

Abstract
Kindlin-2, a widely distributed cytoskeletal protein, has been implicated in integrin activation, and its absence is embryonically lethal in mice and causes severe developmental defects in zebrafish. Knockdown of kindlin-2 levels in endothelial cells resulted in defective adhesive and migratory responses, suggesting that angiogenesis might be aberrant even with partial reduction of kindlin-2. This hypothesis has now been tested in the kindlin-2+/− mice. RM1 prostate tumors grown in kindlin-2+/− mice had fewer blood vessels, which were thinner and shorter and supported less tumor growth compared with wild-type littermates. The vessels that did form in the kindlin-2+/− mice lacked smooth muscle cells and pericytes and had thinner basement membranes, indicative of immature vessels. VEGF-induced angiogenesis in matrigel implants was also abnormal in the kindlin-2+/− mice. Vessels in the kindlin-2+/− mice were leaky, and BM transplantation from kindlin-2+/− to WT mice did not correct this defect. Endothelial cells derived from kindlin-2+/− mice had integrin expression levels similar to WT mice but reduced αVβ3-dependent signaling, migration, adhesion, spreading, and tube formation. Developmental angiogenesis was markedly impaired by kindlin-2 morpholinos in zebrafish. Taken together, kindlin-2 plays an important role in pathologic and developmental angiogenesis, which arises from defective activation of integrin αVβ3.
Introduction
Although endothelial cells (ECs) are the primary cell type that forms blood vessel tubes, other cell types, including BM-derived cells, smooth muscle cells, and pericytes, are all engaged in forming blood-bearing conduits. Growth factor–growth factor receptor interactions are involved in initial recruitment of these cells while other ligand-receptor systems govern migration of the responding cells into angiogenic tissues (reviewed in Folkman1). Among the cellular receptors that specialize in regulating the adhesive and migratory responses of cells during angiogenesis are members of the integrin family.2 Integrins of the β1 and β3 subfamilies on ECs have been implicated in angiogenesis in vivo through knockout3 and inhibitor approaches4 and integrin αVβ3 and α5β1 are targets of antiangiogenic drugs for cancer treatment (reviewed in Avraamides et al5).
The regulation of integrin function in angiogenesis as well as many other responses depends on their ability to undergo activation, a rapid transition from a low- to a high-affinity state for recognition of their ligands. Particularly relevant to angiogenesis is the ability of VEGF to activate integrin αVβ3 and α5β1 by engaging one of its EC receptors, VEGFR2.6 Indeed, VEGFR2 and integrin αVβ3 can form a physical complex in EC and influence the functions of each other.6,7
Two sets of cytoskeletal proteins, the talins and the kindlins, bind to the β cytoplasmic tails and are now known to be involved in integrin activation.8–10 Talin has long been known to be critical for integrin activation11 and more recent studies have shown an obligatory requirement for the kindlins in integrin activation in the context of intact cells and whole organisms.12–16 Three kindlin family members are found in vertebrates, are quite homologous, and are sometimes expressed within the same cell type.9,10,17 However, the kindlins cannot compensate for one another; deficiencies of each kindlin in mice and/or humans gives rise to a distinct phenotype.9,18 Deficiencies of kindlin-1 give rise to similar phenotypes in humans and mice,19,20 and mutations in the Kindlin-3 gene in humans can give rise to symptoms ranging from bleeding, to frequent infections and osteopetrosis, resulting from an inability to activate integrins on blood and BM cells.15,16
Kindlin-2 is the most widely distributed kindlin family member. Inactivation of the Kindlin-2 gene in mice is peri-implantation lethal, because of failed endodermal and epiblast attachment.12,21 In vitro experiments performed with cells derived from kindlin-2 deficient mice12 or siRNA knockdown demonstrated defects in integrin activation despite the presence of talin.13 One cell type in which kindlin-2 knockdown had a profound functional effect was ECs, where knockdown of kindlin-2 led to a defect in adhesion and migration on β3 but not β1 integrin ligands.13,17 These findings suggested that kindlin-2 might regulate angiogenesis. To pursue this hypothesis in vivo, we have tested the effects of down-regulation of Kindlin-2 gene expression in mice and zebrafish. The results establish that even partial inactivation of the Kindlin-2 gene has a profound effect on the formation and function of new blood vessels and assign a previously unrecognized function to kindlin-2.
Methods
Mice
The development of kindlin-2+/− mice was previously described,12,21,22 and 8- to 12-week-old female and male mice were used in the experiments described under animal protocols approved by the Lerner Research Institute Institutional Animal Care and Use Committee.
Tumor angiogenesis
Male mice were injected subcutaneously with 1.5 × 106 RM1 prostate cancer cells. Tumors were collected on day 10, weighed, and processed for immunohistochemical staining with Abs specified in supplemental Methods (available on the Blood Web site; see the Supplemental Materials link at the top of the online article). Stained sections were analyzed using fluorescent or bright-field imaging microscopy (Leica) and ImagePro Plus Capture and Analysis software (Media Cybernetics). Angiogenesis was evaluated using Matrigel plug assay as described in supplemental Methods.
Blood vessel permeability
After intravenous injection of Evans blue dye (100 μL; Sigma-Aldrich) mustard oil was applied to the dorsal and ventral surfaces of one ear, while the other ear served as the control. Five minutes after Evans blue injection, 10 μL of VEGF (100 ng; R&D Systems) or PBS was injected intradermally at adjacent locations in the flanks of the mice. After 30 minutes, skin samples of similar size were removed, weighed, photographed, and Evans blue was extracted with 1 mL of formamide overnight at 56°C with constant shaking. The amount of extracted dye was measured spectrophotometrically at 610 nm.
WT and kindlin-2+/− aortic ECs
Mouse aortic ECs were isolated from aortas of WT and kindlin-2+/− mice as described.23 In adhesion assays, the cells were allowed to adhere to fibrinogen (10 μg/mL for coating), vitronectin (2.5 μg/mL), fibronectin (10 μg/mL), or collagen (10 μg/mL) for 45 minutes at 37°C in serum-free DMEM F-12 medium. EC migration was assessed in serum-free DMEM F-12 medium using Boyden chambers with 8 μm-pore filters (Corning) with immobilized ligands on their lower surface at the concentrations used in adhesion assays, and VEGF (500 ng/mL) in the lower chamber. ECs (2 × 105) were added to the upper chamber and incubated for 6 hours at 37°C in 5% CO2. The number of adherent or migrated cells was quantified using the Cyquant Cell Proliferation Assay Kit (Invitrogen). For spreading assays, the cells adherent to integrin ligands for 1 hour at 37°C were fixed with 4% paraformaldehyde and stained with Alexa 568–phalloidin. Cell area was analyzed with ImagePro Plus Capture and Analysis software (Media Cybernetics). Tube formation by WT and kindlin-2+/− ECs was compared as described in supplemental Methods.
To assess integrin activation, ECs were incubated with selected concentrations of soluble Alexa 488–labeled fibrinogen (Fg) and FITC-labeled collagen Iin HBSS buffer containing 0.1% BSA, 0.5mM CaCl2, MgCl2 for 1 hour at 37°C. To activate the integrins, the cells were pretreated with VEGF (50 or 500 ng/mL) or PMA (100nM) for 20 minutes at 37°C followed by addition of the ligands. Ligand binding was analyzed using a FACSCalibur flow cytometer and CellQuest software (BD Biosciences).
