

Abstract
Neuronal membrane properties dictate neuronal responsiveness. Plasticity of membrane properties alters neuronal function and can arise in response to robust neuronal activity. Despite the potential for great impact, there is little evidence for a rapid effect of activity-dependent changes of membrane properties on many neuronal functions in vivo in mammalian brain. In this study it was tested whether periods of neuronal firing lead to a rapid change of membrane properties in neurons of a rat brain region important for some forms of learning, the lateral nucleus of the amygdala, using in vivo intracellular recordings. Our results demonstrate that rapid plasticity of membrane properties occurs in vivo, in response to action potential firing. This plasticity of membrane properties leads to changes of synaptic integration and subsequent synaptic plasticity. These changes require Ca2+ and hyperpolarization-activated ion channels, but are NMDA independent. Furthermore, the parameters and time course of these changes would not have been predicted from most in vitro studies. The plasticity of membrane properties demonstrated here may represent a basic form of in vivo short-term plasticity that modifies neuronal function.
Introduction
Learning relies upon plasticity in numerous brain regions. A change of synaptic strength is the most commonly described form of neuronal plasticity and may play a role in learning (Rogan et al., 1997; Maren, 1999; Shapiro and Eichenbaum, 1999; Martin and Morris, 2002; Whitlock et al., 2006). However, membrane properties dictate the neuronal response to synaptic inputs and are also modifiable. Chronic treatments that alter neuronal activity will, over time, lead to opposing changes that modify membrane excitability or the ion channels that regulate excitability (Turrigiano et al., 1994; Desai et al., 1999; Golowasch et al., 1999; Baines et al., 2001; Beraneck et al., 2003; Pratt and Aizenman, 2007; Grubb and Burrone, 2010).
Periods of robust synaptic activity or paired presynaptic and postsynaptic activity, as occur during procedures to induce long-term potentiation of synapses (LTP), can also lead to a rapid change of membrane excitability (Aizenman and Linden, 2000; Mahon et al., 2003; Frick et al., 2004; van Welie et al., 2004; Zhang et al., 2004; Fan et al., 2005; Campanac and Debanne, 2007; Narayanan and Johnston, 2007; Campanac et al., 2008; Brown and Randall, 2009; Jung and Hoffman, 2009), including a change in the population spike in early reports of LTP (Bliss and Lomo, 1973). In addition, neuronal action potential (AP) activity or postsynaptic depolarization alone is sufficient to induce a rapid change of membrane excitability in some conditions (Aizenman and Linden, 2000; Nelson et al., 2003; Cudmore and Turrigiano, 2004; Fan et al., 2005; Kim et al., 2007; Narayanan and Johnston, 2007). This rapid plasticity of membrane excitability may play a role in modulation of learning or memory processes in mammals. However, if it is involved in mammalian learning, it must occur in vivo. While studies have examined whether synaptic LTP induction is associated with changes of membrane properties in vivo (Baranyi et al., 1991; King et al., 1999), to date there are very few examples of a change of mammalian membrane properties or membrane excitability measured in vivo in response to learning paradigms (Rosenkranz and Grace, 2002b) or in response to neuronal firing activity (Mahon et al., 2003; Paz et al., 2009). These studies did not examine whether similar requirements underlie rapid in vivo plasticity as have been found in vitro, nor did they examine the consequences on synaptic integration and synaptic plasticity.
The basolateral amygdala (BLA), and in particular the lateral nucleus (LAT), plays a key role in learning (LeDoux, 1993; Fanselow and Poulos, 2005) and its neurons display synaptic plasticity (Maren, 2005; Sigurdsson et al., 2007). However, plasticity of BLA neuronal membrane properties is unknown. A shift in membrane properties that underlie membrane excitability has significant consequences for integration of synaptic inputs. It may also influence subsequent plasticity of other types. In this study it is tested whether activity-dependent plasticity of membrane properties occurs in vivo in the LAT, whether its requirements are similar to in vitro plasticity, and whether this leads to a change of membrane excitability, synaptic integration, and synaptic plasticity.
Materials and Methods
All procedures were performed in accordance with the Institutional Animal Care and Use Committee of Rosalind Franklin University of Medicine and Science and followed the Guide for the Care and Use of Laboratory Animals published by the U.S. National Institutes of Health. Male rats (age 9–12 weeks old) were used for this study. Rats were housed in pairs with a 12:12 h light:dark cycle, and food and water were available ad libitum.
In vivo intracellular electrophysiology.
Recordings were obtained from the BLA of rats, specifically from the LAT (Rosenkranz and Grace, 2002a). Rats were anesthetized with an initial intraperitoneal injection of 8% chloral hydrate (all chemicals were from Sigma-Aldrich unless noted otherwise) and supplemented with intravenous administration of 8% chloral hydrate as necessary. Rats were placed in a stereotaxic device (David Kopf Instruments). Temperature was monitored rectally and maintained near 37°C. Burr holes were drilled in the skull over both amygdalae (coordinates 5.0 mm lateral, 3.0 mm caudal from bregma). A small stainless steel screw was implanted in the skull over one amygdala to record EEG (DAM 50, World Precision Instruments). During experiments, anesthesia depth was monitored with EEG. For some experiments a burr hole was also drilled in the skull overlying the medial geniculate nucleus of the thalamus (MGN; coordinates 3.4 mm lateral, 5.6 mm caudal from bregma). The dura overlying the MGN thalamus was removed and a concentric bipolar stimulation electrode (0.25 mm outer diameter; Rhodes Medical Instruments) was lowered into the thalamus. Recordings did not begin until at least 45 min after implantation of the stimulation electrode. For intracellular recordings, electrodes were filled with 1–2% neurobiotin in 2 m potassium acetate. When indicated, electrodes also included other chemicals: CdCl2 (0.5 mm), NiCl2 (0.5 mm), MK-801 (2 mm), or MgCl2 (20 mm). In experiments that included intracellular dialysis with these chemicals, a minimum of 25 min was used to allow time for the chemical blocker to diffuse into the recorded neuron. These doses ensure blockade of the targeted channels. The dura overlying the amygdala was removed, and electrodes were slowly lowered to record neuronal activity.
Data were collected at 10 kHz on a Mac Pro (Apple), visualized online (AxoGraph X) and saved for later analysis. Series resistance was compensated using built-in amplifier bridge circuitry (IR-183, Cygnus Technology). Several measurements were obtained during the intracellular recordings. Epochs of 60 s were used to measure the resting membrane potential. The input resistance (Rn) was measured as the linear fit of the voltage response to a series of hyperpolarizing current steps (0 to −300 pA, 800 ms, repeated four times at each current step). The membrane time constant, τ, was measured from the fit of the relaxation of the voltage back to rest following a hyperpolarizing current step that evoked a voltage change of 5–10 mV (repeated 5–10 times). Neuronal excitability was measured here as the number of action potentials evoked by a brief current pulse (1 s; amplitude that evokes 6–9 action potentials under baseline conditions). To minimize the impact of membrane potential, which can strongly influence our measure of excitability, the membrane potential was held near the initial resting potential (Vrest) [with constant direct current (DC) if needed] whenever excitability was measured. When indicated, excitability was also quantified as the number of action potentials evoked by a series of depolarizing current steps (0–1000 pA, 800 ms, repeated 4–5 times at each current step). Excitability was measured as the slope of the linear fit to the relationship between the current intensity and the number of action potentials evoked by each depolarizing current step. Input resistance and excitability were measured at both resting membrane potential and at −70 mV to facilitate comparisons between groups.
Synaptic activity was evoked in some experiments by stimulation of the MGN (0.1–0.9 mA, 0.2 ms duration). To examine postsynaptic contributions to EPSP summation, EPSC-shaped currents were injected into the cell body to evoke depolarizations that were similar to EPSPs, termed αPSPs (geometry based on α function: A(t/α)e(−αt), where A is the amplitude of the injected current, t is time, and 1/α is the time to peak). A train of five αPSPs, at 50 ms intervals, and a current amplitude that induces a 4–8 mV peak voltage change were used. Their summation was quantified as the amplitude of (last PSP − first PSP)/first PSP. Spontaneous synaptic activity was measured for 60 s. Spontaneous synaptic events were detected using a sliding template of EPSP shape. The number of events, half-width, and amplitude were measured.
Activity-dependent changes.
To examine activity-dependent effects on membrane properties, neuronal action potential firing was induced in two forms. Initial experiments used depolarization by DC injection to bring the membrane potential past the action potential firing threshold. This depolarization had a duration of 15–60 s and an amplitude that resulted in 0–40 Hz action potential firing (i.e., bringing the neuron membrane potential to approximately −55 to −45 mV). To examine activity-dependent changes in a more accurately controlled manner, the membrane potential was brought near to but below action potential threshold (typically −55 mV), and action potentials were evoked by current steps of 60 ms duration. These current steps repeated at a 5 Hz, a frequency near theta, and resulted in theta burst firing (TBF). The amplitude of the current steps was modulated to evoke a predetermined number of action potentials per current step (from ∼1 to 4 action potentials). The duration of the TBF lasted 15–60 s. Excitability was examined by current step injections (as described above, In vivo intracellular electrophysiology) before and after TBF, at 5–30 s intervals. To estimate baseline excitability, these responses were averaged.
Local microinfusions.
In a subset of experiments, intra-LAT perfusions were performed. Glass pipettes were pulled to fine tip and broken back under microscopic control for microinfusions. The pipette was lowered into the LAT and aimed within 2 mm of the targeted recording area. Pipettes were filled with a HEPES-based aCSF containing the following (in mm): 125 NaCl, 2.5 KCl, 1.25 NaH2PO4, 10 HEPES, 2 CaCl2, 1 MgCl2, 25 dextrose and 2–3% Fast Green to mark the pipette tip. In some experiments, the solution also contained 1 mg/ml ZD7288. Microinfusions were controlled by a nanoliter injector pump (WPI 1400, World Precision Instruments) at a rate of 100 nl/min. Drug or vehicle artificial CSF (aCSF) was infused immediately following the TBF or following a control period of test pulses to measure excitability. Measures of membrane properties were taken periodically before and after infusions. For statistical comparisons, equivalent time points relative to infusions were used.
LTP induction.
To examine the effects of activity-dependent changes on synaptic plasticity, TBF was followed by induction of long-term synaptic plasticity in some experiments (LTP). LTP was induced by pairing constant depolarization (−55 to −50 mV) with a 20 Hz train of EPSPs evoked by the stimulation electrode placed in the MGN (see above, In vivo intracellular electrophysiology). An EPSP amplitude of 5–10 mV was used for induction of LTP. The amplitude of the evoked EPSP was monitored at 0.1 Hz before and after LTP induction. LTP was induced at 3 min after the end of TBF, a period when TBF-induced effects still persist.
At the conclusion of experiments, brains were removed, soaked in 4% paraformaldehyde, and cryoprotected with 25% sucrose. Brains were sliced at 60 μm using a freezing microtome and stained for Neurobiotin using standard ABC peroxidase staining (Vector Laboratories). Sections were counterstained with neutral red and cresyl violet stains. Neurons were excluded from analysis if they were found to lie outside the BLA, their morphology was inconsistent with BLA pyramidal neurons (McDonald, 1982), their action potentials did not overshoot 0 mV, they displayed firing characteristics inconsistent with BLA pyramidal neurons (Lang and Pare, 1997; Rosenkranz and Grace, 2002a), or their resting membrane potential was more depolarized than −60 mV.
Statistical analysis.
When performing planned comparisons between two groups, two-tailed unpaired t tests were used. All comparisons between more than two groups were made with one-way or two-way ANOVAs, depending upon the number of factors. When comparing groups across multiple factors where only one factor was a repeated measure, a mixed model repeated measures two-way ANOVA was used. Post hoc Student's t tests with Bonferroni corrections were used to compare individual groups if significant values were obtained in ANOVAs. An α level of 0.05 was considered significant except after multiple comparisons when the α level was made more stringent based upon the method of Bonferroni. Before parametric comparisons were performed, data were tested for normality of distribution (Kolmgorov and Smirnov test) and for equality of the SD (Bartlett's test). Statistical tests were performed using Igor Pro (Wavemetrics) or Prism 5 software (GraphPad Software). All values are expressed as the mean ± SEM.
