

Abstract
We have employed DFT protocols to calculate the NMR properties of the vannusals, a class of natural products whose structures have been the subject of recent investigations. The originally assigned structure of vannusal B was revised after a long synthetic journey which generated a series of closely related diastereomers. In this work we show how DFT calculations based on density functionals and basis sets designed for the prediction of NMR spectra (M06/pcS-2 level of theory) can be used to reproduce the observed parameters, thereby offering to the synthetic chemist a useful tool to discard or accept putative structures of unknown organic molecules.
Keywords: natural substances, structure determination, vannusals, DFT, NMR spectroscopy
Introduction
The domain of naturally occurring substances is a bottomless trove of intriguing molecules. Many of them are characterized not only by interesting biological activities (which is the reason why they are often chased) but, also from a more fundamental point of view, by their unusual structures. Indeed, the isolation of new molecules from natural extracts marks the starting point of structural investigations which generally culminate with a proposed structure. Such endeavors take advantage of a variety of spectroscopic methods, among which NMR spectroscopy plays an undisputed pivotal role. Thus, the isolated fractions are submitted to the large array of available experimental NMR methods, which ultimately (should) lead to stringent constraints on molecular structure and conformation. Nevertheless, even the wealth of information that can be so gathered may not be sufficient to arrive at an unambiguous structural proposal. It is not infrequent in the literature to see that an original structure is revised to one that better fits further spectroscopic data, or after total synthesis of the proposed structure has revealed that the spectra do not match.1
However, the total synthesis of a natural product can only be undertaken once its structure has been narrowed down to a limited manifold, owing to the substantial cost of such work.
It is then apparent that determining the structure of complex naturally occurring molecules involves a multifaceted investigation that exploits several resources and expertise, and often results in a long and winding path to the final target. Given such complexity, it is desirable to devise novel avenues that help to sort out candidate structures, especially at the spectroscopic stage.
Modern computational chemistry methods, especially DFT, have proven to be excellent tools for determining molecular structures. Recently, such capabilities have been broadened to span spectroscopic properties such as NMR chemical shifts and couplings,2-7 possibly aided by empirical methods.8 These developments have been widely exploited to determine molecular structures, including those of natural substances,9 with little if any recourse to empirical evidence; the latter strength is particularly critical whenever molecules with unusual or unprecedented constituents or connectivities are considered. Thus, high-level DFT calculations have aided the structure determination of arsenicin A10 and clarified issues on the structure of hexacyclinol,11,12 as well as helped in the structural revision of several other natural substances.13
However, such achievements have involved the comparison of constitutional isomers, which is just one of the issues at stake when complex organic molecules are involved. Indeed, most of their complexity arises from the possibility of many stereoisomers having very similar connectivities and magnetic environments of each nucleus. Goodman and co-workers have extensively investigated this issue, defining a novel statistical approach to select the correct diastereomer to be assigned to a single set of experimental NMR shifts, a common occurrence when dealing with natural substances.6b Not surprisingly, the issue of stereoisomerism is precisely the ground where many structural revisions have been undertaken. One such case is provided by the vannusals.
Vannusal B is a marine natural product that was isolated from the tropical interstitial ciliate Euplotes vannus. The originally assigned structure (structure 2-1 in Figure 1), a rather unusual molecular architecture, consisting of a C30 molecular framework, seven rings and thirteen stereogenic centers, was proposed on the basis of spectroscopic data, mainly NMR.14 In this study, we retrace the path that has, with time and effort, led to a revision of the originally proposed structure of vannusal B15 to the correct structure of this natural product.16 During this re-examination, we shall indicate the stages at which DFT calculations would have provided critical information to that effect.
Figure 1.

The eight diastereomeric structures of the vannusals investigated in Ref. 15.
