

Abstract
Progressive cognitive deficits that emerge with aging are a result of complex interactions of genetic and environmental factors. Whereas much has been learned about the genetic underpinnings of these disorders, the nature of “acquired” contributing factors, and the mechanisms by which they promote progressive learning and memory dysfunction, remain largely unknown. Here, we demonstrate that a period of early-life “psychological” stress causes late-onset, selective deterioration of both complex behavior and synaptic plasticity: two forms of memory involving the hippocampus, were severely but selectively impaired in middle-aged, but not young adult, rats exposed to fragmented maternal care during the early postnatal period. At the cellular level, disturbances to hippocampal long-term potentiation paralleled the behavioral changes and were accompanied by dendritic atrophy and mossy fiber expansion. These findings constitute the first evidence that a short period of stress early in life can lead to delayed, progressive impairments of synaptic and behavioral measures of hippocampal function, with potential implications to the basis of age-related cognitive disorders in humans.
Keywords: memory, long-term potentiation, aging, hippocampus, stress, dendritic atrophy
Introduction
Epidemiological and experimental evidence suggest that early-life experience, particularly “social” or “psychological” stress, may predispose to cognitive dysfunction (Ammerman et al., 1986; Meaney et al., 1988; Kaplan et al., 2001), as well as depression (Patchev et al., 1997; Sanchez et al., 2001; Heim et al., 2004), that appears much later in life. This implies that early-life stress can have effects that (although not necessarily latent) begin to impact neuronal function significantly during adulthood and aging. Although enduring effects of early-life experience have been studied in animal models (Meaney et al., 1988; van Oers et al., 1999; Huot et al., 2002; Poeggel et al., 2003), models for delayed consequences of early-life stress on cognitive function, with onset during adulthood, are uncommon.
The hippocampus is critical for various types of memory (Hollup et al., 2001) and is unusually vulnerable to stress (Bremner et al., 1997; McEwen, 1999; Kim and Diamond, 2002; Sapolsky, 2002). Repeated maternal separation, a potent stressor in neonatal rats, results in significant deficits of hippocampal function, but these are apparent already in young adults (Huot et al., 2002). In addition, chronic stress, or long-term elevation of plasma stress hormones in adult and aging rats, affects hippocampus-dependent cognitive function (Landfield et al., 1978; Kerr et al., 1991; Dachir et al., 1993; Luine et al., 1994; Bodnoff et al., 1995; Kim and Diamond, 2002; Alfarez et al., 2003), but these disturbances are generally transient (Pavlides et al., 2002).
Prompted by the evidence for late developing consequences of early stress in humans, here we explicitly tested whether psychological early-life stress in rats causes an enduring deterioration of hippocampus that worsens from young adulthood to middle age. The stressor was chosen to produce neuroendocrine changes that were substantial early in life (Avishai-Eliner et al., 2001) but were fully normalized by adulthood. We found that impairments to memory were minimal in young adulthood but severe in middle age, suggesting that the underlying neurobiological abnormalities were progressive. Accordingly, measures of synaptic function and plasticity, the presumed substrates for learning and memory, declined with age and were accompanied by neuroanatomical evidence for progressive disruption of normal hippocampal circuitry.
Materials and Methods
Animals and general procedures
Experiments were approved by the Institutional Animal Care Committee. Sprague Dawley male rats (Zivic-Miller Laboratories, Zelienople, PA) were born in University of California, Irvine vivaria and maintained in quiet rooms on a 12 h light/dark cycle with access to ad libitum chow and water. Birth of pups was verified at 12 h intervals, and day of birth was considered day 0. Litters were adjusted to 12, if necessary, and mixed so that effects of experimental manipulations were compared among littermates. All experiments, including behavioral/learning memory tests and harvest of plasma for hormonal measures (by decapitation), were conducted from 8:00 A.M. to 10:00 A.M. to minimize diurnal variability of stress hormones. Corticosterone was measured using radioimmunoassay, as described previously (Avishai-Eliner et al., 2001; Brunson et al., 2001).
Early-life chronic stress paradigm was modified from Avishai-Eliner et al. (2001). Briefly, for the stress group, cage environment and maternal behavior were altered by placing pups and dams in cages with limited nesting/bedding material on postnatal day 2 (P2). Cages were fitted with a soft plastic mesh bottom, 2.5 cm from cage floor (allowing collection of droppings); nesting material consisted of one paper towel that was used by the dam to construct a rudimentary nest area. The control group resided in “normal” cages containing ∼0.33 cubic feet of chips. All litters were completely undisturbed (and bedding was not changed) during P2-P9.
Morris water maze
Among the procedures established by Morris (1984), we chose the variant with the least swim time (and potential stress) using 2 training days and testing on the third. Briefly, for the water maze, a circular pool was filled with room temperature water (Morris, 1984; Brunson et al., 2001). A transparent platform was placed in a constant position for each set of trials, in the middle of a quadrant, 1-2 cm below water surface. During the 2 consecutive training days (8 trials/d; platform position fixed for each day), the water was not opacified, and rats learned to associate the location of the platform to visual cues from objects in the testing room. On the test day, the platform was placed in a new location, the water was opacified using powdered milk, and rats were placed in the water facing the pool wall. Starting positions were randomly rotated in different quadrants, and rats were subjected to six trials. Latency to reaching the platform was recorded (Brunson et al., 2001). This, as well as all other functional tests, was performed without knowledge of group (“blindly”).
Object recognition
This memory test, relying on spontaneous exploratory behavior, has been described in detail (Clark et al., 2000; Brunson et al., 2001) and is sensitive to hippocampal (Broadbent et al., 2004), as well as perirhinal (Mumby et al., 2002), lesions. Briefly, adult rats were tested in a quiet room in Plexiglas cages lined with opaque paper, leaving the front panel open to permit observation. Subjects were habituated (five 1 h sessions). Glass and metal test objects (e.g., padlock, light bulb, or hook) and duplicate objects, were used in sample and test trials to avoid odor cues. During the tests, objects were placed ∼6 cm from cage walls. The object recognition memory test consisted of giving each animal a sample trial, during which it was allowed to explore two objects for 5 min. The test trial was given 24 h later and consisted of a 5 min epoch in which animals were presented with a duplicate of an object from the sample trial and a novel object. In both trials, duration of exploration of each object (sniffing with the animal's nose in direct contact <2 cm from the object) was recorded.
Elevated plus maze
The elevated plus maze consisted of two open arms (50 × 10 cm) and two cross-wise closed arms (50 × 10 × 40 cm) with an open roof 50 cm above the floor. Rats were placed in the center of the maze, facing a closed arm. Each rat was tested only once for 5 min, and the maze was wiped with ethanol after each test. Decreased time on open arms relative to open plus closed arms is used as an index of anxiety.
Electrophysiology
All hippocampal physiology experiments were performed and analyzed blindly.
Brain slice preparation. Brains were rapidly removed from 4- and 12-month-old rats and submerged in ice-cold dissection medium. Transverse slices were cut from the middle third of the septotemporal axis of the hippocampus at a thickness of 350 μm. Recordings began after at least 1 h of incubation.
Field potential recording. Electrophysiological recording was performed using standard techniques (Kramár and Lynch, 2003). Slices were recorded in an interface chamber perfused (75 ml/h) with artificial CSF (aCSF) (in mm: 124 NaCl, 3 KCl, 1.25 KH2PO4, 2.5 MgSO4, 3.4 CaCl2, 26 NaHCO3, and 10 glucose) at 31 ± 1°C. Additionally, the slice surface was exposed to warm, humidified 95% O2/5% CO2.
Long-term potentiation protocol. A single recording electrode (2-3 MΩ resistance) filled with 2 m NaCl was positioned extracellularly in the stratum radiatum of the hippocampal pyramidal cell fields to record field EPSPs (fEPSPs). Stimulating pulses were delivered to the commissural/associational fibers in stratum radiatum every 20 s via a concentric bipolar stimulating electrode. Standard input/output curves were generated, and the test stimulation current was determined from that required to elicit a response that was approximately one-half of the maximum fEPSP amplitude. Baseline responses were recorded for at least 20 min to ensure stability before application of high-frequency stimulation (HFS). To induce long-term potentiation (LTP), a single train of HFS was applied at the test-stimulus intensity (frequency, 100 Hz; duration, 1 s), and responses were measured for 30 min during postinduction. Studies using slices from middle-aged rats show that, under control conditions, there is no significant difference in the level of LTP measured at 30 versus 60 min (Rex et al., 2005).