Results
Prolonged action potential firing rapidly reduces membrane excitability in an activity-dependent manner
Excitability is a neuronal feature that reflects the contribution of several membrane properties and was used as the initial measure for an effect of action potential firing on neuronal properties in vivo. Action potential firing caused by neuronal depolarization [10–20 Hz firing, 60 s depolarization by direct current to membrane potential (Vm) −50 to −45 mV] caused a robust decrease of excitability (Fig. 1A) (n = 11/12 neurons, ∼39.4 ± 8.1% of baseline excitability after 1 min; p < 0.001, one-way repeated-measures ANOVA, F(11,32) = 21.27). This decrease of neuronal excitability lasted between 5 and 10 min before returning to baseline levels (Fig. 1B) (baseline significantly different from postfiring values at 0–8.5 min after firing, post hoc two-way t test comparisons with Bonferroni corrections).
Figure 1.

Neuronal activity causes a decrease of membrane excitability. A, Membrane excitability, demonstrated here as the number of action potentials evoked by a current step, was decreased by a 60 s period of neuronal action potential firing (top, voltage trace with current trace underneath depicting a typical experiment; bottom, voltage and current traces enlarged to display the number of action potentials evoked by current steps. B, This change lasted 5–10 min. Overall, action potential frequencies of >10 Hz were required to induce this decrease of membrane excitability (black circle, >10 Hz; white circle, <10 Hz). C, The number of action potentials during the activity is correlated with the change of membrane excitability. D, Shorter periods (15–30 s) of activity did not consistently cause a decrease of membrane excitability that lasted longer than 2 min.
This effect of neuronal firing depended upon the number of action potentials evoked during the period of prolonged action potential firing, as well as the duration of depolarization. A depolarization was less effective if it resulted in fewer action potentials (Fig. 1B) (1–10 Hz; excitability reduced to 87.8 ± 3.6% of baseline 1 min after the end of activity, n = 10; p = 0.556, one-way repeated-measures ANOVA, F(9,32) = 0.95). There was a significant correlation between the number of action potentials evoked during the 60 s depolarization and the decrease in measured excitability (Fig. 1B) (r2 = 0.675, p < 0.01, n = 23; slope = −0.10 ± 0.015; each additional AP leads to approximately −0.1% change of excitability). This effect of action potential firing relies upon concurrent action potential firing and depolarization and is not caused solely by the amplitude of depolarization, as a large depolarization that resulted in initial robust firing (20–40 Hz during the beginning) followed by strong firing rate accommodation (and therefore fewer action potentials, reduced to 0–10 Hz by the end) had a weaker effect on excitability (84.7 ± 10.2% of baseline excitability after 1 min, n = 6; two-way repeated-measures ANOVA comparison between 10 and 20 Hz and 20–40 Hz, p = 0.004, main effect of firing rate F(1,34) = 11.0) than prolonged firing in the presence of a smaller depolarization (as above, 39.4 ± 8.1% after 1 min).
Depolarization produced only transient changes if it was of shorter duration. Thus, 15 s depolarization resulted in decreased excitability only when measured immediately after depolarization (Fig. 1C) (10–20 Hz firing, n = 6; p < 0.01, one-way repeated-measures ANOVA, F(5,32) = 3.09; baseline significantly different from postfiring values at 0–30 s after firing, post hoc two-tailed t test comparisons with Bonferroni corrections), but this quickly returned to baseline and no significant changes were observed when measured at >1 min after depolarization (93.1 ± 7.4% of baseline excitability after 1 min). An intermediate effect was observed after a 30 s depolarization (Fig. 1C) (10–20 Hz firing; 67.9 ± 5.4% of baseline excitability after 1 min, n = 5; p < 0.001, one-way repeated-measures ANOVA, F(4,32) = 6.89; baseline significantly different from postfiring values at 0–2 min after firing, post hoc two-tailed t test comparisons with Bonferroni corrections). This is consistent with an activity-dependent effect on neuronal membrane excitability.
Theta burst firing decreases membrane excitability in vivo
Activity-dependent effects of neuronal firing on excitability can be more precisely measured if the action potentials are evoked in a more predictable and more controllable manner. In addition, continuous firing for 60 s might not be a normal firing pattern in vivo. Therefore, short depolarizing pulses were used to control the timing and frequency of action potentials. It has been shown that the BLA displays rhythmicity near theta frequencies (Pare and Collins, 2000; Pape et al., 2005) and that burst firing near theta frequencies can induce robust plasticity in other brain regions in vitro (Narayanan and Johnston, 2007). Therefore, short depolarizing pulses were used to induce burst firing of LAT neurons near theta rhythmicity, a procedure termed theta burst firing or TBF (Fig. 2A) (60 ms pulses repeated at 5 Hz). The amplitude of each pulse was set to evoke an average of 1–4 APs per burst. When evoked from resting membrane potentials, 60 s of TBF activity produced only a transient decrease of excitability (n = 10, p < 0.01, F(9,32) = 6.68; baseline significantly different from postfiring values at 0–60 s after firing; post hoc two-tailed t test comparisons with Bonferroni corrections) that returned to baseline after 1 min (Fig. 2B) (87.7 ± 8.7% of baseline excitability). However, when the membrane was depolarized to more closely mimic the membrane potential in awake animals (Steriade et al., 2001) (here, approximately −60 mV, subthreshold to significant spontaneous action potential firing) and theta burst firing was applied (5 Hz, 60 s), a robust decrease of excitability was observed (Fig. 2A,B) (30.7 ± 2.6% of baseline excitability after 1 min, n = 11/12 neurons, p < 0.001, F(11,32) = 35.99; baseline significantly different from postfiring values at 0–7 min after firing; post hoc two-tailed t test comparisons with Bonferroni corrections). There was a correlation between the average number of action potentials per burst and the observed decrease of excitability measured after 1 min (Fig. 2C) (r2 = 0.66, p < 0.01, n = 16; slope= −0.189 ± 0.036). However, depolarization to −55 mV alone was insufficient to induce a long-term reduction of membrane excitability (Fig. 2B) (103.2 ± 4.3% of baseline excitability after 1 min, n = 7 neurons, p = 0.007, F(6,32) = 1.85; postfiring significantly greater than baseline values at 0–30 s after depolarization; post hoc two-tailed t test comparisons with Bonferroni corrections). The effects of TBF on excitability are consistent with a rapid in vivo reduction of membrane excitability following relatively short periods of activity, and this change is dependent upon the depolarization state of the neuron.
Figure 2.

Theta burst firing activity causes a decrease of membrane excitability. A, In our protocol to examine the effects of neuronal firing on excitability, membrane excitability is measured before and after theta burst firing for several minutes (top, voltage trace with current trace underneath depicting a typical experiment; bottom, voltage and current traces enlarged to display the number of action potentials evoked by current steps). B, The decrease of membrane excitability caused by TBF lasted >5 min (black circle, TBF - depol), while depolarization alone (white circle, depol only) or TBF without depolarization (gray circle, TBF – no depol) did not cause a prolonged change of excitability. C, There was a correlation between the average number of action potentials evoked per burst and the degree of change in excitability. D, Membrane excitability was still found to be decreased after TBF when measured from a complete input–output curve of the membrane excitability in response to a range of current step amplitude in a subsample of neurons (right; gray line represents the difference between baseline and post-TBF excitability).
In a subset of neurons, a more comprehensive measure of membrane excitability was obtained before and after TBF using a range of current steps (0–1000 pA; see Materials and Methods) to formulate a complete input–output curve of excitability. A reduction of excitability after TBF activity was observed (quantified as the mean number of action potentials/current step; n = 6, p = 0.0003, F(1,56) = 29.13, two-way repeated-measures ANOVA). This reduction was seen across the entire range of measure (Fig. 2D, left) (reflected as a reduction of the slope of the input–output relationship; baseline, 1.47 ± 0.096 AP for each 100 pA increase of current step input; post-TBF activity, 1.06 ± 0.085 AP for each 100 pA increase of current step input; p = 0.0028, F(1,56) = 9.78, two-way repeated-measures ANOVA).
The reduction of membrane excitability caused by TBF can be due to several membrane properties that influence neuronal excitability, including a shift in action potential threshold and input resistance. However, there was no significant change in action potential threshold (baseline, −50.6 ± 1.9 mV; post-TBF, −50.1 ± 1.7 mV; p = 0.07, t = 1.98, df = 15, two-tailed paired t test). There was a significant reduction of input resistance after TBF (Fig. 3A) (baseline, 33.4 ± 1.6 MΩ; post-TBF activity, 30.1 ± 1.0 MΩ; p = 0.014, t = 3.46, df = 7, two-tailed paired t test). This was accompanied by a significant shortening of the membrane time constant (Fig. 3B) (baseline, 17.9 ± 1.7 ms; post-TBF activity, 15.7 ± 1.6 ms; p = 0.015, t = 3.35, df = 7, two-tailed paired t test). A change of the resting membrane potential, Vm, is unlikely to explain the changes induced by TBF because these changes were compensated with offsetting DC before measurement of excitability (following TBF there was typically a small hyperpolarization of the Vm that returned to baseline in 1–2 s). This change of membrane properties has similar requirements as the reduction of membrane excitability induced by TBF. Thus, if the TBF was applied without depolarization, a condition that does not lead to a decrease of membrane excitability (see above), there was no significant change of input resistance (Fig. 3C) (baseline, 35.4 ± 2.24 MΩ; post-TBF, 33.9 ± 2.52 MΩ; n = 10, p = 0.139, t = 1.63, paired t test) or membrane time constant (Fig. 3C) (baseline, 16.9 ± 0.96 ms; post-TBF, 16.7 ± 1.22 ms; n = 8, p = 0.913, t = 0.114, paired t test).
Figure 3.
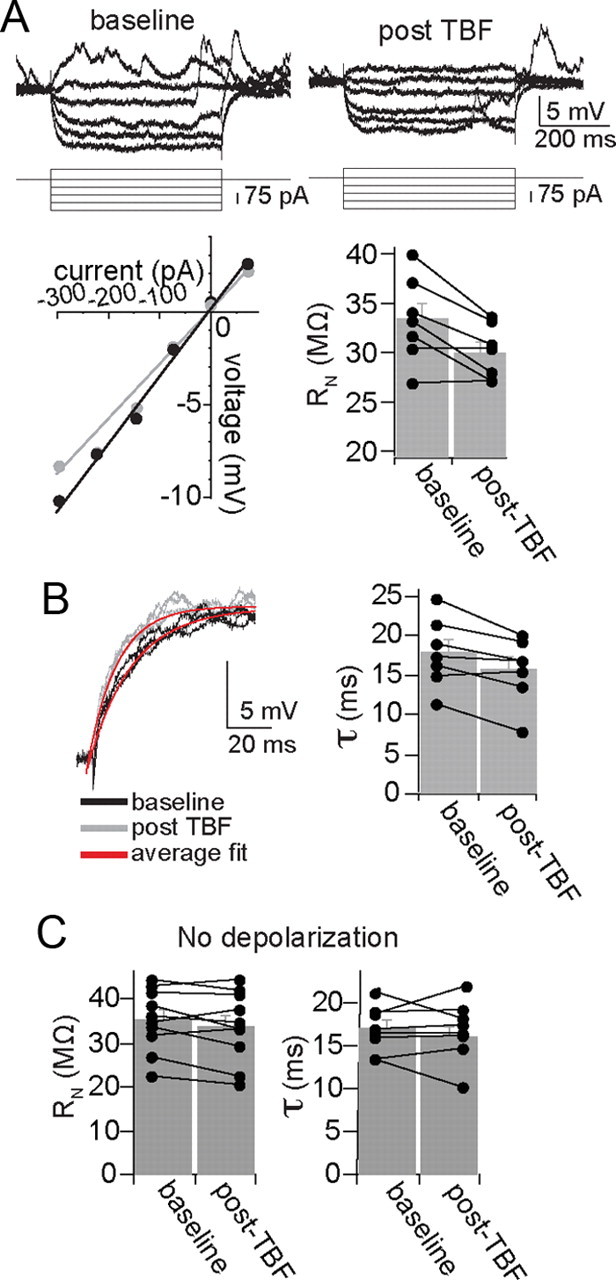
Theta burst firing modifies membrane properties of BLA neurons. A, Neuronal input resistance was decreased after TBF (in the example displayed here, baseline Rn = 39 MΩ, post-TBF Rn = 32 MΩ; black circle, baseline; gray circle, post-TBF). B, TBF also led to a faster time constant (top; baseline, 16.2 ms; post-TBF, 13.7 ms; black line is baseline, gray line is post-TBF, red line is average fit). C, Neither input resistance nor membrane time constant was significantly altered by TBF if performed without depolarization from Vrest.
