

Abstract
Purpose
Recent studies suggest that temozolomide has activity in neuroendocrine tumors. Low levels of the DNA repair enzyme, O6-methylguanine DNA methyltransferase (MGMT), are associated with sensitivity to temozolomide in other tumor types. We evaluated the prevalence of MGMT deficiency in neuroendocrine tumors and correlated MGMT deficiency with treatment response to temozolomide-based regimens.
Experimental Design
The prevalence of MGMT deficiency, measured by immunohistochemistry, was assessed in 97 archival neuroendocrine tumor specimens. Rates of treatment response and survival were next evaluated in a cohort of 101 consecutive neuroendocrine tumor patients who had received treatment with a temozolomide-based regimen at one of three institutions. MGMT expression was directly correlated with treatment response in 21 patients who had available tumor tissue and response data.
Results
In archival specimens, MGMT deficiency was observed in 19 of 37 (51%) pancreatic neuroendocrine tumors and 0 of 60 (0%) carcinoid tumors (P < 0.0001). In the clinical cohort, 18 of 53 (34%) patients with pancreatic neuroendocrine tumors but only 1 of 44 (2%) patients with carcinoid tumors (P < 0.001) experienced a partial or complete response to temozolomide-based therapy. Among 21 patients with evaluable tumor tissue who had also received treatment with temozolomide, 4 of 5 patients with MGMT-deficient tumors (all pancreatic neuroendocrine tumors) and 0 of 16 patients with tumors showing intact MGMT expression responded to treatment (P = 0.001).
Conclusions
MGMT deficiency, measured by immunohistochemistry, is more common in pancreatic neuroendocrine tumors than in carcinoid tumors as is treatment response to temozolomide-based therapy. Absence of MGMT may explain the sensitivity of some pancreatic neuroendocrine tumors to treatment.
The alkylating agents streptozocin or dacarbazine are commonly incorporated in chemotherapy regimens for patients with advanced neuroendocrine tumors (1–7). Temozolomide is an alkylating agent initially developed as an oral and more easily tolerated alternative to dacarbazine. Initial clinical studies done with temozolomide showed clear evidence of activity in both melanoma and glioma (8–10). Recently, temozolomide has also been shown to have moderate activity in patients with advanced neuroendocrine tumors.
In an initial prospective study, treatment with temozolomide and thalidomide was associated with objective responses in 5 of 11 (45%) patients with pancreatic neuroendocrine tumors and 1 of 14 patients with carcinoid tumors (11). In a second prospective study, treatment with temozolomide and bevacizumab was associated with tumor responses in 4 of 17 (24%) patients with pancreatic neuroendocrine tumors and 0 of 12 patients with carcinoid tumors (12). Both regimens incorporated a dose-intense temozolomide regimen of 150 mg/m2/d for 7 days administered on an every other week schedule.
Retrospective series further support the use of temozolomide in neuroendocrine tumors. In a series of 36 patients treated with temozolomide monotherapy, tumor regression was observed in 31% of bronchial carcinoid tumors and 8% of pancreatic neuroendocrine tumors (13). In small, retrospective series of patients with pancreatic neuroendocrine tumors, combination therapy with temozolomide and capecitabine has been associated with a tumor response rates of 59% to 71% (14, 15).
The cytotoxic effect of temozolomide has been attributed to its ability to induce DNA methylation at the O6 position of guanine. Methylation of guanine results in DNA mismatch, ultimately resulting in apoptosis and tumor cell death (16). The sensitivity of tumor cells to alkylating agents, including temozolomide, has been associated with decreased levels of the DNA repair enzyme, O6-methylguanine DNA methyltransferase (MGMT), which, through its ability to restore DNA to its normal form, can prevent chemotherapy-induced cell death (17). Among patients with either advanced melanoma or glioblastoma treated with temozolomide, loss of tumoral MGMT expression was associated with an improvement in survival (18–22).