Whole-cell recording. Hippocampal slices were prepared using a vibrating tissue slicer [Leica (Bannockburn, IL) VT1000] and placed in a holding chamber for > 2 h (Kramár et al., 2004). Slices were recorded submerged in aCSF of the same composition as described above for field potential recording. GABAA currents were blocked with 50 μm picrotoxin. CA1b pyramidal neurons were visualized with an infrared microscope [Olympus (Melville, NY) BX50WI] with differential interference contrast configuration, and whole-cell recordings were made with 3-5 MΩ recording pipettes containing the following (in mm): 140 K+ gluconate, 2 ATP, 0.4 GTP, 8 NaCl, and 10 HEPES, pH 7.4 (290-300 mOsm). Membrane capacitances and resistances were determined by a biphasic test pulse (peak-to-peak height, 20 mV; frequency, 20 Hz). Synaptic responses were evoked every 20 s by a stimulating electrode placed in CA1 stratum radiatum and recorded with a patch amplifier (AxoPatch 1D; Molecular Devices, Union City, CA) with a four-pole low-pass Bessel filter at 2 kHz. Recordings were digitized at 1-10 kHz with pClamp 9 (Molecular Devices). Afterhyperpolarizations (AHPs) were generated by stimulating the Schaffer collateral/commissural projections with a single theta burst (four pulses at 100 Hz) every 4 min. The stimulation intensity was adjusted as needed to reliably elicit action potentials. During current-clamp recordings, the membrane potential was maintained at -60 mV. Input and series resistances were continuously monitored, and recordings were discarded if significant changes occurred. Experimental treatments were initiated after a stable baseline of at least 15 min.
Timm histochemistry and mossy fiber analysis
Rats (12 months of age; n = 6 per group) were deeply anesthetized and perfused transcardially with 200 ml of sodium sulfide perfusion solution, followed by 200 ml of fresh 4% buffered paraformaldehyde. Brains were postfixed for 24 h, cryoprotected (25% sucrose), cut into serial coronal sections (50 μm) throughout dorsal hippocampus, and mounted onto glass slides. For Timm staining, slides were exposed to a solution containing 5.1 g of citric acid, 2.7 g of Na-citrate, 3.4 g of hydroquinone, 0.17 g of silver nitrate, and 120 ml of 50% gum arabic, in 200 ml of double-distilled water. Slides were incubated in the dark at 27°C, and the reaction was stopped by rinsing the slides. Slides were dipped (1 min) in 1% Na thiosulfate, rinsed, counterstained, dehydrated, cleared with xylene, and coverslipped. The extent of aberrant mossy fiber “sprouting” was evaluated using a semiquantitative scale (0-5) for terminal sprouting in the CA3 hippocampal region, as described by Holmes et al. (1999). Scores of 0 indicate no granules in stratum pyramidale (SP) or stratum oriens (SO) of the CA3 region; 1, occasional discrete bundles of granules in SP and SO; 2, occasional-to-moderate granules in SP or SO; 3, prominent granules in SP or SO; 4, prominent near-continuous band of granules in SP or SO of entire CA3; 5, continuous or near-continuous dense band of granules in SP or SO along the entire CA3. Timm scores were determined blindly for three matched dorsal hippocampal sections per animal per group. Both hippocampi were analyzed and averaged to yield final scores.
Quantitative analysis of dendritic trees of labeled CA1 pyramidal neurons
Intracellular labeling was performed using the basic technique described by Horikawa and Armstrong (1988). Briefly, recording electrodes were filled with pipette solution containing neurobiotin (3 mg/ml; Vector Laboratories, Burlingame, CA). After recording, slices were transferred immediately to a fixative of 0.1 m phosphate buffer containing 4% paraformaldehyde, pH 7.4, and stored overnight at 4°C. After sectioning, tissue was washed, quenched, and incubated in avidin-biotin-HRP. Labeled neurons were then treated with DAB solution. The reacted slices were rinsed in PBS-Triton X-100 (0.3%), mounted on slides, dehydrated through graded ethyl alcohol, cleared in xylene, and coverslipped with Permount. Fully outlined individual cells (n = 8-10 per group) were then evaluated by an investigator “blinded” to the experimental-group origin of each neuron. Cells were reconstructed using camera lucida, and total dendritic length was estimated. In addition, Sholl analysis (Chen et al., 2004), a method providing quantitative description of the dendritic tree by evaluating the number of dendrites that cross or branch within virtual concentric circles drawn at fixed distances from the cell body, was used. Numbers of primary dendrites, total dendritic length, and number of dendritic intersections at each concentric circle (20, 40, and 60 μm, etc.) were measured. For these experiments, differences among groups were compared using two-way ANOVA (for treatment and distance from soma), with post hoc analyses (GraphPad Prism; GraphPad Software, San Diego, CA). Dendritic length and primary dendrites were compared using Student's t test. Data are expressed as mean ± SEM.
Statistical considerations
All results were analyzed for statistical significance (p < 0.05) using a one- or two-way ANOVA, Student's t test, or the nonparametric Mann-Whitney U test, as appropriate. The GraphPad Prism software package was used.
Results
Early-life, transient psychological stress results in late-onset, progressive hippocampal dysfunction
Chronic psychological stress was induced in neonatal rats for 1 week (P2-P9) by limiting nesting material in the cages (Avishai-Eliner et al., 2001). This resulted in abnormal nurturing behavior in the dams (Fig. 1A), including reduced and fragmented nursing and grooming of pups (Fig. 1B). On termination of this neonatal stress period, pups (n = 12 per group) had enlarged adrenals, increased corticosterone levels, and modest weight loss (Fig. 1C, left), but these signs of chronic stress were no longer found in middle-aged rats (Fig. 1C, right) (n = 5 controls, 6 early-life stress). The hormonal surge of the early-life stress group to a 20 min restraint stress was also similar to those of controls. Immediately after the stress, plasma corticosterone levels of controls (n = 5) and the early-life stress group (n = 10) were 12. 0 ± 0.8 and 14.1 ± 1.5 μg/dl, respectively. Glucocorticoid receptor mRNA levels in hippocampal CA1 were comparable in 12-month-old rats of both groups [early stress (n = 7), 0.34 ± 0.04; controls (n = 5), 0.28 ± 0.05 relative optical density units; p = 0.37].
Figure 1.
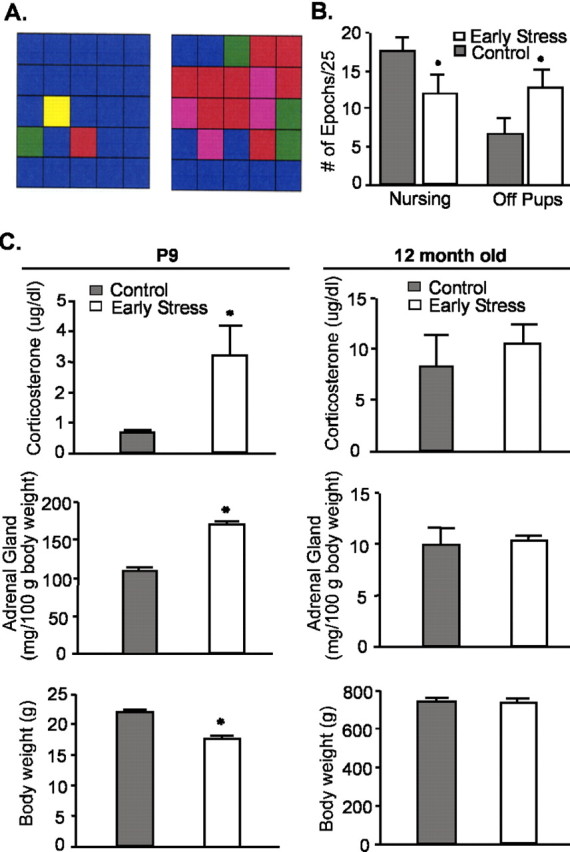
Early-life stress evokes transitory alterations of neuroendocrine parameters. A, Representative analysis grids of maternal behavior. Left, A dam rearing pups in a normal cage environment; right, a dam rearing pups in a cage with reduced nesting material. Grids depict 25 consecutive 3 min epochs and types of “nurturing” behaviors are color coded as follows: blue, nursing; yellow, carrying pups; green, licking/grooming; red, off pups; and pink, self grooming. B, Dams limited in nesting material spend less time nursing and more time off their pups. C, Parameters indicative of stress immediately after the early-life stress period (P9; left column) and in middle-aged rats (12 months of age; right column). Elevated basal corticosterone levels, higher adrenal gland weights, and modestly lower body weight were found in chronically stressed P9 rats (n = 12 per group), but these changes were no longer present in middle-aged rats (n = 5 controls, 6 early stress). All studies and plasma harvest (by decapitation) were performed from 8:00 A.M. to 10:00 A.M. (*p < 0.05; Student's t test). Error bars represent SEM.