In signaling experiments, equal numbers of ECs were treated with VEGF (0-2 μg/mL), placenta growth factor (PLGF; 0-4 μg/mL) or PMA (0-100nM) in serum-free DMEM F-12 for 10 minutes at 37°C and lysed in Laemmli sample-loading buffer containing 1 tablet of phosphatase inhibitor PhosStop and protease inhibitor cocktail (Roche). To analyze integrin-dependent signaling, the cells were pretreated with VEGF (1 μg/mL) for 15 minutes and allowed to adhere to BSA, Fg, or fibronectin (Fn) (100 μg/mL) for 15 minutes at 37°C. Adherent cells were lysed in 100 μL of ice-cold RIPA buffer. Cell lysates were analyzed on Western blots using anti-phospho-ERK1/2, phospho-p90RSK. and anti-total ERK 1/2 Abs (Cell Signaling Technology).
Angiogenesis in zebrafish
AB strain zebrafish embryos were collected immediately after spawning and injected at the 1-2 cell stage with 4.6 nL of morpholino. Control and kindlin-2 morpholinos (GeneTools LLC), described previously,21 were used at a concentration of 0.1mM. The kindlin-3 morpholino is a splice blocking morpholino designed to exon 6 and used at 0.1mM and 0.2mM (sequence = TGCACCTCAAACACTTCTTACCTTAG). Embryos were examined by live microscopy using a Nikon AZ-100 at 24 hours postfertilization (hpf) and 48 hpf. Images from 48 hpf embryos were collected and quantitated using Nikon NIS Elements software in combination with GraphPad Prism 3. For VEGF inducer treatment, embryos were dechorionated at 6 hpf and incubated in GS4012 (Calbiochem) at 5 and 10 μg/mL.24 Angiogenesis of treated embryos was examined at 48 hpf.
Statistical analyses
All data from in vivo and ex vivo assays are presented as means ± SEM using SPSS software (Norusis SPSS Inc) for 2-sample t tests with a 2-tailed significance set at 0.05. Calculated P values are reported for each test.
Results
Abnormal angiogenesis in kindlin-2+/− mice
In prior studies, we reported that 50%-70% knockdown of kindlin-2 in ECs with siRNAs led to significant defects in adhesive and migratory responses of these cells in vitro.13 Based on these observations, we reasoned that kindlin-2+/− mice with partial reduction in kindlin-2 levels might provide an adult organism with postnatal defects in angiogenesis. Accordingly, we injected murine RM1 prostate tumor cells into the flanks of male WT and kindlin-2+/− littermates. RM1 tumors that develop in WT mice at day 10 are known to be highly vascularized, and tumor growth is heavily dependent on angiogenesis.25 Staining of tumor sections for ECs with CD31 showed significantly reduced vessel density and vascular area in tumors grown in kindlin-2+/− mice compared with WT mice (4200 ± 700 μm2 vs 15 700 ± 4000 μm2, P = .0186, n = 6; Figure 1A left and right panels). Blood vessels in tumors within the kindlin-2+/− mice were shorter and thinner than in WT mice (Figure 1A left panel). Consistent with the blunted angiogenic response in the kindlin-2+/− mice, the tumors were significantly smaller than those derived in WT mice: 151 ± 68 mg vs 382 ± 62 mg, P = .042, n = 7 (Figure 1B).
Figure 1.

Abnormal angiogenesis in kindlin-2+/− mice. (A) Representative images of tumor sections stained with CD31 Ab (brown; left panel). Scale bars, 200 μm (top panel), 50 μm (bottom panel). Image analysis shows decreased area of CD31-stained vasculature in tumors grown in kindlin-2+/− mice compared with WT mice (right panel). Data are representative of 3 independent experiments with 6 mice per group. (B) Average weight of prostate tumors grown in kindlin-2+/− mice is lower than in WT mice (left panel) and representative tumors are shown (right panel). Data are means ± SEM, (n = 7 mice per group) and are representative of 3 independent experiments. (C) Angiogenesis in Matrigel implants in the presence of VEGF. (Left panel) Hemoglobin content in Matrigel implants from kindlin-2+/− mice was higher compared with WT mice. (Right panel) Representative images of Matrigel implant sections stained with CD31 Ab (brown). Scale bars, 150 μm. All data are representative of 3 independent experiments including 6 mice per group. CD31-positive areas were quantified in 5-10 independent fields per tumor implant or Matrigel plug and the average area per field was determined from duplicate measurements of each of the fields analyzed.
In a second angiogenesis model, mice were injected subcutaneously with Matrigel or Matrigel + VEGF. After 8 days, there was little angiogenesis in the plugs in either genotype without VEGF. VEGF did induce an angiogenic response in both kindlin-2+/− mice and WT mice, but based on CD31 staining showed the vessels to be thinner and shorter in the kindlin-2+/− mice compared with WT mice (Figure 1C right panel). The VEGF-containing plugs from the kindlin-2+/− mice contained 3.5-fold more hemoglobin compared with WT mice: 4.7 ± 1.15 vs 1.12 ± 0.14, P = .0149, n = 5 (Figure 1C left panel) which seem juxtaposed to the suppression of tumor angiogenesis in the kindlin-2+/− mice. However, if the vessels formed in the kindlin-2+/− mice were impaired, the increased hemoglobin content of the plugs could be a result of blood leakage from ill-formed vessels. Hence, we examined the blood vessels from the kindlin-2+/− and WT mice in the tumor model for differences in maturation and functional integrity.
The neovasculature in kindlin-2+/− mice is immature and leaky
Smooth muscle cells, an indicator of vascular maturity, were reduced considerably in kindlin-2+/− mice as evaluated by smooth muscle cell actin (SMA; Figure 2A top panel) and desmin (Figure 2A bottom panel) staining. In WT mice, 30% of total CD31+ blood vessels stained for SMA, whereas only 8% of blood vessels formed in kindlin-2+/− mice expressed this indicator of maturation (P = .0012, n = 6; Figure 2B). Immunostaining for laminin, an independent indicator of vessel maturation, showed > 50% reduction basement membrane thickness of blood vessels in kindlin-2+/− mice (2.1 ± 0.15 μm) compared with WT mice (5 ± 0.5 μm, P = 0001, n = 6; Figure 2C-D). In addition, the majority of tumor-induced blood vessels in kindlin-2+/− mice were positive for CD105, a marker of newly formed blood vessels (Figure 2E). Costaining of tumor sections for CD31 and NG2 chondroitin sulfate proteoglycan, a marker of pericytes-mural cells that stabilize blood vessels, revealed decreased recruitment of these cells into angiogenic niches; pericytes interacting with blood vessels in tumors in the kindlin-2+/− mice were markedly reduced compared with WT mice (Figure 2F).
Figure 2.

