

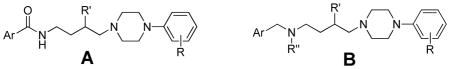
Abstract
N-(3-fluoro-4-(4-(2,3-dichloro- or 2-methoxyphenyl)piperazine-1-yl)-butyl)-aryl carboxamides were prepared and evaluated for binding and function at dopamine D3 (D3R) and D2 receptors (D2R). In this series, we discovered some of the most D3R selective compounds reported to date, (e.g. 8d and 8j >1000-fold D3R-selective over D2R.) In addition, chimeric receptor studies further identified the second extracellular (E2) loop as an important contributor to D3R binding selectivity. Further, compounds lacking the carbonyl group in the amide linker were synthesized and while these amine-linked analogues bound with similar affinities to the amides at D2R, this modification dramatically reduced binding affinities at D3R by >100-fold (e.g. D3RKi for 15b = 393 v. for 8j = 2.6 nM) resulting in compounds with significantly reduced D3R selectivity. This study supports a pivotal role for the D3R E2 loop and the carbonyl group in the 4-phenylpiperazine class of compounds and further reveals a point of separation between structure-activity relationships at D3R and D2R.
The dopamine D2-like receptor family, which is comprised of the D2, D3 and D4 receptor subtypes, has been a target for discovering medications for the treatment of neuropsychiatric and neurodegenerative disorders for decades. Following cloning and in situ studies, dopamine D3 receptors (D3R) were discovered to be primarily localized in limbic regions of the rat and human brain.1,2 Studies have linked changes in the expression of D3R to drug addiction and this has led to an interest in the development of D3R-selective compounds as potential medications.3,4 Because of the high degree of amino acid sequence homology (78% in the transmembrane regions) between D2R and D3R, it has been challenging to obtain subtype selective agents. However, both D2R and D3R selective agonists and antagonists have been described.5–13
Recently, D3R selective agents with varying intrinsic activities have been identified.4 We have previously described novel substituted 4-phenylpiperazines, linked by 2-butenyl- or 3-hydroxy butyl linkers to an extended arylamide moiety, that are highly selective for the D3R.5,6,11 Several of these compounds (e.g. PG 01037, 1) have provided excellent tools for investigating D3R in animal models of psychostimulant abuse.14–20
Further, a recent extension of SAR at D3R has resulted in the discovery of some of the most D3R-selective compounds reported to date with D3R binding affinities in the low nanomolar range and ~400-fold selectivity over the D2R. Importantly, in that study, the first D3R antagonist (R-PG 648; 2) was reported wherein enantioselectivity was more pronounced at D3R than at D2R, and a binding region on the second extracellular loop (E2) was identified as playing a role in both enantioselectivity and D3R vs. D2R binding selectivity. Moreover, molecular models, based on SAR studies using site-directed mutagenesis and computational predictions, differentiate D2R from D3R and D4 receptor binding and also highlight the contributions of E2 to subtype selectivity.21
Structure-activity relationships (SAR) previously developed demonstrate the importance of the length and composition on the linking chain between the aryl amide and the 4-phenylpiperazine in this class of D3R ligands.22,4 Herein, two new series of analogues were synthesized and evaluated for binding at hD3R and hD2R expressed in HEK 293 cells to 1) optimize D3R affinity and selectivity and to 2) further characterize the role of the linking chain in this class of compounds. Based on the success of previously reported 3-OH analogues (e.g. compound 2 in Fig. 1) and SAR that showed sterically bulky substituents in this position were not well tolerated at D3R,6 we replaced the 3-OH with a 3-F group. In addition, as Micheli and colleagues12,13 reported that the amide function in their D3R antagonists, based on SB277011A (3), could be replaced with bioisosteric groups and had beneficial effects in vivo (e.g. 4), we compared analogues with and without the linking chain carbonyl group and further extended N-methyl and N-n-propyl substituents to potentially optimize for D3R affinity.
Figure 1.

Chemical Structures of Representative D3R-selective Ligands
Chemistry
Scheme 1 outlines the synthetic strategy used for the 3-F-substituted arylpiperazine butyl carboxamide derivatives. Compounds 5a and 5b were synthesized by employing an epoxide opening of 2-(2-(oxiran-2-yl)ethyl)isoindoline-1,3-dione with the corresponding arylpiperazines, under reflux conditions.6 These resulting alcohols were treated with N,N-diethylaminosulfur trifluoride (DAST) to give intermediates 6a and 6b, which were subjected to phthalimide deprotection using hydrazine to give the primary amine intermediates 7a and 7b. The desired 3-F-substituted carboxamides were synthesized by coupling these intermediates with the corresponding carboxylic acids or carboxylic acid chlorides.
Scheme 1. Synthesis of 3-F-amide Analoguesa.

a Reagents and conditions: (a) DAST, CH2Cl2, −78 °C to RT, 12 h; (b) hydrazine, EtOH, reflux, 3 h; (c) CDI, THF (Method A); (d) SOCl2 (Method B).
In Scheme 2, compounds 10a–b were prepared by deprotecting the corresponding phthalimide protected amines 9a–b and subjecting them to propionyl chloride in the presence of triethylamine, as the base, to give amides 11a–b. These amides were reduced to the corresponding amines 12a–b using LiAlH4 and then treated with the appropriate aldehyde and subjected to a one-pot imine reduction with Na(OAc)3BH, resulting in the desired n-propyl substituted tertiary amine compounds 13a–f. Compounds 14a–b were synthesized by stepwise imine reduction of 9H-fluorene-2-carbaldehyde with the corresponding amine. These secondary amines were converted into methyl substituted tertiary amines 15a–b by treating with formaldehyde and Na(OAc)3BH. N-n-Propyl substituted tertiary amines were purified by preparative chromatography; all the remaining compounds were purified by flash column chromatography or recrystallization from 2-propanol. Reaction conditions and yields were not optimized, and spectroscopic data refer to the free base.
Scheme 2. Synthesis of Amine-linked Analoguesa.

aReagents and conditions: (a) hydrazine, EtOH, reflux, 3 h; (b) propionylchloride, CH2Cl2, TEA, 0 °C to RT, 4 h; (c) LAH, THF, reflux, 4 h (d) aldehyde, Na(OAc)3BH, DCE, RT, 24 h (Method C); (e) 9H-fluorene-2-carbaldehyde, MeOH, RT, 12h; (f) NaBH4, 30 min; (g) HCHO, Na(OAc)3BH, DCE, 12 h.
Pharmacological Results and Discussion
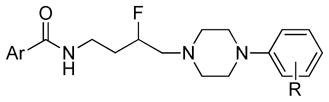
All compounds were evaluated in competition binding assays using stably transfected HEK 293 cells expressing either human D2L, D3, or D4 dopamine receptors.5 The radioligand was the high-affinity D2-like receptor antagonist 2,3-dimethoxy-5-(125I)-iodo-N-(9-benzyl-9-azabicyclo(3.3.1)nonan-3-yl)benzamide ([125I]IABN).23 In Table 1, binding data for the N-(3-fluoro-4-(4-(2-methoxy or 2,3-dichlorophenyl)piperazine-1-yl)-butyl)-aryl carboxamides are shown. None of the compounds tested showed high binding affinities for D4 receptors. In contrast, all compounds shown in Table 1 exhibited high binding affinities for D3R (Ki range = 1.5–28.6 nM) and selectivity over D2R. Notably, compounds 8d and 8j showed nanomolar binding affinities for D3R (Ki = 6.1 and 2.6 nM, respectively) and >1000-fold selectivity over D2R. Among the 2-OCH3- series 8k and 8m showed >60-fold D3R-selectivity over D2R. In the 2,3-diCl-series 8h, 8l and 8n showed >130-fold D3R-selectivity over D2R. Indeed, in this series, all the 2,3-diClphenylpiperazine analogues exhibited greater D3R-selectivity compared to the corresponding 2-OCH3-analogues, although their cLogP values of >5 may be a limitation for in vivo studies. There was no consistent effect on D2R or D3R binding affinities that was dictated by the nature of the terminal aryl amide.
Table 1.
Human D2R-Family Receptor Subtype Binding Data on N-(3-Fluoro-4-(4-(2-methoxy or 2,3-dichlorophenyl) piperazine-1-yl)-butyl)-aryl carboxamidesa
 | ||||||||
|---|---|---|---|---|---|---|---|---|
| compd | R | Ar | clogPb | D2R | D3R | D4R | D2/D3 | D4/D3 |
| Ki±S.E.M, nM | ||||||||
| 8a | 2-OCH3 |
|
3.9 | 390±42 | 20.5±4.3 | ND | 19 | - |
| 8b | 2,3-diCl |
|
5.5 | 378±42 | 6.20±1.5 | 3090±650 | 61 | 498 |
| 8c | 2-OCH3 |
|
3.7 | 329±36 | 28.6±6.0 | ND | 11 | - |
| 8d | 2,3-diCl |
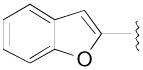
|
5.3 | 14600±5070 (9160±3730) | 6.10±0.8 (5.40±0.6) | 5340±2290 | 2400 (1700)c | 875 |
| 8e | 2-OCH3 |
|
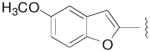
4.0 | 274±13 | 13.9±1.1 | ND | 20 | - |
| 8f | 2,3-diCl |
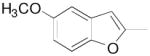
|
4.9 | 279±63 | 8.00±1.4 | ND | 35 | - |
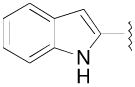
| 8g | 2-OCH3 |
|
3.7 | 421±33 | 9.50±1.9 | 1490±39.6 | 44 | 157 |
| 8h | 2,3-diCl |
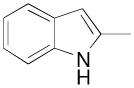
|
5.4 | 526±51 | 4.00±0.9 | 3360±484 | 131 | 841 |
| 8i | 2-OCH3 |
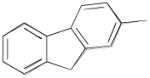
|
5.1 | 2890±1040 | 10.1±1.9 | 3180±922 | 286 | 315 |
| 8j | 2,3-diCl |
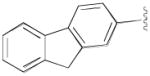
|
6.1 | 4260±1990 | 2.60±0.7 | ND | 1640 | - |
| 8k | 2-OCH3 |
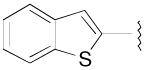
|
3.7 | 241±38 | 3.80±0.8 | ND | 63 | - |
| 8l | 2,3-diCl |
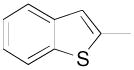
|
5.4 | 472±76 | 2.90±0.4 | ND | 163 | - |
| 8m | 2-OCH3 |
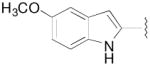
|
3.3 | 302±32 | 4.60±0.4 | ND | 66 | - |
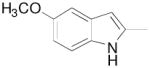
| 8n | 2,3-diCl |
|
4.8 | 230±16 | 1.50±0.3 | ND | 153 | - |
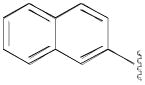
| 8o | 2,3-diCl |
|
5.3 | 195±25 | 6.10±0.9 | ND | 32 | - |
| 1d | - | - | 5.3 | 93.3±12.0 | 0.7±0.1 | 375±18 | 133 | 540 |
| (±)2e | - | - | 4.2 | 502±51.5 | 1.39±0.19 | 4900±1034 | 358 | 5300 |
Binding inhibition values determined using HEK 293 cells transfected with hD2LR or hD3R and125I-IABN radioligand as described.5,6
Partition coefficients (clogP) were calculated using ChemDraw Ultra, Version 11.0, CambridgeSoft 2007;
D2/D3 ratio using Ki values from chimera study in Table 4;
data previously reported in ref. 5;
data previously reported in ref. 11; ND = Not determined.
To determine the importance of the carboxamide moiety on D3R affinity and selectivity, we replaced the amide group linking the extended aryl ring system to the butyl-4-phenylpiperazine with a methylenamine, while preserving the linker length. Further, as the N-n-propyl substitution has demonstrated importance in D3R affinity and selectivity in previously reported and structurally similar compounds,24 we compared this substituent effect to the unsubstituted and N-methyl substituted amines. These results are shown in Table 2. Reference amide-containing and D3R selective compounds 16–20 are included in Table 2 for comparison.
Table 2.