Spatial memory was not significantly disturbed in early-life stressed animals tested at 4-5 months of age in a variant of the Morris water maze that reduces the “stressful” aspects of the task (Morris, 1984; Brunson et al., 2001). When tested after 2 training days, both control (n = 8) and early-life stress (n = 11) groups reached the hidden platform faster in successive trials (Fig. 2A, left), with an insignificant trend toward impaired performance of early-life stress versus controls (F = 2.02; p = 0.16; two-way ANOVA). However, serious impairments occurred in early-life stress rats at 12 months of age (Fig. 2A, right). These animals failed to reduce their platform-finding time on successive trials (effect of treatment, F = 8.8; p < 0.01; two-way ANOVA). Spatial memory deficits of early-life stress rats were also evident from probe trials for memory of platform location from a previous day (Fig. 2B,C): early-life stress rats spent significantly less time in the quadrant housing the platform on the previous day (p < 0.05). Analysis of the interaction of age and treatment effects on the Morris water maze performance demonstrated a significant interaction in the performance on the last trial of the testing day (F(1,24) = 4.23; p = 0.05; two-way ANOVA on the 4- and 12-month-old cohorts) but not the first trial of the testing day (F(1,24) = 1.00; p = 0.91). This is consistent with a progressive effect of the early-life stress. In addition, the pronounced memory problems could not be attributed to different levels of anxiety, because performance of control and early-life stress groups was comparable in the elevated plus maze test for anxiety (Fig. 2D).
Figure 2.

Deficient memory is evident in middle-aged (12 months) but not in younger (4- to 5-month-old) rats exposed to chronic early-life stress. A, At 4-5 months of age (left), both early-life stress (n = 8) and control rats (n = 11) require progressively less time to find a hidden platform in the Morris water-maze test. Escape latencies (time to find the hidden platform) on the testing day (day 3) are shown after 2 training days. By 12 months of age (right), early-life stress rats require significantly longer time (p < 0.001; two-way ANOVA; vs controls) to locate a hidden platform. An interaction of early-life stress and age of testing for the last trial (F(1,24) = 4.23; p = 0.05) suggests that the effects of early-life stress may be progressive. B, A representative probe trial search pattern from a control rat, preferentially exploring the quadrant containing the hidden platform on the previous testing day (left) is compared with that of an early-life stress rat (right) that does not prefer any quadrant. C, Quantitative analysis of the probe trials of middle-aged control and early-life stress rats showing that the latter group spent significantly less time in the quadrant that housed the platform on the previous day (p < 0.05). D, Anxiety level, as determined by elevated plus maze analysis, was not altered in the early-life stressed rats. E, Using the object-recognition paradigm, exploration pattern and duration of two novel objects of early-life stress rats (n = 15) and controls (n = 9) were indistinguishable on day 1 (left). However, 1 d later (day 2), controls discriminated between familiar and novel objects [remembering the familiar object and exploring it for a significantly (asterisk) shorter time], whereas early-life stress rats did not, indicating impairment of recognition memory. As shown on the right, the mean total exploration time on day 2 of the object recognition test did not differ between the groups (p > 0.05). Note that tests were performed and analyzed blindly. Error bars represent SEM.
To further exclude potential subtle, group-specific differences in the impact of the stressful aspects of the water-maze, we also used the object recognition procedure, which is sensitive to hippocampal lesions (Broadbent et al., 2004) (but see Mumby et al., 2002; Forwood et al., 2005). Middle-aged, early-life stressed rats (n = 15) failed to distinguish a novel object from one that they had seen the previous day, whereas age-matched controls (n = 9) spent approximately twice as long exploring the novel test object (p < 0.05). Notably, early-life stress and control rats interacted with both objects on the first day to approximately the same degree (Fig. 2E, left) and were also comparable in total time spent exploring both objects (novel and familiar) on day 2 (Fig. 2E, middle and right) (p > 0.05).
Synaptic plasticity defects parallel the behavioral memory deficits
Potential mechanisms for the learning/memory deficits were investigated by measuring synaptic plasticity in acute hippocampal slices from 12-month-old rats (note that all of the studies were performed by investigators unaware of the treatment group). Initial tests were performed in field CA3, because of reports that it is particularly vulnerable to chronic stress in adults (McEwen, 1999; Kim and Diamond, 2002), and focused on LTP, a form of synaptic facilitation implicated in memory formation (Roman et al., 1987; Bliss and Collingridge, 1993; Malenka and Nicoll, 1999; Silva, 2003). Single stimulation pulses within a narrow current range delivered to the commissural/associational projections were used to elicit field EPSPs in the apical dendrites, and LTP was induced using HFS. As shown in Figure 3, A and C (left), LTP (amplitude of the field EPSP as percentage of baseline) was significantly reduced in early-life stress slices compared with controls at 20 min after induction (early stress, 6.3 ± 6.4% vs control, 19 ± 9%; mean ± SD; p < 0.05). These synaptic plasticity defects might provide a plausible mechanism for the observed behavioral memory deficits. If so, the LTP impairments should be substantially less pronounced in young adults in which memory impairments were less evident. We tested this prediction and found that the slices from early-life stress rats had normal appearing LTP in field CA3 when evaluated at 4-5 months of age (Fig. 3B,C, right). As shown, 30 min after HFS, potentiated response amplitude of the field EPSP did not differ in early-stress and control groups (21 ± 7 and 26 ± 12% increase above baseline for early stress and control, respectively; p > 0.05). Thus, the physiological changes related to plasticity after early-life stress follow the same progressive time course observed for the functional learning and memory tests, and both emerge relatively late in adult life.
Figure 3.

Synaptic plasticity defects and physiological abnormalities in field CA3 arise in middle-aged rats subjected to early-life stress. A, LTP instratum radiatum of field CA3 in slices from middle-aged rats. A single HFS train was delivered at the time point indicated by the arrow. Potentiation (illustrated as percentage of average baseline field EPSP amplitude) decayed to near baseline in the stress group but not in age-matched controls (mean ± SEM). B, LTP in field CA3 of slices prepared from young adult rats. There were no evident differences between early stress and control groups 30 min after induction (p > 0.05). C, Representative EPSPs before and after HFS illustrate that LTP was deficient in the early-stress group compared with controls in slices prepared from middle-aged rats (left). There were no detectable differences between potentiated EPSPs in control and stress groups when slices were prepared from young adult rats (right). Calibration: 5 ms, 1 mV. D, Amplitude of field EPSPs in CA3 as a function of stimulus pulse duration (mean ± SEM); the input/output curves were noticeably flatter in slices from early-stress rats. E, Traces are the first 25 responses to the HFS used to induce LTP in area CA3; although initially similar, responses in the early-stress slices exhaust much more quickly than those in control slices. Calibration: 50 ms, 0.5 mV. F, In field CA3, mean amplitude of the fiber volleys (left) and evoked responses (right) were significantly reduced in early stress compared with control slices prepared from middle-aged rats (*p < 0.05). G, Input/output curves for antidromic responses in field CA3. Antidromic responses to the same stimulation currents were larger in early-stress slices compared with controls, indicating that the stress episode caused CA3 pyramidal cells to be more excitable. Error bars represent SEM.