Neovasculature in kindlin-2+/− mice is immature. Immunohistochemistry and image analyses of RM1 prostate tumors implanted into WT and kindlin-2+/− mice. (A) Costaining for SMA (green) and CD31 (red; top panel) and for desmin (green) and CD31 (red; bottom panel) in tumors from WT and kindlin-2+/− mice. Nuclei are stained with DAPI. Arrowheads indicate SMA-positive vascular structures. Scale bars, 150 μm (top panel) and 50 μm (bottom panel). (B) Quantification of the data presented in the bottom panel of panel A. (C) Laminin-stained blood vessels in tumors grown in WT and kindlin-2+/− mice. Scale bars, 19 μm. (D) Thickness of laminin-positive basement membrane in microvessels formed in WT and kindlin-2+/− mice. Data are expressed as mean ± SEM and are representative of 3 independent experiments including 6-8 mice per group as indicated. (E) Costaining for CD105 (red) and CD31 (green). Overlay of the images reveals increased percent of CD105-positive blood vessels (yellow fluorescence) in tumors grown in kindlin-2+/− mice compared with WT mice. Scale bars, 150 μm. (F) Costaining for CD31 (red) and the pericyte marker, NG2 (green), shows reduced recruitment of pericytes and their interactions with blood vessels in tumors grown in kindlin-2+/− mice compared with WT mice. Scale bars, 50 μm. Five to 10 independent fields per tumor were examined. The images are representative of 3 independent experiments including 6-8 mice per group.
With defective pericyte recruitment, reduced maturation, and decreased laminin deposition, we considered whether the blood vessels in the kindlin-2+/− mice might be defective. Plasma leakage measured as an area positive for plasma-derived fibrin was twice as great in tumors grown in kindlin-2+/− mice compared with WT mice (7.8% ± 2.0% vs 17.2% ± 2.2%, P = .0006, n = 5; Figure 3A-B). Moreover, tumors grown in kindlin-2+/− animals had a substantially larger necrotic core (∼ 25% ± 4.5% of the total tumor area), compared with tumors in WT mice (12.3% ± 4.3%, P = .0413, n = 8), suggesting that the vasculature formed does not deliver bloodborne nutrients to support tumor growth in kindlin-2+/− mice (Figure 3C-D). The permeability of preexisting blood vessels in WT and kindlin-2+/− mice was examined. Baseline permeability of vasculature in ear skin (without mustard oil) was enhanced by ∼ 70% in kindlin-2+/− mice compared WT mice (P < .01). Application of mustard oil, a proinflammatory stimulus, on ear skin, enhanced permeability in WT mice by ∼ 75% (P < .05) while it had a negligible effect in the kindlin-2+/− mice (P = .159; Figure 3E). Similarly, when PBS was administered on dorsal skin as a control, blood vessel permeability in kindlin-2+/− mice was more than twice as great compared with WT mice (P < .01). However, VEGF, a potent inducer of vascular permeability, increased blood vessel permeability in WT mice by ∼ 80% (P < .05), while the already highly permeable vessels in kindlin-2+/− mice did not respond to VEGF (P = .78; Figure 3F). Thus, baseline vascular permeability is high in kindlin-2+/− mice and unresponsive to VEGF, indicating that not only newly formed but also preexisting blood vessels are defective in kindlin-2+/− mice.
Figure 3.

New and preexisting vasculature is leaky in kindlin-2+/− mice. (A) Fibrin content (brown) in tumors grown in WT and kindlin-2+/− mice. Scale bars, 30 μm. (B) Quantification of fibrin-positive area shown in panel A. (C) Representative photographs of necrotic areas in tumors grown in WT and kindlin-2+/− mice. Sections were stained with hematoxylin and eosin. Scale bars, 200 μm (top panel) and 150 μm (bottom panel). (D) Quantification of necrotic areas showed increased necrosis in tumors grown in kindlin-2+/− mice. All data are mean ± SEM and are representative of 3 independent experiments, including 5-8 mice per group as indicated. (E) Comparison of leakage from ear blood vessels in WT and kindlin-2+/− mice on stimulation with mustard oil. Blue ears of kindlin-2+/− mice indicate enhanced leakage of Evans Blue dye from blood vessels (left panel). Quantitative analysis revealed increased leakage from ear blood vessels in kindlin-2+/− mice compared with WT mice (right panel). (F) Representative photographs of Evans blue leakage from dorsal skin vasculature of WT and kindlin-2+/− mice on application of PBS or VEGF (left panel). Quantification of dorsal skin vasculature permeability (right panel). All vascular permeability data are expressed as mean ± SEM and are representative of 3 independent experiments with 5-6 mice per group as indicated.
BM-derived cells are important contributors to angiogenesis. Although kindlin-2 expression in hematopoietic cells is very low compared with kindlin-3,15,26 we investigated whether BM-derived cells might account for impaired angiogenesis and vascular leakiness in kindlin-2+/− mice. BM was transplanted from kindlin-2+/− mice to WT recipients (kindlin-2+/−→WT); and, as a control, from WT to WT mice (WT→WT). After 6-8 weeks, angiogenesis in prostate tumors and vascular permeability were assessed in both groups of recipient mice. Vascular area, plasma-derived fibrin content, and tumor growth were similar in (kindlin-2+/−→WT) and (WT→WT) mice (supplemental Figure 1A-C). In addition, vascular permeability in ear and dorsal skin was similar in (kindlin-2+/−→WT) and (WT→WT) mice in the absence or presence of permeability stimuli (supplemental Figure 1D-E). Thus, dysfunctional EC rather than BM-derived cells appear to contribute to abnormal angiogenesis and enhanced vascular permeability in the kindlin-2+/− mice.
Knockdown of zebrafish kindlin-2 results in aberrant angiogenesis
We next sought to understand the role of kindlin-2 during developmental angiogenesis and turned to the zebrafish as a facile model. We had previously reported on the ability of 2 independent morpholinos to knockdown kindlin-2 expression in the developing zebrafish and showed that kindlin-2 knockdown resulted in severe cardiac and skeletal muscle abnormalities.21,22 To study the effect of morpholino knockdown on vascular development, we used live imaging of a special marker zebrafish line that expresses GFP in all vascular ECs (GFP fused to the VEGF receptor promoter) and DsRed in all blood cells (DsRed fused to GATA-1 promoter).27 Injection of a control morpholino into 1-2 cell stage embryos resulted in normal vascular development at 24 and 48 hpf (Figure 4A). Injection of kindlin-2 morpholinos resulted in severe disturbances in embryonic vascular formation (Figure 4B-C). As depicted from 48 hpf morphant embryos, multiple abnormalities were noted, including thin, foreshortened intersegmental vessels (ISVs), aberrant branching of ISVs (double asterisks in Figure 4B-C), and failure to form a continuous dorsal longitudinal anastomotic vessel (DLAV). These abnormalities ranged in severity between morphant embryos (Figure 4B-C), but 90% of embryos injected with kindlin-2 morpholinos exhibited some degree of abnormal vessel formation (n = 100). We quantitated the degree of vasculature abnormality by measuring the length of ISVs at 48 hpf in control and kindlin-2 morphant embryos and found that ISVs from kindlin-2 embryos were significantly shorter (Figure 4D). In contrast, dorsal arteries (DAs) and posterior cardinal veins (PCVs) of kindlin-2 morphants were morphologically normal. Given that these vessels form by vasculogenesis and not angiogenesis,28 these findings indicate that the primary defect associated with kindlin-2 knockdown is in angiogenesis.
Figure 4.

Morpholino mediated knockdown of zebrafish kindlin-2 results in abnormal developmental angiogenesis. (A) Control morphant (CTL MO) embryo examined by live microscopy at 48 hpf. Proper vessel formation was observed in all control morphant embryos. ISV indicates intersegmental vessel; DLAV, longitudinal anastamotic vessel; DA, dorsal aorta; and PCV, posterior cardinal vein. (B-C) Two examples of kindlin-2 morphants (KIND2 MO). DLAVs were discontinuous and absent in many places (**). ISVs were thin and short. Aberrant horizontal interconnecting vessels were also frequently observed (arrows). Scale bar, 50 μm. (D) Quantitation of total ISV length. ISVs were measured at 48 hpf in control and kindlin-2 morphants. Average values (in micrometers) were (CTL vs KIND2): 114 ± 14 versus 71 ± 10; P = .02. Four independent experiments were performed with 10 fish per group.