Theta burst firing decreases synaptic integration in vivo
Modification of membrane properties by TBF, such as input resistance and membrane time constant, should lead to a change of the integrative properties of these neurons. To test this, parameters of spontaneous synaptic events that are sensitive to changes of membrane properties were measured. TBF caused a reduction in the amplitude of spontaneous synaptic events (Fig. 4A) (baseline, 3.81 ± 0.20 mV; post-TBF activity, 3.32 ± 0.18 mV; p = 0.0029, t = 3.80, df = 11, two-tailed paired t test) and a decrease in the half-width of events (Fig. 4B) (baseline, 11.98 ± 1.12 ms; post-TBF activity, 10.64 ± 0.77 ms; p = 0.015, t = 2.87, df = 11, two-tailed paired t test), without a change in the frequency of events (Fig. 4B) (inter-event interval: baseline, 0.59 ± 0.03 s; post-TBF activity, 0.58 ± 0.04 s; p = 0.329, t = 1.022, df = 11, two-tailed paired t test). These spontaneous synaptic events often occur in groups, leading to summation (Fig. 4C). TBF caused a reduction of the peak amplitude of these summated events (Fig. 4C) (baseline, 7.54 ± 0.40 mV; post-TBF activity, 6.19 ± 0.33 mV; p = 0.002, t = 4.02, df = 11, two-tailed paired t test). As a measure of postsynaptic activity that is independent of event detection thresholds, the SD of the mean membrane potential was also measured and was significantly decreased after TBF activity (baseline, 2.71, ± 0.24 mV; post-TBF activity, 2.42 ± 0.29 mV; p = 0.020, t = 2.71, df = 11, two-tailed paired t test), observable as a narrowing of the distribution of the membrane potential (Fig. 4D). This is consistent with a change in postsynaptic properties that influences integration of synaptic activity. However, these measured changes may be caused by a retrograde modulation of presynaptic function. Therefore, the voltage response to EPSC-shaped current injection was also measured, a parameter strongly dependent upon postsynaptic membrane properties (αPSPs; see Materials and Methods). TBF caused a reduction in the summation of αPSPs (Fig. 4E) (baseline, 0.77 ± 0.10; post-TBF activity, 0.67 ± 0.12; p = 0.041, t = 2.60, df = 6, two-tailed paired t test; there was no significant difference in the amplitude of the first PSP despite the previously noted reduction of input resistance, probably because the time course of the PSC is rapid, and the peak of the PSP occurs more rapidly than the membrane time constant in response to a square step: baseline, 5.90 ± 0.59 mV; post-TBF activity, 5.78 ± 0.57 mV; p = 0.767, t = 0.311, df = 6, two-tailed paired t test). These data point to a postsynaptic change of synaptic integration caused by TBF.
Figure 4.
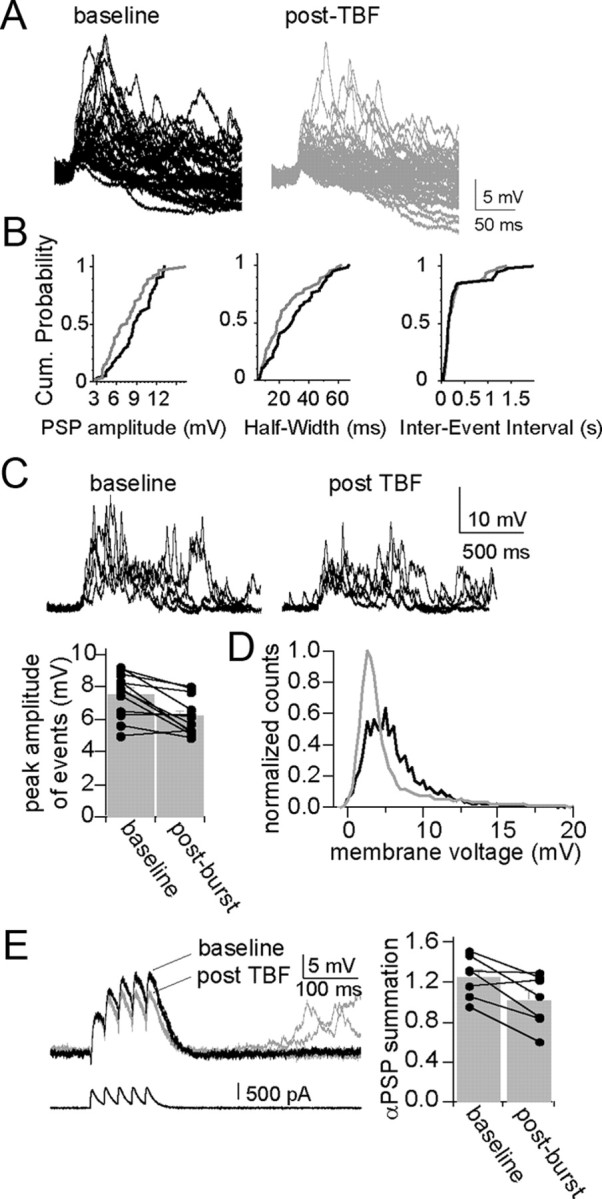
Theta burst firing decreases synaptic integration. A, Theta burst firing modulates the properties of spontaneous synaptic events. Spontaneous synaptic events displayed a range of amplitudes in vivo. B, Several parameters of spontaneous synaptic events are modified after theta burst firing, including amplitude and half-width, depicted in these and cumulative probability plots from a single example. The inter-event interval did not change. C, D, Spontaneous synaptic events occur in bursts in vivo in BLA neurons (baseline) (C). TBF results in a decrease in the average peak amplitude of these bursts of events (post-TBF) (C) and a leftward shift in the distribution of the membrane potential (black line is baseline, gray line is post-TBF) (D). E, This can be accounted for with a postsynaptic mechanism, as there is also a decrease in the summation of αPSPs after TBF.
Plasticity of membrane excitability does not require NMDA receptors in vivo
Previous studies in vitro have demonstrated that many forms of plasticity rely upon activation of postsynaptic NMDA receptors and Ca2+ influx through these channels or other sources. To determine what factors contribute to change of excitability caused by TBF, chemical blockers were included in the intracellular pipette to target the recorded neuron. In these experiments, a minimum of 25 min was used to allow time for the chemical blocker to diffuse into the recorded neuron before neurons were exposed to a strong 5 Hz bursting protocol (2–4 APs per burst) paired with depolarization. Intracellular blockade of NMDA channels with administration of high concentrations of Mg2+ (20 mm) (Mayer et al., 1984; Nowak et al., 1984; Johnson and Ascher, 1990) or MK801 (2 mm) (Berretta and Jones, 1996; Humeau et al., 2003) did not block TBF-dependent reduction of membrane excitability, though the effectiveness of TBF activity was reduced (Fig. 5A,B) (intracellular MK801, 56.1 ± 5.3% of baseline excitability 1 min after TBF activity, n = 6; intracellular Mg2+, 48.3 ± 5.8%, n = 6, control 30.7 ± 2.6%; significant effect of NMDA blockade compared to control, p = 0.0068, F(2,21) = 6.39, two-way repeated-measures ANOVA). It is possible that MK801 failed to block the effects of TBF due to lack of sufficient intracellular diffusion of this drug, resulting in suboptimal blockade of NMDA channels. Therefore, experiments were replicated with systemic administration of a fairly high dose of MK-801 (0.2 mg/kg i.p.; dose was higher than effective doses for blockade of amygdala-dependent behaviors) (Venable and Kelly, 1990; Baker and Azorlosa, 1996; Langton et al., 2007), and similar results were observed (systemic MK801, 62.2 ± 7.4% of baseline excitability 1 min after TBF activity, n = 6; significant effect of NMDA blockade compared to control, p = 0.022, F(1,16) = 5.12, two-way repeated measures ANOVA). Thus, when NMDA channels are blocked, TBF still induces a change of membrane excitability. Although it should be noted that while TBF still induced a reduction of excitability in the presence of MK801, the effectiveness of TBF was reduced.
Figure 5.

Plasticity of membrane excitability is not dependent upon NMDA receptors. A, In the presence of MK-801 intracellular (depicted here) or extracellular or high intracellular levels of Mg2+ to block NMDA receptors, TBF still caused a decrease of membrane excitability. B, Plotted over time, neither Mg2+ nor MK801 fully blocked TBF-induced reductions of membrane excitability. C, Blockade of NMDA receptors does not block changes of membrane properties caused by theta burst firing. When NMDA channels are blocked (in this instance with intracellular MK-801), theta burst firing still effectively reduces input resistance and membrane time constant.
In addition to a reduction of membrane excitability after TBF in the presence of NMDA blockers, TBF still induced a change of membrane properties when NMDA channels were blocked (Fig. 5C), including a reduction of Rn (baseline Rn, 30.2 ± 2.7 MΩ; post-TBF, 26.7 ± 2.6 MΩ; p < 0.001, t = 9.56, df = 5, two-tailed paired t test) and a faster membrane Tau (baseline, 19.7 ± 1.4 ms; post-TBF, 17.3 ± 1.3 ms; p = 0.009, t = 4.14, df = 5, two-tailed paired t test).
Plasticity of membrane excitability requires Ca2+ channel activity in vivo
The decrease of excitability observed after TBF was prevented by blockade of Ca2+ channels with Ni2+ (0.5 mm) or Cd2+ (0.5 mm) in the intracellular pipette (Fig. 6A,B) (intracellular Cd2+, 86.6 ± 2.0% of baseline excitability 1 min after TBF activity, n = 7; intracellular Ni2+, 97.7 ± 2.4%, n = 6; significant effect of Ca2+ channel blockade compared to control, p < 0.001, F(2,20) = 10.22, two-way repeated-measures ANOVA). When examined with a more comprehensive measure of membrane excitability using a full input–output measurement (0–1000 pA) to verify the effectiveness of Ca2+ channel blockade, it was found that blockade of Ca2+ channels with Cd2+ prevented the impact of TBF on membrane excitability over the entire input–output curve (Fig. 6C, left) (change in the slope of the input-output relationship, baseline 1.31 ± 0.14 AP for each 100 pA increase of current step input; post-TBF activity, 1.47 ± 0.15 AP for each 100 pA increase of current step input, p = 0.71, t = 0.38, two-tailed unpaired t test).
Figure 6.