In vitro Binding of Amine-linked Analogs at D2R and D3Ra
 | ||||||||
|---|---|---|---|---|---|---|---|---|
| compound | temp | R | Ar | R′ | R″ | D2R Ki ± S.E.M., nM | D3R Ki ± S.E.M., nM | D2/D3 |
| 13a | B | 2-OCH3 |
|
H | n-propyl | 69.9±7.3 | 67.9±2.4 | 1 |
| 13b | B | 2,3-diCl |
|
H | n-propyl | 162.0±1.2 | 101±27 | 2 |
| 13c | B | 2-OCH3 |
|
H | n-propyl | 196±30 | 71.5±2.0 | 3 |
| 13d | B | 2,3-diCl |
|
H | n-propyl | 269±24 | 129.±30 | 2 |
| 13e | B | 2-OCH3 |
|
H | n-propyl | 119 ± 11 | 80.8±32 | 1.5 |
| 13f | B | 2,3-diCl |
|
H | n-propyl | 313 ± 8.4 | 104±21 | 3 |
| 14a | B | 2,3-diCl |
|
OH | H | 2090 ± 370 | 134±23 | 16 |
| 14b | B | 2,3-diCl |
|
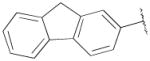
F | H | 5270±1900 | 467±60 | 11 |
| 15a | B | 2,3-diCl |
|
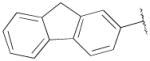
OH | Me | 869±140 | 162±32 | 5 |
| 15b | B | 2,3-diCl |
|
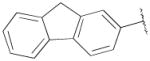
F | Me | 2060±320 | 393±38 | 5 |
| 16e | A | 2-OCH3 |
|
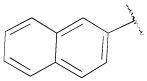
H | - | 33.6±5.9 | 0.30±0.06 | 112 |
| 17b,f | A | 2,3-diCl |
|
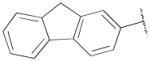
H | - | 112 ± 22 | 2.00±0.4 | 56 |
| 18c,g | A | 2-OCH3 |
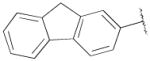
|
H | - | 55.4±5.3 | 0.30±0.1 | 185 |
| 19b,h | A | 2,3-diCl |
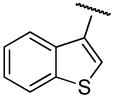
|
H | - | 64.7±8.9 | 0.80±0.2 | 81 |
| 20b,i | A | 2,3-diCl |
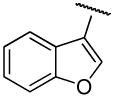
|
H | - | 44.8±11 | 0.80±0.3 | 56 |
Binding inhibition values determined using HEK 293 cells transfected with hD2LR or hD3R dopamine receptors and125I-IABN radioligand as described.5,6
data previously reported in ref. 5;
data previously reported in ref. 6.
BP 897 (N-(4-(4-(2-methoxyphenyl)piperazin-1-yl)butyl)-2-naphthamide);
NGB 2904 (N-(4-(4-(2,3-dichlorophenyl)piperazin-1-yl)butyl)-9H-fluorene-2-carboxamide);
PG 576 (N-(4-(4-(2-methoxyphenyl)piperazin-1-yl)butyl)-9H-fluorene-2-carboxamide);
PG01032 (Benzo[b]thiophene-2-carboxylic acid {4-[4-(2,3-dichloro-phenyl)-piperazin-1-yl]-butyl}-amide);
PG01059 (N-(4-(4-(2,3-dichlorophenyl)piperazin-1-yl)butyl)benzofuran-2-carboxamide).
In general, when the amide was replaced by a methyleneamine, D2R binding was either unaffected or slightly changed, whereas D3R binding affinity was substantially decreased. For example, when the amide in 19 was replaced by an N-n-propyl amine linker (13b), D2R binding affinity was reduced by 2.5 fold, whereas D3R binding affinity was decreased by 126-fold. This resulted in decreasing D3R-selectivity from 81-fold to 2-fold. Another example is with compound 8j compared to 15b, wherein D3R binding affinity was reduced from 2.6 nM to 393 nM (150-fold), and D2R binding affinity was increased by a modest ~2-fold. Thus in this set of compounds, D3R-selectivity was reduced from 1640- to 5-fold by eliminating the carboxamide function and replacing it with a tertiary methyleneamine. This trend was apparent throughout the series. These results suggest that the amide group is necessary in this series of analogues for high affinity binding to D3R and provides a point of separation between the D2R and D3R pharmacophores. This SAR may appear to contrast with the recently described Glaxo D3R-selective antagonists wherein the amide group was not necessary for D3R binding.13 However, in the Glaxo compounds, an isosteric replacement of the amide group is present, in contrast to the amine analogues reported herein. The N-n-propyl substituted amines did not restore D3R binding affinity in this set of analogues.
In Table 3, additional in vitro binding data for selected compounds is shown. With few exceptions, the F-analogues did not bind appreciably to these off-target sites or dopamine D1 receptors (data not shown). Moreover, the only compounds that showed moderate binding affinities at 5HT1A (e.g. Ki<100 nM, 8a, 8c, 8e) were less D3R selective over D2R, and therefore would not be candidates for further in vivo investigation. In contrast, the most D3/D2-selective compounds also showed selectivity over D1, 5HT1A and 5HT2A receptors (e.g. 8d, 8i and 8j).
Table 3.
Additional In Vitro Binding and Functional Data for Selected Compounds.
| compd | D3 mitogenesis IC50±S.E.M., nMa | D2 mitogenesis IC50±S.E.M., nMa | D3 beta arrestin IC50 ± S.E.M., nM | 5-HT1A [3H]-8-OH-DPAT Ki ± S.E.M., nMa | 5HT1A [35S]GTPγS EC50 ± S.E.M., nM (% agonist)a | 5-HT2A [125I]DOI Ki ± S.E.M., nMa |
|---|---|---|---|---|---|---|
| 8a | 390±130 | NDe | 699±18 | 49.0±13 | 71.6±1.3 (73) | 2060±460 |
| 8b | 256±79 | NDe | IAf | 159±37 | 1080±380 (123) | 98±34 |
| 8c | 440±130 | NDe | NDe | 37.9±9.4 | 134±44 (60) | 4800±1300 |
| 8d | 430±120 | NDe | IAf | 1150±340 | NDe | >8800 |
| 8e | 780±170 | NDe | 1240±220 | 73.0±17 | 600±170 (76) | 3000±1100 |
| 8f | 1430±480 | 1600±300 | IAf | 501±65 | NDe | 274±56 |
| 8g | 820±300 | NDe | IAf | 101±19 | 1430±670 (83) | 3240±840 |
| 8h | 520±170 | NDe | 1220±310 | 223±60 | 2140±510 (88) | 212±77 |
| 8i | 214±47 | NDe | IAf | 299±30 | NDe | >10,000 |
| 8j | 800±310 | NDe | IAf | 6000±1400 | ND | 4800±1200 |
| 8k | 83±30 | 421±47 | 3210±140 | 139±33 | 440±150 (57%) | 3030±400 |
| 8l | 17.0±5.2 | NDe | 3770±2900 | 2140±640 | NDe | 3290±480 |
| 8m | 1140±310 | NDe | 2990±100 | 1170±190 | NDe | 3540±910 |
| 8n | 320±100; 4.9±1.3 (14.9)b | NDe | IAf | IAf | NDe | 272±79 |
| 8o | 255±43 | NDe | IAf | IAf | NDe | 1910±350 |
| 15b | 918±61 | NDe | NDe | IAf | NDe | 2580±400 |
| (±)2 | 29.9±5.76c | 4370±59.4 171.2±60.5 (22%)b,c |
495 (n=2) | 104±23.5c | NDe | ND |
| sulpiride | 66.22±10.89 | 10.28±1.23 | 560±170 | NDe | NDe | NDe |
Data were obtained through the NIDA Addiction Treatment Discovery Program contract with Oregon Health & Science University (Y1 DA 5007-05).
The first value is the antagonist IC50, the second value is the agonist EC50 with % stimulation in parentheses.
data previously reported in ref. 11.
data previously reported in ref. 6.
ND = Not determined;
IA= <50% inhibition at a concentration of 10 μM.
In addition to binding data at D2R and D3R, selected analogues were evaluated in the quinpirole-induced mitogenesis assay in hD2- or hD3-transfected CHOp cells and in a dopamine-induced beta-arrestin translocation assay in hD3-transfected CHO-K1 cells. These data are shown in Table 3 and compared to the D3R-selective antagonist (±)2 and the D2R-family nonselective antagonist sulpiride. Most of the analogues were antagonists in both the D3R mitogenesis and beta arrestin translocation assays although 8n was a more potent partial agonist than an antagonist in the mitogenesis assay. In general, functional potencies of these analogues were lower than their binding affinities and the compounds were less potent in the beta arrestin assay than in the mitogenesis assay resulting in some compounds showing <50% inhibition at a concentration of 10 μM. Unfortunately, due to solubility limitations, higher concentrations could not be tested. Although we cannot fully explain the disparity in absolute potencies across in vitro assays at this time, these functional assays were not run under steady state conditions as were the binding assays and were not optimized for this class of compounds. Further investigation into this is underway, as a consistent relationship between cell-based D3R functional assay potencies and behavior in vivo has yet to be established. 4 for review
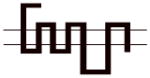
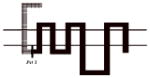
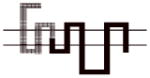
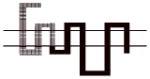
Compound 8d exhibited the greatest D3R versus D2R binding selectivity (2400-fold) of all the 4-phenylpiperazines that we have evaluated to date. Representative competitive radioligand binding curves for the inhibition of the binding of 125I-IABN to human D2L and D3 receptors by compound 8d are shown in Figure 2. To begin to define the molecular basis for this receptor subtype binding selectivity, we performed a series of radioligand binding studies using D3/D2 chimeric receptor proteins to identify what binding domains in the D3R as compared to the D2R were interacting with compound 8d to achieve this magnitude of selectivity. Table 4 shows a schematic representation of the wild type human D2R (all black) and D3R (all grey) compared to the chimeric receptors. Table 4 also shows the affinity of the wild type and chimeric receptors for 125I-IABN and 8d. IABN binds non-selectively at D2R (Kd = 0.03 nM) and D3R (Kd = 0.4 nM) and it binds with similar affinity at each of the chimeric proteins (Ki values range from 0.02 to 0.08 nM).
Figure 2.

Representative Radioligand Binding Competition Curve for the Binding of Compound 8d to human D2R and D3R
Table 4.
Binding Affinities at hD2R, hD3R and Chimeric Receptors for Compound 8d
| IABN | Compound 8d | ||
|---|---|---|---|
|
D2 Wild type | 0.03 ± 0.004 | 9,163 ± 3731 |
|
Chimera A | 0.05 ± 0.009 | 2,369 ± 945 |
|
Chimera B | 0.02 ± 0.004 | 13.9 ± 1.8 |
|
Chimera C | 0.02 ± 0.006 | 20.8 ± 3.0 |
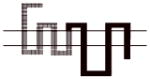
|
Chimera D | 0.01 ± 0.003 | 11.5 ± 2.1 |
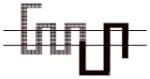
|
Chimera E | 0.03 ± 0.001 | 1.7 ± 0.1 |
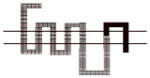
|
Chimera F | 0.03 ± 0.011 | 2.9 ± 0.3 |
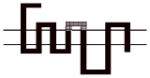
|
D2/D3 E2 loop | 0.08 ± 0.007 | 245 ± 37 |
|
D3/D2 E2 loop | 0.03 ± 0.004 | 31.5 ± 3.0 |
|
D3 Wild type | 0.04 ± 0.003 | 5.4 ± 0.6 |
All of the dissociation constants are expressed as nM and are the mean± S.E.M. For IABN Kd values were obtained by Scatchard analysis of direct binding studies and values for the other compounds are Ki values obtained from competitive radioligand binding analysis. The number of independent experiments performed to obtain the mean values is n ≥ 3 for the chimeric receptors. For the binding of 125I-IABN to D2R wild type n = 10 and for the D3R wild type n = 13. For the binding of 8d to D2R wild type n = 7 and for the D3R wild type n = 5.
A comparison of binding between the D2R with chimera A for 8d indicated that substitution of the first transmembrane spanning (TMS I) region from the D3R onto the D2R backbone resulted in a 4-fold increase in affinity. Previous studies have suggested that the extracellular portion of the TMS I region does not have H2O accessible residues and is, therefore, unlikely to make direct contact with bound ligand.25 Differences in amino acid sequence between the TMS I regions of the D2R and the D3R might contribute to topographical differences between the two binding sites. However, the observed low affinity of 8d at D2R (Figure 1) makes it problematic to determine the reliability of this difference in Ki values.
A more dramatic difference in the affinity (>400-fold) of 8d was observed between wild type D2R and chimeras B, C and D (Table 4). The affinities of 8d at these three chimeric receptors are similar. These results implicate TMS II and TMS III and the first extracellular (E1) loop as important structural elements in the binding selectivity of 8d at D2R and D3R. An additional 5- to 10-fold increase in binding selectivity was found for chimeras E and F compared to the wild type D2R suggesting that difference in the structure of TMS IV and V between D2R and D3R also contribute to the observed binding selectivity of 8d. It is interesting to note that the affinity of 8d for Chimera E was actually 3-fold higher than at the D3R wild type, emphasizing the importance of these regions to D3R binding affinities for the 4-phenylpiperazines.