We postulated that differences in MGMT expression might explain the sensitivity of some neuroendocrine tumors to temozolomide-based therapy. Previous studies evaluating the prognostic or predictive value of immunohistochemical MGMT expression have used various criteria to categorize tumors as having absent, low, or intact of MGMT (19, 23–26). To minimize potential subjectivity in our analysis, we used a prospective classification scheme describing tumors as either MGMT deficient (no detectable expression of MGMT in tumor cells) or MGMT intact. We first evaluated the prevalence of MGMT deficiency in a cohort of 97 archival tissue specimens comprising carcinoid and pancreatic neuroendocrine tumors. We next evaluated whether patterns of treatment response in 101 neuroendocrine tumor patients treated with temozolomide-based regimens at our institutions matched the observed patterns of MGMT deficiency in these tumor subtypes. Finally, we correlated MGMT expression with treatment response in a subset of 21 of these patients with available neuroendocrine tumor tissue specimens.
Materials and Methods
Evaluation of MGMT status in archival tissue specimens
Archival neuroendocrine tumor tissue specimens were identified through a review of pathology records at Brigham and Women's Hospital. Additional tumor blocks were requested for consenting patients who had received temozolomide-based therapy using an institutional review board-approved protocol. Paraffin sections (4 μm) were used for immunohistochemical staining. Tissue sections were incubated for 60 min at 60°C, deparaffinized, and rehydrated in graded ethanol solutions. Endogenous peroxidase activity was blocked by incubating the slides in 3% H2O2 for 10 min. The slides were then rinsed under running water for 5 min. Heat-induced epitope retrieval was done using a microwave oven at 199°F for 30 min in preheated 10 mmol/L citrate buffer (pH 6.0). The slides were then transferred to PBS. The tissue sections were then blocked with 1.5% horse serum for 15 min and incubated for 1 h at room temperature in a humid chamber with mouse monoclonal antibody to MGMT (1:25 dilution; clone MT 3.1; Lab Vision), a biotinylated secondary antibody (mouse IgG), and then avidin-horseradish peroxidase (Vectastain Elite ABC Kit; Vector Laboratories) according to the manufacturer's instructions. The slides were washed in PBS between incubations. Tissue sections were developed using 3,3′-diaminobenzidine (Sigma) as a substrate and counterstained with Gill's hematoxylin (Fisher Scientific) according to the manufacturers' instructions.
Immunohistochemical MGMT expression was measured in a blinded fashion by two pathologists (M.S.R. and J.L.H.) who reviewed all cases concurrently at a multiheaded microscope. Nuclear MGMT expression was scored as either “intact” or “deficient” in tumor cells using a prospective classification scheme. Tumors were scored as “intact” when there was nuclear staining for MGMT in any tumor cells. Tumors were scored as “deficient” when there was a complete absence of nuclear staining for MGMT in all tumor cells. Nonneoplastic cells (lymphocytes, stromal cells, and endothelial cells) served as an internal positive control in all tissue sections. The MGMT expression status was then correlated with tumor type and treatment outcome.
Identification of neuroendocrine patients who had received temozolomide-based therapy
We examined patients with locally advanced or metastatic neuroendocrine tumors who received temozolomide-based therapy either as part of prospectively conducted clinical trials or off-protocol at the discretion of the treating physician. Patients were treated at one of three institutions: Dana-Farber Cancer Institute, Massachusetts General Hospital, or Beth Israel Deaconess Medical Center. Patients were identified either through review of two clinical trials that included temozolomide or through an institutional review board-approved protocol in which patients provide informed consent for the use of medical records, biospecimens, and clinical outcome data for medical research purposes. Medical records and clinical trial records were used to obtain demographic and treatment information as well as to assess response to temozolomide-based therapy.