We further examined the dynamics of the abnormal vasculature in kindlin-2 morphants using live imaging of DsRed-labeled blood cells. In both control and kindlin-2 morphants, blood flow was observed in the DA and the PCV (supplemental Videos 1 and 2). However, little if any blood flow was observed in ISVs from kindlin-2 morphants (supplemental Video 2), indicating that the vasculature is not only morphologically but also functionally abnormal. In the small number of kindlin-2 morphants with normal cardiac appearance and heart rate, we found similar aberrations in circulation through their ISVs (data not shown), indicating a primary defect in angiogenesis independent of cardiac abnormalities.
In view of the abnormal angiogenic response of the kindlin-2+/− mice to VEGF, we sought to address the effect of VEGF in the zebrafish on kindlin-2 morphants. The VEGF inducer GS4012 has been shown to rescue vasculature defects in certain zebrafish genetic models with abnormal blood vessel formation.24 We treated control and kindlin-2 morphant embryos using the previously published methodology for this inducer.24 Unlike what has been observed for these other models of abnormal vessel formation, GS4012 did not alter the angiogenesis in the kindlin-2 morphants (supplemental Figure 2), suggesting a role for kindlin-2 “downstream” of VEGF signaling.
To determine whether the abnormalities in angiogenesis were specific for kindlin-2, we also examined the effect of kindlin-3 knockdown on vascular formation in zebrafish. Morpholinos to kindlin-3 were injected in 1-2 cell stage embryos. General embryonic development was unaltered in kindlin-3 morphants. Analysis of vasculature using the VEGFR-GFP/GATA1-DsRed zebrafish line in kindlin-3 morphants did not reveal any abnormalities (supplemental Figure 3). Thus, the defects observed are specific for kindlin-2 knockdown.
Kindlin-2+/− endothelial cells exhibit impaired VEGF-mediated signaling, integrin activation, and integrin-dependent functions
To consider the mechanisms resulting in abnormal blood vessel formation and function in the kindlin-2+/− mice, we characterized intracellular signaling induced by growth factors and phorbol esters in ECs isolated from aortas of WT and kindlin-2+/− mice. The kindlin-2+/− ECs grew slowly compared with WT cells but showed no obvious morphologic differences. We verified that kindlin-2 expression was reduced in kindlin-2+/− ECs on Western blots of EC lysates with anti-kindlin-2 and anti-actin Abs (Figure 5A). When normalized to actin loading, kindlin-2 levels in the EC from the kindlin-2+/− mice were ∼ 52% of those in WT ECs based by densitometry. FACS analysis of WT and kindlin-2+/− ECs revealed similar expression levels of β3 and β1 integrins and VEGFR2 (Table 1). Cells were exposed to soluble VEGF, a ligand of both VEGFR2 and VEGFR1, to PLGF that interacts only with VEGFR1,29 or to PMA, and ERK1/2 phosphorylation, which is known to be induced by these activators in ECs, was assessed. VEGF, PLGF, and PMA induced activation of ERK1/2 and its downstream target p-90RSK in dose-dependent manner in WT ECs while they failed to activate ERK1/2 in kindlin-2+/− ECs (Figure 5B). Although decreased kindlin-2 expression does not affect VEGFR2 levels, it clearly impaired VEGFR2-dependent signaling. Moreover, cellular signaling in kindlin-2+/− ECs was also blunted to other stimuli tested. In these experiments, the cells were treated in suspension with soluble growth factors. Because there is little opportunity to integrins to engage ligand under these conditions, these results indicate that ERK1/2 phosphorylation occurs downstream of growth factor receptors and suggest functions of kindlin-2 in this signaling that are independent of integrin activation, This is consistent with the observation that VEGF-dependent ERK1/2 phosphorylation still occurs in β3-deficient ECs.30
Figure 5.

Comparison of receptor expression and intracellular signaling in WT and kindlin-2+/− aortic endothelial cells. (A) Kindlin-2 expression is reduced by ∼ 50% in kindlin-2+/− compared with WT ECs. Representative Western blots of EC lysates probed with anti-kindlin-2 and anti-actin (loading control) Abs are shown. Each band represents kindlin-2 or actin expression in ECs isolated from single individual mouse. ECs from 5 WT and 7 kindlin-2+/− mice were analyzed in 1 experiment. (B) VEGF, PMA, and PLGF induce phosphorylation of ERK1/2 and p90RSK in WT, but not in kindlin-2+/− ECs. The cells were incubated with increasing concentrations of soluble activators for 10 minutes at 37°C and lysed as described in “WT and kindlin-2+/− aortic ECs.” Western blots were probed with Abs to phospho-ERK1/2, phospho-p90RSK, or total ERK1/2 (loading control). The images are representative of 2 independent experiments.
Table 1.
FACS analysis revealed similar expression levels of VEGFR2 and the β3 and β1 integrins on WT and kindlin-2+/− ECs
| Receptor | WT ECs, MFI | Kindlin-2+/− ECs, MFI |
|---|---|---|
| Integrin β3 subunit | 105.58 ± 22.12 | 113.57 ± 23.12 |
| Integrin β1 subunit | 364.66 ± 33.6 | 330.5 ± 30.5 |
| VEGFR2 | 32.2 ± 7 | 25.4 ± 5.8 |
To test the role of kindlin-2 in integrin-dependent responses of ECs, we compared the adhesion of ECs from kindlin-2+/− and WT to ligands recognized by β3 integrins, fibrinogen (Fg) and vitronectin (Vn), or β1 ligands, collagen, and Fn. As shown in Figure 6A, adhesion of the EC from the kindlin-2+/− mice was significantly blunted on β3 but not β1 integrin substrates. In addition, migration of the EC from the kindlin-2+/− mice in response to VEGF was blunted on β3 but not β1 substrates (Figure 6B). When integrin dependent-ERK1/2 and p90RSK phosphorylation were compared in adherent ECs, they were robustly activated in WT cells adherent to Fg on VEGF stimulation while no increase was observed in kindlin-2+/− EC (Figure 6C). In contrast, on Fn, a β1 ligand, and control BSA, ERK1/2 activation was similar in ECs isolated from both genotypes. The kindlin-2+/− ECs also showed reduced spreading on Fg and Vn (by 50%-70%), whereas on collagen and Fn it was similar to that observed with WT ECs (Figure 6D). Of note, overexpression of kindlin-2 in the kindlin-2+/− EC overcame the defective spreading on Vn (supplemental Figure 4) while expression of a double mutant (QW/AA) form of kindlin-2, which does not bind integrins, did not. Thus, in examining 3 integrin-mediated responses, ECs derived from the kindlin-2+/− exhibited a selective reduction in integrin β3- but not β1-mediated responses, and the abnormalities in angiogenesis observed in the kindlin-2+/− mice in vivo were also observed with ECs isolated from these mice. Tube formation by kindlin-2+/− ECs was significantly impaired compared with those developed by WT ECs. Although kindlin-2+/− cells migrated into the substratum and accomplished the early steps in tube formation, that is, they organized into structures resembling tubes, the kindlin-2+/− EC failed to complete these tube-like structures (Figure 6E).
Figure 6.