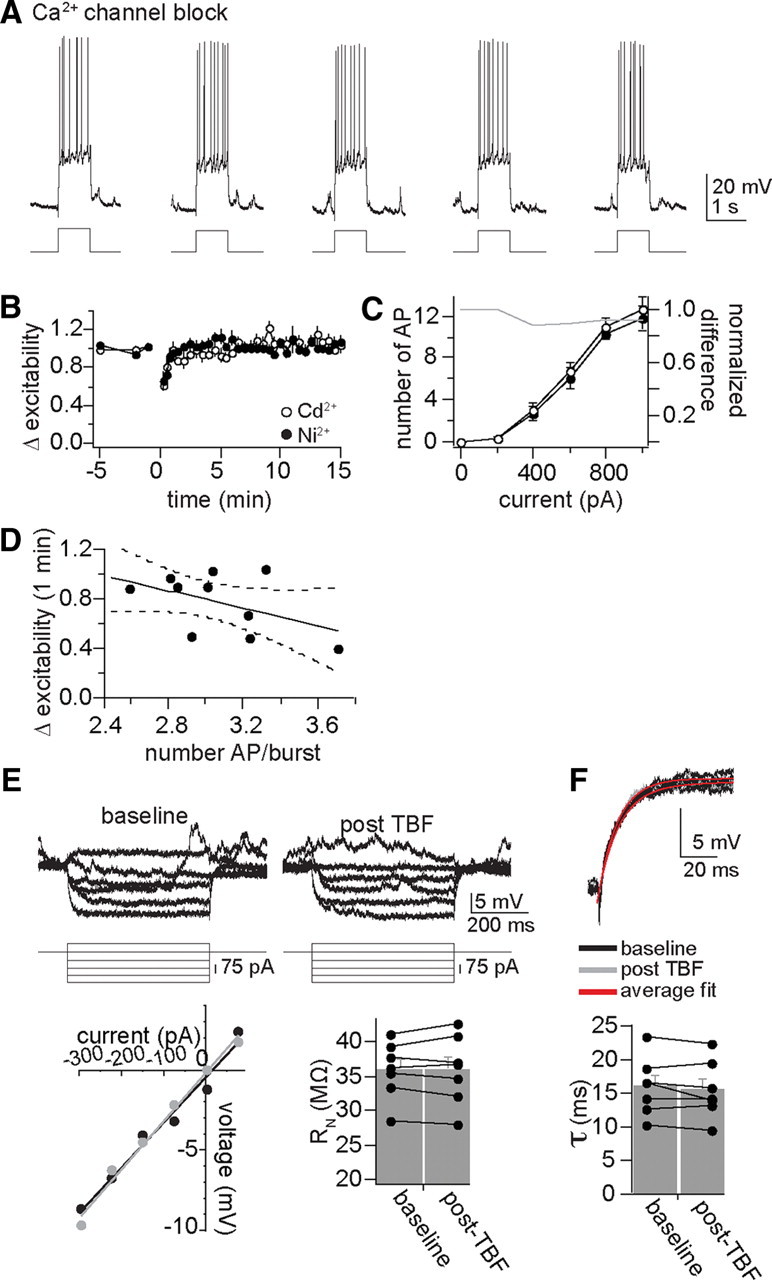
Plasticity of membrane excitability is dependent upon calcium channels. A, With intracellular blockade of Ca2+ channels with Cd2+ (depicted here) or Ni2+, TBF was not able to cause a decrease of membrane excitability. B, Plotted over time, intracellular application of Cd2+ or Ni2+ blocked TBF-induced reductions of membrane excitability. C, Even when membrane excitability was measured over a range of current steps to produce a full input–output curve, TBF did not decrease excitability if Ca2+ channels were blocked (plotted here is intracellular Cd2+; gray line represents the difference between baseline and post-TBF excitability). D, This lack of effectiveness of TBF is not caused by insufficient action potential firing during TBF, as most of the experiments were performed with >2.5 action potentials per burst, and there was still minimal plasticity. E, The effects of TBF on neuronal input resistance were blocked by intracellular blockade of calcium channels with Cd2+ or Ni2+ (displayed in this figure are examples with Cd2+, baseline Rn = 40 MΩ, post-TBF = 42 MΩ; black circle, baseline; gray circle, post-TBF). F, The effects of TBF on membrane time constant were also blocked by intracellular blockade of calcium channels (bottom; baseline, 12.7 ms; post-TBF, 13.1 ms). In almost all control neurons tested (see above), TBF decreased Rn and the time constant, while this effect of TBF was rarely observed if calcium channels were blocked (plotted here with intracellular Cd2+).
In addition to disruption of the effects of TBF on excitability, intracellular Ni2+ or Cd2+ blocked the effects of TBF activity on input resistance (Fig. 6E) (Cd2+: n = 7; baseline, 35.9 ± 1.6 MΩ; post-TBF, 36.0 ± 1.9 MΩ; Ni2+: n = 6; baseline, 38.22 ± 1.61 MΩ; post-TBF, 39.49 ± 1.65 MΩ) and membrane time constant (Fig. 6F) (Cd2+: n = 7; Tau baseline, 16.07 ± 1.63 ms; post-TBF, 15.50 ± 1.62 ms; Ni2+: n = 6; baseline Tau, 17.7 ± 1.45 ms; post-TBF, 17.5 ± 1.61 ms).
In the presence of intracellular Ni2+ or Cd2+ there was also no observed effect of TBF on spontaneous EPSPs summation (Fig. 7A) (Cd2+: n = 7; barrage peak amplitude, 7.38 ± 0.69 mV; post-TBF, 7.49 ± 0.54 mV; Ni2+: n = 6; baseline peak amplitude, 7.11 ± 0.81 mV; post-TBF, 7.35 ± 0.82 mV) or αPSP summation (Fig. 7B) (Cd2+: n = 7; baseline, 0.85 ± 0.14; post-TBF activity, 0.81 ± 0.15; Ni2+: n = 6; baseline, 0.83 ± 0.17; post-TBF, 0.86 ± 0.17). In addition, with intracellular Cd2 or Ni2 there was no significant effect of TBF on parameters of single EPSPs that are sensitive to a change of membrane properties (Fig. 7C,D) such as amplitude (Cd2+: n = 7; baseline, 3.94 ± 0.26 mV; post-TBF, 4.15 ± 0.24 mV, p > 0.05, paired t test; Ni2+: n = 6; baseline, 4.08 ± 0.29 mV; post-TBF, 4.18 ± 0.29 mV, p > 0.05, paired t test) and half-width (Cd2+: n = 7; baseline, 12.58 ± 1.37 ms; post-TBF, 11.82 ± 1.21 ms, p > 0.05, paired t test; Ni2+: n = 6; baseline, 13.73 ± 1.22 ms; post-TBF, 12.58 ± 1.21 ms, p > 0.05, paired t test). The blockade of the effects of TBF on membrane properties, membrane excitability, and synaptic integration by blockers of Ca2+ channels indicates a common associated trigger for these effects of TBF activity on neuronal function in vivo. These data indicate that periods of robust in vivo firing lead to a change of membrane properties that is associated with decreased excitability and decreased synaptic integration and that these changes are induced by Ca2+ influx through voltage-gated Ca2+ channels.
Figure 7.
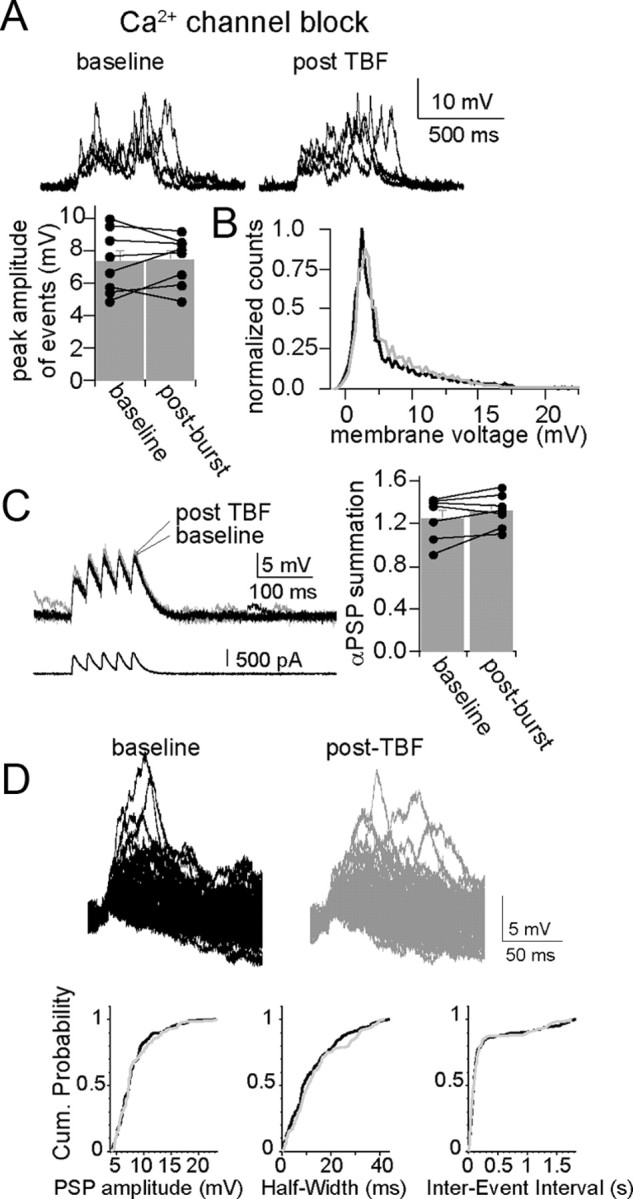
Plasticity of synaptic integration is dependent upon calcium channels. A, B, Blockade of Ca2+ channels with intracellular Cd2+ (depicted here) or Ni2+ blocks the effects of TBF on integration of spontaneous synaptic events as demonstrated by the absence of a change in the average peak amplitude of bursts of synaptic events (A) and by the absence of a shift in the distribution of the membrane potential (black line is baseline, gray line is post-TBF) (B). C, Consistent with this, the effect of TBF on summation of αPSPs was blocked by intracellular blockade of Ca2+ channels (Cd2+ depicted here). D, Modification of spontaneous single synaptic events was also absent after TBF when intracellular blockers of Ca2+ channels were present (in this case Cd2+); neither the amplitude, half-width, nor inter-event interval was modified by theta burst firing.
One potential concern is that intracellular blockade of Ca2+ channels can exert actions that indirectly influence the induction of TBF plasticity, such as an increased basal firing rate leading to a ceiling effect for TBF induction or a decreased number of action potential firing during the TBF activity leading to reduced TBF induction. The basal spontaneous firing rate was very low under our recording conditions. Only 1/13 neurons with intracellular Ni2+/Cd2+ displayed spontaneous firing (0.067 Hz). The firing during the TBF activity was not disrupted (total number of action potentials during TBF: Cd2+, 956.2 ± 45.4, n = 7; Ni2+, 863.3 ± 37.4, n = 6; equivalent control, 841.8 ± 56.0, n = 12; p = 0.41, F(2,20) = 0.94, one-way ANOVA).
Hyperpolarization-activated ion channels contribute to expression
The observed reduction of excitability, input resistance, and integration of synaptic inputs following TBF activity is consistent with an increase of hyperpolarization-activated ion channel (h channel) function. To test whether h channels are involved in the expression of TBF-induced plasticity, h channels were pharmacologically blocked. To do this, microinfusions of ZD7288 (1 mg/ml) into the LAT were performed. All recording sites included for analysis were within 1.5 mm of the infusion site. To confirm the effectiveness of ZD7288 and the time course of its actions, ZD7288 infusions were performed in control experiments following a baseline period. The baseline period consisted of the usual repeated testing of membrane excitability (Fig. 8A). ZD7288, but not aCSF, caused a rapid increase of excitability that did not reverse over the time course tested (Fig. 8B,C, right) (two-way repeated-measures ANOVA, significant effect of time F(1,32) = 4.42, p < 0.001; significant effect of infusion, F(1,32) = 21.34, p < 0.01; ZD7288, n = 6; aCSF, n = 4).
Figure 8.