Computational Section
All structures were optimized at the B3LYP/6-31G(d,p) level of theory, which we found to be adequate for organic molecules.3,9 NMR chemical shifts were calculated using the recently introduced hybrid M06 functional17 with the pcS-2 basis set, specifically designed for the calculations of NMR shielding constants.18 13C chemical shifts were calculated as δ = σref − σ, where σref is the shielding constant of TMS calculated at the same level of theory (σref = 176.225 ppm). For spin-spin coupling constants on model systems we used the hybrid B97-2 functional19 with the pcJ-2 basis set, specifically designed for the calculations of NMR scalar couplings20 for consistency with our previous studies.12 Test calculations to estimate long-range solvent effects on the NMR properties were conducted using the PCM model with methanol as a solvent. All optimizations were run using the software package Gaussian 0321 while NMR properties were calculated using Gaussian 09.22
We optimized only a single conformation for each one of the eight vannusals, where the hydroxyl groups were arranged so as to form intramolecular hydrogen bonds. It is unlikely that this conformation is highly populated in methanol solution, where hydroxyl groups will be involved in hydrogen bonding with the solvent; nevertheless, it is a consistent way to treat the molecules in the gas phase. It is expected that the OH orientations will only slightly affect 13C resonances.23 On the other hand, it would be impractical to account for the conformational population in methanol of the vannusals by computer simulation, since a force field having the necessary accuracy for such natural substances is not available.
The results have been statistically analyzed by linear regression of calculated shifts (δcalc) against experimental ones (δexp). The results were then evaluated in terms of the maximum absolute error MaxErr and the corrected mean absolute error (CMAE).4a,9 Both parameters are calculated with respect to the value predicted by the linear fit rather than to the experimental value, so as to avoid the possible bias introduced by a systematic error in the correlation, e.g. caused by an inaccurate evaluation of the reference shielding. Thus, MaxErr = max(|δcalc − δfit|) and CMAE = (Σi|δcalc − δfit|/n)/b, where δcalc is the calculated chemical shift, δfit is the chemical shift obtained from the linear fit: δfit = (δexp − a)/b, a and b are the intercept and the slope of the fitting line comprising n data points.
We have excluded from the correlations the resonances of olefinic and carbonyl carbons (C1, C2, C11, C12, C27, C31; see Supporting Information for full correlation graphs) which would flatten all statistical parameters by widening the range of chemical shifts up to about 200 ppm; focussing on a narrower range allows to highlight the differences in the region of interest for the comparison.
We also remark that the vannusals have conformational degrees of freedom in the hydroxyisopropyl, acetate, aldehyde and olefinic groups attached to the main carbon skeleton. Also important, as already mentioned, is the flexibility of the hydroxyl groups and their interaction with the protic solvent (methanol) in which the NMR spectra have been collected. We have neglected all these additional sources of variance in the calculated chemical shifts but, as we will see, the resulting 13C shifts are not significantly affected by these contributions.
Results and Discussion
Calibration of the computational protocol
Before discussing in detail the results obtained for vannusals, we present the results of a calibration of the computational protocol used (see Computational Section). To this end we have selected strychnine as a test case since it is a rather rigid molecule, with several functional groups, and for which accurate NMR properties were recently calculated.9 In Figure 2 we compare the results obtained for 13C chemical shifts of strychnine using the previously tested protocol (B3LYP/cc-pVTZ) against the new one used here (M06/pcS-2).
Figure 2.
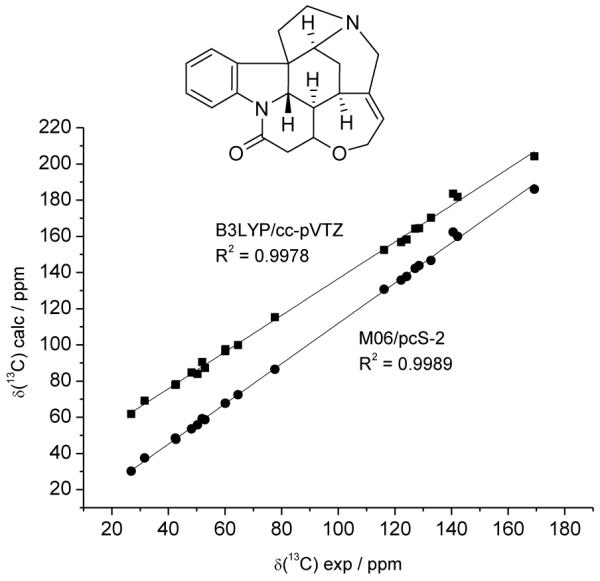
Correlation between calculated and experimental 13C chemical shifts of strychnine. Calculated data with the B3LYP/cc-pVTZ protocol are displaced by 30 ppm along the y axis for clarity. Fitting parameters of δcalc = a + bδexp are: a = 5.3 ppm, b = 1.0127 (B3LYP/cc-pVTZ); a = 0.7 ppm, b = 1.1117 (M06/pcS-2). The structure of strychnine is shown in the inset.