The location and nature of the disrupted synaptic physiology in CA3
In examining the nature of LTP disruption, we found that differences between the groups in the magnitude of potentiation were already evident during the first 2 min after HFS, suggesting that a failure of induction (as opposed to stabilization) was responsible for the absence of LTP. We then found that the amplitudes of baseline synaptic responses in CA3 stratum radiatum were significantly smaller in the early-stress slices prepared from middle-aged rats than in matched controls (early stress, 0.5 ± 0.18 mV; control, 1.3 ± 0.3 mV; mean ± SD; p < 0.05). In addition, CA3 commissural/associational responses to the same magnitude of stimulation currents were significantly smaller in the early-life stress slices compared with those from controls, a point that was clearly confirmed in additional experiments using input/output curves (Fig. 3D). Additional evaluation of single HFS trains demonstrated that, at the same stimulation current, the amplitudes of successive field EPSPs within the HFS train decreased dramatically in the early-stress slices (Fig. 3E). These latter results are suggestive of a deficit in axon terminals and, in particular, of transmitter mobilization or in the frequency-following characteristics of the presynaptic axons. We further investigated the point by comparing the amplitudes of the fiber volleys [extracellular reflections of synchronously discharging axons (Andersen et al., 1978)] in the two groups. As shown in Figure 3F, the volleys were smaller (p < 0.05) in the early-life stress slices, suggesting that axonal excitability, number of axons, or both, were affected by early stress. However, the size of the field EPSPs in the slices used for fiber volley measurements were reduced to a greater degree by the early stress exposure than the volleys themselves (Fig. 3F); the ratio of EPSP to fiber volley was reduced from a control value of 7.1 ± 4.5 to 3.4 ± 2.1 (p < 0.03). This result again points to an additional disturbance in neurotransmitter release, the numbers of synapses generated by individual fibers, or the postsynaptic events that generate EPSPs. Finally, we used antidromic activation of pyramidal neurons to ask whether early stress affects the excitability of CA3 cells. Slices were incubated in CNQX (10 μm) and APV (100 μm) to block synaptic responses, and the short latency anti-dromic response triggered by single pulse stimulation of the Schaffer collaterals in field CA3 was measured in CA3b stratum pyramidale. Surprisingly, the input/output curve for the antidromic population spike was significantly elevated in the case of middle-aged animals exposed to early stress (Fig. 3G) (F = 51.91 for group effect; p < 0.0001). Given that we found no evidence of increased fiber excitability in the commissural/associational axons (see above), or their extension as Schaffer collaterals into field CA1 (see below), this result can be attributed to a change in the firing thresholds of the pyramidal neurons.
In summary, in field CA3, early stress causes a modest decrease in axonal excitability, a more massive depression of synaptic responses and their ability to follow high-frequency stimulation, and an increase in the excitability of the pyramidal neurons. Several mechanisms could contribute to these deficits. These include degeneration of CA3 pyramidal cells, resulting in a reduced number of axons in the commissural/associational projections, a pruning of axon collateral branches, or reduced excitability of axons. To narrow the range of potential mechanisms, systematic cell counting was undertaken and indicated that CA3 cell loss did not occur in the early-life stress rats (data not shown), thus excluding the first possibility. With the data presented thus far, we cannot distinguish between the second and third possibilities. However, a loss of synapses in stratum radiatum, either because of decreased periterminal collateralization or atrophy of spines, would account for most of the data, particularly given that deafferented/de-efferented neurons tend to be more excitable (Spehlmann et al., 1970; Buzsaki et al., 1989; Scharfman et al., 1998). Additional support for this idea is discussed below.
Structural foundation of the disrupted CA3 circuitry
What could be the neurobiological basis for the trigger of the striking disruption of basic synaptic physiology in CA3 of middle-aged rats after early-life stress and for the progressive nature of the corresponding LTP deficits? Field CA3 is the sole target of the peculiar mossy fiber system, which, unlike other hippocampal pathways, generates primary axons throughout the period during which psychological stress was induced in the present study (Amaral and Dent, 1981; Henze et al., 2000). Because of its delayed development, this system may be particularly affected by early-life stress, an event that could secondarily disturb the CA3 pyramidal neurons it innervates. Indeed, previous work showed that repeated, long (180 min) episodes of maternal separation/stress reduced the density of mossy fibers in 4-monthold rats (Huot et al., 2002). We tested for such effects using comparable histochemical methods (Timm's stain) in middle-aged animals and found that projections of the mossy fibers in contact with the dendritic branches of the pyramidal cells extended farther into the stratum oriens (basal dendrites) of CA3 in the early-stress animals than in matched controls (n = 6 per group) (Fig. 4A, arrows). In addition, there was an overall expansion of the mossy fibers in slices from early-stress rats compared with controls (Fig. 4B).
Figure 4.

Mossy fiber anatomy and physiology in slices from middle-aged rats subjected to early-life stress compared with age-matched controls. A, Sections of CA3 pyramidal cell fields from control and early-life stress (killed at 12 months; n = 6 in each group) rats, subjected to Timm's stain for visualizing the high zinc content of mossy fiber terminals (axons of the CA3-innervating granule cells). In early-life stressed rats, these terminals were abnormally abundant with in CA3 stratum oriens (so; arrow). Scale bar, 50 μm. sp, Stratum pyramidale. B, Quantification of the Timm's stained sections confirmed that mossy fiber sprouting was significantly increased in early-life stress animals (*p < 0.0001; Student's t test). C, The amplitudes (in millivolts) of mossy fiber synaptic responses, recorded from stratum lucidum, in control (white; n = 10) and stress (gray; n = 11) groups were not significantly different in CA3a or CA3b (n = 10 in each group). D, The degree of paired-pulse facilitation (i.e., the ratio of the size of the second pulse over the size of the first pulse in a pair of pulses separated by 50 ms) observed at the mossy fiber synapse in early-stress slices (gray; n = 11) was not different from control values (white; n = 10). Typical mossy fiber paired-pulse responses from CA3b are illustrated for control (left) and early-stress (right) slices from middle-aged rats (inset). Calibration: 20 ms, 1 mV. E, The metabotropic glutamate agonist DCG-IV (1 μm) depressed mossy fiber responses in control and early-stress slices (n = 4 in each group). Response amplitudes were normalized to predrug baseline values. Error bars represent SEM.
Physiological analyses of the mossy fibers were conducted first to determine whether aberrant growth was accompanied by disturbances in synaptic transmission and, second, to provide a test of whether the physiological disturbances established for the commissural/associational system are reflective of the entire field CA3. Stimulation electrodes were positioned in the hilus of the dentate gyrus proximal to the granule cell layer, and recording pipettes were placed in the stratum lucidum of fields CA3b and CA3a. Because the mossy fibers disperse from their origins in the granule cells and, with distance, “leave the slice” in increasing numbers, recording sites closer to the dentate gyrus (CA3b in the present case) have greater axonal continuity with the stimulation site than do more distal sites (i.e., CA3a); as a result, stimulation pulses delivered to the infragranular region elicit larger responses in the former than in the latter loci. The results summarized in Figure 4C indicate that, in marked contrast to the findings for the commissural/associational projections, mossy fiber synaptic responses were essentially the same in middle-aged slices from control and early-stress rats. This was the case for responses recorded from both CA3a and CA3b. We then used paired-pulse facilitation to determine whether release characteristics of the terminals vary between the groups; the results described in Figure 4D indicate that major differences were not in evidence. Finally, we infused the agonist 2S, 2′R, 3′R,2-(2′,3′-dicarboxycyclopropyl) glycine (DCG-IV) to assess the status of type II metabotropic glutamate receptors located on mossy fiber terminals (Shigemoto et al., 1997). Previous studies established that the compound depresses mossy fiber, but not commissural/associational, responses (Kamiya et al., 1996; Yeckel et al., 1999). In accord with this, DCG-IV caused a rapid reduction in the potentials recorded in the stratum lucidum of CA3b in the early-stress group and their age-matched controls (EPSP amplitude depressed by 31 ± 10 and 50 ± 6% below baseline values during the last 10 min of drug infusion for control and early-stress groups, respectively) (Fig. 4E). It should be noted that the effects of the drug were actually slightly larger in the early-stress group, although this difference was small and did not reach statistical significance (p < 0.08). Together, these results indicate that the massive disturbances to the commissural/associational projections are not accompanied by gross changes in the proximal dendrites of the CA3 pyramidal cells and that stress-related sprouting does not detectably change the basic physiology of the mossy fiber system. In addition, these data indicate that deficits in the mossy fiber system are not likely the direct cause of the profound deficits of LTP observed after early-life stress.