Studies on the three-dimensional structure of bovine rhodopsin,26 and more recently on the adrenergic receptors,27,28 have indicated that the conserved disulfide bond, which joins the conserved cysteine residue located within the extracellular E2 loop with the top of the third (TMS III) region, brings the E2 loop in close proximity to the extracellular portion of the helical TMS regions of the G-protein coupled receptors. In addition, several studies have suggested that amino acid residues within the E2 loop of the dopamine receptors can interact with ligands positioned in the neurotransmitter (orthosteric) binding site, thereby influencing binding affinity.29, 30 Recently, this region of D3R was also implicated in the binding of a novel allosteric antagonist SB269,652 (N-((1R,4R)-4-(2-(7-cyano-3,4-dihydroisoquinolin-2(1H)-yl)ethyl)cyclohexyl)-1H-indole-2-carboxamide), which bears structural similarity to compounds reported previously and herein.31
To further investigate the possible contribution of the D3R E2 loop in the binding selectivity of 8d, two additional chimeric receptors were constructed (D2/D3 E2 loop and D3/D2 E2 loop) and evaluated for affinity for 8d. The D2/D3 E2 loop chimera contains the wild type D2R structure but the E2 loop is identical to the amino acid sequence found in the D3R subtype. D3/D2 E2 loop chimera is the reciprocal mutant receptor. A 37-fold increase in affinity was observed by substituting the E2 loop of the D3R on the D2R scaffold and a >5-fold decrease in affinity was observed when the E2 loop of the D2 receptor was substituted for the E2 loop of the D3R on the D3R scaffold. The results of these binding studies suggest that 8d interacts with the E2 loop and that interaction contributes to the D3R binding selectivity. We hypothesize that the 3-F substitution on the four carbon linking chain of 8d is the moiety in contact with the E2 loop. These data further support SAR described in our previous study with the 3-OH-substituted phenylpiperazines.11 However, the dramatic improvement in binding observed in chimera B, as compared to A, suggests that the E1 loop may also be involved in the differential binding of these compounds to D3R compared to D2R. Of note, while this manuscript was in preparation, the crystal structure of the human D3R binding in complex with the D2-like receptor antagonist eticlopride was published.32 In addition to characterizing the eticlopride binding domain, a second binding pocket was defined with compound 2 (R-22 = R-PG 648), wherein the 2,3-diCl-phenylpiperazine overlapped with eticlopride in the orthosteric site, and the 3-OH-butylamide-linked indole portion of the molecule occupied a second domain that was differentially engaged by nonconserved residues in D3R and D2R.32 These studies provided further evidence of differential binding domains that exist for D2R and D3R subtypes and highlight the importance of both the E1 and E2 loops that interface with TMS I, II and VII. Hence, additional chimera and single point mutation studies, along with extensive molecular dynamics studies are underway to further characterize the binding domains of the D3R and D2R subtypes and to further define these interactions of the D3R-selective ligands at the molecular level.
In summary, earlier SAR studies with prototypic 2,3-dichloro or 2-methoxy-4-phenylpiperazine analogues included varying substituents on the butyl amide linker that exhibited high affinity and D3R selectivity. Furthermore, enantioselectivity was achieved with the 3-OH-substituted analogue 2, with enantioselectivity being more pronounced (15-fold) at D3R than D2R (<2-fold).11 In the present study, a series of 3-F-carboxamides also showed high D3R affinity and selectivity, including two of the most D3R selective compounds (8d and 8j) reported to date. Chimera studies confirmed the pivotal contribution of the E2 loop in the D3R binding selectivity of 8d suggesting that the 3-F, like the 3-OH group in compound 2, possesses physicochemical properties that are well tolerated in this region of D3R and likely influences how the rest of the molecule resides in the binding site. Further, the contribution of the E1 extracellular loop was also revealed and future chimera studies that explicitly test this are underway. In addition, the arylamide clearly extends beyond the orthosteric site that is indistinguishable in D2R and D3R, to a binding domain that only tolerates the D3R pharmacophore exemplified by compounds such as those reported herein and shown recently.32 Finally, the carbonyl of the carboxamide moiety in these molecules is essential for high affinity binding at D3R, but not D2R, further separating SAR between these homologous proteins. Structure-based protein modeling based on the D3R crystal structure will undoubtedly identify critical amino acid residues that are different in these regions between D3R and D2R and will provide further rationale for the design of novel D3R-selective agents.
Experimental Methods
1H and 13C NMR spectra were acquired using a Varian Mercury Plus 400 spectrometer. Chemical shifts are reported and referenced according to deuterated solvent for 1H spectra (CDCl3, 7.26), 13C spectra (CDCl3, 77.2), 19F spectra (CFCl3, 0). Combustion analysis was performed by Atlantic Microlab, Inc. (Norcross, GA) and agrees within 0.4% of calculated values. Melting point determinations were conducted using a Thomas-Hoover melting point apparatus and are uncorrected. Anhydrous solvents were purchased from Aldrich were used without further purification, except for tetrahydrofuran, which was freshly distilled from sodium-benzophenone ketyl. All other chemicals and reagents were purchased from Aldrich Chemical Co., Combi-Blocks, TCI, America., Matrix Scientific; Lancaster Synthesis, Inc. (Alfa Aesar) and AK Scientific, Inc. Final compounds (free base) were purified by column chromatography (EMD Chemicals, Inc.; 230–400 mess, 60 Å) or preparative thin layer chromatography (silica gel, Analtech, 1000 μm). The final products were converted into oxalate salts, typically by treating the free base with 1:1 molar ratio of oxalic acid in acetone followed by precipitation from a combination of organic solvents. Yields and reaction conditions are not optimized. Generally, yields and spectroscopic data refer to the free base. On the basis of NMR, GC-MS (where obtainable), and combustion analysis data, all final compounds are >95% pure. The procedures to determine the binding affinities at the human D2-like receptors have been described.
Synthesis. 2-(3-fluoro-4-(4-(2,3-dichlorophenyl)piperazin-1-yl)butyl)isoindoline-1,3-dione (6a)
To a solution of compound 5a (1.8 g, 4.4 mmol) in dry CH2Cl2 (50 mL) cooled at −78 °C was added a solution of DAST (1.06 g, 6.6 mmol) in dry CH2Cl2 (25 mL) over a period of 30 min. This reaction mixture was continuously stirred for 12 h at room temperature. The reaction mixture was diluted with CH2Cl2 and washed with ice-H2O and 5% NaHCO3 solution, and the organic layer was dried over anhydrous Na2SO4. The solvent was removed under vacuum to afford the crude product. This was further purified by column chromatography (30% EtOAc-hexane) afford the compound 6a (1.09 g) in 60% yield. Mp 132–134 °C; 1H NMR (400 MHz, CDCl3) δ ppm 1.94–2.136 (m, 2H), 2.53–2.80 (m, 7H), 3.05 (brs, 4H), 3.86–3.91 (m, 1H), 6.94 (dd, J = 6.4, 2.8 Hz, 1H), 7.13–7.15 (m, 2H), 7.72 (dd, J = 5.6, 3.2 Hz, 2H), 7.86 (dd, J = 5.6, 3.2 Hz); 13C NMR (100 MHz, CDCl3) δ 168.451, 151.43, 134.24, 132.31, 127.73, 127.67, 124.81, 123.52, 118.82, 90.76 (d, J = 170 Hz, 1C), 62.17 (d, J = 20.6 Hz, 1C), 54.10, 51.44, 34.72 (d, J = 4.6 Hz, 1C), 32.46 (d, J = 20.6 Hz, 1C); 19F NMR (376 MHz, CDCl3/CFCl3) δ −183.0 to −183.39 (m, 1F).
2-(3-Fluoro-4-(4-(2,3-dichlorophenyl)piperazin-1-yl)butyl)isoindoline-1,3-dione (6b)
This compound was prepared by the method described for compound 6a, employing 5b (1.6 g, 5 mmol) and DAST (870 mg, 5.4 mmol), afford 6b (940 mg) in 58% yield. Mp 97–99 °C; 1H NMR (400 MHz, CDCl3) δ ppm 1.95–2.13 (m, 2H), 2.50–2.78 (m, 7H), 3.07 (brs, 4H), 3.85 (m, 3H), 3.86–3.90 (m, 1H), 4.70–4.88 (m, 1H), 6.83–7.01 (m, 4H), 7.71 (dd, J = 5.6, 3.2 Hz), 7.85 (dd, J = 5.6, 2.8 Hz); 13C NMR (100 MHz, CDCl3) δ 168.40, 152.37, 141.38, 134.11, 132.23, 123.41, 123.03, 121.08, 118.31, 111.23, 90.68 (d, J = 170 Hz, 1C), 62.27 (d, J = 20.6 Hz, 1C), 55.45, 54.16, 50.69, 34.64 (d, J = 5.3 Hz, 1C), 32.41 (d, J = 20.6 Hz, 1C); 19F NMR (376 MHz, CDCl3/CFCl3) δ − 182.85 to −183.30 (m, 1F).
3-Fluoro-4-(4-(2-methoxyphenyl)piperazin-1-yl)butan-1-amine (7a)
To a solution of 6a (1.14 g, 2.8 mmol) in EtOH was added hydrazine (270 mg, 8.4 mmol) and stirred at reflux for 3 h. The cooled reaction mixture was evaporated in vacuo, the solid residue was partitioned between CHCl3 and 20% K2CO3 solution, and the organic layer was collected and dried over Na2SO4. The solvent was removed under vaccum to afford the 7a, which was used for next step without further purification. Yield: 95% (748 mg). 1H NMR (400 MHz, CDCl3) δ ppm 1.62–1.86 (m, 2H), 2.48–2.95 (m, 5H), 3.11 (brs, 4H), 3.86 (s, 3H), 4.76–4.96 (m, 1H), 6.84–7.02 (m, 4H); 13C NMR (100 MHz, CDCl3) δ 152.48, 141.51, 123.15, 121.20, 118.42, 11.36, 91.06 (d, J = 168.4 Hz, 1C), 62.9 (d, J = 21.4 Hz, 1C), 55.57, 54.26, 50.81, 38.51 (d, J = 4.6 Hz, 1C), 37.63 (d, J = 19.8 Hz, 1C); 19F NMR (376 MHz, CDCl3/CFCl3) δ −182.5 to −182.9 (m, 1F); GC/MS RT(RIMS): 14.1 min (281 [M]+ m/z).
4-(4-(2,3-dichlorophenyl)piperazin-1-yl)-3-fluorobutan-1-amine (7b)
This compound was prepared by the method described for compound 7a, employing 6b (700 mg, 1.56 mmol) and hydrazine (164 mg, 4.7 mmol), afford 7b (480 mg) in 96% yield. 1H NMR (400 MHz, CDCl3) δ ppm 1.62–1.91 (m, 2H), 1.49–1.94 (m, 7H), 3.07 (brs, 4H), 4.75–4.94 (m, 1H), 6.95 (dd, J = 6.4, 2.8 Hz, 1H), 7.13–7.15 (m, 2H), 7.72 (dd, J = 5.6, 3.2 Hz, 2H), 7.11–7.16 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 151.36, 134.14, 127.64, 127.58, 124.71, 118.72, 90.9 (d, J = 168.5 Hz, 1C), 62.64 (d, J = 20.6 Hz, 1C), 53.99, 53.97, 51.38, 38.38 (d, J = 4.6 Hz, 1C), 37.45 (d, J = 19.8 Hz, 1C); 19F NMR (376 MHz, CDCl3/CFCl3) δ −182.61 to −183.01 (m, 1F); GC/MS RT(RIMS): 15.5 min (299 [M-HF]+ m/z).
General Amidation Procedures
Procedure A
CDI (1 equiv) was added to a solution of the carboxylic acid (1 equiv) in THF (10 mL/mmol). The reaction mixture was stirred at room temperature for 2 h. The solution was cooled to 0 °C and the appropriate amine (1 equiv) was added dropwise. The reaction mixture was allowed to warm up to room temperature and stirred for 2–3 h. The solvent was removed in vacuo. The residue was diluted in CHCl3 (30 mL) and washed with saturated aq NaHCO3 solution (2 × 10 mL). The organic layer was dried with Na2SO4 and concentrated in vacuo. The crude product was purified by crystallization from 2-PrOH or flash column chromatography, as indicated.
Procedure B
Thionyl chloride (SOCl2; 2 mL/mmol) was added to the carboxylic acid (1 equiv). The solution was stirred at reflux for 3h and concentrated in vacuo. Residual SOCl2 was removed by azeotropic distillation in dry benzene. The resulting solid was dissolved in amylene stabilized CHCl3 (5 mL). To a stirred solution of the amine (1 equiv) in amylene stabilized CHCl3 (20 mL) and 0.5 M aq NaOH (8 mL) cooled to 0 °C, was added the acid chloride solution drop wise. The solution was stirred vigorously for 3 h at room temperature. The organic layer was separated, dried with Na2SO4 and concentrated in vacuo. The crude product was purified by crystallization from 2-PrOH or flash column chromatography, as indicated.