Clearly, using the newest functional and basis set increases the quality of the correlation. It is important to note that this excellent agreement is also a result of some advantageous occurrences. First, strychnine is fairly rigid and thus no conformational averaging of chemical shifts takes place. Second, the molecule is relatively nonpolar and the experimental data are collected in chloroform. Thus, external perturbations which affect the chemical shift to some degree are excluded and this allows to better appreciate the performance of the various protocols. Therefore, we expect the agreement for vannusals (which are more flexible and possess many hydroxyl groups involved in hydrogen bonds with methanol) to be lower.9,23
13C NMR chemical shifts of vannusals
The structures of the previously synthesized vannusals15,16 are shown in Figure 1. The original structure (2-1) proposed by Guella and co-workers14 features a carbon skeleton where an ethylene bridge (C14-C17) is arranged on the top side of the molecule. This arrangement is common to the four structures n-1 displayed on top of Figure 1. The four molecules differ in the stereochemistry of C21 and C25: (S,S), (R,S), (R,R) and (S,R) for 2-1, 3-1, 4-1 and 5-1, respectively. The other four structures at the bottom of Figure 1 (n-2), are epimeric to the corresponding top structures at the carbons indicated. These involve the whole “northeast” region of the molecule. The “southwest” region of the molecule has, instead, the same configuration for all compounds. Thus, the four bottom structures in Figure 1 can be viewed as the “northeast” enantiomers of the four top structures.
The reassignment of the structure of vannusal B from the originally proposed architecture 2-1 to the correct structure 5-2 thus followed a two-station path: firstly it was necessary to realize that the true structure of vannusal B is a “northeast” enantiomer of the originally proposed molecule; then it had to be determined which of the four possible configurations at C21 and C25 was the correct one.
In Figure 3 we show the correlation between calculated 13C NMR chemical shifts of stereoisomer 2-1 (the originally proposed structure) with the experimental values of natural vannusal B (5-2) and the experimental values of the same compound (2-1).
Figure 3.

Calculated 13C chemical shifts of the originally proposed structure 2-1 plotted against (a) experimental values of natural vannusal B, 5-2; (b) experimental values of structure 2-1. The data point for C21 (a major outlier) is labelled.
This presentation showcases the virtue of the correlation when a given putative structure is compared with the experimental values of the natural substance and underscores the performance of the DFT protocol when the correct experimental values are used. The bottom panel of Figure 3 can then be used as a sort of reference for the predictive power of the computational protocol. The calculated values are in rather good agreement with the experimental values of 2-1, highlighting the reliability of the protocol for this type of molecules even when solvent and conformational effects are neglected.
In Table 1 we report the statistical parameters for all correlations presented. For structure 2-1 the R2 coefficient is close to one and both the maximum absolute error (MaxErr) and the corrected mean absolute error (CMAE) are small. If the correlation is done using the experimental values of vannusal B (Figure 3a) all statistical parameters drop substantially; in particular C21 is largely in error and lies off its correlation line by more than 16 ppm. Had DFT-NMR shifts been available when the assignments were done, this observation would have suggested that the proposed structure was questionable.
Table 1.
Statistical parameters for the correlations of 13C chemical shifts.
| Calc. vs Expt. | a | b | R2 | MaxErr | CMAE |
|---|---|---|---|---|---|
| 2-1 vs 5-2 | 0.15 | 1.0983 | 0.9580 | 16.2 | 2.5 |
| 2-1 vs 2-1 | 1.03 | 1.0934 | 0.9877 | 4.5 | 1.7 |
|
| |||||
| 2-2 vs 5-2 | 0.06 | 1.1104 | 0.9662 | 11.9 | 2.3 |
| 2-2 vs 2-2 | 0.84 | 1.1087 | 0.9819 | 8.0 | 2.0 |
|
| |||||
| 3-1 vs 5-2 | −1.00 | 1.1331 | 0.9499 | 11.2 | 3.2 |
| 3-1 vs 3-1 | 0.66 | 1.0933 | 0.9846 | 6.0 | 1.8 |
|
| |||||
| 3-2 vs 5-2 | −0.90 | 1.1066 | 0.9631 | 14.4 | 2.4 |
| 3-2 vs 3-2 | 0.52 | 1.0722 | 0.9852 | 7.9 | 1.7 |
|
| |||||
| 4-1 vs 5-2 | 0.42 | 1.0895 | 0.9670 | 11.7 | 2.4 |
| 4-1 vs 4-1 | 0.79 | 1.0845 | 0.9830 | 10.0 | 1.7 |
|
| |||||
| 4-2 vs 5-2 | 0.04 | 1.1012 | 0.9802 | 8.9 | 1.8 |
| 4-2 vs 4-2 | 0.24 | 1.1007 | 0.9847 | 9.3 | 1.5 |
|
| |||||
| 5-1 vs 5-2 | 1.48 | 1.0737 | 0.9927 | 3.7 | 1.3 |
| 5-1 vs 5-1 | 1.89 | 1.0644 | 0.9948 | 3.3 | 1.0 |
|
| |||||
| 5-2 vs 5-2 | 0.26 | 1.0898 | 0.9948 | 3.0 | 1.1 |
a and b are the intercept and slope of the linear fitting line, respectively, and R2 its correlation coefficient. MaxErr is the maximum absolute error with respect to the linear fit. CMAE is the corrected mean absolute error (see text for the definition of both parameters).