Comparison of integrin-mediated functions in WT and kindlin-2+/− ECs. The kindlin-2+/− aortic ECs show suppressed adhesion (A) and migration (B) on fibrinogen and vitronectin compared with WT ECs while these responses are the same in both kindlin-2+/− and WT EC on collagen I and fibronectin. Data are expressed as means ± SEM of quadruple samples and are representative of at least 3 independent experiments of pooled EC of 3-6 mice. (C) Reduced ERK1/2 and p90RSK phosphorylation in VEGF-stimulated kindlin-2+/− cells adherent to fibrinogen compared with WT ECs while it was similar for both cell types on fibronectin and BSA. Western blots were reprobed with Ab to total ERK1/2 to demonstrate equal loading of proteins. The images are representative of 2 independent experiments. (D right panel) Representative images of WT and kindlin-2+/− ECs spread on various integrin ligands for 1 hour at 37°C (right panel). The cells are stained with Alexa 568–phalloidin. Scale bar, 50 μm. (D) Quantitative analysis of the data (left panel); reduced spreading of kindlin-2+/− ECs on fibrinogen and vitronectin (right panel). The areas of ∼ 200 cells per group from 2 independent experiments were measured. (E) Impaired tube formation by kindlin-2+/− ECs compared with WT ECs. Images of ECs forming tubes taken at 1, 3, and 6 hours are shown and are representative of 2 independent experiments including 6 individual EC samples per group (6 mice per group).
To assess integrin-dependent functions of kindlin-2+/− ECs, we quantified the ability of EC to bind soluble integrin ligands, an interaction dependent on integrin activation/clustering. The kindlin-2+/− and WT ECs did bind soluble Fg, a β3 integrin ligand, in a dose-dependent fashion, but the kindlin-2+/− ECs bound 2-fold less Fg than WT cells. Stimulation of the cells with PMA, a potent inducer of integrin activation/clustering, increased Fg binding to WT EC, but EC from the kindlin-2+/− mice showed no increase in Fg binding in response to PMA (Figure 7A). Moreover, VEGF stimulated soluble Fg binding to WT ECs, whereas it failed to enhance Fg binding to kindlin-2+/− cells. In the presence of this stimulator, 3- to 4-fold less Fg bound to kindlin-2+/− cells than to WT ECs (Figure 7B). Mn2+, which activates integrins independent of inside-out signaling, induced robust and similar soluble Fg binding to both WT and kindlin-2+/− ECs, indicating that the β3 integrins in the ECs from the kindlin-2+/− mice were not intrinsically defective in their ability to bind ligand (Figure 7B). In contrast to the deficits in Fg binding to nonstimulated or VEGF-stimulated ECs from the kindlin-2+/− mice, these cells did bind collagen I to a similar extent as WT ECs, suggesting that the ∼ 50% decrease in kindlin-2 expression does not alter activation of β1 integrins (Figure 7C).
Figure 7.
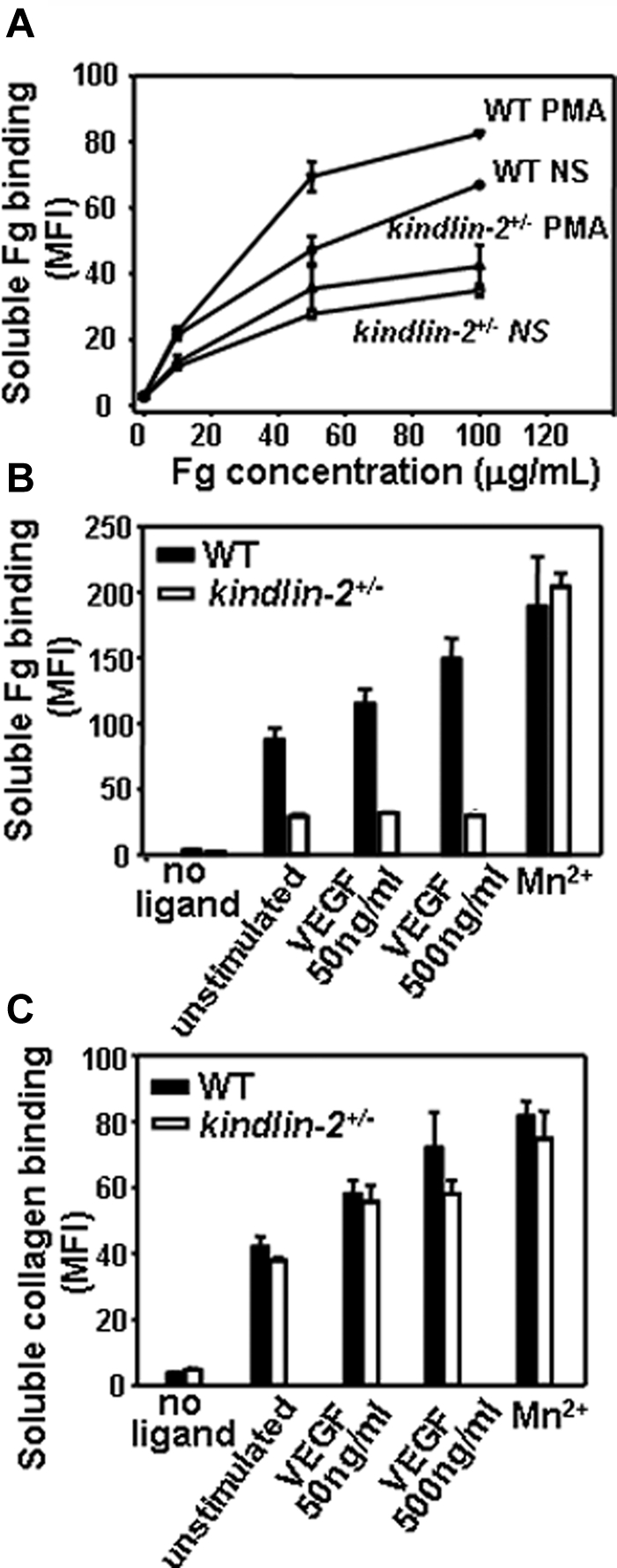
Comparison of integrin activation in WT and kindlin-2+/− aortic endothelial cells. (A) Dose-dependence of soluble Alexa 488–labeled Fg binding to resting or PMA-stimulated ECs. Soluble Fg binding to kindlin-2+/− ECs is decreased and independent on PMA stimulation. (B) Soluble Fg (50 μg/mL) binding to WT and kindlin-2+/− ECs in the presence of VEGF and Mn2+ ions. Fg binding to kindlin-2+/− ECs is significantly reduced and independent on VEGF stimulation. (C) Soluble FITC-collagen I (50 μg/mL) binding to WT and kindlin-2+/− ECs in the presence of VEGF and Mn2+ ions. Collagen I binding to WT and kindlin-2+/− ECs is the same in the absence or presence of VEGF. All data are means ± SEM of triplicate samples and are representative of 3 independent experiments with pooled ECs from 4-6 mice.
Discussion
Our studies show that the integrin coactivator, kindlin-2,12,13,31 is essential in pathologic and developmental angiogenesis. This conclusion is supported by in vivo experiments performed in kindlin-2+/− mice and in kindlin-2 morphant zebrafish. In kindlin-2+/− mice, we observed impaired angiogenesis in tumor and Matrigel models, and the few vessels that did form were disorganized, immature, and leaky. These impairments led to smaller RM1 prostate tumors in kindlin-2+/− mice. Preexisting vessels also showed increased permeability in the kindlin-2+/− mice and were unresponsive to VEGF. Our studies also indicate that zebrafish with a knockdown of kindlin-2 are unable to correct their defect in angiogenesis in response to VEGF. Thus, the dependence of angiogenesis on kindlin-2 has been documented in 2 difference species, and, in both organisms, angiogenesis in response to VEGF was defective.