Assessment of response and survival
For all patients in this analysis, radiologic response was measured using Response Evaluation Criteria in Solid Tumors. Patients who had enrolled on prospective, phase II studies underwent baseline staging computed tomography scans within 4 weeks of treatment initiation, and every 8 weeks thereafter. Response measurements for patients who received temozolomide-based therapy outside of a study setting were obtained using the nearest pretreatment computed tomography scan and subsequent scans obtained as part of routine clinical care. Biochemical response was measured based on baseline chromogranin A levels obtained before initiation of temozolomide-based therapy. Patients were considered to have a partial biochemical response if there was a ≥50% reduction in plasma chromogranin A from the baseline level on two successive measurements. Overall survival (OS) was defined as the time from initiation of temozolomide-based treatment until death from any cause. Progression-free survival (PFS) was defined as the time from initiation of temozolomide therapy to the date of documented progression or death from any cause. OS and PFS were calculated using the Kaplan-Meier method.
Results
We first evaluated MGMT expression in a cohort of 97 archival neuroendocrine tumor specimens and compared the prevalence of MGMT deficiency in pancreatic neuroendocrine and carcinoid samples (Table 1; Fig. 1). Among 37 pancreatic neuroendocrine tumors, 19 (51%) were MGMT deficient. Absence of MGMT was observed in 13 of 24 nonfunctional pancreatic neuroendocrine tumors, 3 of 10 insulinomas, 2 of 2 gastrinomas, and 1 of 1 glucagonoma. In contrast, MGMT staining was intact in all 60 carcinoid tumors, comprising 20 typical bronchial carcinoid tumors, 20 atypical bronchial carcinoid tumors, and 20 small intestine carcinoid tumors (P < 0.0001). Heterogeneous staining for MGMT was observed in our study; tumors with heterogeneous staining were classified as MGMT “intact” according to the classification scheme. We noted particularly prominent heterogeneity in three atypical bronchial carcinoid tumors, suggesting that a significant subpopulation of cells in these tumors was MGMT deficient.
Table 1. Immunohistochemical MGMT expression in neuroendocrine tumors.
| Tumor type | n | MGMT deficient, n (%) | MGMT intact, n (%) |
|---|---|---|---|
| Pancreatic neuroendocrine | 37 | 19 (51) | 17 (49) |
| Nonfunctional | 24 | 13 | 11 |
| Insulinoma | 10 | 3 | 7 |
| Gastrinoma | 2 | 2 | 0 |
| Glucagonoma | 1 | 1 | 0 |
| Carcinoid | 60 | 0 | 60 (100)* |
| Lung | 40 | 0 | 40 |
| Typical | 20 | 0 | 20 |
| Atypical | 20 | 0 | 20 |
| Small intestine | 20 | 0 | 20 |
P < 0.0001.
Fig. 1.
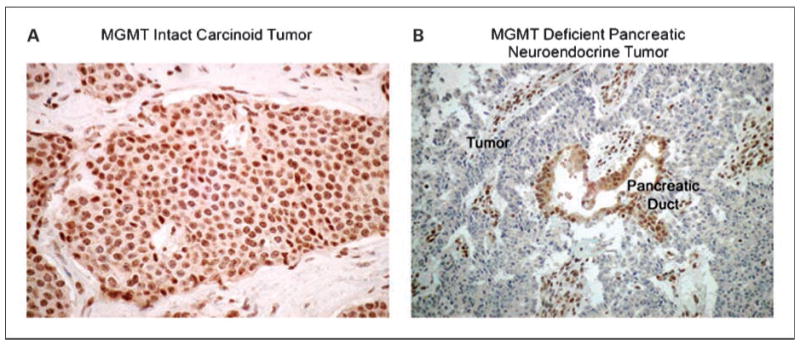
Representative MGMT staining in carcinoid and pancreatic neuroendocrine tumors.
To evaluate whether patterns of treatment response might mirror the prevalence of MGMT deficiency in these tumor types, we next identified 101 patients who had received temozolomide-based therapy for neuroendocrine tumors and recorded treatment outcome according to tumor type (Table 2). The patient cohort had a median age of 57 years and had been diagnosed a median of 19.5 months before initiating treatment with temozolomide. Fifty-three patients had pancreatic neuroendocrine tumors, 44 had carcinoid tumors, and 4 had pheochromocytoma/paraganglioma. The majority of patients had received one or more systemic treatments for their malignancy before receiving treatment with temozolomide.