The graphs of the correlations for the other structures are reported in the Supporting Information; it suffices here to analyze the statistical parameters of Table 1. Intercepts and slopes of the fitting lines are very similar to what is generally observed for 13C chemical shift correlations using DFT protocols.4-9 The R2 coefficient is quite instructive: when the correlation is done with respect to the experimental values of that particular structure R2 is well above 0.98, confirming the generally good performance of the DFT protocol. In contrast, when the calculated values are correlated with the experimental values of vannusal B the quality of the correlation is much lower (except, obviously, for 5-2 which is the true structure of vannusal B), indicating that the computational protocol is capable of distinguishing among the different vannusal structures. In fact, R2 rises systematically as we move from the originally proposed structure 2-1 to the correct structure 5-2, following the same path that was walked through during the quest for the true structure of vannusal B, i.e. 2-1, 2-2, 3-1, 3-2, 4-1, 4-2, 5-1 and 5-2, gathering new intelligence as a new diastereomeric structure was synthesized and its NMR spectra were compared with those of the natural substance. Similarly, MaxErr and CMAE decrease, confirming that the synthetic route followed was converging toward the correct assignment.
Finally, in Figure 4 we show the correlation between experimental and calculated 13C chemical shifts obtained for the true structure of vannusal B. The statistical parameters, reported in Table 1 (last entry), are very good, particularly MaxErr and CMAE which are the best of all.
Figure 4.
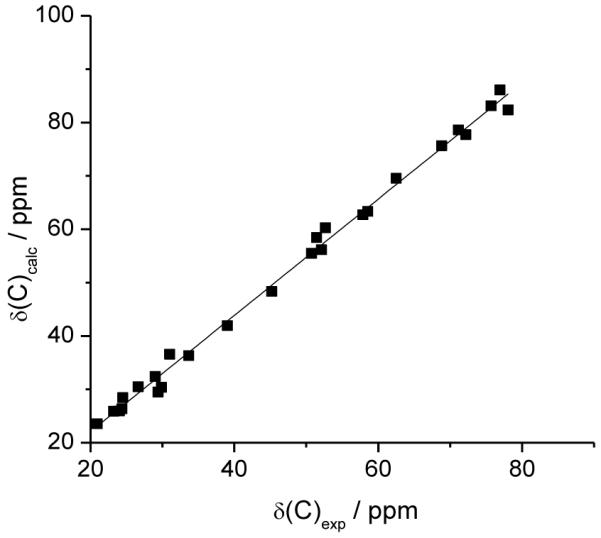
Correlation between experimental and calculated 13C chemical shifts of vannusal B, 5-2.
We note that also the “northeast” enantiomer of vannusal B (5-1) has a very good correlation with the experimental values of the natural substance. Thus, it would have been rather difficult to distinguish between 5-1 and 5-2 based only on 13C chemical shifts.
As a final test we have estimated the long-range solvent effects on the carbon shielding repeating the calculations for vannusal B (5-2) but including the self-consistent solvent reaction field of methanol by means of the PCM method. Calculated shieldings were hardly distinguishable from the results obtained in the gas phase, thus confirming the weak dependence of carbon resonances on dielectric polarization9 (see results in the Supporting Information).