The most extensively characterized function of kindlin-2 and the other 2 kindlin family members are their roles in integrin activation.9,10,12 The defects in angiogenesis observed in the kindlin-2+/− mice are consistent with defective activation of αVβ3. The proangiogenic functions of αVβ3 on ECs, include its role in mediating adhesion, spreading, migration, and tube formation, are well-documented5,32 and these functions were significantly reduced in ECs derived from the kindlin-2+/− mice. Soluble Fg binding, an interaction directly mediated by αVβ3 and dependent on its activation,32 was significantly blunted. Together, these observations all point to a role of kindlin-2 in regulating αVβ3 activation. The pivotal role of kindlin-2–dependent β3 integrin activation in angiogenesis is consistent with extensive evidence demonstrating the importance of αVβ3 in angiogenesis (reviewed in Avraamides et al5) using Abs, peptides, small molecules, or antisense approaches in vitro33,34 and in vivo.4,35 Angiogenesis induced by growth factors or tumors is also suppressed in knock-in mice expressing Y747F/Y759F mutations in the β3 cytoplasmic tail of αVβ3.36
Despite these interrelationships between kindlin-2, αVβ3 and angiogenesis, the phenotype of the kindlin-2+/− mice does not overlap with the β3-KO mice. The β3-deficient mice displayed enhanced angiogenesis and increased tumor growth.30,37,38 This phenotype has been ascribed to increased expression of VEGFR2 and enhanced VEGF-dependent signaling in the β3-KO ECs.30,38 In contrast, in kindlin-2+/− ECs, VEGFR2 expression does not differ from that in WT ECs. The defect in angiogenesis in the kindlin-2+/− mice also appears to be more pronounced than observed in the Y747F/Y759F knock-in mice, where angiogenesis is defective but not as severely impaired. Despite similar VEGFR2 expression levels in WT and kindlin-2+/− mice, downstream signaling from occupancy of VEGFR2 was defective in kindlin-2+/− ECs and vasculature as they are unresponsive to VEGF, indicating that kindlin-2 plays a crucial role in VEGF-mediated integrin activation. This defect was corroborated in zebrafish, where a VEGF inducer failed to rescue abnormal developmental angiogenesis in the kindlin-2–deficient fish. These observations are in accord with our previous studies demonstrating that VEGF is a potent stimulator of the αVβ3 activation and αVβ3-dependent EC functions via VEGFR2-dependent inside-out signaling.6,7 Thus, the kindlin-2+/− phenotype demonstrates a critical role of β3 integrins in angiogenesis in vivo in the absence of compensatory increases in VEGFR2. However, kindlin-2 also promotes the β3 integrin functions via other inside-out signaling pathways, for example, PKC-dependent signaling.13 Because PMA, a potent PKC stimulator, enhances integrin clustering, impaired PMA-induced Fg binding to kindlin-2+/− ECs raises the possibility that kindlin-2 influences not only integrin activation but also clustering; that is, affects affinity and/or avidity modulation39 and might do so by direct binding to the β-cytoplasmic tail or by participating in cytoskeletal rearrangements.
In the β3-KO mice, macrophage recruitment into tumors was significantly reduced, which attenuated the immune response and led to enhanced tumor growth.37 However, transplantation of kindlin-2+/− BM cells into WT mice did not affect tumor growth or angiogenesis indicating that abnormal neovascularization in kindlin-2+/− mice is attributed to dysfunctional endothelium/subendothelial matrix. In addition, neutrophil and macrophage content in tumors and in tumor-surrounding tissues was similar in WT and kindlin-2+/− mice (data not shown). Indeed, kindlin-2 expression both at mRNA and protein level is very low in cells of hematopoietic origin where kindlin-3 appears to be the primary kindlin involved in integrin activation.15,26
Additional prominent features of the kindlin-2+/− vasculature were its immaturity and leakiness, which may also be attributable to dysfunctional αVβ3. Although maintenance of endothelial barrier function is a complex process regulated by multiple receptors and signaling molecules, that build tight and adherens junctions (reviewed in Mehta and Malik40), numerous lines of evidence point to the importance of endothelial focal adhesions containing β3 integrins in forming these junctions.41,42 Focal adhesions not only provide structures for anchoring the endothelium to its surrounding matrices in the vascular wall, but also transmit forces and biochemical signals between the cells and their matrix. Direct evidence that underscores the physiologic significance of integrin-matrix interactions in vascular integrity comes from the studies showing that disruption of integrin-ECM interaction with RGD peptides increases venular permeability by 2- to 3-fold in vivo43 and reduces the barrier properties of endothelial monolayers.44 In mature EC cultures, αVβ3 also localizes to and maintains the integrity of the junctions45 and decreased β3 expression with siRNA increases permeability.46 Mechanistically, it has been suggested that this integrin collaborates with other intercellular molecules to form lateral junctions and specifically participates in formation of adherens junctions. In addition, integrin-associated signaling molecules, such as FAK, ILK, paxillin, and Rac-1, increase expression and assembly of E- and N-cadherins in various cells, which mediate cell-cell adhesions.47 Junctional adhesion molecule C (JAM-C) associates with the β3 integrins and inhibits its activation resulting in enhanced permeability.46 However, while there are many interconnections between known functions of αVβ3 and the ability of kindlin-2 to control activation of this integrin in EC, at this point we do not exclude that integrin-independent mechanisms of kindlin-2 may also contribute to control of intercellular permeability. In this regard, it is noteworthy that kindlin-2 has been shown to colocalize with E-cadherin along the lateral and apical plasma membrane of basal keratinocytes.48,49
Recruitment and adherence of mural cells to forming vessels were severely impaired in kindlin-2+/− mice. Pericytes interact with ECs, stabilize newly formed tubes, modulate blood flow and vascular permeability, and regulate EC functions.50 Pericyte abnormalities, especially the partial dissociation of pericytes from endothelium, lead to vascular fragility and leakiness, tumor hypoxia, apoptosis, and death (reviewed in Lu and Sood50). PDGFs secreted by ECs and PDGFR-β on pericytes are key players in pericyte recruitment, and EC:pericyte contacts are mediated by the interactions between EC α5β1 and pericyte VCAM-1. However, we found no evidence for abnormal expression or activation of the β1 integrins on kindlin-2+/− ECs. Further studies are needed to delineate mechanism by which kindlin-2 regulates recruitment and adhesive interactions of mural cells.
Our ex vivo data indicate that kindlin-2+/− ECs show suppressed αVβ3 activation and responses to the αVβ3-specific substrates, whereas activation of the β1 integrins and β1-dependent EC functions are normal. Although kindlin-2 has been shown to have differential effects on the activation of β1 and the β3 integrins,31 at this juncture we cannot exclude that 50% expression of kindlin-2 is sufficient to support β1 integrin activation and the role of these integrins in angiogenesis. Similarly, our recent studies show that kindlin-2 knockdown in HUVECs reduced cellular responses to the β3-integrin–specific ligands while inhibition of kindlin-3 attenuated β1-mediated cell spreading. In addition, kindlin-2 colocalized with the β3 integrin in focal adhesions, while kindlin-3 did not, suggesting distinct functional specificities.17,13 However, in platelets it is kindlin-3 that interacts and regulates integrin αIIbβ3 activation.15,51 Interestingly, knockdown of kindlin-2, but not of kindlin-3, resulted in abnormal angiogenesis in zebrafish. Thus, the present study supports the emerging concept that different kindlin family members exhibit selectivity in their support of different integrins in specific cell types.
