drocourt L, Gerbal-Chaloin S, Fabre JM, Maurel P, Vilarem MJ. Dual effect of dexamethasone on CYP3A4 gene expression in human hepatocytes. Sequential role of glucocorticoid receptor and pregnane X receptor. Eur J Biochem. 2001;268:6346–58. doi: 10.1046/j.0014-2956.2001.02540.x. [DOI] [PubMed] [Google Scholar]
- 33.Mulatero C, McClaren BR, Mason M, Oliver RT, Gallagher CJ. Evidence for a schedule-dependent deleterious interaction between paclitaxel, vinblastine and cisplatin (PVC) in the treatment of advanced transitional cell carcinoma. Br J Cancer. 2000;83:1612–6. doi: 10.1054/bjoc.2000.1480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Teunissen MW, Willemse PH, Sleijfer DT, Sluiter WJ, Breimer DD. Antipyrine metabolism in patients with disseminated testicular cancer and the influence of cytostatic treatment. Cancer Chemother Pharmacol. 1984;13:181–5. doi: 10.1007/BF00269025. [DOI] [PubMed] [Google Scholar]
- 35.Engel G, Hofmann U, Heidemann H, Cosme J, Eichelbaum M. Antipyrine as a probe for human oxidative drug metabolism: identification of the cytochrome P450 enzymes catalyzing 4-hydroxyantipyrine, 3-hydroxymethylantipyrine, and norantipyrine formation. Clin Pharmacol Ther. 1996;59:613–23. doi: 10.1016/S0009-9236(96)90001-6. [DOI] [PubMed] [Google Scholar]
- 36.Harris JW, Rahman A, Kim BR, Guengerich FP, Collins JM. Metabolism of taxol by human hepatic microsomes and liver slices: participation of cytochrome P450 3A4 and an unknown P450 enzyme. Cancer Res. 1994;54:4026–35. [PubMed] [Google Scholar]
- 37.Rahman A, Korzekwa KR, Grogan J, Gonzalez FJ, Harris JW. Selective biotransformation of taxol to 6 alpha-hydroxytaxol by human cytochrome P450 2C8. Cancer Res. 1994;54:5543–6. [PubMed] [Google Scholar]