Early-life stress affects CA3 and CA1 synaptic function differentially
The selective perturbations to the commissural/associational system in the early-stress rats could arise, in part, from impaired conduction in the axons of CA3 pyramidal cells. If so, the collateral branches of these axons that are directed to field CA1 (i.e., the Schaffer collaterals) should have the same defects as the branches terminating within field CA3. We tested this point and obtained the results illustrated in Figure 5. LTP was clearly impaired in the early-stress, middle-aged rats to approximately the same degree as that found in field CA3; mean percentage potentiation at 20 min after induction was 38 ± 19% for the middle-aged controls and 14 ± 16% for the early-stress group (p < 0.03). However, in marked contrast to field CA3, the mean size of the baseline (pre-LTP) field EPSPs did not differ between control and early-stress slices (Fig. 5B). This also held for input/output curves for orthodromic and antidromic responses; as shown in Figures 5, C and D, there were no evident group differences across a broad range of stimulation intensities. Whole-cell recording was performed in an additional effort to isolate the factors responsible for the LTP deficit. Membrane properties of the cells (resting membrane potential, capacitance, resistance, and spike threshold) were not measurably different for the early-stress and control groups (Table 1), and intracellular responses recorded in the presence of picrotoxin to block GABAA receptor-mediated currents were of comparable size and shape (Fig. 5E,G, Table 1). Input/output curves for CA1 pyramidal cells proved equivalent in current-clamped neurons (Fig. 5E,F), again indicating that early stress did not significantly affect axonal excitability in CA1. NMDA receptor functioning was estimated by measuring the voltage-sensitive component of the EPSC (in the presence of picrotoxin) at 25 ms after the time point corresponding to the peak of the EPSC (i.e., the peak of the primarily AMPA-mediated component of the EPSC, measured at a holding potential of -80 mV), as illustrated in Figure 5G. Figure 5H summarizes the effects of membrane potential on the size of this EPSC component; in this figure, all values are expressed as a fraction of the maximal amplitude recorded across the range of clamped voltages. As shown, there were no apparent differences between middle-aged control and early-stressed cases with regard to the voltage sensitivity of the NMDA receptor-mediated synaptic currents. Note also that the absolute sizes of the AMPA- and NMDA-mediated components of the EPSC (recorded at a membrane potential of 0 mV) were similar in control and early-stress groups (Fig. 5H, right). Finally, previous work has shown that the AHP that follows theta bursts and high-frequency trains can exert a substantial influence on the magnitude of LTP (Arai and Lynch, 1992; Sah and Bekkers, 1996; Stackman et al., 2002; Kramár et al., 2004). We tested for early stress-related differences in AHPs and found no evidence for an increase over control in either the maximum amplitude or area of the AHP in slices prepared from early-stress animals (Fig. 5I).
Figure 5.

Deficits in LTP are not accompanied by disturbances in basic physiology in field CA1 of middle-aged rats subjected to early-life stress. A, Percentage of LTP at the Schaffer collateral/commissural synapse in stratum radiatum of field CA1 of middle-aged rats. Potentiation was equivalent in control and early-stress slices immediately after HFS but decayed much more quickly in the latter group and was significantly lower than control values at 30 min after HFS (p < 0.05; data illustrated as mean ± SEM). Typical EPSPs from early-stress (left) and control (right) slices before HFS illustrate that response size and waveform were comparable between groups. Calibration: 5 ms, 1 mV. B, The average amplitude of Schaffer collateral responses was not different between control (cont; white) and early-stress (gray) groups. C, Input/output curves for EPSPs in CA1 stratum radiatum indicate that an identical magnitude of stimulation current elicited responses in early-stress slices (n = 5) that were not measurably different from those recorded in control slices (n = 5). D, Input/output curves for antidromic responses in field CA1. Changes in response amplitude as a function of stimulation pulse duration were not significantly different in slices from early-life stress rats compared with age-matched controls. E, Current-clamp recordings from CA1 pyramidal cells in the presence of picrotoxin (50 μm). EPSPs were similar in control (left) and stressed (right) slices across a range (0.06-0.11 ms) of stimulus pulse durations. Calibration: 60 ms, 4 mV. F, Grouped current-clamp data for the effect depicted in E. Responses in CA1 pyramidal cells to the same level of stimulation current were not detectably different in control (white circles) and stress (gray circles) slices. G, In voltage-clamp experiments, the voltage-sensitive NMDA current was measured in the presence of 50 μm picrotoxin at the time point indicated by the dotted line. Small NMDA currents begin to emerge by -40 mV, whereas more prominent NMDA currents are apparent at -20 and 0 mV. No obvious abnormalities in NMDA currents were seen in the early-stress slices (right) when compared with controls (left). Calibration: 20 ms, 100 pA. H, The NMDA contribution to EPSCs in CA1 pyramidal cells is expressed as a percentage of the overall response across a range of holding potentials (left) and is not detectably different in recordings from control (white; n = 6) and early-stress (gray; n = 10) slices. The size of the AMPA- and NMDA-mediated currents are plotted for control (white) and early-stress (gray) groups at a holding potential of 0 mV (right). These quantifications reveal no differences in the NMDA-mediated component of EPSCs in CA1 pyramidal cells between the early-stress and control groups. I, A train of theta bursts (indicated by an arrow) was delivered to current-clamped CA1 pyramidal neurons to elicit an AHP; the top trace illustrates an example recorded in control tissue. Calibration bars for top trace: 0.3 s, 10 mV. Magnification of the AHP in representative traces from control and early-stress slices shows that AHPs were similar in both groups. The depolarizing responses to the stimulation train (indicated with an arrow on the control trace) were cutoff for illustration purposes in the magnified traces. The similarity between groups was quantified (right). Neither the amplitude nor the area of AHPs in CA1 neurons was significantly affected by early-life exposure to stress. Calibration for magnified, bottom traces: 0.3 s, 3 mV. Error bars represent SEM.
Table 1.
Membrane properties of CA1 pyramidal cells from control and early-stress groups
|
|
Control (n = 8) |
Early stress (n = 10) |
|---|---|---|
| VR (mV) | −65 ± 2 | −61 ± 2 |
| Cm (pF) | 19 ± 1 | 16 ± 1 |
| Rm (MΩ) | 32 ± 6 | 25 ± 2 |
| IAP | 230 ± 17 | 217 ± 16 |
| τEPSC(ms) |
14 ± 2 |
12 ± 1 |
VR, Resting membrane potential; Cm, membrane capacitance; Rm, membrane resistance; IAP, action potential current threshold; τEPSC, decay τ constant of EPSC. Data are shown as mean ± SEM.
Having found no substantial physiological disturbances that might account for the loss of LTP in area CA1 of the middle-aged early-stress slices, we investigated possible perturbations to pyramidal cell anatomy. Because chronic stress during adulthood may result in dendritic atrophy (Magarinos and McEwen, 1995a), we evaluated the dendritic length and branching patterns in CA1 pyramidal cells. Biotin-filled whole-cell recording electrodes were used to clamp and label neurons in field CA1 of control and early-stress slices, after which the grid intersection technique was used to measure the extent of the dendritic tree. Photomicrographs of typical pyramidal cells are shown in Figure 6; the dendritic tree of the neuron from the early-stress animal is noticeably impoverished relative to its age-matched control. Quantitative comparisons substantiated this impression, in that both the total dendritic length (p < 0.018) and the number of intersections produced by the apical and basal dendritic trees (two-way ANOVA for both experimental group and distance from soma; p < 0.001) were significantly reduced in the early-stress neurons. This significant atrophy of the CA1 pyramidal cell dendrites is consonant with the argument (see above) that early stress ultimately results in a reduction in the density of collateral branches generated by CA3 pyramidal cell axons. Given the multiple reports supporting the conclusion that LTP is accompanied by changes in the anatomy of dendritic spines (Lee et al., 1980; Chang and Greenough, 1984; Desmond and Levy, 1986; Popov et al., 2004; Segal, 2005), disturbances to the morphological properties of the dendritic ramification also explains the loss of potentiation seen in field CA1.
Figure 6.

Dendritic atrophy of CA1 pyramidal cells from early-life stressed middle-aged rats. Photomicrographs of biocytin-labeled pyramidal cells in field CA1 (left) illustrate the reductions in total dendritic length and in dendritic arborization in the early-stress group. Scale bar, 80 μm. These observations were quantified in the graphs on the right. The number of dendritic intersections, a measure of arborization, was lower in CA1 apical dendrites (right, top panel) of middle-aged early-stress rats. This effect diminished with increasing distance from the soma, indicating that the more distally situated dendrites in stratum lacunosum/moleculare, the region in which the perforant path axons from the entorhinal cortex terminate, were not affected. The number of intersections in the basal dendritic field of CA1 was also reduced (right, middle panel) in the early-stress group (gray circles) compared with age-matched controls (white circles). The total length of dendritic processes in CA1 (bottom right) was also lower in the early-stressed compared with the control group. *p = 0.018; n = 8-10 per group. Error bars represent SEM.
Discussion
The principal conclusions from these results are as follows: (1) progressive deficits in hippocampus-involving cognitive functions emerge relatively late during adulthood after a relatively short period of early-life psychological stress; (2) learning and memory impairments are accompanied by disturbed LTP in CA3 and CA1, which follows a parallel time course; (3) synaptic plasticity in CA3 stratum radiatum is likely disrupted by decreased afferent activation of the region, as evidenced by reductions in sizes of synaptic responses and fiber volleys. Progressive aberrant growth and innervation by the mossy fibers may ultimately lead to reduced axonal excitability and/or collateral branching of the commissural/associational fibers. (4) In contrast to CA3, basic synaptic physiology is preserved in CA1 of aging rats after early-life stress, although LTP is disrupted. The latter effect may be attributable to a reduction in input from CA3 pyramidal cell axons, evident from dendritic atrophy of CA1 pyramidal cells. Together, these findings provide a unique neuroanatomical and functional signature for the neurobiological processes initiated by early-life stress that ultimately result in hippocampal dysfunction.