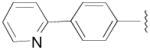
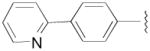
N-(3-fluoro-4-(4-(2-methoxyphenyl)piperazin-1-yl)butyl)-6-phenylnicotinamide (8a)
CDI (908 mg, 5.6 mmol) was added to a solution of 4-(pyridine-2-yl)benzoic acid hydrochloride (658 mg, 2.8 mmol) in pyridine (10 mL). The reaction mixture was stirred at room temperature for 2 h. The solution was cooled to 0 °C and the amine 7a (787 mg, 2.8 mmol) in pyridine (5 mL) was added dropwise. The reaction was allowed to warm to room temperature and stirred overnight. The resulting solution was evaporated, diluted in CHCl3 (30 mL) and washed with saturated aq. NaHCO3 (2 × 10 mL). The organic layer was dried over Na2SO4 and evaporated under reduced pressure. The crude product was purified by column chromatography using EtOAc/CHCl3/MeOH (5:5:1) and 1% triethylamine afford 8a (470 mg) in 36% yield. The oxalate salt was precipitated from 2-PrOH. Mp 170–172 °C; 1H NMR (400 MHz, CDCl3) δ ppm 1.95–2.17 (m, 2H), 2.56–2.83 (m, 6H), 3.09 (brs, 4H), 3.59–3.76 (m, 2H), 3.86 (s, 3H), 4.82–5.0 (m, 1H), 6.7 (brs, 1H), 6.84–7.02 (m, 4H), 7.27–7.30 (m, 1H), 7.77–7.79 (m, 2H), 7.86–7.89 (m, 2H), 8.05–8.09 (m, 2H), 8.71–8.73 (m, 1H); 13C NMR (100 MHz, CDCl3) δ 167.48, 156.45, 152.47, 150.01, 142.45, 141.38, 137.13, 134.91, 127.63, 127.27, 123.21, 122.98, 121.20, 121.08, 118.43, 111.35, 92.0 (d, J = 167.7 Hz, 1C), 62.37 (d, J = 21.3 Hz, 1C), 55.58, 54.30, 50.74, 37.05, 33.17(d, J = 19.8 Hz, 1C); 19F NMR (376 MHz, CDCl3/CFCl3) δ −181.38 to −181.82 (m, 1F); Anal. (C27H31FN4O2· C2H2O4·1/2H2O) C, H, N.
N-(4-(4-(2,3-Dichlorophenyl)piperazin-1-yl)-3-fluorobutyl)-6-phenylnicotinamide (8b)
This compound was prepared by the method described for compound 8a, employing 4-(pyridine-2-yl)benzoic acid hydrochloride (367 mg, 1.6 mmol), 7b (480 mg, 1.5 mmol), and CDI (243 mg, 1.5 mmol), afford 8b (406 mg) in 54% yield. Mp 163–165 °C; 1H NMR (400 MHz, CDCl3) δ ppm 1.95–2.17 (m, 2H), 2.56–2.84 (m, 6H), 3.07 (brs, 4H), 3.60–3.76 (m, 2H), 4.81–5.0 (m, 1H), 6.7 (brs, 1H), 6.93 (dd, J = 7.6, 2.0 Hz, 1H), 7.12–7.17 (m, 2H), 7.26–7.30 (m, 1H), 7.75–7.81 (m, 2H), 7.86–7.89 (m, 2H), 8.06–8.09 (m, 2H), 8.71–8.73 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 167.48, 156.41, 151.34, 150.09, 142.47, 137.15, 134.88, 134.24, 127.73, 127.68, 127.61, 127.28, 124.86, 122.99, 121.07, 118.82, 91.95 (d, J = 167.7 Hz, 1C), 62.24 (d, J = 21.4 Hz, 1C), 54.13, 51.41, 37.04 (d, J = 3.8 Hz, 1C), 33.18 (d, J = 19.9 Hz, 1C); 19F NMR (376 MHz, CDCl3/CFCl3) δ −181.6 to −182.06 (m, 1F); Anal. (C26H27Cl2FN4O) C, H, N.
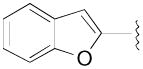
N-(3-Fluoro-4-(4-(2-methoxyphenyl)piperazin-1-yl)butyl)benzofuran-2-carboxamide (8c)
Prepared from benzofuran-2-carboxylic acid and 7a according to general procedure B. The crude compound was purified by column chromatography using EtOAc/CHCl3/MeOH (20:20:1) and 1% triethylamine to afford 8c in 73% yield. The oxalate salt was precipitated from EtOAc. Mp 168–170 °C; 1H NMR (400 MHz, CDCl3) δ ppm 1.99–2.12 (m, 2H), 2.56–2.83 (m, 6H), 3.12 (brs, 4H), 3.63–3.71 (m, 2H), 3.86 (s, 3H), 4.82–5.0 (m, 1H), 6.85–7.06 (m, 5H), 7.27–7.31 (m, 1H), 7.38–7.5 (m, 3H), 7.67 (d, J = 8.0 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 159.26, 154.94, 152.48, 148.92, 141.39, 127.84, 127.11, 123.93, 123.24, 122.96, 121.20, 118.44, 111.98, 111.36, 110.63, 91.47 (d, J = 183.1 Hz, 1C), 62.35 (d, J = 21.3 Hz, 1C), 55.58, 54.30, 50.76, 36.12 (d, J = 4.6 Hz, 1C), 33.29 (d, J = 20.5 Hz, 1C); 19F NMR (376 MHz, CDCl3/CFCl3) δ −182.11 to −182.62 (m, 1F); Anal. (C24H28FN3O3· C2H2O4· 1/2H2O) C, H, N.
N-(4-(4-(2,3-Dichlorophenyl)piperazin-1-yl)-3-fluorobutyl)benzofuran-2- carboxamide (8d)
Prepared from benzofuran-2-carboxylic acid and 7b according to general procedure B. The crude compound was purified by column chromatography using EtOAc/CHCl3/MeOH (20:20:1) and 1% triethylamine afford 8d in 82% yield. Mp 164–166 °C; 1H NMR (400 MHz, CDCl3) δ ppm 1.98–2.14 (m, 2H), 2.57–2.84 (m, 6H), 3.09 (brs, 4H), 3.63–3.73 (m, 2H), 4.81–5.0 (m, 1H), 6.94 (dd, J = 6.8, 2.4 Hz, 1H), 7.0 (brs, 1H), 7.11–7.18 (m, 2H), 7.28–7.50 (m, 3H), 7.66–7.69 (m, 1H); 13C NMR (100 MHz, CDCl3) δ 159.26, 154.94, 151.38, 148.89, 134.27, 127.83, 127.75, 127.69, 127.15, 124.87, 123.96, 122.99, 118.82, 111.95, 110.67, 91.47 (d, J = 168.4 Hz, 1C), 62.23 (d, J = 20.6 Hz, 1C), 54.15, 51.46, 36.14 (d, J = 4.5 Hz, 1C), 33.29 (d, J = 20.5 Hz, 1C); 19F NMR (376 MHz, CDCl3/CFCl3) δ −182.36 to −182.75 (m, 1F); Anal. (C23H24Cl2FN3O2· 1/5H2O) C, H, N.
N-(3-Fluoro-4-(4-(2-methoxyphenyl)piperazin-1-yl)butyl)-5-methoxybenzofuran-2-carboxamide (8e)
Prepared from 5-methoxybenzofuran-2-carboxylic acid and 7a according to general procedure B. The crude compound was purified by column chromatography using EtOAc/CHCl3/MeOH (20:20:1) and 1% triethylamine afford 8e in 82% yield. Mp 118–120 °C; 1H NMR (400 MHz, CDCl3) δ ppm 1.97–2.12 (m, 2H), 2.56–2.83 (m, 6H), 3.12 (brs, 4H), 3.62–3.72 (m, 2H), 3.85 (s, 3H), 3.86 (s, 3H), 4.82–5.0 (m, 1H), 6.85–7.09 (m, 7H), 7.35–7.41 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 159.25, 156.68, 152.48, 149.97, 141.41, 128.36, 123.22, 121.20, 118.43, 116.77, 112.55, 111.37, 110.71, 104.18, 91.48 (d, J = 167.6 Hz, 1C), 62.35 (d, J = 21.3 Hz, 1C), 55.58, 54.32, 50.77, 36.11, 33.29 (d, J = 20.6 Hz, 1C); 19F NMR (376 MHz, CDCl3/CFCl3) δ −182.1 to −182.6 (m, 1F); Anal. (C25H30FN3O4 ·1/2H2O) C, H, N.
N-(4-(4-(2,3-Dichlorophenyl)piperazin-1-yl)-3-fluorobutyl)-5-methoxybenzofuran-2-carboxamide (8f)
Prepared from 5-methoxybenzofuran-2-carboxylic acid and 7b according to general procedure B. The crude compound was recrystallized from 2-PrOH. Yield: 65%. The oxalate salt was precipitated from CHCl3. Mp 128–130 °C; 1H NMR (400 MHz, CDCl3) δ ppm 1.96–2.14 (m, 2H), 2.56–2.83 (m, 6H), 3.08 (brs, 4H), 3.62–3.72 (m, 2H), 3.85 (s, 3H), 4.79–4.99 (m, 1H), 6.92–6.98 (m, 2H), 7.02 (dd, J = 8.8, 2.8 Hz, 1H), 7.08 (d, J = 2.4 Hz, 1H), 7.11–7.17 (m, 2H), 7.35–7.41 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 159.26, 156.70, 151.39, 149.96, 149.55, 134.27, 128.37, 127.69, 124.87, 118.82, 116.80, 112.53, 110.74, 104.20, 91.47 (d, J = 168.4 Hz, 1C), 62.23 (d, J = 20.6 Hz, 1C), 56.09, 54.17, 51.47, 36.11 (d, J = 4.6 Hz, 1C), 36.97, 33.3(d, J = 19.8 Hz, 1C); 19F NMR (376 MHz, CDCl3/CFCl3) δ −181.53 to − 181.92 (m, 1F); Anal. (C24H26Cl2FN3O· C2H2O4·3/4H2O) C, H, N.
N-(3-Fluoro-4-(4-(2-methoxyphenyl)piperazin-1-yl)butyl)-1H-indole-2-carboxamide (8g)
Prepared from 1H-indole-2-carboxylic acid and 7a according to general procedure A. The crude compound was purified by column chromatography using EtOAc/CHCl3/MeOH (20:20:1) and 1% triethylamine afford 8g in 62% yield. Mp 148–150 °C; 1H NMR (400 MHz, CDCl3) δ ppm 1.90–2.15 (m, 2H), 2.56–2.84 (m, 6H), 3.11 (brs, 4H), 3.61–3.77 (m, 2H), 3.86 (s, 3H), 4.81–5.02 (m, 1H), 6.60 (brs, 1H), 6.83–7.04 (m, 5H), 7.14 (t, J = 7.6 Hz, 1H), 7.29 (t, J = 7.6 Hz, 1H), 7.44 (d, J = 8.0 Hz, 1H), 7.65 (d, J = 8.0 Hz, 1H), 9.31 (brs, 1H); 13C NMR (100 MHz, CDCl3) δ 161.94, 152.48, 141.40, 136.47, 130.86, 127.87, 124.75, 123.24, 122.18, 121.22, 120.91, 118.43, 112.17, 111.37, 102.19, 91.9 (d, J = 167.7 Hz, 1C), 62.24 (d, J = 21.3 Hz, 1C), 55.58, 54.31, 50.77, 36.65, 33.3 (d, J = 19.8 Hz, 1C); 19F NMR (376 MHz, CDCl3/CFCl3) δ −181.8 to − 181.26 (m, 1F); Anal. (C24H29FN4O2· 1/4H2O) C, H, N.
N-(4-(4-(2,3-Dichlorophenyl)piperazin-1-yl)-3-fluorobutyl)-1H-indole-2-carboxamide (8h)
Prepared from 1H-indole-2-carboxylic acid and 7b according to general procedure A. The crude compound was purified by column chromatography using EtOAc/CHCl3/MeOH (20:20:1) and 1% triethylamine afford 8h in 65% yield. The oxalate salt was precipitated from CHCl3. Mp 203–205°C; 1H NMR (400 MHz, CDCl3) δ ppm 1.94–2.10 (m, 2H), 2.56–2.82 (m, 6H), 3.07 (brs, 4H), 3.61–3.76 (m, 2H), 4.81–5.0 (m, 1H), 6.60 (t, J = 5.2 Hz, 1H), 6.83–6.92 (m, 1H), 6.93 (dd, J = 7.2, 2.4 Hz, 1H), 7.11–7.26 (m, 3H), 7.27–7.31 (m, 2H), 7.45 (d, J = 8.0 Hz, 1H), 7.64 (d, J = 8.4 Hz, 1H), 9.47 (brs, 1H); 13C NMR (100 MHz, CDCl3) δ 161.97, 151.36, 136.49, 134.27, 130.83, 127.86, 127.70, 124.88, 124.78, 122.17, 120.94, 118.12, 112.19, 102.20, 91.85 (d, J = 168.4 Hz, 1C), 62.26 (d, J = 21.4 Hz, 1C), 54.14, 51.43, 36.62, 33.31 (d, J = 19.8 Hz, 1C); 19F NMR (376 MHz, CDCl3/CFCl3) δ −182.0 to −182.5 (m, 1F); Anal. (C23H25Cl2FN4O· C2H2O4) C, H, N.