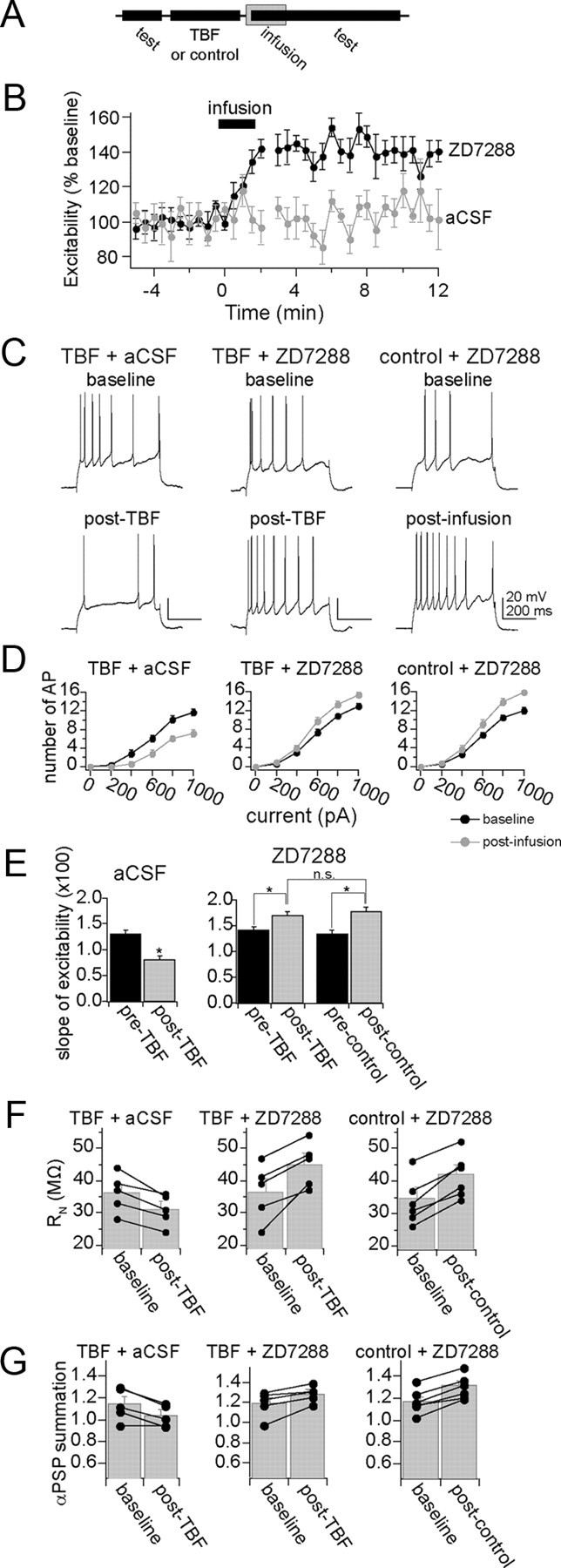
Blockade of h channels disrupts the expression of plasticity of membrane properties. A, A procedure was designed to specifically test the role of h channels in the expression, not induction, of membrane plasticity caused by TBF. In this design, membrane properties are tested, followed by the TBF (or control) activity. This is immediately followed by local infusion of ZD7288 or aCSF. During and following this infusion, membrane properties are tested again. B, Infusion of ZD7288, but not aCSF vehicle, caused an increase of membrane excitability. C, TBF caused a decrease of membrane excitability, even after aCSF infusion (left panel). However, the decreased excitability caused by TBF is reversed by local infusion of ZD7288 (middle panel). ZD7288 also causes increased excitability when it follows a control period (right panel, as in B). D, The effects on membrane excitability are observed over a range of current step amplitudes (0–1000 pA). E, To further quantify the impact of membrane excitability, the slope of the input–output relationship was measured (“slope of excitability”). As above, TBF caused a reduction of membrane excitability when aCSF was infused. The effect of TBF on excitability was reversed by ZD7288. Importantly, the final slope of the input-output relationship was similar after ZD7288 in both the control and the TBF group (right). F, Infusion of ZD7288 reversed the effects of TBF on Rn, such that the final value was similar to Rn values following ZD7288 infusion after a control procedure. G, Infusion of ZD7288 reversed the effects of TBF on summation of αPSPs, such that the final value was similar to αPSPs values following ZD7288 infusion after a control procedure.
To test whether blockade of h channels disrupts plasticity induced by TBF, ZD7288 was infused immediately following a strong 5 Hz TBF protocol (3–4 APs per burst) (Fig. 8A). Infusion of ZD7288 following TBF blocked TBF-induced changes of membrane excitability. Thus, infusion of ZD7288 rapidly induced an increase of excitability above the diminished post-TBF levels and above baseline levels (Fig. 8C, middle), quantified as the increased excitability in response to a range of current steps (Fig. 8D, middle) (0–1000 pA; quantified as the mean number of action potentials/current step, n = 5, p < 0.01, F(1,24) = 13.6, two-way repeated-measures ANOVA). Infusions of aCSF did not block TBF-induced plasticity (n = 5, p < 0.01, F(1,24) = 15.8, two-way repeated-measures ANOVA), and TBF resulted in a decrease of membrane excitability when aCSF was infused (Fig. 8C,D, left). Furthermore, ZD7288 infusion resulted in a similar final membrane excitability in control and TBF conditions, measured as the slope of the input–output relationship (Fig. 8E) (ZD7288 + TBF, 1.69 ± 0.078 AP for each 100 pA increase of current step input, n = 5; ZD7288 + control activity, 1.73 ± 0.093 AP for each 100 pA increase of current step input, n = 6, p = 0.53, F(1,62) = 0.39, two-way repeated measure ANOVA). Because TBF decreases membrane excitability, this is consistent with a greater degree of h channel activity following TBF activity. It further demonstrates that when the influence of h channels is removed, control and TBF groups are equal. In addition, after ZD7288 was infused, input resistance and integration of αPSPs were similar between control and TBF groups (Fig. 8F,G), demonstrating that h channel activity underlies the expression of TBF-induced reduction of excitability, input resistance, and PSP integration.
Activation of h channels leads to a relatively slow voltage sag during a hyperpolarizing pulse. To further verify a role for h channels in the expression of TBF-induced plasticity, the amplitude of the voltage sag was measured before and after a strong 5 Hz TBF protocol (3–4 APs per burst), following aCSF infusion. TBF caused an increase in the amplitude of the voltage sag (Fig. 9A,B) calculated as (peak amplitude − steady state)/peak (baseline, 0.094 ± 0.0075; post-TBF, 0.11 ± 0.0050; p = 0.0032, t = 6.33, n = 5, two-tailed paired t test) after TBF activity. The voltage sag was blocked by infusions of ZD7288, confirming the h channel origin of the voltage sag in the neurons in vivo (Fig. 9C,D).
Figure 9.
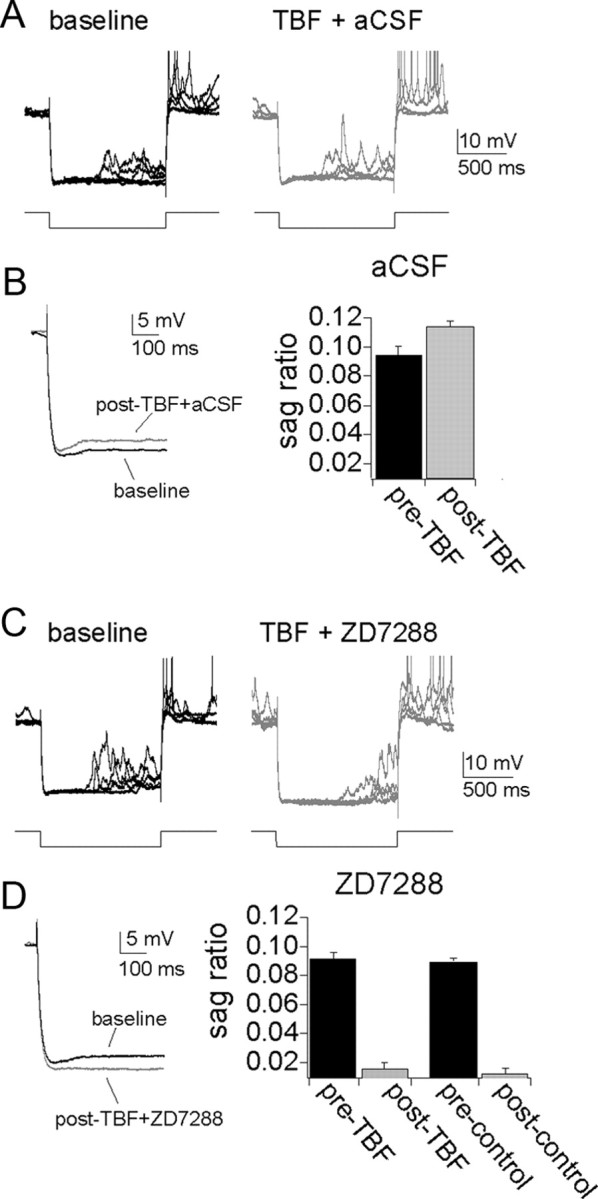
Theta burst firing leads to an increase of voltage sag, an indicator of h channel activity. A, There is a small but consistent voltage sag during the response to large hyperpolarizing voltage deflections. The amplitude of this voltage sag is increased after TBF coupled with aCSF infusion. B, The amplitude of the voltage sag is quantified by the sag ratio, which increases after TBF. C, D, The voltage sag is blocked by infusion of ZD7288 following either TBF or control periods. Action potentials in A and C are truncated for clarity.
Theta burst firing reduces synaptic plasticity in vivo
An important feature of neuronal function that contributes to learning is the capacity for activity-dependent changes in synaptic strength, exemplified as LTP of synaptic inputs. If TBF exerts a significant modulatory influence in vivo, it is expected to impact LTP. TBF was followed by an LTP protocol (Materials and Methods). In control experiments, TBF was performed without concomitant depolarization from the resting membrane potential (which does not lead to a long-term change of membrane properties or excitability; see above). Under these control conditions, LTP of synaptic inputs was readily observed (Fig. 10A, top, B) 147.4 ± 5.4% of baseline after 5 min; n = 7, p < 0.001, F(2,108) = 20.62, two-way repeated-measures ANOVA). However, if parameters for effective TBF were used with concomitant depolarization from the resting membrane potential (which leads to a change of membrane properties and reduction of excitability), subsequent LTP induction was much less effective (Fig. 10A, bottom, B) (97.7 ± 2.3% of baseline after 5 min; n = 7, p = 0.51, F(2,108) = 0.861, two-way repeated-measures ANOVA). This result does not appear to depend upon EPSP size before LTP (mean EPSP amplitude control TBF, 7.7 ± 0.9 mV, n = 7; TBF with depolarization, 7.1 ± 1.0 mV, n = 7), and a similar number of action potentials were evoked during the TBF protocols (control TBF, 2.8 ± 0.4 action potentials per burst, n = 7; TBF with depolarization, 2.6 ± 0.4 action potentials per burst, n = 7). If TBF truly has a modulatory effect on LTP induction, it is expected that TBF procedures that lead to only moderate changes of excitability would have a smaller effect on LTP induction. A weaker 30 s TBF protocol (see above) had a smaller impact on LTP induction (Fig. 10B) (120.6 ± 5.7% of baseline after 5 min; n = 5, p < 0.001, F(2,72) = 8.97, two-way repeated-measures ANOVA). This association between the effectiveness of the TBF protocol on excitability and on subsequent LTP indicates that these changes may share a common mechanism. To further examine the link between the effects of TBF, associations between the various TBF-induced effects were examined within single neurons.
Figure 10.
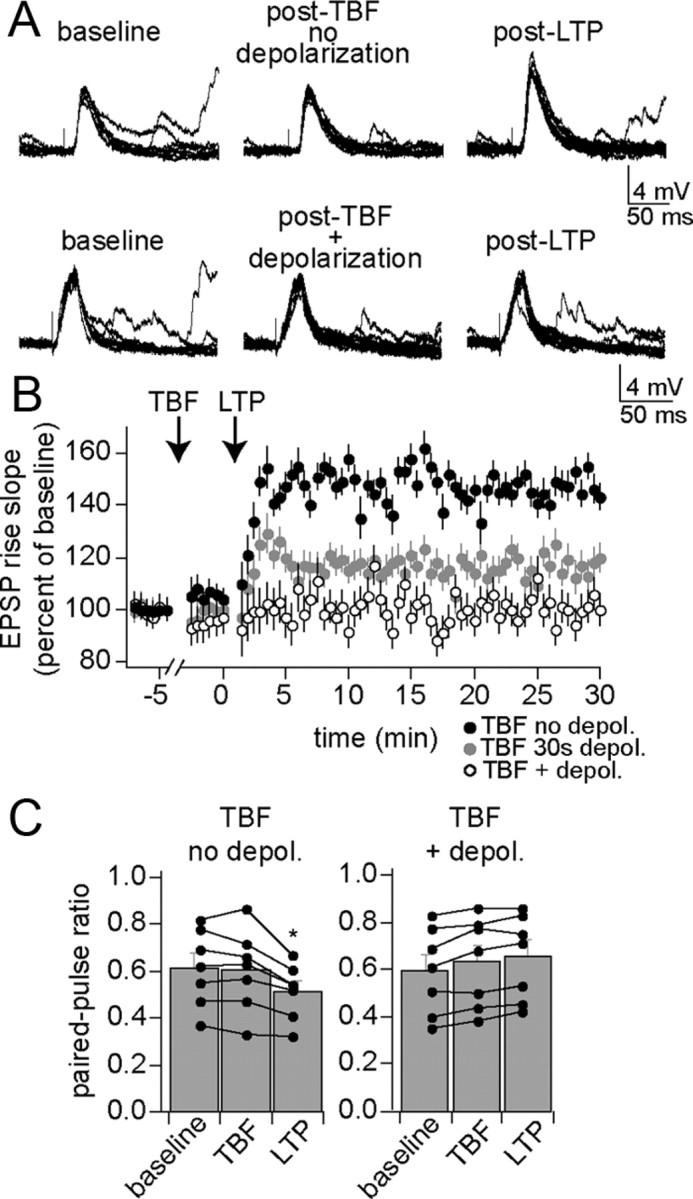
Theta burst firing decreases synaptic plasticity. Synaptic plasticity, measured here as LTP of a thalamic evoked EPSP, can be induced in the BLA. A, This LTP is observed in conditions when TBF is ineffective at changing membrane properties (top; TBF without being paired with depolarization). In conditions that promote TBF effects on membrane properties (TBF paired with depolarization), LTP is absent (bottom). B, When TBF is only moderately effective at inducing a change of membrane properties [30 s TBF paired with depolarization (depol)], LTP is attenuated but not absent. C, LTP induced after the weak TBF protocol is accompanied by a change of paired-pulse ratio, indicative of a presynaptic change during LTP. When LTP is blocked by previous TBF, a change of paired-pulse ratio is also blocked.