Late-onset disruption of hippocampal function after early-life stress
Progressive cognitive deficits emerging with aging are a result of complex interactions of genetic and environmental factors (Bi et al., 2001; Mattson and Chan, 2001). Although much has been learned about the genetic underpinnings of these disorders (St George-Hyslop and Petit, 2005; Tanzi and Bertram, 2005), the nature of “acquired” contributing factors, and the mechanisms by which they promote progressive learning and memory dysfunction, remain largely unknown. Here, we demonstrate that a period of early-life psychological stress causes late-onset, selective deterioration of both complex behavior and synaptic plasticity.
The role of stress in influencing the structure and function of hippocampal neurons has been a focus of a significant body of research (McEwen, 1999; Brunson et al., 2001, 2003; Joels et al., 2003; Blank et al., 2004). Persistent stress may contribute to deficits in hippocampus-dependent learning and memory (Luine et al., 1994), including those found during senescence (McEwen, 1999; Yau et al., 2002). Chronic stress in adults may negatively affect LTP in several hippocampal subfields (Pavlides et al., 2002; Alfarez et al., 2003), including commissural/associational projections in CA3, in which, however, mossy fiber potentiation remains intact (Pavlides et al., 2002). Notably, these stress-evoked LTP changes reverse with time. Lifetime exposure to stress and the associated elevation in plasma glucocorticoids has also been suggested to underlie progressive excitotoxicity and learning/memory deficits in the aging hippocampus (Landfield et al., 1978; Hibberd et al., 2000).
In addition to functional disturbances, stress in adults causes atrophy of the apical dendritic field in CA3 (Magarinos and McEwen, 1995a), which could reduce measures of afferent excitability. The mechanisms for the structural remodeling and functional deficits in CA3 involve glucocorticoids and glutamate-mediated excitotoxicity (Magarinos and McEwen, 1995b; Joels et al., 2003). However, both the structural and functional (impaired LTP) stress-evoked changes reverse with time (Pavlides et al., 2002; Alfarez et al., 2003). Thus, although enduring stress during adulthood and adolescence (Isgor et al., 2004) may impair hippocampus-dependent learning and cause prominent deficits in aged animals, the results reported here constitute the first evidence that a short period of early-life stress can impair synaptic and behavioral measures of hippocampal function in a progressive manner and with onset relatively late during adulthood.
Early-life stress evoked by maternal separation (Sanchez et al., 2001; Avishai-Eliner et al., 2002) or seizures (Holmes and Ben-Ari, 1998) can negatively influence hippocampal function during adulthood, and mild learning and memory deficits in 4-monthold rats separated repeatedly from their mothers during the first postnatal weeks have been reported (Huot et al., 2002). However, the nature of the synaptic perturbations responsible for these deficits, and the means by which they are generated, have remained unclear. The present study, using a different early-stress paradigm, found minimal memory disturbances in early adult-hood but found severe deficits in middle-age. This progressive functional decline raises the possibility that the deterioration of brain systems responsible for encoding memory either commences or continues during adulthood in rats that experienced stress-evoking fragmented maternal care. Indeed, this was confirmed here, because there were few LTP effects of early stress in young adults but prominent impairments in middle-aged animals. Notably, memory problems were selective, because exploratory activity and anxiety measures were not appreciably different in early-stress rats and controls.
Mechanisms for stress-induced late-onset hippocampal deficits: age-dependent disturbances in area CA3
What mechanisms could account for the apparently late-arising progressive dysfunction of the hippocampus at both behavioral and cellular levels? If the mechanisms by which early-life stress influences hippocampal neurons are similar to those found during adulthood, early stress might set in motion changes that gradually increase the vulnerability of the hippocampus to glucocorticoids, eventually eliciting conditions in which normal stress becomes pathogenic (McEwen, 1999). Alternatively, stress during periods of hippocampal development could influence permanently growth and maturation of hippocampal systems that are particularly vulnerable during these periods (Avishai-Eliner et al., 2002; Khalilov et al., 2003). Indeed, the combined neuroanatomical and physiological evidence in this study support this latter possibility.
Disturbance of the mossy fibers could contribute to CA3 deficits because the development of the mossy fibers is delayed, rendering them more sensitive to postnatal manipulations (Henze et al., 2000). Indeed, the mossy fiber projection was significantly expanded in middle-aged rats that had experienced early-life stress. However, synaptic transmission at mossy fiber synapses was not impaired: the only physiological result involving mossy fibers that approached statistical significance was a slightly greater sensitivity to a group II metabotropic glutamate receptor agonist, DCG-IV. Group II metabotropic glutamate receptors are located presynaptically at mossy fiber synapses (Shigemoto et al., 1997), and their activation suppresses glutamate release from mossy fiber terminals (Kamiya et al., 1996; Yeckel et al., 1999). These receptors may be facilitated in some manner in early-stress rats so as to counteract the excessive innervation of CA3 by the mossy fibers, thereby masking any physiological effects.
Several measures of basic CA3 synaptic physiology were detrimentally affected in the early-stress middle-aged rats. Amplitudes of EPSPs evoked by stimulating the CA3 commissural/associational fibers in stratum radiatum were significantly smaller in early-stress slices, and, furthermore, the size of corresponding fiber volleys was depressed. Additionally, antidromic responses were unusually large, suggesting that pyramidal cells were more excitable. This pattern of results indicates that innervation of CA3 stratum radiatum by axon collaterals from other CA3 cells is reduced in early-stress middle-aged rats, reflecting either reduced numbers or decreased excitability of the axons. Approximately 75% of the input to CA3 pyramidal neurons comes from CA3 associational axons (Amaral et al., 1990); thus, their decline would constitute a major input loss. Such deafferentation/de-efferentation of CA3 would likely increase cell excitability (Buzsaki et al., 1989; Scharfman et al., 1998), as was indeed observed here.
This hypothesis raises the question of the origin of the progressive deficits in CA3 commissural/associational axons. Expansion of mossy fiber innervation, accompanied by increased cell excitability, could promote excitatory “sharp waves” (Buzsaki, 1986) that are intrinsically generated within CA3. Chronic enhancement of this activity, which is propagated throughout CA3 via the associational axons (Kubota et al., 2003), could lead to progressive decline of the associational system via mechanisms similar to those evoked by chronic stress in adult rats [i.e., excitotoxicity (Magarinos and McEwen, 1995b)]. The resulting reduced output of the commissural/associational axons and correspondingly lower response sizes would explain the LTP deficits observed in CA3 stratum radiatum. Specifically, they would decrease the postsynaptic depolarization generated by a given stimulation current and hence the likelihood of unblocking the voltage-sensitive NMDA-type glutamate receptors that trigger LTP.
Enduring changes in CA3 influence neuronal structure and synaptic function in CA1
The CA3 associational system enables pyramidal cells to activate their neighbors and thereby amplifies the output of CA3. Therefore, a depressed CA3 associational system as outlined above should result in reduced activation of CA1. Chronic reduction of afferent input to CA1 from CA3 could account for the impoverished dendritic trees of CA1 pyramidal cells found in middle-aged, early-stress rats in the present study. This CA1 atrophied dendritic arbor is likely responsible for the LTP impairments of the region, considering that postsynaptic changes are required for LTP formation (Lee et al., 1980; Chang and Greenough, 1984; Desmond and Levy, 1986; Luscher et al., 2000; Popov et al., 2004; Lin et al., 2005).
In conclusion, the present studies constitute the first evidence in animals that a period of early-life moderate stress causes late-onset, gradual deterioration of hippocampus-mediated learning and memory function during late adulthood and early aging. These effects may relate to cognitive deficits and psychiatric disorders suffered by humans who experienced severe stress (e.g., neglect/abuse) as children (Ammerman et al., 1986; Kaplan et al., 2001) and suggest a neurobiological basis for the components of these disorders not related to established genetic predispositions. Indeed, the anatomical and physiological underpinnings of the early stress-evoked behavioral changes described here point to novel targets for therapeutics, with potential implications to the prevention and treatment of age-related cognitive disorders in humans.
Footnotes
This work was supported by National Institutes of Health (NIH) Grants NS39307, NS28912 (T.Z.B.), and NS045260 (G.L.). L.L.C. was supported by NIH Postdoctoral Training Grant T32 AG00096. We appreciate K. LeBlanc's and K. Fenoglio's constructive assistance and M. Hinojosa's excellent editorial help.