Table 2. (A) Patient characteristics and treatment response.
| Characteristics | n | Radiologic response, n (%) | Biochemical response (baseline elevated), n (%) |
|---|---|---|---|
| Tumor type | |||
| Pancreatic neuroendocrine | 53 | 18/53 (34) | 16/32 (50) |
| Carcinoid tumors | 44 | 1/44 (2) | 6/27 (22) |
| Lung | 8 | 1/8 (13) | 3/8 (11) |
| Small bowel | 19 | 0 | 1/19 (4) |
| Other/unknown | 17 | 0 | 0 |
| Paraganglioma/pheochromocytoma | 4 | 1/4 (25) | 2/2 (100) |
| Gender | |||
| Male | 51 | 10/51 (20) | 9/31 (30) |
| Female | 50 | 10/50 (20) | 15/30 (50) |
| Median age | 57 | ||
| Treatment regimen | |||
| Temozolomide/thalidomide | 44 | 8/44 (18) | 14/25 (56) |
| Temozolomide/bevacizumab | 52 | 11/52 (21) | 9/33 (27) |
| Temozolomide/xeloda | 1 | 1/1 (100) | 1/1 (100) |
| Temozolomide alone | 4 | 0/4 (0) | 0/4 (0) |
| Treatment status | |||
| Phase II study | 63 | 12/63 (19) | 11/37 (30) |
| Off-study | 38 | 8/38 (21) | 13/24 (54) |
| Median time from diagnosis (mo) | 19.5 | ||
| No. prior systemic antitumoral treatments* | |||
| 0 | 44 | 12/44 (27) | 11/30 (37) |
| 1 | 35 | 3/35 (8) | 10/19 (53) |
| 2 | 6 | 2/6 (33) | 2/5 (40) |
| 3 | 6 | 2/6 (33) | 1/5 (20) |
| 4 | 1 | 1/1 (100) | NA |
| (B) MGMT status and treatment response | |||
|---|---|---|---|
| MGMT status | n | Radiologic response,n (%) | Biochemical response (baseline elevated), n (%) |
| MGMT intact† | 16 | 0/16 (0) | 0/10 (0) |
| MGMT deficient‡ | 5 | 4/5 (80) | 4/5 (80) |
Prior treatment data not available for 9 patients.
Thirteen of 16 tumors with intact MGMT expression were carcinoids; 3 of 16 were pancreatic neuroendocrine tumors.
All 5 MGMT-deficient tumors were pancreatic neuroendocrine tumors.
Of the 101 patients who received temozolomide-based therapy, 63 were treated as part of one of two prospective, single-arm, phase II clinical trials. These trials examined either the combination of temozolomide and thalidomide or temozolomide and bevacizumab. Within the clinical trials, temozolomide was administered at a dose of 150 mg/m2/d in both regimens; thalidomide was administered at doses ranging from 200 to 400 mg/d, and bevacizumab at a dose of 5 mg/kg intravenously every other week. Similar regimens and starting doses were used in the majority of patients receiving temozolomide-based treatment outside of the formal study setting.
No significant differences in tumor response rates were observed based on the type of temozolomide regimen administered. Moreover, patients who received temozolomide as part of a clinical trial appeared to experience a similar objective response rate when compared with those who were treated outside of a clinical trial. A marked difference in response rates was observed, however, between pancreatic neuroendocrine tumors and carcinoid tumors. Among 53 patients with pancreatic neuroendocrine tumors, 18 (34%) experienced partial responses to therapy as defined by Response Evaluation Criteria in Solid Tumors. In contrast, only 1 of 44 (2%) patients with carcinoid tumors experienced an objective response (P < 0.001); the single responder had metastatic well-differentiated bronchial carcinoid tumor. One of 4 patients with pheochromocytoma/paraganglioma responded to treatment.
The median PFS was 13.6 months for pancreatic neuroendocrine tumor patients and 9.6 months for patients with carcinoid tumors who received temozolomide (P = 0.12; Fig. 2A). Median OS was 35.3 months for patients with pancreatic neuroendocrine tumors and 19.4 months for patients with carcinoid tumors (P = 0.07; Fig. 2B).