Associated changes in excitability, synaptic integration and LTP
In a subset of experiments, the association between the effectiveness of TBF on excitability and synaptic integration and excitability and LTP was examined. There was a strong correlation between the magnitude of the effects of TBF on excitability and on input resistance (Fig. 11A) (Rn measured after 3 and 5 min, excitability measured after 2.5 min; r2 = 0.91, significantly different from 0, F = 49.0, p < 0.001, n = 5) and between the effects of TBF on excitability and synaptic integration as measured by summation of αPSPs (Fig. 11B) (r2 = 0.73, significantly different from 0, F = 13.2, p < 0.05, n = 7). Combined with the similar dependence on Ca2+ channel activity, this implies a strong relationship between these two outcomes of TBF. There was also a strong correlation between the magnitude of the effects of TBF on excitability and LTP (Fig. 11C) (LTP measured after 10 min, excitability measured immediately before LTP; r2 = 0.79, significantly different from 0, F = 37.9, p < 0.001, n = 12). In particular, in those instances where TBF was only weakly effective, LTP was more robust.
Figure 11.
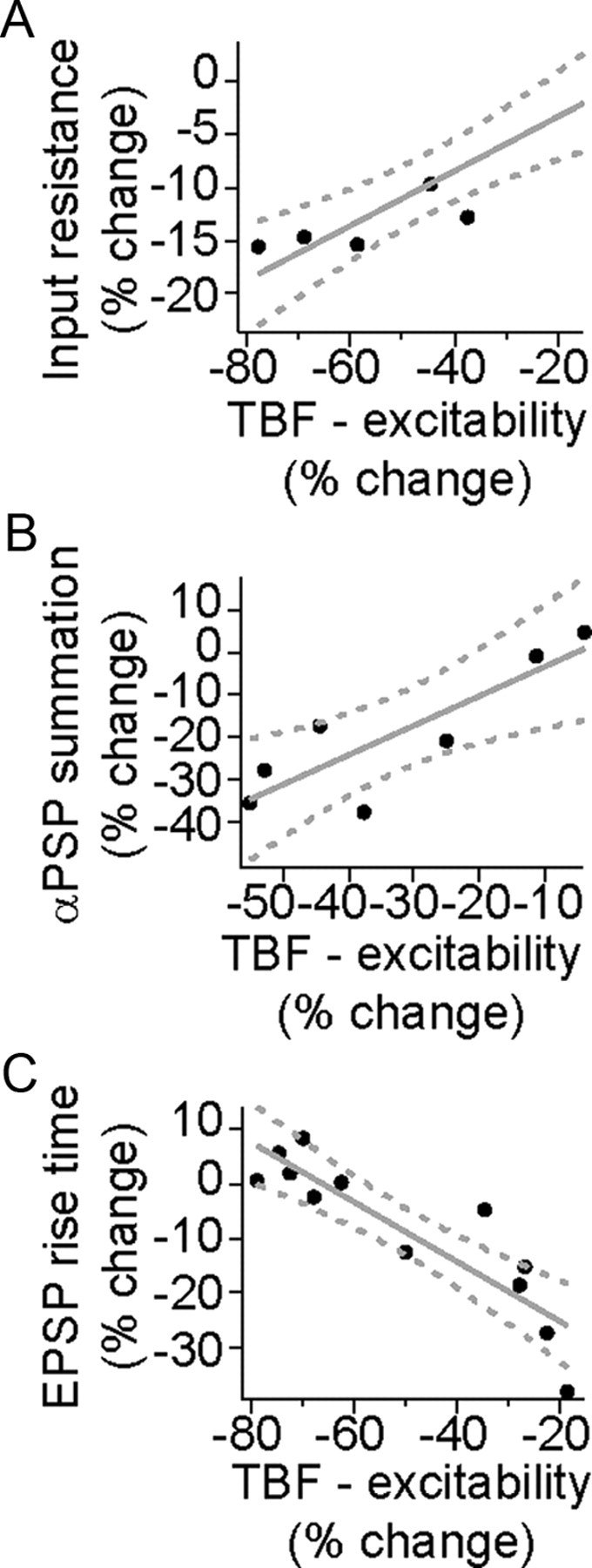
Correlation between the change of excitability induced by theta burst firing and other measures. TBF induced associated changes between excitability and several other measures. A, There was a correlation between the effects of TBF on excitability and input resistance (r2 = 0.91), B, C, Excitability and αPSP summation (r2 = 0.73) (B) and excitability and subsequent LTP (r2 = 0.79) (C).
Discussion
A change in neuronal properties is fundamental to adaptation, learning, and other behaviors. One particular property, membrane excitability, dictates the vigor of action potential firing and output of a neuron. While there is evidence for long-term changes of excitability in many conditions (Turrigiano et al., 1994; Desai et al., 1999; Golowasch et al., 1999; Baines et al., 2001; Beraneck et al., 2003; Pratt and Aizenman, 2007; Beck and Yaari, 2008; Grubb and Burrone, 2010), less is known about rapid in vivo changes of excitability that can accompany periods of robust neuronal firing activity and whether this is associated with changes in synaptic integration or synaptic plasticity. Periods of neuronal firing led to an in vivo reduction of BLA neuronal synaptic integration, reduction of synaptic plasticity, and change in membrane properties that contribute to neuronal excitability. This change required activation of Ca2+ channels but not NMDA receptors. The reduction of excitability was temporary, returning to baseline within 10 min. Plasticity of membrane properties that can be induced rapidly and reverses on the order of minutes instead of seconds or hours may play a significant role in allowing the animal to more effectively filter out less important information during critical behavioral periods. Consistent with this, the impact of TBF on membrane excitability was weaker when induced from more hyperpolarized membrane potentials. Perhaps during states when the amygdala is not particularly recruited and neurons are at a less depolarized membrane potential, this plasticity of membrane excitability will not be triggered.
The requirements for triggering this type of plasticity of membrane properties are likely to occur in the BLA under certain restricted circumstances. One factor that promotes this form of plasticity is depolarization of the membrane potential. Although not examined in the BLA, studies that have examined cortex and other cortical-like structures indicate that neurons do tend to be more depolarized during wakefulness (Baranyi et al., 1993; Steriade et al., 2001; Chen and Fetz, 2005; Rudolph et al., 2007; Poulet and Petersen, 2008; Lee et al., 2009). In our experience, the membrane potential of BLA neurons is more depolarized when anesthesia levels fluctuate toward lighter but still anesthetized states. Another promoting factor is prolonged firing at 5 Hz or greater. While the basal firing rate of LAT neurons is low in awake cats (Gaudreau and Pare, 1996), it appears to be higher in awake rats in some studies (populations of neurons firing between 2 and 5 Hz, even when very rapidly firing potential interneurons are excluded) (Bordi et al., 1993; Maren, 2000; Shabel and Janak, 2009). Furthermore, while many studies demonstrate a transient increase in firing to conditioned stimuli (Quirk et al., 1997), several studies suggest that the baseline firing of BLA neurons can increase for longer durations in the presence of more prolonged stimuli (Uwano et al., 1995; Nishijo et al., 1998; Shabel and Janak, 2009). Several studies demonstrate that the baseline firing of BLA neurons increases over the course of, or for long periods after, a fear conditioning procedure (Ben-Ari and Le Gal la Salle, 1972; Uwano et al., 1995; Maren, 2000; Pare and Collins, 2000; Pelletier et al., 2005). In such circumstances, it may be maladaptive to encode all the environmental sensory cues that are available. Instead, by dampening excitability and subsequent LTP, excessive neuronal firing may select for the more prominent cues or for the cues that were present at the beginning of the onset of the changes.
The activity-dependent decrease of membrane excitability did not rely upon activation of NMDA receptors, although they do modulate its amplitude. However, the membrane plasticity observed here does rely upon influx of Ca2+ through voltage-gated sources, as the plasticity of membrane excitability was blocked by Cd2+ or Ni2+. Many forms of BLA-dependent learning require NMDA receptors, although at least one form of associative synaptic plasticity (Weisskopf et al., 1999; Bauer et al., 2002; Fourcaudot et al., 2009), as well as one form of nonassociative synaptic plasticity (Schroeder and Shinnick-Gallagher, 2004) in the BLA, do not. In addition, the necessity of voltage-gated Ca2+ channels in BLA-dependent forms of fear learning may be limited to long-term memory of conditioned associations (Bauer et al., 2002; McKinney and Murphy, 2006) (but see Cain et al., 2002). In other brain regions there are reports of NMDA-dependent (e.g., Fan et al., 2005; Xu et al., 2005) or NMDA-independent (Aizenman and Linden, 2000; Tsubokawa et al., 2000) changes of membrane excitability following synaptic or firing activity. While our results support an NMDA-independent mechanism for the in vivo change of membrane excitability following theta burst firing, it is possible that NMDA receptors are required for in vivo plasticity that is induced by periods of synaptic activity.
The reduction of excitability after action potential firing was accompanied by a change of membrane properties that decreased the integration of spontaneous synaptic events. This appears to be mediated, at least in part, by a post-synaptic effect, as TBF caused a decrease in the summation of αEPSPs (Fan et al., 2005), which are entirely postsynaptic. A reduction of input resistance and faster membrane time constant could cause this reduction of synaptic integration. Functionally, this would result in a decrease of neuronal output in response to synaptic inputs. The impact of this change is expected to be important for sensory integration within the BLA. A reduction of the neuronal subthreshold membrane response to synaptic inputs, coupled with a reduction in excitability, leads to a reduced responsitivity of the LAT to inputs. These properties should lead to a reduction in the drive of the LAT by sensory inputs and a subsequent reduction of the impact of sensory inputs on BLA-driven affective behaviors.
Previous studies have found evidence for a change of dendritic A-type K+ channels (Frick et al., 2004), h channels (Fan et al., 2005; Brager and Johnston, 2007; Campanac et al., 2008), Na+ channels (Xu et al., 2005), or calcium-activated K+ channels (Sourdet et al., 2003) following robust excitatory synaptic or action potential firing activity. The decrease of input resistance and membrane time constant observed in this study is consistent with an activity-dependent change in conductances that are active near resting membrane potentials. The blockade of expression of TBF-induced plasticity by ZD7288 and the TBF-induced increased in voltage sag strongly indicate a prominent role for h channels in the expression of this form of activity-dependent plasticity. The necessity for Ca2+ in these changes may be related to Ca2+-sensitive second messenger systems (Fan et al., 2005), the role of dendritic Ca2+ spikes in induction of plasticity (e.g., Humeau and Luthi, 2007), or perhaps in the expression of the changes. Subthreshold Ca2+ oscillations are present in BLA neurons (Pape and Driesang, 1998) and likely facilitate firing. However, it is unlikely that a decrease of neuronal firing in the presence of intracellular Cd2+ or Ni2+ underlies the absence of membrane plasticity in these conditions, as a range of levels of action potential firing was evoked, some that equaled or exceeded firing in control conditions.
It has been predicted that a change of membrane properties should modulate the potential for plasticity (for review, Abraham, 2008), sometimes called metaplasticity. The activity-dependent reduction of excitability was associated with a decrease in the induction of synaptic LTP. This finding indicates that metaplasticity can be induced in vivo in a short time scale triggered by neuronal activity, independently of high-frequency synaptic input. It is anticipated that this form of metaplasticity may be able to reduce subsequent associative learning while leaving previous plasticity intact.