Correspondence should be addressed to Dr. Tallie Z. Baram, Departments of Anatomy/Neurobiology and Pediatrics, University of California, Irvine, Medical Sciences I, ZOT 4475, Irvine, CA 92697-4475. E-mail: tallie@uci.edu.
Copyright © 2005 Society for Neuroscience 0270-6474/05/259328-11$15.00/0
References
- Alfarez D, Joels M, Krugers H (2003) Chronic unpredictable stress impairs long-term potentiation in rat hippocampal CA1 area and dentate gyrus in vitro. Eur J Neurosci 17: 1928-1934. [DOI] [PubMed] [Google Scholar]
- Amaral D, Dent J (1981) Development of the mossy fibers of the dentate gyrus. I. A light and electron microscopy study of the mossy fibers and their expansions. J Comp Neurol 195: 51-86. [DOI] [PubMed] [Google Scholar]
- Amaral DG, Ishizuka N, Claiborne B (1990) Neurons, numbers and the hippocampal network. Prog Brain Res 83: 1-11. [DOI] [PubMed] [Google Scholar]
- Ammerman R, Cassisi J, Hersen M, Hasselt VV (1986) Consequences of physical abuse and neglect in children. Clin Psychol Rev 6: 291-310. [Google Scholar]
- Andersen P, Silfvenius H, Sundberg SH, Sveen O, Wigstrom H (1978) Functional characteristics of unmyelinated fibres in the hippocampal cortex. Brain Res 144: 11-18. [DOI] [PubMed] [Google Scholar]
- Arai A, Lynch G (1992) Factors regulating the magnitude of long-term potentiation induced by theta pattern stimulation. Brain Res 598: 173-184. [DOI] [PubMed] [Google Scholar]
- Avishai-Eliner S, Gilles E, Eghbal-Ahmadi M, Bar-El Y, Baram T (2001) Altered regulation of gene and protein expression of hypothalamic-pituitary-adrenal axis components in an immature rat model of chronic stress. J Neuroendocrinol 13: 799-807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avishai-Eliner S, Brunson K, Sandman C, Baram T (2002) Stressed-out, or in (utero)? Trends Neurosci 25: 518-524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bi X, Yong AP, Zhou J, Ribak CE, Lynch G (2001) Rapid induction of intraneuronal neurofibrillary tangles in apolipoprotein E-deficient mice. Proc Natl Acad Sci USA 98: 8832-8837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blank T, Nijholt I, Spiess J (2004) Molecular determinants mediating effects of acute stress on hippocampus-dependent synaptic plasticity and learning. Mol Neurobiol 29: 131-138. [DOI] [PubMed] [Google Scholar]
- Bliss T, Collingridge G (1993) A synaptic model of memory: long-term potentiation in the hippocampus. Nature 361: 31-39. [DOI] [PubMed] [Google Scholar]
- Bodnoff SR, Humphreys AG, Lehman JC, Diamond DM, Rose GM, Meaney MJ (1995) Enduring effects of chronic corticosterone treatment on spatial learning, synaptic plasticity, and hippocampal neuropathology in young and mid-aged rats. J Neurosci 15: 61-69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bremner JD, Randall P, Vermetten E, Staib L, Bronen RA, Mazure C, Capelli S, McCarthy G, Innis RB, Charney DS (1997) Magnetic resonance imaging-based measurement of hippocampal volume in posttraumatic stress disorder related to childhood physical and sexual abuse—a preliminary report. Biol Psychiatry 41: 23-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broadbent NJ, Squire LR, Clark RE (2004) Spatial memory, recognition memory, and the hippocampus. Proc Natl Acad Sci USA 101: 14515-14520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunson K, Eghbal-Ahmadi M, Bender R, Chen Y, Baram T (2001) Long-term, progressive hippocampal cell loss and dysfunction induced by early-life administration of corticotropin-releasing hormone reproduce the effects of early-life stress. Proc Natl Acad Sci USA 98: 8856-8861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunson K, Chen Y, Avishai-Eliner S, Baram TZ (2003) Stress and the developing hippocampus a double-edged sword? Mol Neurobiol 27: 121-136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buzsaki G (1986) Hippocampal sharp waves: their origins and significance. Brain Res 398: 242-252. [DOI] [PubMed] [Google Scholar]
- Buzsaki G, Bayardo F, Miles R, Wong RK, Gage FH (1989) The grafted hippocampus: an epileptic focus. Exp Neurol 105: 10-22. [DOI] [PubMed] [Google Scholar]
- Chang FL, Greenough WT (1984) Transient and enduring morphological correlates of synaptic activity and efficacy change in the rat hippocampal slice. Brain Res 309: 35-46. [DOI] [PubMed] [Google Scholar]
- Chen Y, Bender RA, Brunson KL, Pomper JL, Grigoriadis DE, Wurst W, Baram TZ (2004) Modulation of dendritic differentiation by corticotropin-releasing factor in the developing hippocampus. Proc Natl Acad Sci USA 101: 15782-157827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark R, Zola S, Squire L (2000) Impaired recognition memory in rats after damage to the hippocampus. J Neurosci 20: 8853-8860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dachir S, Kadar T, Robinzon B, Levy A (1993) Cognitive deficits induced in young rats by long-term corticosterone administration. Behav Neural Biol 60: 103-109. [DOI] [PubMed] [Google Scholar]
- Desmond NL, Levy WB (1986) Changes in the postsynaptic density with long-term potentiation in the dentate gyrus. J Comp Neurol 253: 476-482. [DOI] [PubMed] [Google Scholar]
- Forwood SE, Winters BD, Bussey TJ (2005) Hippocampal lesions that abolish spatial maze performance spare object recognition memory at delays of up to 48 hours. Hippocampus 15: 347-355. [DOI] [PubMed] [Google Scholar]
- Heim C, Plotsky PM, Nemeroff CB (2004) Importance of studying the contributions of early adverse experience to neurobiological findings in depression. Neuropsychopharmacology 29: 641-648. [DOI] [PubMed] [Google Scholar]
- Henze D, Urban N, Barrionuevo G (2000) The multifarious hippocampal mossy fiber pathway: a review. Neuroscience 98: 407-427. [DOI] [PubMed] [Google Scholar]
- Hibberd C, Yau JL, Seckl JR (2000) Glucocorticoids and the ageing hippocampus. J Anat 197: 553-562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollup S, Kjelstrup K, Hoff J, Moser M, Moser E (2001) Impaired recognition of the goal location during spatial navigation in rats with hippocampal lesions. J Neurosci 21: 4505-4513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes G, Ben-Ari Y (1998) Seizures in the developing brain perhaps not so benign after all. Neuron 21: 1231-1234. [DOI] [PubMed] [Google Scholar]
- Holmes GL, Sarkisian M, Ben-Ari Y, Chevassus-Au-Louis N (1999) Mossy fiber sprouting after recurrent seizures during early development in rats. J Comp Neurol 404: 537-553. [DOI] [PubMed] [Google Scholar]
- Horikawa K, Armstrong WE (1988) A versatile means of intracellular labeling injection of biocytin and its detection with avidin conjugates. J Neurosci Methods 25: 1-11. [DOI] [PubMed] [Google Scholar]
- Huot R, Plotsky P, Lenox R, McNamara R (2002) Neonatal maternal separation reduces hippocampal mossy fiber density in adult Long Evans rats. Brain Res 950: 52-63. [DOI] [PubMed] [Google Scholar]
- Isgor C, Kabbaj M, Akil H, Watson SJ (2004) Delayed effects of chronic variable stress during peripubertal-juvenile period on hippocampal morphology and on cognitive and stress axis functions in rats. Hippocampus 14: 636-648. [DOI] [PubMed] [Google Scholar]
- Joels M, Verkuyl J, Riel EV (2003) Hippocampal and hypothalamic function after chronic stress. Ann NY Acad Sci 1007: 367-378. [DOI] [PubMed] [Google Scholar]
- Kamiya H, Shinozaki H, Yamamoto C (1996) Activation of metabotropic glutamate receptor type 2/3 suppresses transmission at rat hippocampal mossy fiber synapses. J Physiol (Lond) 493: 447-455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan Z, Iancu I, Bodner E (2001) A review of psychological debriefing after extreme stress. Psychiatr Serv 52: 824-827. [DOI] [PubMed] [Google Scholar]