Vicinal coupling constants in models of the “northeast” region of vannusal B
In the experimental revision of the structure of vannusal B, an important role was played by the analysis of 3J(H,H) coupling constants, particularly for the couplings between H6 and H7 and between H21 and H25. For the first pair, the experimental value15a of 3J(H6,H7) (10.0 Hz) was in agreement with a trans arrangement of the two protons. This observation, together with others, confirmed the correct assignment of the “southwest” region of vannusal B. In contrast, the experimental coupling 3J(H21,H25) of vannusal B (2.0 Hz) was somewhat too small for the proposed configuration in 2-1, where both protons are on the same side and the dihedral angle is expected to be close to 0°, corresponding to a maximum in the Karplus curve. However, a simple Karplus approach may be questionable for vicinal couplings in a cyclic system with several substituents.4c Thus, Nicolaou and co-workers embarked on the synthesis of the four model systems of the “northeast” region, displayed in Figure 5.
Figure 5.

Model systems of the “northeast” region of vannusals. TIPS, triisopropylsilyl; Bz, benzyl. For the sake of comparison, numbering is the same as in vannusals. Configuration labels refer to C21 and C25.
These diastereoisomers correspond to the four possible configurations of C21 and C25: (S,S), (R,S), (R,R) and (S,R). It is straightforward to discard the (R,S) and (R,R) configurations since the experimental values of 3J(H21,H25) (3-10 Hz) are too large compared with that of vannusal B (2.0 Hz).14a The measured couplings (Table 2) suggest that natural vannusal B has the same relative configuration as the (S,R) model system. The vicinal coupling in model (S,S), corresponding to the originally proposed structure (2-1), must also be rather small, the signal appearing as a singlet. Hence, this configuration could not be discarded based only on the determination of the 3J(H21,H25) coupling constant.
Table 2.
| model | Expt. | Calc. | φ b |
|---|---|---|---|
| S,S | < 1 | 3.5 | 49.9 ° |
| R,S | 10.0 | 6.4 | 142.0° |
| R,R | 3.5 | 5.5 | 22.4° |
| S,R | 1.0 | 1.8 | 122.2° |
The same coupling in vannusal B is 2.0 Hz.14a
H21–C21–C25–H25 dihedral angle of the optimized model system.
Proton-proton couplings are more sensitive than 13C shifts to conformational equilibria of the five-membered ring; relatively small changes in the geometry may alter significantly the dihedral angle between the two coupled nuclei, resulting in large variations in the coupling constant. A proper account of all conformations of the five-membered ring would be required in order to attain a close agreement between calculations and experiments,24,25 but this is beyond the scope of the present investigation. Moreover, the hydroxyl groups of the ring or next to it are close to each other, so that their hydrogen bond network, and in turn the five-membered ring conformation, will be strongly influenced by the protic solvent. For these reasons we expect the agreement between calculations and experiments to be, at best, semi-quantitative.
The calculated couplings for models (S,S) and (S,R) are the smallest in the series (2-3 Hz), while larger values of ca. 6 Hz are obtained for the other two models, (R,S) and (R,R), and are in fair agreement with experimental values. Therefore, a preliminary calculation of that coupling constant would have at least correctly suggested that the configurations with a relatively large coupling constant should have been discarded.
Conclusions
DFT calculations can predict 13C NMR chemical shifts to a degree of accuracy that has enabled researchers to sort out many issues in the structural elucidation of complex organic molecules such as natural products. In most cases the issues revolved around constitutional isomers differing in their connectivity – i.e., situations where nuclear spins would experience a different magnetic environment and associated chemical shift differences. A more challenging situation may arise when the comparison concerns stereoisomers having very similar connectivities, with diversity arising from different configurations at stereocenters and possibly leading to small relative shifts in each stereoisomer. Moreover, despite being more sensitive to conformational degrees of freedom which, if not properly accounted for, may diminish the agreement with experimental values, calculated 1H-1H couplings often provide insightful clues concerning the structure of the molecule under investigation.
The case history of vannusal B has proven very informative in this respect. The availability of experimental NMR spectra of all its stereoisomers, independently synthesized, has allowed us to carry out a direct comparison with 13C NMR shifts and J(1H,1H) coupling constants predicted from DFT calculations without any empirical assumption. In this study we have shown that all stereoisomers show subtle, but significant differences in their 13C NMR spectra that can be exploited for their structural assignment. Indeed, the structural revision of the originally assigned structure of vannusal B could have been greatly aided and simplified by a prior knowledge of the relevant NMR parameters, highlighting viable targets and, thereby, allowing synthetic efforts to be concentrated on the most likely structures.