Activity-dependent changes in neuronal properties have been proposed play a role in learning (LeMasson et al., 1993; Turrigiano et al., 1994; Turrigiano and Nelson, 2000; Marder and Goaiilard, 2006). Ex vivo studies have found that learning is associated with changes of excitability in a range of brain regions (Disterhoft et al., 1986; Saar et al., 1998; Schreurs et al., 1998; Oh et al., 2003). Rapid in vivo learning-related changes of membrane properties or currents that contribute to membrane properties have been rarely studied in mammals, although it has been found to exist (Rosenkranz and Grace, 2002b), and has been repeatedly observed in invertebrates (Alkon, 1979; Frysztak and Crow, 1997; Antonov et al., 2001; Lorenzetti et al., 2006). The current study indicates that behavioral conditions that are associated with high periods of neuronal firing are sufficient to induce in vivo rapid plasticity of membrane properties in the LAT and that this modulates subsequent synaptic integration and synaptic plasticity. Behaviorally, this may become very important during the formation of amygdala-dependent memories and may be recruited when the amygdala undergoes heightened periods of activity.
Footnotes
Research support was provided by National Institutes of Health Grant MH084970. I thank Mallika Padival for histological processing of tissue and Dr. Daniel Johnston, Dr. Raymond Chitwood, and Dr. Rishikesh Narayanan for discussions and ideas that underlie the study.
References
- Abraham WC. Metaplasticity: tuning synapses and networks for plasticity. Nat Rev Neurosci. 2008;9:387. doi: 10.1038/nrn2356. [DOI] [PubMed] [Google Scholar]
- Aizenman CD, Linden DJ. Rapid, synaptically driven increases in the intrinsic excitability of cerebellar deep nuclear neurons. Nat Neurosci. 2000;3:109–111. doi: 10.1038/72049. [DOI] [PubMed] [Google Scholar]
- Alkon DL. Voltage-dependent calcium and potassium ion conductances: a contingency mechanism for an associative learning model. Science. 1979;205:810–816. doi: 10.1126/science.223244. [DOI] [PubMed] [Google Scholar]
- Antonov I, Antonova I, Kandel ER, Hawkins RD. The contribution of activity-dependent synaptic plasticity to classical conditioning in Aplysia. J Neurosci. 2001;21:6413–6422. doi: 10.1523/JNEUROSCI.21-16-06413.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baines RA, Uhler JP, Thompson A, Sweeney ST, Bate M. Altered electrical properties in Drosophila neurons developing without synaptic transmission. J Neurosci. 2001;21:1523–1531. doi: 10.1523/JNEUROSCI.21-05-01523.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker JD, Azorlosa JL. The NMDA antagonist MK-801 blocks the extinction of Pavlovian fear conditioning. Behav Neurosci. 1996;110:618–620. doi: 10.1037//0735-7044.110.3.618. [DOI] [PubMed] [Google Scholar]
- Baranyi A, Szente MB, Woody CD. Properties of associative long-lasting potentiation induced by cellular conditioning in the motor cortex of conscious cats. Neuroscience. 1991;42:321–334. doi: 10.1016/0306-4522(91)90378-2. [DOI] [PubMed] [Google Scholar]
- Baranyi A, Szente MB, Woody CD. Electrophysiological characterization of different types of neurons recorded in vivo in the motor cortex of the cat. II. Membrane parameters, action potentials, current-induced voltage responses and electrotonic structures. J Neurophysiol. 1993;69:1865–1879. doi: 10.1152/jn.1993.69.6.1865. [DOI] [PubMed] [Google Scholar]
- Bauer EP, Schafe GE, LeDoux JE. NMDA receptors and L-type voltage-gated calcium channels contribute to long-term potentiation and different components of fear memory formation in the lateral amygdala. J Neurosci. 2002;22:5239–5249. doi: 10.1523/JNEUROSCI.22-12-05239.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck H, Yaari Y. Plasticity of intrinsic neuronal properties in CNS disorders. Nat Rev Neurosci. 2008;9:357–369. doi: 10.1038/nrn2371. [DOI] [PubMed] [Google Scholar]
- Ben-Ari Y, Le Gal la Salle G. Plasticity at unitary level. II. Modifications during sensory-sensory association procedures. Electroencephalogr Clin Neurophysiol. 1972;32:667–679. doi: 10.1016/0013-4694(72)90103-4. [DOI] [PubMed] [Google Scholar]
- Beraneck M, Hachemaoui M, Idoux E, Ris L, Uno A, Godaux E, Vidal PP, Moore LE, Vibert N. Long-term plasticity of ipsilesional medial vestibular nucleus neurons after unilateral labyrinthectomy. J Neurophysiol. 2003;90:184–203. doi: 10.1152/jn.01140.2002. [DOI] [PubMed] [Google Scholar]
- Berretta N, Jones RS. Tonic facilitation of glutamate release by presynaptic N-methyl-d-aspartate autoreceptors in the entorhinal cortex. Neuroscience. 1996;75:339–344. doi: 10.1016/0306-4522(96)00301-6. [DOI] [PubMed] [Google Scholar]
- Bliss TV, Lomo T. Long-lasting potentiation of synaptic transmission in the dentate area of the anaesthetized rabbit following stimulation of the perforant path. J Physiol. 1973;232:331–356. doi: 10.1113/jphysiol.1973.sp010273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bordi F, LeDoux J, Clugnet MC, Pavlides C. Single-unit activity in the lateral nucleus of the amygdala and overlying areas of the striatum in freely behaving rats: rates, discharge patterns, and responses to acoustic stimuli. Behav Neurosci. 1993;107:757–769. doi: 10.1037/0735-7044.107.5.757. [DOI] [PubMed] [Google Scholar]
- Brager DH, Johnston D. Plasticity of intrinsic excitability during long-term depression is mediated through mGluR-dependent changes in Ih in hippocampal CA1 pyramidal neurons. J Neurosci. 2007;27:13926–13937. doi: 10.1523/JNEUROSCI.3520-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown JT, Randall AD. Activity-dependent depression of the spike after-depolarization generates long-lasting intrinsic plasticity in hippocampal CA3 pyramidal neurons. J Physiol. 2009;587:1265–1281. doi: 10.1113/jphysiol.2008.167007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cain CK, Blouin AM, Barad M. L-type voltage-gated calcium channels are required for extinction, but not for acquisition or expression, of conditional fear in mice. J Neurosci. 2002;22:9113–9121. doi: 10.1523/JNEUROSCI.22-20-09113.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campanac E, Debanne D. Plasticity of neuronal excitability: Hebbian rules beyond the synapse. Arch Ital Biol. 2007;145:277–287. [PubMed] [Google Scholar]
- Campanac E, Daoudal G, Ankri N, Debanne D. Downregulation of dendritic Ih in CA1 pyramidal neurons after LTP. J Neurosci. 2008;28:8635–8643. doi: 10.1523/JNEUROSCI.1411-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen D, Fetz EE. Characteristic membrane potential trajectories in primate sensorimotor cortex neurons recorded in vivo. J Neurophysiol. 2005;94:2713–2725. doi: 10.1152/jn.00024.2005. [DOI] [PubMed] [Google Scholar]
- Cudmore RH, Turrigiano GG. Long-term potentiation of intrinsic excitability in LV visual cortical neurons. J Neurophysiol. 2004;92:341–348. doi: 10.1152/jn.01059.2003. [DOI] [PubMed] [Google Scholar]
- Desai NS, Rutherford LC, Turrigiano GG. Plasticity in the intrinsic excitability of cortical pyramidal neurons. Nat Neurosci. 1999;2:515–520. doi: 10.1038/9165. [DOI] [PubMed] [Google Scholar]
- Disterhoft JF, Coulter DA, Alkon DL. Conditioning-specific membrane changes of rabbit hippocampal neurons measured in vitro. Proc Natl Acad Sci U S A. 1986;83:2733–2737. doi: 10.1073/pnas.83.8.2733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan Y, Fricker D, Brager DH, Chen X, Lu HC, Chitwood RA, Johnston D. Activity-dependent decrease of excitability in rat hippocampal neurons through increases in I(h) Nat Neurosci. 2005;8:1542–1551. doi: 10.1038/nn1568. [DOI] [PubMed] [Google Scholar]
- Fanselow MS, Poulos AM. The neuroscience of mammalian associative learning. Annu Rev Psychol. 2005;56:207–234. doi: 10.1146/annurev.psych.56.091103.070213. [DOI] [PubMed] [Google Scholar]
- Fourcaudot E, Gambino F, Casassus G, Poulain B, Humeau Y, Lüthi A. L-type voltage-dependent Ca(2+) channels mediate expression of presynaptic LTP in amygdala. Nat Neurosci. 2009;12:1093–1095. doi: 10.1038/nn.2378. [DOI] [PubMed] [Google Scholar]
- Frick A, Magee J, Johnston D. LTP is accompanied by an enhanced local excitability of pyramidal neuron dendrites. Nat Neurosci. 2004;7:126–135. doi: 10.1038/nn1178. [DOI] [PubMed] [Google Scholar]
- Frysztak RJ, Crow T. Synaptic enhancement and enhanced excitability in presynaptic and postsynaptic neurons in the conditioned stimulus pathway of Hermissenda. J Neurosci. 1997;17:4426–4433. doi: 10.1523/JNEUROSCI.17-11-04426.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaudreau H, Paré D. Projection neurons of the lateral amygdaloid nucleus are virtually silent throughout the sleep–waking cycle. J Neurophysiol. 1996;75:1301–1305. doi: 10.1152/jn.1996.75.3.1301. [DOI] [PubMed] [Google Scholar]
- Golowasch J, Abbott LF, Marder E. Activity-dependent regulation of potassium currents in an identified neuron of the stomatogastric ganglion of the crab Cancer borealis. J Neurosci. 1999;19:RC33. doi: 10.1523/JNEUROSCI.19-20-j0004.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grubb MS, Burrone J. Activity-dependent relocation of the axon initial segment fine-tunes neuronal excitability. Nature. 2010;465:1070–1074. doi: 10.1038/nature09160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humeau Y, Lüthi A. Dendritic calcium spikes induce bi-directional synaptic plasticity in the lateral amygdala. Neuropharmacology. 2007;52:234–243. doi: 10.1016/j.neuropharm.2006.07.010. [DOI] [PubMed] [Google Scholar]
- Humeau Y, Shaban H, Bissière S, Lüthi A. Presynaptic induction of heterosynaptic associative plasticity in the mammalian brain. Nature. 2003;426:841–845. doi: 10.1038/nature02194. [DOI] [PubMed] [Google Scholar]
- Johnson JW, Ascher P. Voltage-dependent block by intracellular Mg2+ of N-methyl-d-aspartate-activated channels. Biophys J. 1990;57:1085–1090. doi: 10.1016/S0006-3495(90)82626-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung SC, Hoffman DA. Biphasic somatic A-type K channel downregulation mediates intrinsic plasticity in hippocampal CA1 pyramidal neurons. PLoS One. 2009;4:e6549. doi: 10.1371/journal.pone.0006549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Jung SC, Clemens AM, Petralia RS, Hoffman DA. Regulation of dendritic excitability by activity-dependent trafficking of the A-type K+ channel subunit Kv4.2 in hippocampal neurons. Neuron. 2007;54:933–947. doi: 10.1016/j.neuron.2007.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King C, Henze DA, Leinekugel X, Buzsáki G. Hebbian modification of a hippocampal population pattern in the rat. J Physiol. 1999;521:159–167. doi: 10.1111/j.1469-7793.1999.00159.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang EJ, Paré D. Similar inhibitory processes dominate the responses of cat lateral amygdaloid projection neurons to their various afferents. J Neurophysiol. 1997;77:341–352. doi: 10.1152/jn.1997.77.1.341. [DOI] [PubMed] [Google Scholar]
- Langton JM, Kim JH, Nicholas J, Richardson R. The effect of the NMDA receptor antagonist MK-801 on the acquisition and extinction of learned fear in the developing rat. Learn Mem. 2007;14:665–668. doi: 10.1101/lm.692407. [DOI] [PubMed] [Google Scholar]