N-(3-Fluoro-4-(4-(2-methoxyphenyl)piperazin-1-yl)butyl)-9H-fluorene-2-carboxamide (8i)
Prepared from 9H-fluorene-2-carboxylic acid and 7a according to general procedure A. The crude compound was purified by column chromatography using EtOAc/CHCl3/MeOH (20:20:1) and 1% triethylamine afford 8i in 75% yield. Mp 175–178 °C; 1H NMR (400 MHz, CDCl3) δ ppm 1.96–2.13 (m, 2H), 2.58–2.84 (m, 6H), 3.10 (brs, 4H), 3.59–3.76 (m, 2H), 3.85 (s, 3H), 3.93 (s, 2H), 4.84–5.03 (m, 1H), 6.75 (brs, 1H), 6.84–6.91 (m, 3H), 6.97–7.02 (m, 1H), 7.34–7.42 (m, 2H), 7.57 (d, J = 7.2 Hz, 1H), 7.79–7.83 (m, 3H), 7.97 (s, 1H); 13C NMR (100 MHz, CDCl3) δ 168.15, 152.46, 145.13, 144.25, 143.67, 141.31, 140.88, 133.01, 127.89, 127.22, 126.03, 125.44, 124.05, 123.27, 121.20, 120.79, 119.97, 118.41, 111.37, 91.96 (d, J = 167.7 Hz, 1C), 62.34 (d, J = 21.4 Hz, 1C), 55.58, 54.27, 50.67, 37.07 (d, J = 10.7 Hz, 1C), 33.2 (d, J = 19.8 Hz, 1C); 19F NMR (376 MHz, CDCl3/CFCl3) δ −181.04 to −181.54 (m, 1F); Anal. (C29H32FN3O2) C, H, N.
N-(4-(4-(2,3-Dichlorophenyl)piperazin-1-yl)-3-fluorobutyl)-9H-fluorene-2-carboxamide (8j)
Prepared from 9H-fluorene-2-carboxylic acid and 7b according to general procedure B. The crude product was recrystallized from 2-PrOH afford 8j in 87% yield. The oxalate salt was precipitated from EtOAc. Mp 189–191 °C; 1H NMR (400 MHz, CDCl3) δ ppm 1.95–2.17 (m, 2H), 2.58–2.85 (m, 6H), 3.07 (brs, 4H), 3.60–3.77 (m, 2H), 3.94 (s, 2H), 4.82–5.02 (m, 1H), 6.55 (brs, 1H), 6.90 (dd, J = 7.6, 1.6 Hz, 1H), 7.08–7.16 (m, 2H), 7.34–7.43 (m, 2H), 7.56–7.59 (m, 1H), 7.76–7.84 (m, 3H), 7.97 (s, 1H); 13C NMR (100 MHz, CDCl3) δ 168.12, 151.33, 145.17, 144.23, 143.69, 140.86, 134.27, 133.0, 127.92, 127.74, 127.67, 127.24, 125.99, 125.46, 124.88, 124.04, 120.80, 119.97, 118.79, 92.0 (d, J = 167.6 Hz, 1C), 62.26 (d, J = 20.6 Hz, 1C), 54.14, 51.39, 37.10 (d, J = 6.1 Hz, 1C), 33.24 (d, J = 19.9 Hz, 1C); 19F NMR (376 MHz, CDCl3/CFCl3) δ −181.52 to −181.9 (m, 1F); Anal. (C28H28Cl2FN3O· C2H2O4) C, H, N.
N-(3-Fluoro-4-(4-(2-methoxyphenyl)piperazin-1-yl)butyl)benzo[b]thiophene-2-carboxamide (8k)
Prepared from benzo[b]thiophene-2-carboxylic acid and 7a according to general procedure B. The crude product was purified by column chromatography using EtOAc/CHCl3/MeOH (20:20:1) and 1% triethylamine afford 8k in 81% yield. The oxalate salt was precipitated from CHCl3. Mp 150–153 °C; 1H NMR (400 MHz, CDCl3) δ ppm 1.94–2.14 (m, 2H), 2.54–2.82 (m, 6H), 3.1 (brs, 4H), 3.58–3.74 (m, 2H), 3.86 (s, 3H), 4.81–5.0 (m, 1H), 6.69 (brs, 1H), 6.85 (d, J = 7.6 Hz, 1H), 6.91 (d, J = 7.6 Hz, 2H), 6.98–7.02 (m, 1H), 7.36–7.44 (m, 2H), 7.76 (s, 1H), 7.80–7.86 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 162.69, 152.48, 141.40, 141.03, 139.30, 138.66, 126.55, 125.38, 125.25, 125.16, 123.22, 122.96, 121.21, 118.41, 111.38, 91.94 (d, J = 168.4 Hz, 1C), 62.36 (d, J = 20.6 Hz, 1C), 55.58, 54.31, 50.77, 37.13 (d, J = 3.8 Hz, 1C), 33.15 (d, J = 20.6 Hz, 1C); 19F NMR (376 MHz, CDCl3/CFCl3) δ −181.68 to −182.08 (m, 1F); Anal. (C24H28FN3O2S· C2H2O4) C, H, N.
N-(4-(4-(2,3-Dichlorophenyl)piperazin-1-yl)-3-fluorobutyl)benzo[b]thiophene -2-carboxamide (8l)
Prepared from benzo[b]thiophene-2-carboxylic acid and 7b according to general procedure B. The crude product was recrystallized from 2-PrOH afford 8l in 80% yield. The oxalate salt was precipitated from EtOAc. Mp 185–187 °C; 1H NMR (400 MHz, CDCl3) δ ppm 1.94–2.16 (m, 2H), 2.56–2.83 (m, 6H), 3.07 (brs, 4H), 3.59–3.76 (m, 2H), 4.81–5.0 (m, 1H), 6.57 (brs, 1H), 6.92 (dd, J = 7.6, 2.4 Hz, 1H), 7.11–7.17 (m, 2H), 7.37–7.45 (m, 2H), 7.76 (s, 1H), 7.81–7.87 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 162.65, 151.36, 141.01, 139.28, 138.53, 134.27, 127.75, 127.69, 126.59, 125.43, 125.26, 125.19, 124.88, 122.96, 118.80, 91.95 (d, J = 168.5 Hz, 1C), 62.23 (d, J = 20.6 Hz, 1C), 54.14, 51.44, 37.15 (d, J = 3.8 Hz, 1C), 33.15 (d, J = 19.8 Hz, 1C); 19F NMR (376 MHz, CDCl3/CFCl3) δ −181.8 to −182.3 (m, 1F); Anal. (C23H24Cl2FN3OS· C2H2O4) C, H, N.
N-(3-Fluoro-4-(4-(2-methoxyphenyl)piperazin-1-yl)butyl)-5-methoxy-1H-indole-2-carboxamide (8m)
Prepared from 5-methoxy-1H-indole-2-carboxylic acid and 7a according to general procedure A. The crude product was purified by column chromatography using EtOAc/CHCl3/MeOH (20:20:1) and 1% triethylamine afford 8m in 64% yield. The oxalate salt was precipitated from CHCl3. Mp 140–142 °C; 1H NMR (400 MHz, CDCl3) δ ppm 1.96–2.13 (m, 2H), 2.55–2.83 (m, 6H), 3.11 (brs, 4H), 3.60–3.76 (m, 2H), 3.84 (s, 3H), 3.86 (s, 3H), 4.81–5.0 (m, 1H), 6.57 (brs, 1H), 6.74–6.76 (m, 1H), 6.85–7.04 (m, 5H), 7.33 (d, J = 8.8 Hz, 1H), 9.32 (brs, 1H); 13C NMR (100 MHz, CDCl3) δ 161.67, 154.67, 152.25, 141.17, 131.57, 131.06, 128.0, 123.01, 120.98, 118.20, 115.81, 112.82, 111.14, 102.30, 101.60, 91.65 (d, J = 167.7 Hz, 1C), 62.16 (d, J = 20.6 Hz, 1C), 55.71, 55.35, 54.08, 50.54, 36.35, 33.08 (d, J = 19.8 Hz, 1C); 19F NMR (376 MHz, CDCl3/CFCl3) δ −181.84 to −182.29 (m, 1F); Anal. (C25H31FN4O3· C2H2O4· H2O) C, H, N.
N-(4-(4-(2,3-Dichlorophenyl)piperazin-1-yl)-3-fluorobutyl)-5-methoxy-1H-indole-2-carboxamide (8n)
Prepared from 5-methoxy-1H-indole-2-carboxylic acid and 7b according to general procedure A. The crude product was purified by column chromatography using EtOAc/CHCl3/MeOH (20:20:1) and 1% triethylamine afford 8n in 46% yield. The oxalate salt was precipitated from CHCl3. Mp 170–172 °C; 1H NMR (400 MHz, CDCl3) δ ppm 1.96–2.15 (m, 2H), 2.55–2.83 (m, 6H), 3.07 (brs, 4H), 3.61–3.77 (m, 2H), 3.84 (s, 3H), 4.81–5.0 (m, 1H), 6.56 (t, J = 5.6 Hz, 1H), 6.75 (d, J = 2.0 Hz, 1H), 6.91–6.98 (m, 2H), 7.03 (d, J = 2.4 Hz, 1H), 7.11–7.17 (m, 2H), 7.34 (d, J = 9.2 Hz, 1H), 9.48 (brs, 1H); 13C NMR (100 MHz, CDCl3) δ 161.98, 154.90, 151.37, 134.27, 131.88, 131.25, 128.21, 127.75, 127.69, 124.88, 118.82, 116.06, 113.10, 102.51, 101.84, 91.84 (d, J = 168.4 Hz, 1C), 62.27 (d, J = 20.6 Hz, 1C), 55.94, 54.14, 51.44, 36.62 (d, J = 4.6 Hz, 1C), 33.33 (d, J = 20.6 Hz, 1C); 19F NMR (376 MHz, CDCl3/CFCl3) δ −182.10 to − 182.54 (m, 1F); Anal. (C24H27Cl2FN4O2· C2H2O4·1/2H2O) C, H, N.
N-(4-(4-(2,3-Dichlorophenyl)piperazin-1-yl)-3-fluorobutyl)-2-naphthamide (8o)
Prepared from 2-naphthoic acid and 7b according to general procedure B. The crude compound was recrystallized from 2-PrOH. Yield: 72%. The oxalate salt was precipitated from CHCl3. Mp 171–173 °C; 1H NMR (400 MHz, CDCl3) δ ppm 1.98–2.20 (m, 2H), 2.58–2.85 (m, 6H), 3.05 (brs, 4H), 3.63–3.79 (m, 2H), 4.84–5.01 (m, 1H), 6.76 (brs, 1H), 6.88 (dd, J = 7.6, 2.0 Hz, 1H), 7.01–7.17 (m, 2H), 7.52–7.59 (m, 2H), 7.81–7.93 (m, 4H), 8.28 (s, 1H); 13C NMR (100 MHz, CDCl3) δ 167.92, 151.35, 134.97, 134.27, 132.85, 131.99, 129.14, 128.75, 128.01, 127.89, 127.75, 127.67, 127.56, 127.04, 124.88, 123.77, 118.79, 92.05 (d, J = 167.7 Hz, 1C), 62.24 (d, J = 21.3 Hz, 1C), 54.15, 51.42, 37.12 (d, J = 3.8 Hz, 1C), 36.97, 33.2(d, J = 19.8 Hz, 1C); 19F NMR (376 MHz, CDCl3/CFCl3) δ −181.53 to −181.92 (m, 1F); Anal. (C25H26Cl2FN3O· C2H2O4· 1/2H2O) C, H, N.
N-(4-(4-(2-Methoxyphenyl)piperazin-1-yl)butyl)propionamide (11a)
Propionyl chloride (0.45 g, 4.8 mmol) was added to a solution of compound 10a (1.07 g, 4 mmol) and triethylamine (1.7 mL, 12mmol) in anhydrous CH2Cl2 (10 mL) at 0 °C under argon atmosphere and then stirred at room temperature for 4 h. The reaction was diluted with CH2Cl2 and washed with H2O and brine, and the organic layer was dried over Na2SO4. The solvent was removed under vacuum afford compound 11a (0.76 g) in 59% yield. 1H NMR (400 MHz, CDCl3) δ ppm 1.15 (t, J = 7.2 Hz, 3H), 1.55–1.61 (m, 4H), 2.18 (q, J = 8 Hz, 2H), 2.43 (t, J = 7.2 Hz, 2H), 2.65 (brs, 4H), 3.10 (brs, 4H), 3.25–3.30 (m, 2H), 3.86 (s, 3H), 6.04 (brs, 1H), 6.84–7.02 (m, 4H); 13C NMR (100 MHz, CDCl3) δ 173.78, 152.33, 141.28, 123.07, 121.06, 118.23, 111.23, 58.12, 55.43, 53.47, 50.07, 39.43, 29.91, 27.60, 24.47, 10.10.