Supplementary Material
Acknowledgment
We gratefully acknowledge CNR for financial support through a Short Term Mobility Grant (STM2010) to GS. We acknowledge the CINECA Award No. HP10CVNM0F, 2010 for the availability of high performance computing resources and support and the Laboratorio Interdipartimentale di Chimica Computazionale (LICC) at the Department of Chemistry of the University of Padova. KCN, AO and HZ thank Dr. D. H. Huang for NMR spectroscopic assistance. Financial support for the work in the Nicolaou laboratories was provided by the Skaggs Institute for Research, the National Science Foundation (NSF fellowship to AO), and the National Institutes of Health (USA) (grants GM063752 and CA100101).
Footnotes
Supporting Information Available. Complete refs 21 and 22, table of calculated data, Cartesian coordinates of optimized geometries, correlation of experimental vs calculated data for all systems investigated. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- (1).Nicolaou KC, Snyder SA. Angew. Chem. Int. Ed. 2005;44:1012–1044. doi: 10.1002/anie.200460864. [DOI] [PubMed] [Google Scholar]
- (2).Kaupp M, Bühl M, Malkin VG, editors. Calculation of NMR and EPR Parameters. Wiley-VCH; Weinheim: 2004. [Google Scholar]
- (3).Bagno A, Saielli G. Theor. Chem. Acc. 2007;117:603–619. [Google Scholar]
- (4).(a) Cimino P, Gomez-Paloma L, Duca D, Riccio R, Bifulco G. Magn. Reson. Chem. 2004;42:S26–S33. doi: 10.1002/mrc.1410. [DOI] [PubMed] [Google Scholar]; (b) Di Micco S, Chini MG, Riccio R, Bifulco G. Eur. J. Org. Chem. 2010:1411–1434. [Google Scholar]; (c) Palermo G, Riccio R, Bifulco G. J. Org. Chem. 2010;75:1982–1991. doi: 10.1021/jo902704u. [DOI] [PubMed] [Google Scholar]; (d) Bifulco G, Dambruoso P, Gomez-Paloma L, Riccio R. Chem. Rev. 2007;107:3744–3779. doi: 10.1021/cr030733c. [DOI] [PubMed] [Google Scholar]
- (5).Sarotti AM, Pellegrinet SC. J. Org. Chem. 2009;74:7254–7260. doi: 10.1021/jo901234h. [DOI] [PubMed] [Google Scholar]
- (6).(a) Smith SG, Goodman JM. J. Org. Chem. 2009;74:4597–4607. doi: 10.1021/jo900408d. [DOI] [PubMed] [Google Scholar]; (b) Smith SG, Goodman JM. J. Am. Chem. Soc. 2010;132:12946–12959. doi: 10.1021/ja105035r. [DOI] [PubMed] [Google Scholar]; (c) Smith SG, Paton RS, Burton JW, Goodman JM. J. Org. Chem. 2008;73:4053–4062. doi: 10.1021/jo8003138. [DOI] [PubMed] [Google Scholar]
- (7).(a) Forsyth DA, Sebag AB. J. Am. Chem. Soc. 1997;119:9483–9494. [Google Scholar]; (b) Sebag AB, Forsyth DA, Plante MA. J. Org. Chem. 2001;66:7967–7973. doi: 10.1021/jo001720r. [DOI] [PubMed] [Google Scholar]
- (8).(a) Elyashberg M, Blinov K, Smurnyy Y, Churanova T, Williams A. Magn. Reson. Chem. 2010;48:219–229. doi: 10.1002/mrc.2571. [DOI] [PubMed] [Google Scholar]; (b) Elyashberg ME, Williams AJ, Martin GE. Prog. NMR Spectrosc. 2008;53:1–104. [Google Scholar]; (c) Stappen I, Buchbauer G, Robien W, Wolschann P. Magn. Reson. Chem. 2009;47:720–726. doi: 10.1002/mrc.2452. [DOI] [PubMed] [Google Scholar]
- (9).Bagno A, Saielli G, Rastrelli F. Chem. Eur. J. 2006;12:5514–5525. doi: 10.1002/chem.200501583. [DOI] [PubMed] [Google Scholar]