- LeDoux JE. Emotional memory systems in the brain. Behav Brain Res. 1993;58:69–79. doi: 10.1016/0166-4328(93)90091-4. [DOI] [PubMed] [Google Scholar]
- Lee AK, Epsztein J, Brecht M. Head-anchored whole-cell recordings in freely moving rats. Nat Protoc. 2009;4:385–392. doi: 10.1038/nprot.2009.5. [DOI] [PubMed] [Google Scholar]
- LeMasson G, Marder E, Abbott LF. Activity-dependent regulation of conductances in model neurons. Science. 1993;259:1915–1917. doi: 10.1126/science.8456317. [DOI] [PubMed] [Google Scholar]
- Lorenzetti FD, Mozzachiodi R, Baxter DA, Byrne JH. Classical and operant conditioning differentially modify the intrinsic properties of an identified neuron. Nat Neurosci. 2006;9:17–19. doi: 10.1038/nn1593. [DOI] [PubMed] [Google Scholar]
- Mahon S, Casassus G, Mulle C, Charpier S. Spike-dependent intrinsic plasticity increases firing probability in rat striatal neurons in vivo. J Physiol. 2003;550:947–959. doi: 10.1113/jphysiol.2003.043125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marder E, Goaillard JM. Variability, compensation and homeostasis in neuron and network function. Nat Rev Neurosci. 2006;7:563–574. doi: 10.1038/nrn1949. [DOI] [PubMed] [Google Scholar]
- Maren S. Long-term potentiation in the amygdala: a mechanism for emotional learning and memory. Trends Neurosci. 1999;22:561–567. doi: 10.1016/s0166-2236(99)01465-4. [DOI] [PubMed] [Google Scholar]
- Maren S. Auditory fear conditioning increases CS-elicited spike firing in lateral amygdala neurons even after extensive overtraining. Eur J Neurosci. 2000;12:4047–4054. doi: 10.1046/j.1460-9568.2000.00281.x. [DOI] [PubMed] [Google Scholar]
- Maren S. Synaptic mechanisms of associative memory in the amygdala. Neuron. 2005;47:783–786. doi: 10.1016/j.neuron.2005.08.009. [DOI] [PubMed] [Google Scholar]
- Martin SJ, Morris RG. New life in an old idea: the synaptic plasticity and memory hypothesis revisited. Hippocampus. 2002;12:609–636. doi: 10.1002/hipo.10107. [DOI] [PubMed] [Google Scholar]
- Mayer ML, Westbrook GL, Guthrie PB. Voltage-dependent block by Mg2+ of NMDA responses in spinal cord neurones. Nature. 1984;309:261–263. doi: 10.1038/309261a0. [DOI] [PubMed] [Google Scholar]
- McDonald AJ. Neurons of the lateral and basolateral amygdaloid nuclei: a Golgi study in the rat. J Comp Neurol. 1982;212:293–312. doi: 10.1002/cne.902120307. [DOI] [PubMed] [Google Scholar]
- McKinney BC, Murphy GG. The L-type voltage-gated calcium channel Cav1.3 mediates consolidation, but not extinction, of contextually conditioned fear in mice. Learn Mem. 2006;13:584–589. doi: 10.1101/lm.279006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narayanan R, Johnston D. Long-term potentiation in rat hippocampal neurons is accompanied by spatially widespread changes in intrinsic oscillatory dynamics and excitability. Neuron. 2007;56:1061–1075. doi: 10.1016/j.neuron.2007.10.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson AB, Krispel CM, Sekirnjak C, du Lac S. Long-lasting increases in intrinsic excitability triggered by inhibition. Neuron. 2003;40:609–620. doi: 10.1016/s0896-6273(03)00641-x. [DOI] [PubMed] [Google Scholar]
- Nishijo H, Uwano T, Tamura R, Ono T. Gustatory and multimodal neuronal responses in the amygdala during licking and discrimination of sensory stimuli in awake rats. J Neurophysiol. 1998;79:21–36. doi: 10.1152/jn.1998.79.1.21. [DOI] [PubMed] [Google Scholar]
- Nowak L, Bregestovski P, Ascher P, Herbet A, Prochiantz A. Magnesium gates glutamate-activated channels in mouse central neurones. Nature. 1984;307:462–465. doi: 10.1038/307462a0. [DOI] [PubMed] [Google Scholar]
- Oh MM, Kuo AG, Wu WW, Sametsky EA, Disterhoft JF. Watermaze learning enhances excitability of CA1 pyramidal neurons. J Neurophysiol. 2003;90:2171–2179. doi: 10.1152/jn.01177.2002. [DOI] [PubMed] [Google Scholar]
- Pape HC, Driesang RB. Ionic mechanisms of intrinsic oscillations in neurons of the basolateral amygdaloid complex. J Neurophysiol. 1998;79:217–226. doi: 10.1152/jn.1998.79.1.217. [DOI] [PubMed] [Google Scholar]
- Pape HC, Narayanan RT, Smid J, Stork O, Seidenbecher T. Theta activity in neurons and networks of the amygdala related to long-term fear memory. Hippocampus. 2005;15:874–880. doi: 10.1002/hipo.20120. [DOI] [PubMed] [Google Scholar]
- Paré D, Collins DR. Neuronal correlates of fear in the lateral amygdala: multiple extracellular recordings in conscious cats. J Neurosci. 2000;20:2701–2710. doi: 10.1523/JNEUROSCI.20-07-02701.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paz JT, Mahon S, Tiret P, Genet S, Delord B, Charpier S. Multiple forms of activity-dependent intrinsic plasticity in layer V cortical neurones in vivo. J Physiol. 2009;587:3189–3205. doi: 10.1113/jphysiol.2009.169334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelletier JG, Likhtik E, Filali M, Paré D. Lasting increases in basolateral amygdala activity after emotional arousal: implications for facilitated consolidation of emotional memories. Learn Mem. 2005;12:96–102. doi: 10.1101/lm.88605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poulet JF, Petersen CC. Internal brain state regulates membrane potential synchrony in barrel cortex of behaving mice. Nature. 2008;454:881–885. doi: 10.1038/nature07150. [DOI] [PubMed] [Google Scholar]
- Pratt KG, Aizenman CD. Homeostatic regulation of intrinsic excitability and synaptic transmission in a developing visual circuit. J Neurosci. 2007;27:8268–8277. doi: 10.1523/JNEUROSCI.1738-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quirk GJ, Armony JL, LeDoux JE. Fear conditioning enhances different temporal components of tone-evoked spike trains in auditory cortex and lateral amygdala. Neuron. 1997;19:613–624. doi: 10.1016/s0896-6273(00)80375-x. [DOI] [PubMed] [Google Scholar]
- Rogan MT, Stäubli UV, LeDoux JE. Fear conditioning induces associative long-term potentiation in the amygdala. Nature. 1997;390:604–607. doi: 10.1038/37601. [DOI] [PubMed] [Google Scholar]
- Rosenkranz JA, Grace AA. Cellular mechanisms of infralimbic and prelimbic prefrontal cortical inhibition and dopaminergic modulation of basolateral amygdala neurons in vivo. J Neurosci. 2002a;22:324–337. doi: 10.1523/JNEUROSCI.22-01-00324.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenkranz JA, Grace AA. Dopamine-mediated modulation of odour-evoked amygdala potentials during pavlovian conditioning. Nature. 2002b;417:282–287. doi: 10.1038/417282a. [DOI] [PubMed] [Google Scholar]
- Rudolph M, Pospischil M, Timofeev I, Destexhe A. Inhibition determines membrane potential dynamics and controls action potential generation in awake and sleeping cat cortex. J Neurosci. 2007;27:5280–5290. doi: 10.1523/JNEUROSCI.4652-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saar D, Grossman Y, Barkai E. Reduced after-hyperpolarization in rat piriform cortex pyramidal neurons is associated with increased learning capability during operant conditioning. Eur J Neurosci. 1998;10:1518–1523. doi: 10.1046/j.1460-9568.1998.00149.x. [DOI] [PubMed] [Google Scholar]
- Schreurs BG, Gusev PA, Tomsic D, Alkon DL, Shi T. Intracellular correlates of acquisition and long-term memory of classical conditioning in Purkinje cell dendrites in slices of rabbit cerebellar lobule HVI. J Neurosci. 1998;18:5498–5507. doi: 10.1523/JNEUROSCI.18-14-05498.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroeder BW, Shinnick-Gallagher P. Fear memories induce a switch in stimulus response and signaling mechanisms for long-term potentiation in the lateral amygdala. Eur J Neurosci. 2004;20:549–556. doi: 10.1111/j.1460-9568.2004.03517.x. [DOI] [PubMed] [Google Scholar]
- Shabel SJ, Janak PH. Substantial similarity in amygdala neuronal activity during conditioned appetitive and aversive emotional arousal. Proc Natl Acad Sci U S A. 2009;106:15031–15036. doi: 10.1073/pnas.0905580106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro ML, Eichenbaum H. Hippocampus as a memory map: synaptic plasticity and memory encoding by hippocampal neurons. Hippocampus. 1999;9:365–384. doi: 10.1002/(SICI)1098-1063(1999)9:4<365::AID-HIPO4>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- Sigurdsson T, Doyère V, Cain CK, LeDoux JE. Long-term potentiation in the amygdala: a cellular mechanism of fear learning and memory. Neuropharmacology. 2007;52:215–227. doi: 10.1016/j.neuropharm.2006.06.022. [DOI] [PubMed] [Google Scholar]
- Sourdet V, Russier M, Daoudal G, Ankri N, Debanne D. Long-term enhancement of neuronal excitability and temporal fidelity mediated by metabotropic glutamate receptor subtype 5. J Neurosci. 2003;23:10238–10248. doi: 10.1523/JNEUROSCI.23-32-10238.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steriade M, Timofeev I, Grenier F. Natural waking and sleep states: a view from inside neocortical neurons. J Neurophysiol. 2001;85:1969–1985. doi: 10.1152/jn.2001.85.5.1969. [DOI] [PubMed] [Google Scholar]
- Tsubokawa H, Offermanns S, Simon M, Kano M. Calcium-dependent persistent facilitation of spike backpropagation in the CA1 pyramidal neurons. J Neurosci. 2000;20:4878–4884. doi: 10.1523/JNEUROSCI.20-13-04878.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turrigiano GG, Nelson SB. Hebb and homeostasis in neuronal plasticity. Curr Opin Neurobiol. 2000;10:358–364. doi: 10.1016/s0959-4388(00)00091-x. [DOI] [PubMed] [Google Scholar]
- Turrigiano G, Abbott LF, Marder E. Activity-dependent changes in the intrinsic properties of cultured neurons. Science. 1994;264:974–977. doi: 10.1126/science.8178157. [DOI] [PubMed] [Google Scholar]
- Uwano T, Nishijo H, Ono T, Tamura R. Neuronal responsiveness to various sensory stimuli, and associative learning in the rat amygdala. Neuroscience. 1995;68:339–361. doi: 10.1016/0306-4522(95)00125-3. [DOI] [PubMed] [Google Scholar]
- van Welie I, van Hooft JA, Wadman WJ. Homeostatic scaling of neuronal excitability by synaptic modulation of somatic hyperpolarization-activated Ih channels. Proc Natl Acad Sci U S A. 2004;101:5123–5128. doi: 10.1073/pnas.0307711101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venable N, Kelly PH. Effects of NMDA receptor antagonists on passive avoidance learning and retrieval in rats and mice. Psychopharmacology. 1990;100:215–221. doi: 10.1007/BF02244409. [DOI] [PubMed] [Google Scholar]
- Weisskopf MG, Bauer EP, LeDoux JE. L-type voltage-gated calcium channels mediate NMDA-independent associative long-term potentiation at thalamic input synapses to the amygdala. J Neurosci. 1999;19:10512–10519. doi: 10.1523/JNEUROSCI.19-23-10512.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitlock JR, Heynen AJ, Shuler MG, Bear MF. Learning induces long-term potentiation in the hippocampus. Science. 2006;313:1093–1097. doi: 10.1126/science.1128134. [DOI] [PubMed] [Google Scholar]
- Xu J, Kang N, Jiang L, Nedergaard M, Kang J. Activity-dependent long-term potentiation of intrinsic excitability in hippocampal CA1 pyramidal neurons. J Neurosci. 2005;25:1750–1760. doi: 10.1523/JNEUROSCI.4217-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Shin JH, Linden DJ. Persistent changes in the intrinsic excitability of rat deep cerebellar nuclear neurones induced by EPSP or IPSP bursts. J Physiol. 2004;561:703–719. doi: 10.1113/jphysiol.2004.071696. [DOI] [PMC free article] [PubMed] [Google Scholar]