N-(4-(4-(2,3-Dichlorophenyl)piperazin-1-yl)butyl)propionamide (11b)
This compound was prepared by the method described for compound 11a, employing 10b (2.3 g, 7.6 mmol), propionyl chloride (0.84 g, 4.8 mmol) and triethylamine (3.2 mL, 23 mmol), afford 11b (1.5 g) in 56% yield. 1H NMR (400 MHz, CDCl3) δ ppm 1.15 (t, J = 7.2 Hz, 3H), 1.56–1.60 (m, 4H), 2.19 (q, J = 8 Hz, 2H), 2.44 (t, J = 7.2 Hz, 2H), 2.64 (brs, 4H), 3.07 (brs, 4H), 3.25–3.31 (m, 2H), 5.88 (brs, 1H), 6.93–6.96 (m, 1H), 7.11–7.17 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 173.81, 151.31, 134.19, 127.64, 127.61, 124.77, 118.67, 58.09, 53.40, 51.39, 39.46, 29.98, 27.68, 24.46, 10.12.
4-(4-(2-Methoxyphenyl)piperazin-1-yl)-N-propylbutan-1-amine (12a)
Compound 11a (0.76 g, 2.4 mmol) in anhydrous THF (6 mL) was added dropwise into a suspension of lithium aluminum hydride (LiAlH4; 0.46 g, 12 mmol) in anhydrous THF (20 mL) at 0 °C under an argon atmosphere. The reaction mixture was stirred at reflux for 4 h, cooled to room temperature, and then cooled further to 0 °C. Saturated NaOH/H2O (5 mL) was added drop wise to quench the excess LiAlH4. The mixture was filtered, and the reaction mixture was dried over Na2SO4. The solvent was removed under vacuum afford compound 12a (0.52 g) in 71 % yield. 1H NMR (400 MHz, CDCl3) δ ppm 0.92 (t, J = 7.6 Hz, 3H), 1.49–1.57 (m, 6H), 2.42 (t, J = 7.2 Hz, 2H), 2.56–2.66 (m, 6H), 3.09 (brs, 4H), 6.84–7.01 (m, 4H); 13C NMR (100 MHz, CDCl3) δ 152.38, 141.45, 123.02, 121.10, 118.31, 111.23, 58.75, 55.46, 53.59, 51.93, 50.77, 49.91, 28.19, 24.92, 23.21, 11.94.
4-(4-(2,3-Dichlorophenyl)piperazin-1-yl)-N-propylbutan-1-amine (12b)
This compound was prepared by the method described for compound 12a, employing 11b (1.4 g, 3.9 mmol) and lithium aluminum hydride (LiAlH4) (0.74 g, 19.5 mmol), afford 12b (1.1 g) in 82% yield. 1H NMR (400 MHz, CDCl3) δ ppm 0.93 (t, J = 7.6 Hz, 3H), 1.50–1.60 (m, 6H), 2.39–2.46 (m, 2H), 2.58–2.66 (m, 6H), 3.07 (brs, 4H), 6.94–6.98 (m, 1H), 7.13–7.16 (m, 2H).
General procedure C for imine reduction
4-(4-(2-methoxy or 2,3-dichlorophenyl)piperazin-1-yl)-N-propylbutan-1-amine (1 equiv) and appropriate aldehyde (1 equiv) were mixed in 1,2-dichloroethane (5 mL) and then treated with sodium triacetoxyborohydride (1.5 equiv) and AcOH (0.2 g). The mixture was stirred at room temperature under an argon atmosphere for 24 h. The reaction mixture was quenched by adding 1 N NaOH, and the product was extracted with EtOAc. The EtOAc extract was washed with brine, dried over Na2SO4 and concentrated in vacuo. The crude product was purified by preparative thin layer chromatography using EtOAc/CHCl3/MeOH (5:5:1).
N-(Benzo[b]thiophen-2-ylmethyl)-4-(4-(2-methoxyphenyl)piperazin-1-yl)-N-propylbutan-1-amine (13a)
Prepared from 4-(4-(2-methoxyphenyl)piperazin-1-yl)-N-propylbutan-1-amine(12a) and benzo[b]thiophene-2-carbaldehyde according to general procedure C in 29% yield. The oxalate salt was precipitated from EtOAc. Mp 122–126 °C; 1H NMR (400 MHz, CDCl3) δ ppm 0.87 (t, J = 7.2 Hz, 3H), 1.46–1.58 (m, 6H), 2.33 (t, J = 7.2 Hz, 2H), 2.42–2.50 (m, 4H), 2.57 (brs, 4H), 3.07 (brs, 4H), 3.78 (d, J = 1.2 Hz, 2H), 3.86 (s, 3H), 6.84–7.02 (m, 4H), 7.28 (s, 1H), 7.32–7.38 (m, 2H), 7.82–7.85 (m, 2H), 7.94–7.98 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 152.4, 141.5, 140.8, 139.1, 135.2,124.3, 123.8, 123.7, 123.0, 122.9, 122.8, 121.1, 118.4, 111.3, 58.7, 56.5, 56.5, 55.5, 54.0, 53.5, 53.3, 50.7, 25.2, 24.8, 20.4, 12.1; Anal. (C27H37N3OS· 2C2H2O4·3/2H2O) C, H, N.
N-(Benzo[b]thiophen-2-ylmethyl)-4-(4-(2,3-dichlorophenyl)piperazin-1-yl)-N-propylbutan-1-amine (13b)
Prepared from 4-(4-(2,3-dichlorophenyl)piperazin-1-yl)-N-propylbutan- 1-amine (12b) and benzo[b]thiophene-2-carbaldehyde according to general procedure C in 22 % yield. The oxalate salt was precipitated from EtOAc. Mp 168–171 °C; 1H NMR (400 MHz, CDCl3) δ ppm 0.88 (t, J = 7.2 Hz, 3H), 1.40–1.60 (m, 6H), 2.31 (t, J = 7.2 Hz, 2H), 2.42–2.45 (m, 4H), 2.52 (brs, 4H), 3.03 (brs, 4H), 3.78 (s, 2H), 6.92–6.95 (m, 1H), 7.28 (s, 1H), 7.31–7.38 (m, 2H), 7.82–7.85 (m, 1H), 7.95–7.98 (m, 1H); 13C NMR (100 MHz, CDCl3) δ 151.5, 140.8, 139.1, 135.2, 134.1, 127.6, 124.6, 124.3, 123.8, 123.7, 122.9, 122.8, 118.7, 116.1; Anal. (C23H35Cl2N3· 2C2H2O4) C, H, N.
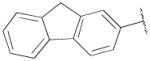
N-((9H-Fluoren-2-yl)methyl)-4-(4-(2-methoxyphenyl)piperazin-1-yl)-N-propyl butan-1-amine (13c)
Prepared from 4-(4-(2-methoxyphenyl)piperazin-1-yl)-N-propylbutan-1-amine (12a) and 9H-fluorene-2-carbaldehyde according to general procedure C in 56 % yield. The oxalate salt was precipitated from EtOAc. Mp 112–114 °C; 1H NMR (400 MHz, CDCl3) δ ppm 0.87 (t, J = 7.2 Hz, 3H), 1.48–1.58 (m, 6H), 2.36–2.50 (m, 6H), 2.62 (brs, 4H), 3.07 (brs, 4H), 3.62 (s, 2H), 3.85 (s, 3H), 3.88 (s, 2H), 6.84–7.02 (m, 4H), 7.21–7.38 (m, 3H), 7.50–7.54 (m, 2H), 7.71 (d, J = 8 Hz, 1H), 7.76 (d, J = 7.6 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 152.4, 143.4, 141.8, 141.5, 127.7, 126.8, 126.5, 125.7, 125.1, 123.0, 121.1, 119.8, 119.5, 118.3, 111.3, 59.0, 58.8, 56.1, 55.5, 53.8, 53.6, 50.7, 37.0, 24.8, 20.3, 12.1; Anal. (C32H41N3O· 9/4C2H2O4· 3/4H2O) C, H, N.
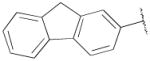
N-((9H-Fluoren-2-yl)methyl)-4-(4-(2,3-dichlorophenyl)piperazin-1-yl)-N-propylbutan-1-amine (13d)
Prepared from 4-(4-(2,3-dichlorophenyl)piperazin-1-yl)-N-propylbutan-1-amine (12b) and 9H-fluorene-2-carbaldehyde according to general procedure C in 40% yield. The oxalate salt was precipitated from EtOAc. Mp 136–140 °C; 1H NMR (400 MHz, CDCl3) δ ppm 0.88 (t, J = 7.2 Hz, 3H), 1.49–1.55 (m, 6H), 2.35–2.47 (m, 6H), 2.59 (brs, 4H), 3.03 (brs, 4H), 3.62 (s, 2H), 3.89 (s, 2H), 6.91–6.94 (m, 1H), 7.12–7.15 (m, 2H), 7.27–7.38 (m, 3H), 7.51–7.54 (m, 2H), 7.71 (d, J = 8.0 Hz, 1H), 7.76 (d, J = 7.8 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 151.5, 143.4, 141.8, 140.5, 134.1, 127.7, 127.6, 126.8, 126.5, 125.7, 125.2, 124.6, 119.8, 119.5, 118.7, 59.0, 58.7, 56.1, 53.8, 53.4, 51.4, 37.0, 25.2, 24.8, 20.3, 12.1; Anal. (C31H37Cl2N3· 2C2H2O4· 1/2H2O) C, H, N.
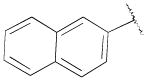
4-(4-(2-Methoxyphenyl)piperazin-1-yl)-N-(naphthalen-2-ylmethyl)-N-propylbutan-1-amine (13e)
Prepared from 4-(4-(2-methoxyphenyl)piperazin-1-yl)-N-propylbutan-1-amine (12a) and 2-naphthaldehyde according to general procedure C in 17% yield. The oxalate salt was precipitated from EtOAc. Mp 151–154 °C; 1H NMR (400 MHz, CDCl3) δ ppm 0.90 (t, J = 7.2 Hz, 3H), 1.50–1.60 (m, 6H), 2.39 (t, J = 6.8 Hz, 2H), 2.45 (t, J = 7.2 Hz, 2H), 2.50 (t, J = 6.2 Hz, 2H), 2.63 (brs, 4H), 3.10 (brs, 4H), 3.73 (s, 2H), 3.87 (s, 3H), 6.85–7.04 (m, 4H), 7.43–7.50 (m, 2H), 7.50 (dd, J = 10.4, 1.6 Hz, 1H), 7.76 (s, 1H), 7.80–7.85 (m, 3H); 13C NMR (100 MHz, CDCl3) δ 152.3, 141.4, 138.0, 133.4, 132.7, 127.7, 127.6, 127.4, 127.1, 125.8, 125.4, 122.9,121.0, 118.2, 111.2, 59.0, 58.8, 56.0, 55.4, 53.8, 53.5, 50.7, 25.2, 24.8, 20.3, 12.0; Anal. (C29H29N3O· 2C2H2O4·H2O) C, H, N.
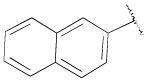
4-(4-(2,3-Dichlorophenyl)piperazin-1-yl)-N-(naphthalen-2-ylmethyl)-N-propylbutan-1-amine (13f)
Prepared from 4-(4-(2,3-dichlorophenyl)piperazin-1-yl)-N-propylbutan-1-amine (12b) and 2-naphthaldehyde according to general procedure C in 21% yield. The oxalate salt was precipitated from EtOAc. Mp 158–161 °C; 1H NMR (400 MHz, CDCl3) δ ppm 0.88 (t, J = 7.2 Hz, 3H), 1.49–1.56 (m, 6H), 2.36 (t, J = 7.2 Hz, 2H), 2.41–2.48 (m, 4H), 2.58 (brs, 4H), 3.04 (brs, 4H), 3.70 (s, 2H), 3.86 (s, 3H), 6.92–6.95 (m, 1H), 7.13–7.15 (m, 2H), 7.32–7.38 (m, 2H), 7.42–7.46 (m, 2H), 7.50 (dd, J = 10.0, 1.6 Hz, 1H), 7.74 (s, 1H), 7.78–7.83 (m, 3H); 13C NMR (100 MHz, CDCl3) δ 151.6, 138.2, 134.2, 133.6, 132.9, 127.9, 127.8, 127.6, 127.3, 126.0, 125.6, 124.7, 118.8, 59.2, 58.8, 56.2, 53.9, 53.4, 51.5, 25.3, 24.9, 20.5, 12.2; Anal. (C23H35Cl2N3· 5/2C2H2O4· H2O) C, H, N.