- Kerr DS, Campbell LW, Applegate MD, Brodish A, Landfield PW (1991) Chronic stress-induced acceleration of electrophysiologic and morphometric biomarkers of hippocampal aging. J Neurosci 11: 1316-1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khalilov I, Holmes G, Ben-Ari Y (2003) In vitro formation of a secondary epileptogenic mirror focus by interhippocampal propagation of seizures. Nat Neurosci 6: 1079-1085. [DOI] [PubMed] [Google Scholar]
- Kim J, Diamond D (2002) The stressed hippocampus, synaptic plasticity and lost memories. Nat Rev Neurosci 3: 453-462. [DOI] [PubMed] [Google Scholar]
- Kramár EA, Lynch G (2003) Developmental and regional differences in the consolidation of long-term potentiation. Neuroscience 118: 387-398. [DOI] [PubMed] [Google Scholar]
- Kramár EA, Lin B, Lin C, Arai A, Gall C, Lynch G (2004) A novel mechanism for the facilitation of theta-induced long-term potentiation by brain-derived neurotrophic factor. J Neurosci 24: 5151-5161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubota D, Colgin L, Casale M, Brucher F, Lynch G (2003) Endogenous waves in hippocampal slices. J Neurophysiol 89: 81-89. [DOI] [PubMed] [Google Scholar]
- Landfield PW, Waymire JC, Lynch G (1978) Hippocampal aging and adrenocorticoids: quantitative correlations. Science 202: 1098-1102. [DOI] [PubMed] [Google Scholar]
- Lee KS, Schottler F, Oliver M, Lynch G (1980) Brief bursts of high-frequency stimulation produce two types of structural change in rat hippocampus. J Neurophysiol 44: 247-258. [DOI] [PubMed] [Google Scholar]
- Lin B, Kramár EA, Bi X, Brucher FA, Gall CM, Lynch G (2005) Theta stimulation polymerizes actin in dendritic spines of hippocampus. J Neurosci 25: 2062-2069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luine V, Villegas M, Martinez C, McEwen B (1994) Repeated stress causes reversible impairments of spatial memory performance. Brain Res 639: 167-170. [DOI] [PubMed] [Google Scholar]
- Luscher C, Nicoll RA, Malenka RC, Muller D (2000) Synaptic plasticity and dynamic modulation of the postsynaptic membrane. Nat Neurosci 3: 545-550. [DOI] [PubMed] [Google Scholar]
- Magarinos A, McEwen B (1995a) Stress-induced atrophy of apical dendrites of hippocampal CA3c neurons comparison of stressors. Neuroscience 69: 83-88. [DOI] [PubMed] [Google Scholar]
- Magarinos A, McEwen B (1995b) Stress-induced atrophy of apical dendrites of hippocampal CA3c neurons involvement of glucocorticoid secretion and excitatory amino acid receptors. Neuroscience 69: 89-98. [DOI] [PubMed] [Google Scholar]
- Malenka R, Nicoll R (1999) Long-term potentiation—a decade of progress? Science 285: 1870-1874. [DOI] [PubMed] [Google Scholar]
- Mattson MP, Chan SL (2001) Dysregulation of cellular calcium homeostasis in Alzheimer's disease: bad genes and bad habits. J Mol Neurosci 17: 205-224. [DOI] [PubMed] [Google Scholar]
- McEwen B (1999) Stress and hippocampal plasticity. Annu Rev Neurosci 22: 105-122. [DOI] [PubMed] [Google Scholar]
- Meaney M, Aitken D, Berkel CV, Bhatnager S, Sapolsky R (1988) Effect of neonatal handling on age-related impairments associated with the hippocampus. Science 239: 766-768. [DOI] [PubMed] [Google Scholar]
- Morris R (1984) Developments of a water-maze procedure for studying spatial learning in the rat. J Neurosci Methods 11: 47-60. [DOI] [PubMed] [Google Scholar]
- Mumby DG, Glenn MJ, Nesbitt C, Kyriazis DA (2002) Dissociation in retrograde memory for object discriminations and object recognition in rats with perirhinal cortex damage. Behav Brain Res 132: 215-226. [DOI] [PubMed] [Google Scholar]
- Patchev VK, Montkowski A, Rouskova D, Koranyi L, Holsboer F, Almeida OF (1997) Neonatal treatment of rats with the neuroactive steroid tetrahydrodeoxycorticosterone (THDOC) abolishes the behavioral and neuroendocrine consequences of adverse early life events. J Clin Invest 99: 962-996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavlides C, Nivon L, McEwen B (2002) Effects of chronic stress on hippocampal long-term potentiation. Hippocampus 12: 245-257. [DOI] [PubMed] [Google Scholar]
- Poeggel G, Helmeke C, Abraham A, Schwabe T, Friedrich P, Braun K (2003) Juvenile emotional experience alters synaptic composition in the rodent cortex, hippocampus, and lateral amygdala. Proc Natl Acad Sci USA 100: 16137-16142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popov VI, Davies HA, Rogachevsky VV, Patrushev IV, Errington ML, Gabbott PL, Bliss TV, Stewart MG (2004) Remodelling of synaptic morphology but unchanged synaptic density during late phase long-term potentiation (LTP): a serial section electron micrograph study in the dentate gyrus in the anaesthetised rat. Neuroscience 128: 251-262. [DOI] [PubMed] [Google Scholar]
- Rex CS, Kramár EA, Colgin LL, Lin B, Gall CM, Lynch G (2005) Long-term potentiation is impaired in middle-aged rats: Regional specificity and reversal by adenosine receptor antagonists. J Neurosci 25: 5956-5966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roman F, Staubli U, Lynch G (1987) Evidence for synaptic potentiation in a cortical network during learning. Brain Res 418: 221-226. [DOI] [PubMed] [Google Scholar]
- Sah P, Bekkers JM (1996) Apical dendritic location of slow afterhyperpolarization current in hippocampal pyramidal neurons: implications for the integration of long-term potentiation. J Neurosci 16: 4537-4542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez M, Ladd C, Plotsky P (2001) Early adverse experience as a developmental risk factor for later psychopathology evidence from rodent and primate models. Dev Psychopathol 13: 419-449. [DOI] [PubMed] [Google Scholar]
- Sapolsky RM (2002) Chickens, eggs and hippocampal atrophy. Nat Neurosci 5: 1111-1113. [DOI] [PubMed] [Google Scholar]
- Scharfman HE, Goodman JH, Du F, Schwarcz R (1998) Chronic changes in synaptic responses of entorhinal and hippocampal neurons after amino-oxyacetic acid (AOAA)-induced entorhinal cortical neuron loss. J Neurophysiol 80: 3031-3046. [DOI] [PubMed] [Google Scholar]
- Segal M (2005) Dendritic spines and long-term plasticity. Nat Rev Neurosci 6: 277-284. [DOI] [PubMed] [Google Scholar]
- Shigemoto R, Kinoshita A, Wada E, Nomura S, Ohishi H, Takada M, Flor PJ, Neki A, Abe T, Nakanishi S, Mizuno N (1997) Differential presynaptic localization of metabotropic glutamate receptor subtypes in the rat hippocampus. J Neurosci 17: 7503-7522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva A (2003) Molecular and cellular cognitive studies of the role of synaptic plasticity in memory. J Neurobiol 54: 224-237. [DOI] [PubMed] [Google Scholar]
- Spehlmann R, Chang CM, Daniels JC (1970) Excitability of partially deafferented cortex. I. Macroelectrode studies. Arch Neurol 22: 504-509. [DOI] [PubMed] [Google Scholar]
- Stackman RW, Hammond RS, Linardatos E, Gerlach A, Maylie J, Adelman JP, Tzounopoulos T (2002) Small conductance Ca2+-activated K+ channels modulate synaptic plasticity and memory encoding. J Neurosci 22: 10163-10171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- St George-Hyslop PH, Petit A (2005) Molecular biology and genetics of Alzheimer's disease. C R Biol 328: 119-130. [DOI] [PubMed] [Google Scholar]
- Tanzi RE, Bertram L (2005) Twenty years of the Alzheimer's disease amyloid hypothesis: a genetic perspective. Cell 120: 545-555. [DOI] [PubMed] [Google Scholar]
- van Oers HJ, de Kloet ER, Levine S (1999) Persistent effects of maternal deprivation on HPA regulation can be reversed by feeding and stroking, but not by dexamethasone. J Neuroendocrinol 11: 581-588. [DOI] [PubMed] [Google Scholar]
- Yau J, Noble J, Hibberd C, Rowe W, Meaney M, Morris R (2002) Chronic treatment with the antidepressant amitriptyline prevents impairments in water maze learning in aging rats. J Neurosci 22: 1436-1442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeckel MF, Kapur A, Johnston D (1999) Multiple forms of LTP in hippocampal CA3 neurons use a common postsynaptic mechanism. Nat Neurosci 2: 625-633. [DOI] [PMC free article] [PubMed] [Google Scholar]