Supplementary Material
Acknowledgments
This work was supported by National Institutes of Health grants from the National Institute of Arthritis and Musculoskeletal and Skin Diseases (1K08AR054835, J.J.D.), and the Heart, Lung, and Blood Institute (P01 HL 073311 and R01 HL096062, E.F.P.).
Footnotes
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: E.P. and J.J.D. designed and performed experiments, analyzed and interpreted data, and wrote the paper; J.A.G. provided the kindlin-2+/− and WT mice; N.G., D.S., X.Z.W., and K.B. performed experiments and analyzed data; C.N. was responsible for animal husbandry, coordinated animal experimentation, and performed experiments; Y.-Q.M. contributed ideas and provided essential reagents; T.B. interpreted data and assisted as an expert in angiogenesis; E.F.P. designed, coordinated, and supervised research and wrote the paper; and all authors critically reviewed the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Edward F. Plow, PhD, Department of Molecular Cardiology, Lerner Research Institute, Cleveland Clinic, 9500 Euclid Ave, NB50, Cleveland, OH 44195; e-mail: plowe@ccf.org.
References
- 1.Folkman J. Angiogenesis. Annu Rev Med. 2006;57:1–18. doi: 10.1146/annurev.med.57.121304.131306. [DOI] [PubMed] [Google Scholar]
- 2.Hynes RO. Integrins: bidirectional, allosteric signaling machines. Cell. 2002;110(6):673–687. doi: 10.1016/s0092-8674(02)00971-6. [DOI] [PubMed] [Google Scholar]
- 3.Hynes RO. A reevaluation of integrins as regulators of angiogenesis. Nat Med. 2002;8(9):918–921. doi: 10.1038/nm0902-918. [DOI] [PubMed] [Google Scholar]
- 4.Friedlander M, Brooks PC, Shaffer RW, Kincaid CM, Varner JA, Cheresh DA. Definition of two angiogenic pathways by distinct alphav integrins. Science. 1995;270(5241):1500–1502. doi: 10.1126/science.270.5241.1500. [DOI] [PubMed] [Google Scholar]
- 5.Avraamides CJ, Garmy-Susini B, Varner JA. Integrins in angiogenesis and lymphangiogenesis. Nat Rev Cancer. 2008;8(8):604–617. doi: 10.1038/nrc2353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Byzova TV, Goldman CK, Pampori N, et al. A mechanism for modulation of cellular responses to VEGF: activation of the integrins. Mol Cell. 2000;6(4):851–860. [PubMed] [Google Scholar]
- 7.De S, Razorenova O, McCabe NP, O'Toole T, Qin J, Byzova TV. VEGF-integrin interplay controls tumor growth and vascularization. Proc Natl Acad Sci U S A. 2005;102(21):7589–7594. doi: 10.1073/pnas.0502935102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Moser M, Legate KR, Zent R, Fassler R. The tail of integrins, talin, and kindlins. Science. 2009;324(5929):895–899. doi: 10.1126/science.1163865. [DOI] [PubMed] [Google Scholar]
- 9.Plow EF, Qin J, Byzova T. Kindling the flame of integrin activation and function with kindlins. Curr Opin Hematol. 2009;16(5):323–328. doi: 10.1097/MOH.0b013e32832ea389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Malinin NL, Plow EF, Byzova TV. Kindlins in FERM adhesion. Blood. 2010;115(20):4011–4017. doi: 10.1182/blood-2009-10-239269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Calderwood DA. Integrin activation. J Cell Sci. 2004;117(Pt 5):657–666. doi: 10.1242/jcs.01014. [DOI] [PubMed] [Google Scholar]
- 12.Montanez E, Ussar S, Schifferer M, et al. Kindlin-2 controls bidirectional signaling of integrins. Genes Dev. 2008;22(10):1325–1330. doi: 10.1101/gad.469408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ma YQ, Qin J, Wu C, Plow EF. Kindlin-2 (Mig-2): a co-activator of beta3 integrins. J Cell Biol. 2008;181(3):439–446. doi: 10.1083/jcb.200710196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ussar S, Moser M, Widmaier M, et al. Loss of Kindlin-1 causes skin atrophy and lethal neonatal intestinal epithelial dysfunction. PLoS Genet. 2008;4(12):e1000289. doi: 10.1371/journal.pgen.1000289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Malinin NL, Zhang L, Choi J, et al. A point mutation in kindlin-3 ablates activation of three integrin subfamilies in humans. Nat Med. 2009;15(3):313–318. doi: 10.1038/nm.1917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Svensson L, Howarth K, McDowall A, et al. Leukocyte adhesion deficiency-III is caused by mutations in KINDLIN3 affecting integrin activation. Nat Med. 2009;15(3):306–312. doi: 10.1038/nm.1931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bialkowska K, Ma YQ, Bledzka K, et al. The integrin coactivator kindlin-3 is expressed and functional in a non-hematopoietic cell, the endothelial cell. J Biol Chem. 2010;285(24):18640–18649. doi: 10.1074/jbc.M109.085746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Meves A, Stremmel C, Gottschalk K, Fassler R. The Kindlin protein family: new members to the club of focal adhesion proteins. Trends Cell Biol. 2009;19(10):504–513. doi: 10.1016/j.tcb.2009.07.006. [DOI] [PubMed] [Google Scholar]
- 19.Kindler T. Congenital poikiloderma with traumatic bulla formation and progressive cutaneous atrophy. Br J Dermatol. 1954;66(3):104–111. doi: 10.1111/j.1365-2133.1954.tb12598.x. [DOI] [PubMed] [Google Scholar]
- 20.Ussar S, Moser M, Widmaier M, et al. Loss of Kindlin-1 causes skin atrophy and lethal neonatal intestinal epithelial dysfunction. PLoS Genet. 2008;4(12):e1000289. doi: 10.1371/journal.pgen.1000289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dowling JJ, Gibbs E, Russell M, et al. Kindlin-2 is an essential component of intercalated discs and is required for vertebrate cardiac structure and function. Circ Res. 2008;102(4):423–431. doi: 10.1161/CIRCRESAHA.107.161489. [DOI] [PubMed] [Google Scholar]
- 22.Dowling JJ, Vreede AP, Kim S, Golden J, Feldman EL. Kindlin-2 is required for myocyte elongation and is essential for myogenesis. BMC Cell Biol. 2008;9:36. doi: 10.1186/1471-2121-9-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mahabeleshwar GH, Somanath PR, Byzova TV. Methods for isolation of endothelial and smooth muscle cells and in vitro proliferation assays. Methods Mol Med. 2006;129:197–208. doi: 10.1385/1-59745-213-0:197. [DOI] [PubMed] [Google Scholar]
- 24.Peterson RT, Shaw SY, Peterson TA, et al. Chemical suppression of a genetic mutation in a zebrafish model of aortic coarctation. Nat Biotechnol. 2004;22(5):595–599. doi: 10.1038/nbt963. [DOI] [PubMed] [Google Scholar]
- 25.Huang X, Raskovalova T, Lokshin A, et al. Combined antiangiogenic and immune therapy of prostate cancer. Angiogenesis. 2005;8(1):13–23. doi: 10.1007/s10456-005-2893-y. [DOI] [PubMed] [Google Scholar]