4-(((9H-Fluoren-2-yl)methyl)amino)-1-(4-(2,3-dichlorophenyl)piperazin-1-yl)butan-2-ol (14a)
To a solution of compound 10c (318 mg, 1 mmol) in MeOH (10 mL) was added 9H-fluorene-2-carbaldehyde (194 mg, 1 mmol) and the reaction mixture was stirred under argon for 12 h. NaBH4 (75 mg, 2 mmol) was added to the reaction mixture and stirred for 30 min. The reaction was quenched by adding 1 N NaOH (15 mL). The resulting mixture was filtered through celite, and the residue was washed with CHCl3 (50 mL). The organic layer was separated and dried over Na2SO4. The solvent was removed under reduced pressure. The crude product was purified by column chromatography using CHCl3/MeOH (9:1) to afford the compound 14a (455 mg) in 92% yield. The oxalate salt was precipitated from CHCl3. Mp 168–170 °C; 1H NMR (400 MHz, CDCl3) δ ppm 1.58–1.71 (m, 2H), 2.36–3.0 (m, 8H), 3.07 (brs, 4H), 3.88 (s, 4H), 3.91–3.98 (m, 1H), 6.94 (dd, J = 6.8, 2.8 Hz, 1H), 7.11–7.16 (m, 1H), 7.27–7.39 (m, 3H), 7.51–7.55 (m, 2H), 7.71–7.78 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 151.38, 143.71, 143.41, 141.62, 140.88, 138.69, 134.14, 127.61, 127.57, 127.03, 126.86, 126.70, 125.16, 125.06, 124.69, 119.92, 119.87, 118.72, 67.81, 64.71, 54.39, 53.68, 51.49, 47.50, 36.96, 34.34; Anal. (C28H31Cl2N3O· 2C2H2O4· 4/5H2O) C, H, N.
N-((9H-Fluoren-2-yl)methyl)-4-(4-(2,3-dichlorophenyl)piperazin-1-yl)-3-fluorobutan-1-amine (14b)
This compound was prepared by the method described for compound 14a, employing 7b (320 mg, 1 mmol), 9H-fluorene-2-carbaldehyde (194 mg, 1 mmol) and NaBH4 (75 mg, 2 mmol), afford 14b (448 mg) in 90% yield. The oxalate salt was precipitated from CHCl3. Mp 202–204 °C; 1H NMR (400 MHz, CDCl3) δ ppm 1.74–1.98 (m, 2H), 2.49–2.87 (m, 8H), 3.05 (brs, 4H), 3.87 (s, 2H), 3.89 (s, 2H), 4.76–4.95 (m, 1H), 6.92 (dd, J = 8.0, 2.4 Hz, 1H), 7.09–7.17 (m, 2H), 7.27–7.39 (m, 3H), 7.52–7.55 (m, 2H), 7.72–7.78 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 151.36, 143.71, 143.41, 141.66, 140.82, 139.14, 134.14, 127.63, 127.57, 126.97, 126.87, 126.69, 125.17, 125.01, 124.70, 119.91, 119.87, 118.71, 91.25 (d, J = 168.4 Hz, 1C), 62.56 (d, J = 21.3 Hz, 1C), 54.46, 53.98, 51.37, 45.49 (d, J = 4.6 Hz, 1C), 36.97, 34.18 (d, J = 10.2 Hz, 1C); 19F NMR (376 MHz, CDCl3/CFCl3) δ −181.98 to −181.59 (m, 1F); Anal. (C28H30Cl2FN3· 2C2H2O4) C, H, N.
4-(((9H-Fluoren-2-yl)methyl)(methyl)amino)-1-(4-(2,3-dichlorophenyl)piperazin-1-yl)butan-2-ol (15a)
14a (124 mg, 0.25 mmol) and formaldehyde 37 % in H2O (61μL) were mixed in 1,2-dichloroethane (5 mL) and then treated with sodium triacetoxyborohydride (159 mg, 0.75 mmol). The mixture was stirred at room temperature for 12 h. The reaction mixture was quenched by adding 1N NaOH (5 mL), and the product was extracted with EtOAc. The organic extract was washed with brine, dried over Na2SO4 and concentrated in vacuo. The crude product was purified by column chromatography using CHCl3/MeOH (9:1) to afford 15a (103 mg) in 81 % yield. The oxalate salt was precipitated from CHCl3. Mp 155–157 °C; 1H NMR (400 MHz, CDCl3) δ ppm 1.58–1.78 (m, 2H), 2.25 (s, 3H), 2.30–2.80 (m, 8H), 3.04 (brs, 4H), 3.47–3.72 (m, 2H), 3.87 (s, 2H), 3.91–3.98 (m, 1H), 6.90–6.95 (m,1H), 7.09–7.16 (m, 2H), 7.25–7.37 (m, 3H), 7.49–7.53 (m, 2H), 7.71–7.80 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 151.40, 143.57, 143.39, 141.53, 140.99, 136.96, 134.06, 127.98, 127.50, 126.81, 126.69, 125.86, 125.12, 124.56, 119.88, 119.71, 118.66, 68.94, 64.79, 63.0, 56.10, 53.79, 51.38, 42.05, 36.92, 31.34; Anal. (C29H33Cl2N3O· 2C2H2O4· H2O) C, H, N.
N-((9H-Fluoren-2-yl)methyl)-4-(4-(2,3-dichlorophenyl)piperazin-1-yl)-3-fluoro-N-methylbutan-1-amine (15b)
This compound was prepared by the method described for compound 15a, employing 14b (160 mg, 0.32 mmol), formaldehyde 37 % in H2O (78μL) and sodium triacetoxyborohydride (203 mg, 0.96 mmol), afford 15b (121 mg) in 74% yield. The oxalate salt was precipitated from CHCl3. Mp 173–175 °C; 1H NMR (400 MHz, CDCl3) δ ppm 1.78–1.98 (m, 2H), 2.25 (s, 3H), 2.48–2.77 (m, 8H), 3.05 (brs, 4H), 3.57 (s, 2H), 3.89 (s, 2H), 4.75–4.94 (m, 1H), 6.92 (dd, J = 7.6, 2.4 Hz, 1H), 7.10–7.17 (m, 2H), 7.26–7.39 (m, 3H), 7.51–7.55 (m, 2H), 7.71–7.78 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 151.37, 143.54, 143.42, 141.68, 140.86, 137.83, 134.13, 127.86, 127.62, 127.57, 126.85, 126.67, 125.83, 125.16, 124.67, 119.88, 119.66, 118.70, 91.05 (d, J = 168.4 Hz, 1C), 62.80, 62.45 (d, J = 20.6 Hz, 1C), 53.97, 53.24 (d, J = 4.6 Hz, 1C), 51.37, 42.42, 36.96, 31.65 (d, J = 20.6 Hz, 1C); 19F NMR (376 MHz, CDCl3/CFCl3) δ −181.67 to −181.28 (m, 1F); Anal. (C29H32Cl2FN3· 2C2H2O4·5/4H2O) C, H, N.
Radioligand Dopamine Receptor Binding Assays
A filtration binding assay was used to characterize the binding properties of membrane-associated receptors. For human D2L, D3, and D4 dopamine receptors expressed in HEK 293 cells, tissue homogenates (50 mL) were suspended in 50 mM Tris-HCl/150 mM NaCl/10 mM EDTA buffer pH 7.5 and incubated with 50 μL of 125I-IABN at 37 °C for 60 min. Nonspecific binding was determined using 25 μM (+)-butaclamol. For competition experiments, the radioligand concentration was generally equal to 0.5 times the Kd value, and the concentration of the competitive inhibitor ranged over 5 orders of magnitude. Binding was terminated by the addition of cold wash buffer (10 mM Tris-HCl/150 mM NaCl, pH 7.5) and filtration over glass-fiber filters (Schleicher and Schuell No. 32). Filters were washed with 10 mL of cold buffer, and the radioactivity was measured using a Packard Cobra gamma counter. Estimates of the equilibrium dissociation constant and maximum number of binding sites were obtained using unweighted nonlinear regression analysis of data modeled according to the equation describing mass action binding. Data from competitive inhibition experiments were modeled using nonlinear regression analysis to determine the concentration of inhibitor that displaced 50% of the specific binding of the radioligand. Competition curves were modeled for a single site, and the IC50 values were converted to equilibrium dissociation constants (Ki values) using the Cheng-Prusoff correction. Mean Ki values SEM are reported for at least three independent experiments.
Preparation of chimeric receptors
Four methods were utilized to construct the chimeras discussed in this paper. The first method exploits a Pst1 restriction site in the first intracellular loop (I1) of the receptor that is common to both the human D2 and human D3R cDNAs. Digested fragments were gel purified, ligated into the pIRESneo2 expression vector and transformed into competent DH5-alpha E. coli cells. Individual colonies were picked and plasmid DNA was purified. Constructs were sequenced to verify the chimeras and then transfected into HEK 293 cells. The second method involved making chimeric D2/D3Rs using the method of Moore and Blakely (1994). A human D3R cDNA and a human D2 receptor cDNA were cloned in tandem into the pIRESneo2 vector with a unique restriction site (Hpa1) located between the two receptor cDNAs. The construct was digested at this site and the linear construct was transformed into competent cells. DNA was prepared from individual colonies and sequenced to verify the chimera and then transfected into HEK-293 cells for expression. The third method was devised to create chimeras that the previous methods had not produced, specifically in TMS4 and TMS5. Site-directed mutagenesis (Quick-Change Site-Directed Mutagenesis Kit, Stratagene) was performed on D2 and D3 clones in pGEM7Z to create a unique restriction site to both cDNAs at the two different TMS regions (Xho1 at TMS4 and Xba1 at TMS5). Clones were then digested with the appropriate enzyme, fragments were gel purified and ligated to create chimeras that transitioned at TMS4 and TMS5 locations. Site-directed mutagenesis was performed on these new chimeras to delete the previously added restriction sites and the DNA sequence was verified for each mutant. Chimeric receptors were then cloned into the pIRES vector and were transfected into HEK-293 cells. Expression of the receptor construct was verified using a radioligand binding assay. Finally, The D2/D3E2 and D3/D2E2 receptor chimeras were prepared using a PCR based site-directed mutagenesis (Quick-Change Site-Directed Mutagenesis Kit, Stratagene) strategy with synthetic oligonucleotides encoding the E2 loop with the appropriate 5′ and 3′ flanking regions. Both wild type receptor genes were in the pIRES expression vector (Clontech). The size of the oligonucleotide for preparation of the D2/D3E2 chimera was 69 bases and for the D3/D2E2 loop was 66 bases. The chimeric receptors were transfected into HEK-293 cells. The authenticity of the chimeric receptor was verified by DNA sequencing and the expression of the receptor construct in HEK 293 was verified by radioligand binding using [125I]IABN. Tables S2 and S3 in the Supplementary Information provide the sequence transition points for the chimeric receptor proteins and amino acid sequence of wild type and chimeric human D3/D2 dopamine receptors, respectively.
Beta Arrestin Assay
The ability of the agonist-activated D3R to recruit beta-arrestin-2 is based on the DiscoverRx PathHunter technology which involves complementation of beta-galactosidase. The amino-terminal fragment of beta-galactosidase is fused to the D3R while the carboxyl-terminal fragment of the enzyme is fused to beta-arrestin-2. Upon agonist activation of the receptor, beta-arrestin is recruited to the receptor resulting in the formation of an active beta-galactosidase. Beta-galactosidase activity is then detected through the addition of a luminogenic substrate to the transfected cells as follows. CHO-K1 D3R cells (DiscoveRx) were seeded at a density of 5,000 cells/well in 384-well black, clear-bottom plates. Following 24-hour incubation, the cells were treated with multiple concentrations of the antagonist under study (0 – 10 μM) in Hanks’ Balanced Buffer Solution containing 2% DMSO. Following a 10 minute preincubation, the cells were next treated with an EC95 dose of Dopamine (100 nM) and then incubated at 37°C for 90 minutes. DiscoveRx reagent (2.5X) was then added to cells followed by a 60 minute incubation at room temperature. Luminescence was measured on a Hamamatsu FDSS μ-cell reader.
Supplementary Material
Acknowledgments
This work was supported by the NIDA (A.H.N.) and NINDS (D.R.S.) Intramural Research Programs, NIH Grants DA13584-03S1 and DA13584-01 (RL) and NIDA Addiction Treatment Discovery Program contract with SRI (N01DA-1-8816). BAK was supported by an NIH Postdoctoral Visiting Fellowship, BAL was supported by an NIH Summer Student Fellowship, and SAK was supported by an NIH Post-baccalaureate Intramural Research Training Award (IRTA) Fellowship. The authors would like to thank Drs. Robert H. Mach and Zhude Tu in Department of Radiology at Washington University School of Medicine in St. Louis, MO for generously providing us with the precursor that was used for the radioiodination of 125I-IABN and Dr. Aaron Janowski and Robert Johnson, Oregon Health & Science University for obtaining the mitogenesis data for sulpiride. We would also like to thank Drs. Jonathan Javitch and Lei Shi for helpful discussions during the preparation of this manuscript.