- (10).Tähtinen P, Saielli G, Guella G, Mancini I, Bagno A. Chem. Eur. J. 2008;14:10445–10452. doi: 10.1002/chem.200801272. [DOI] [PubMed] [Google Scholar]
- (11).Rychnovsky SD. Org. Lett. 2006;8:2895–2898. doi: 10.1021/ol0611346. [DOI] [PubMed] [Google Scholar]
- (12).Bagno A, Saielli G. Org. Lett. 2009;11:1409–1412. doi: 10.1021/ol900164a. [DOI] [PubMed] [Google Scholar]
- (13).(a) Timmons C, Wipf P. J. Org. Chem. 2008;73:9168–9170. doi: 10.1021/jo801735e. [DOI] [PubMed] [Google Scholar]; (b) Wipf P, Kerekes D. J. Nat. Prod. 2003;66:716–718. doi: 10.1021/np0300277. [DOI] [PubMed] [Google Scholar]; (c) White KN, Amagata T, Oliver AG, Tenney K, Wenzel PJ, Crews P. J. Org. Chem. 2008;73:8719–8722. doi: 10.1021/jo800960w. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Yang J, Huang S-X, Zhao Q-S. J. Phys. Chem. A. 2008;112:12132–12139. doi: 10.1021/jp8072415. [DOI] [PubMed] [Google Scholar]; (e) Braddock DC, Rzepa HS. J. Nat. Prod. 2008;71:728–730. doi: 10.1021/np0705918. [DOI] [PubMed] [Google Scholar]
- (14).(a) Guella G, Dini F, Pietra F. Angew. Chem. Int. Ed. 1999;38:1134–1136. doi: 10.1002/(SICI)1521-3773(19990419)38:8<1134::AID-ANIE1134>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]; (b) Guella G, Callone E, Di Giuseppe G, Frassanito R, Frontini FP, Mancini I, Dini F. Eur. J. Org. Chem. 2007:5226–5234. [Google Scholar]
- (15).(a) Nicolaou KC, Zhang H, Ortiz A, Dagneau P. Angew. Chem. Int. Ed. 2008;47:8605–8610. doi: 10.1002/anie.200804228. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Nicolaou KC, Zhang H, Ortiz A. Angew. Chem. Int. Ed. 2009;48:5642–5647. doi: 10.1002/anie.200902028. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Nicolaou KC, Zhang H, Ortiz A. Angew. Chem. Int. Ed. 2009;48:5648–5652. doi: 10.1002/anie.200902029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).(a) Nicolaou KC, Ortiz A, Zhang H, Dagneau P, Lanver A, Jennings MP, Arseniyadis S, Faraoni R, Lizos DE. J. Am. Chem. Soc. 2010;132:7138–7152. doi: 10.1021/ja100740t. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Nicolaou KC, Ortiz A, Zhang H, Guella G. J. Am. Chem. Soc. 2010;132:7153–7176. doi: 10.1021/ja100742b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Truhlar DG, Zhao Y. Theor. Chem. Acc. 2008;120:215–241. [Google Scholar]
- (18).Jensen F. J. Chem. Theory Comp. 2008;4:719–727. doi: 10.1021/ct800013z. [DOI] [PubMed] [Google Scholar]
- (19).Wilson PJ, Bradley TJ, Tozer DJ. J. Chem. Phys. 2001;115:9233–9242. [Google Scholar]
- (20).Jensen F. J. Chem. Theory Comput. 2006;2:1360–1369. doi: 10.1021/ct600166u. [DOI] [PubMed] [Google Scholar]
- (21).Frisch MJ, et al. Gaussian 03, Revision D.02. Gaussian, Inc.; Wallingford CT: 2004. [Google Scholar]
- (22).Frisch MJ, et al. Gaussian 09, Revision B.01. Gaussian, Inc.; Wallingford CT: 2009. [Google Scholar]
- (23).Bagno A, Rastrelli F, Saielli G. J. Org. Chem. 2007;72:7373–7381. doi: 10.1021/jo071129v. [DOI] [PubMed] [Google Scholar]
- (24).Wu A, Cremer D. Int. J.Mol. Sci. 2003;4:158–192. [Google Scholar]
- (25).Casella G, Ferrante F, Saielli G. Org. Biomol. Chem. 2010;8:2711–2718. doi: 10.1039/c000679c. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.