- 26.Moser M, Bauer M, Schmid S, et al. Kindlin-3 is required for beta2 integrin-mediated leukocyte adhesion to endothelial cells. Nat Med. 2009;15(3):300–305. doi: 10.1038/nm.1921. [DOI] [PubMed] [Google Scholar]
- 27.Buchner DA, Su F, Yamaoka JS, et al. pak2a mutations cause cerebral hemorrhage in redhead zebrafish. Proc Natl Acad Sci U S A. 2007;104(35):13996–14001. doi: 10.1073/pnas.0700947104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.San Antonio JD, Zoeller JJ, Habursky K, et al. A key role for the integrin alpha2beta1 in experimental and developmental angiogenesis. Am J Pathol. 2009;175(3):1338–1347. doi: 10.2353/ajpath.2009.090234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Clauss M, Weich H, Breier G, et al. The vascular endothelial growth factor receptor Flt-1 mediates biological activities. Implications for a functional role of placenta growth factor in monocyte activation and chemotaxis. J Biol Chem. 1996;271(30):17629–17634. doi: 10.1074/jbc.271.30.17629. [DOI] [PubMed] [Google Scholar]
- 30.Reynolds LE, Wyder L, Lively JC, et al. Enhanced pathological angiogenesis in mice lacking beta3 integrin or beta3 and beta5 integrins. Nat Med. 2002;8(1):27–34. doi: 10.1038/nm0102-27. [DOI] [PubMed] [Google Scholar]
- 31.Harburger DS, Bouaouina M, Calderwood DA. Kindlin-1 and -2 directly bind the C-terminal region of beta integrin cytoplasmic tails and exert integrin-specific activation effects. J Biol Chem. 2009;284(17):11485–11497. doi: 10.1074/jbc.M809233200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Byzova TV, Rabbani R, D'Souza S, Plow EF. Role of integrin alphaVbeta3 in vascular biology. Thromb Haemost. 1998;80(5):726–734. [PubMed] [Google Scholar]
- 33.Stromblad S, Becker JC, Yebra M, Brooks PC, Cheresh DA. Suppression of p53 activity and p21WAFI/CIPI expression by vascular cell integrin alphaVbeta3 during angiogenesis. J Clin Invest. 1996;98(2):426–433. doi: 10.1172/JCI118808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Leavesley DI, Schwartz MA, Rosenfeld M, Cheresh DA. Integrin beta1- and beta3-mediated endothelial cell migration is triggered through distinct signaling mechanisms. J Cell Biol. 1993;121(1):163–170. doi: 10.1083/jcb.121.1.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Brooks PC, Clark RA, Cheresh DA. Requirement of vascular integrin alphavbeta3 for angiogenesis. Science. 1994;264(5158):569–571. doi: 10.1126/science.7512751. [DOI] [PubMed] [Google Scholar]
- 36.Mahabeleshwar GH, Feng W, Phillips DR, Byzova TV. Integrin signaling is critical for pathological angiogenesis. J Exp Med. 2006;203(11):2495–2507. doi: 10.1084/jem.20060807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Taverna D, Moher H, Crowley D, Borsig L, Varki A, Hynes RO. Increased primary tumor growth in mice null for beta3- or beta3/beta5- integrins or selectins. Proc Natl Acad Sci U S A. 2004;101(3):763–768. doi: 10.1073/pnas.0307289101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Reynolds AR, Reynolds LE, Nagel TE, et al. Elevated Flk1 (vascular endothelial growth factor receptor 2) signaling mediates enhanced angiogenesis in beta3-integrin-deficient mice. Cancer Res. 2004;64(23):8643–8650. doi: 10.1158/0008-5472.CAN-04-2760. [DOI] [PubMed] [Google Scholar]
- 39.Buensuceso C, de Virgilio M, Shattil SJ. Detection of integrin alpha IIbbeta 3 clustering in living cells. J Biol Chem. 2003;278(17):15217–15224. doi: 10.1074/jbc.M213234200. [DOI] [PubMed] [Google Scholar]
- 40.Mehta D, Malik AB. Signaling mechanisms regulating endothelial permeability. Physiol Rev. 2006;86(1):279–367. doi: 10.1152/physrev.00012.2005. [DOI] [PubMed] [Google Scholar]
- 41.Wu MH. Endothelial focal adhesions and barrier function. J Physiol. 2005;569(Pt 2):359–366. doi: 10.1113/jphysiol.2005.096537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Romer LH, Birukov KG, Garcia JG. Focal adhesions: paradigm for a signaling nexus. Circ Res. 2006;98(5):606–616. doi: 10.1161/01.RES.0000207408.31270.db. [DOI] [PubMed] [Google Scholar]
- 43.Wu MH, Ustinova E, Granger HJ. Integrin binding to fibronectin and vitronectin maintains the barrier function of isolated porcine coronary venules. J Physiol. 2001;532(Pt 3):785–791. doi: 10.1111/j.1469-7793.2001.0785e.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schlegel N, Waschke J. Impaired integrin-mediated adhesion contributes to reduced barrier properties in VASP-deficient microvascular endothelium. J Cell Physiol. 2009;220(2):357–366. doi: 10.1002/jcp.21772. [DOI] [PubMed] [Google Scholar]
- 45.Lampugnani MG, Resnati M, Dejana E, Marchisio PC. The role of integrins in the maintenance of endothelial monolayer integrity. J Cell Biol. 1991;112(3):479–490. doi: 10.1083/jcb.112.3.479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Li X, Stankovic M, Lee BP, et al. JAM-C induces endothelial cell permeability through its association and regulation of beta3 integrins. Arterioscler Thromb Vasc Biol. 2009;29(8):1200–1206. doi: 10.1161/ATVBAHA.109.189217. [DOI] [PubMed] [Google Scholar]
- 47.Yano H, Mazaki Y, Kurokawa K, Hanks SK, Matsuda M, Sabe H. Roles played by a subset of integrin signaling molecules in cadherin-based cell-cell adhesion. J Cell Biol. 2004;166(2):283–295. doi: 10.1083/jcb.200312013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ussar S, Wang HV, Linder S, Fassler R, Moser M. The Kindlins: subcellular localization and expression during murine development. Exp Cell Res. 2006;312(16):3142–3151. doi: 10.1016/j.yexcr.2006.06.030. [DOI] [PubMed] [Google Scholar]
- 49.Lai-Cheong JE, Ussar S, Arita K, Hart IR, McGrath JA. Colocalization of kindlin-1, kindlin-2, and migfilin at keratinocyte focal adhesion and relevance to the pathophysiology of Kindler syndrome. J Invest Dermatol. 2008;128(9):2156–2165. doi: 10.1038/jid.2008.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lu C, Sood AK. Role of pericytes in angiogenesis. In: Teicher BA, Ellis LM, editors. Antiangiogenic Agents in Cancer Therapy. 2nd ed. Humana Press: Totowa, NJ; 2008. pp. 117–132. [Google Scholar]
- 51.Moser M, Nieswandt B, Ussar S, Pozgajova M, Fassler R. Kindlin-3 is essential for integrin activation and platelet aggregation. Nat Med. 2008;14(3):325–330. doi: 10.1038/nm1722. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.