Abbreviations
- DAST
N,N-diethylaminosulfur trifluoride
- D2R
dopamine D2 receptor
- D3R
dopamine D3 receptor
- E2
second extracellular loop
- BBB
blood-brain barrier
- TMS
transmembrane spanning
- D2/D3E2
a human D2 receptor with the E2 loop of the D3 receptor
- D3/D2E2
a human D3 receptor with the E2 loop of the D2 receptor
- hD2 or hD3
human D2 or D3 receptor proteins
Footnotes
Supporting Information Available: Elemental analysis results and Chimera sequence data. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Sokoloff P, Martres MP, Andrieux M, Beasancon R, Pilon C, Bouthenet ML, Souil E, Schwartz JC. Localization and function of the D3 dopamine receptor. Arzneimittelforschung. 1992;42(2A):224–230. [PubMed] [Google Scholar]
- 2.Mengod G, Villaró MT, Landwehrmeyer GB, Martinez-Mir MI, Niznik HB, Sunahara RK, Seeman P, O’Dowd BF, Probst A, Palacios JM. Visualization of dopamine D1, D2 and D3 receptor mRNAs in human and rat brain. Neurochem Int. 1992;20(Suppl):33S–43S. doi: 10.1016/0197-0186(92)90208-9. [DOI] [PubMed] [Google Scholar]
- 3.Joyce JN, Millan MJ. Dopamine D-3 receptor antagonists as therapeutic agents. Drug Discovery Today. 2005;10:917–925. doi: 10.1016/S1359-6446(05)03491-4. [DOI] [PubMed] [Google Scholar]
- 4.Heidbreder CA, Newman AH. Current perspectives on selective dopamine D3 receptor antagonists as pharmacotherapeutics for addictions and related disorders. Ann NY Acad Sci. 2010;1187:4–34. doi: 10.1111/j.1749-6632.2009.05149.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Grundt P, Carlson EE, Cao J, Bennett CJ, McElveen E, Taylor M, Luedtke RR, Newman AH. Novel Heterocyclic Trans Olefin Analogues of N-{4-[4-(2,3-Dichlorophenyl)piperazin-1-yl]butyl}arylcarboxamides as Selective Probes with High Affinity for the Dopamine D3 Receptor. J Med Chem. 2005;48:839–848. doi: 10.1021/jm049465g. [DOI] [PubMed] [Google Scholar]
- 6.Grundt P, Prevatt KM, Cao J, Taylor M, Floresca CZ, Choi JK, Jenkins BG, Luedtke RR, Newman AH. Heterocyclic analogues of N-(4-(4-(2,3-dichlorophenyl)piperazin-1-yl)butyl)arylcarboxamides with functionalized linking chains as novel dopamine D3 receptor ligands: potential substance abuse therapeutic agents. J Med Chem. 2007;50:4135–4146. doi: 10.1021/jm0704200. [DOI] [PubMed] [Google Scholar]
- 7.Grundt P, Husband SL, Luedtke RR, Taylor M, Newman AH. Analogues of the dopamine D2 receptor antagonist L741,626: Binding, function, and SAR. Biorg Med Chem Lett. 2007;17(3):745–749. doi: 10.1016/j.bmcl.2006.10.076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chu W, Tu Z, McElveen E, Xu J, Taylor M, Luedtke RR, Mach RH. Synthesis and in vitro binding of N-phenyl piperazine analogs as potential dopamine D3 receptor ligands. Bioorg Med Chem. 2005;13:77–87. doi: 10.1016/j.bmc.2004.09.054. [DOI] [PubMed] [Google Scholar]
- 9.Vangveravong S, Xu J, Zeng C, March RH. Synthesis of N-substituted 9-azabicyclo[3.3.1]nonan-3alpha-yl carbamate analogs as sigma2 receptor ligands. Bioorg Med Chem. 2006;14(20):6988–6997. doi: 10.1016/j.bmc.2006.06.028. [DOI] [PubMed] [Google Scholar]
- 10.Vangveravong S, Taylor M, Xu J, Cui J, Calvin W, Babic S, Luedtke RR, March RH. Synthesis and characterization of selective dopamine D(2) receptor antagonists. 2. Azaindole, benzofuran, and benzothiophene analogs of L-741,626. Bioorg Med Chem. 2010;18(14):5291–5300. doi: 10.1016/j.bmc.2010.05.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Newman AH, Grundt P, Cyriac G, Deschamps JR, Taylor M, Kumar R, Ho D, Luedtke RR. N-(4-(4-(2,3-Dichloro- or 2-methoxyphenyl)piperazin-1-yl)butyl)heterobiarylcarboxamides with functionalized linking chains as high affinity and enantioselective D3 receptor antagonists. J Med Chem. 2009;52:2559–2570. doi: 10.1021/jm900095y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Micheli F, Bonanomi G, Blaney FE, Braggio S, Capelli AM, Checchia A, Curcuruto O, Damiani F, Fabio RD, Donati D, Gentile G, Gribble A, Hamprecht D, Tedesco G, Terreni S, Tarsi L, Lightfoot A, Stemp G, Macdonald G, Smith A, Pecoraro M, Petrone M, Perini O, Piner J, Rossi T, Worby A, Pilla M, Valerio E, Griffante C, Mugnaini M, Wood M, Scott C, Andreoli M, Lacroix L, Schwarz A, Gozzi A, Bifone A, Ashby CR, Jr, Hagan JJ, Heidbreder C. 1,2,4-triazol-3-ylthiopropyltetrahydrobenzazepines: a series of potent and selective dopamine D(3) receptor antagonists. J Med Chem. 2007;50:5076–5089. doi: 10.1021/jm0705612. [DOI] [PubMed] [Google Scholar]
- 13.Michelli F, Arista L, Bonanomi G, Blaney FE, Braggio S, Capelli AM, Checchia A, Damiani F, Di-Fabio R, Fontana S, Gentile G, Griffante C, Hamprecht D, Marachioro C, Mugnaini M, Piner J, Ratti E, Tedesco G, Tarsi L, Terreni S, Worby A, Ashby CR, jr, Heidbreder C. 1,2,4-Triazolyl azabicyclo[3.1.0]hexanes: a new series of potent and selective dopamine D3 receptor antagonists. J Med Chem. 2010;53:374–391. doi: 10.1021/jm901319p. [DOI] [PubMed] [Google Scholar]
- 14.Choi JK, Mandeville JB, Chen YI, Grundt P, Sarkar SK, Newman AH, Jenkins BG. Imaging brain regional and cortical laminar effects of selective D3 agonists and antagonists. Psychopharmacology. 2010;212:59–72. doi: 10.1007/s00213-010-1924-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Achat-Mendes C, Grundt P, Cao J, Platt DM, Newman AH, Spealman RD. Dopamine D3 and D2 receptor mechanisms in the abuse-related behavioral effects of cocaine: studies with preferential antagonists in squirrel monkeys. J Pharmacol Exp Ther. 2010;334(2):556–565. doi: 10.1124/jpet.110.167619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Orio L, Wee S, Newman AH, Pulvirenti L, Koob GF. The dopamine D3 receptor partial agonist CJB090 and antagonist PG01037 decrease progressive ratio responding for methamphetamine in rats with extended-access. Addict Biol. 2010;15(3):312–323. doi: 10.1111/j.1369-1600.2010.00211.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Higley MJ, Sabatini BL. Competitive regulation of synaptic Ca(2+) influx by D2 dopamine and A2A adenosine receptors. Nat Neurosci. 2010;13:958–966. doi: 10.1038/nn.2592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Baladi MG, Newman AH, France CP. Dopamine D3 receptors mediate the discriminative stimulus effects of quinpirole in free-feeding rats. J Pharmacol Exp Ther. 2010;332(1):308–315. doi: 10.1124/jpet.109.158394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kumar R, Riddle LR, Griffin SA, Chu W, Vangveravong S, Neisewander J, Mach RH, Luedtke RR. Evaluation of D2 and D3 dopamine receptor selective compounds on L-dopa-dependent abnormal involuntary movements in rats. Neuropharmacology. 2009;56(6–7):956–969. doi: 10.1016/j.neuropharm.2009.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Collins GT, Witkin JM, Newman AH, Svensson KA, Grundt P, Cao J, Woods JH. Dopamine agonist-induced yawning in rats: a dopamine D3 receptor-mediated behavior. J Pharmacol Exp Ther. 2005;314(1):310–319. doi: 10.1124/jpet.105.085472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ehrlich K, Götz A, Bollinger S, Tschammer N, Bettinetti L, Härterich S, Hübner H, Lanig H, Gmeiner P. Dopamine D2, D3, and D4 selective phenylpiperazines as molecular probes to explore the origins of subtype specific receptor binding. J Med Chem. 2009;52(15):4923–4935. doi: 10.1021/jm900690y. [DOI] [PubMed] [Google Scholar]
- 22.Boeckler F, Gmeiner P. The structural evolution of dopamine D3 receptor ligands: structure-activity relationships and selected neuropharmacological aspects. Pharmacol Ther. 2006;112(1):281–333. doi: 10.1016/j.pharmthera.2006.04.007. [DOI] [PubMed] [Google Scholar]
- 23.Luedtke RR, Freeman RA, Boundy VA, Martin MV, Huang YS, Mach RH. Characterization of I-125-IABN a novel azabicyclononane benzamide selective for D2-like dopamine receptors. Synapse. 2000;38:438–449. doi: 10.1002/1098-2396(20001215)38:4<438::AID-SYN9>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 24.Biswas A, Zhang S, Fernandez F, Gosh B, Zhen J, Kuzhikandathil E, Reith MEA, Dutta AK. Further structure–activity relationships study of hybrid 7-{[2-(4-phenylpiperazin-1-yl)ethyl]propylamino}-5,6,7,8-tetrahydronaphthalen-2-ol analogues: identification of a high-affinity D3-preferring agonist with potent in vivo activity with long duration of action. J Med Chem. 2008;51:101–117. doi: 10.1021/jm070860r. [DOI] [PubMed] [Google Scholar]
- 25.Shi L, Simpson MM, Ballesteros JA, Javitch JA. The first transmembrane segment of the dopamine D2 receptor: accessibility in the binding-site crevice and position in the transmembrane bundle. Biochemistry. 2001;40:12339–12348. doi: 10.1021/bi011204a. [DOI] [PubMed] [Google Scholar]
- 26.Palczewski K, Kumasaka T, Hori T, Behnke CA, Motoshima H, Fox BA, Le Trong I, Teller DC, Okada T, Stenkamp RE, Yamamoto M, Miyano M. Crystal structure of rhodopsin: A G protein-coupled receptor. Science. 2000;289(5480):739–745. doi: 10.1126/science.289.5480.739. [DOI] [PubMed] [Google Scholar]
- 27.Cherezov V, Rosenbaum DM, Hanson MA, Rasmussen SG, Thian FS, Kobilka TS, Choi HJ, Kuhn P, Weis WI, Kobilka BK, Stevens RC. High-resolution crystal structure of an engineered human beta2-adrenergic G protein-coupled receptor. Science. 2007;318(5854):1258–1265. doi: 10.1126/science.1150577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Warne T, Serrano-Vega MJ, Baker JG, Moukhametzianov R, Edwards PC, Henderson R, Leslie AG, Tate CG, Schertler GF. Structure of a beta1-adrenergic G-protein-coupled receptor. Nature. 2008;454(7203):486–491. doi: 10.1038/nature07101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shi L, Javitch JA. The second extracellular loop of the dopamine D2 receptor lines the binding-site crevice. Proc Natl Acad Sci U S A. 2004;101(2):440–445. doi: 10.1073/pnas.2237265100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lan H, Durand CJ, Teeter MM, Neve KA. Structural determinants of pharmacological specificity between D(1) and D(2) dopamine receptors. Mol Pharmacol. 2006;69(1):185–194. doi: 10.1124/mol.105.017244. [DOI] [PubMed] [Google Scholar]
- 31.Silvano E, Millan MJ, Mannoury la Cour C, Han Y, Duan L, Griffin SA, Luedtke RR, Aloisi G, Rossi M, Zazzeroni F, Javitch JA, Maggio R. The Tetrahydroisoquinoline Derivative SB269,652 is an Allosteric Antagonist at Dopamine D3 and D2 Receptors. Mol. Pharmacol. 2010;78:925–934. doi: 10.1124/mol.110.065755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chien EYT, Liu W, Zhao Q, Katritch V, Hanson MA, Shi L, Newman AH, Javitch JA, Cherezov V, Stevens RC. Structure if the Human Dopamine D3 Receptor in Complex with a D2/D3 Selective Antagonist. Science. 2010;3330:1091–1095. doi: 10.1126/science.1197410. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.