Fig. 2.
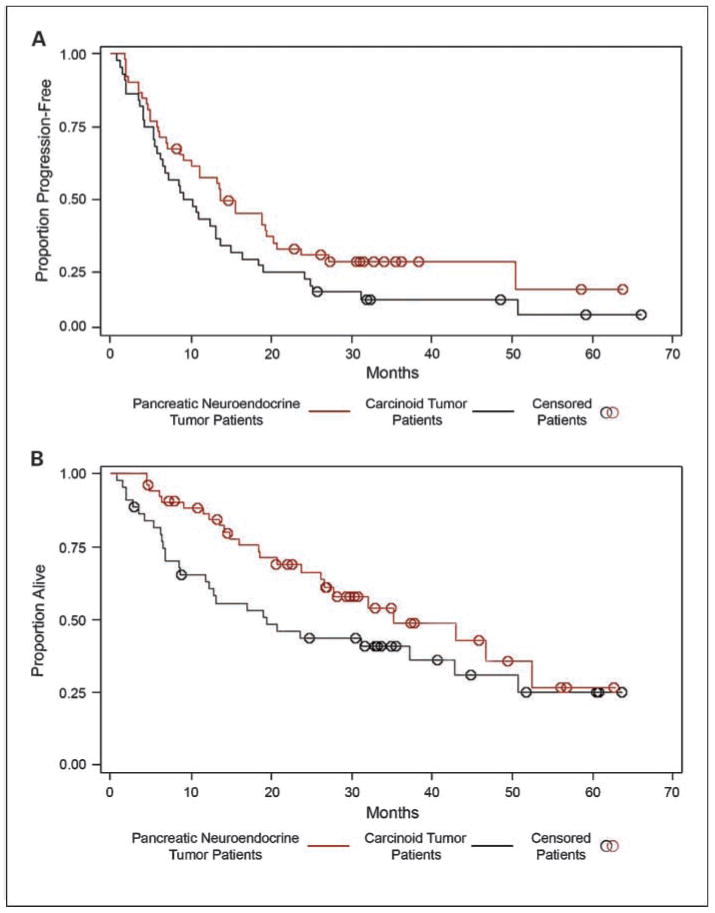
PFS and OS for carcinoid and pancreatic neuroendocrine tumor patients treated with temozolomide-based therapy. A, median PFS was 13.6 mo for pancreatic neuroendocrine tumor patients and 9.6 mo for carcinoid tumor patients (P = 0.12). B, median OS was 35.3 mo for pancreatic neuroendocrine tumor patients and 19.4 mo for carcinoid tumor patients (P = 0.07).
In light of the parallel patterns of MGMT deficiency and treatment response among carcinoid and pancreatic neuroendocrine tumors, we postulated that MGMT expression might directly correlate with response to temozolomide therapy. We therefore examined the effect of immunohistochemical MGMT expression on clinical outcomes among 21 temozolomide-treated patients, comprising all patients for whom both clinical data and archival, paraffin-embedded specimens were available. Tumors from 16 of the treated patients (13 carcinoid tumors and 3 pancreatic neuroendocrine tumors) showed intact MGMT expression. None of these 16 patients experienced radiologic or biochemical responses to temozolomide. Five patients had tumors that were MGMT deficient; all five tumors were pancreatic neuroendocrine tumors. Four of these 5 (80%) patients experienced partial radiologic responses to treatment (P = 0.001); 4 of 5 also experienced biochemical (chromogranin A) responses. One patient who did not experience a radiologic response experienced a chromogranin A response; conversely, one of the radiologic responders did not have a chromogranin A response.
Among those patients who received temozolomide-based therapy, the median PFS for patients whose tumors showed intact MGMT expression was 9.3 months compared with 19.2 months for patients with MGMT-deficient tumors (Fig. 3A; P = 0.11). The median OS for patients whose tumors showed intact MGMT expression was 19.1 months; the median OS for patients with MGMT deficient tumors has not been reached (Fig. 3B).
Fig. 3.
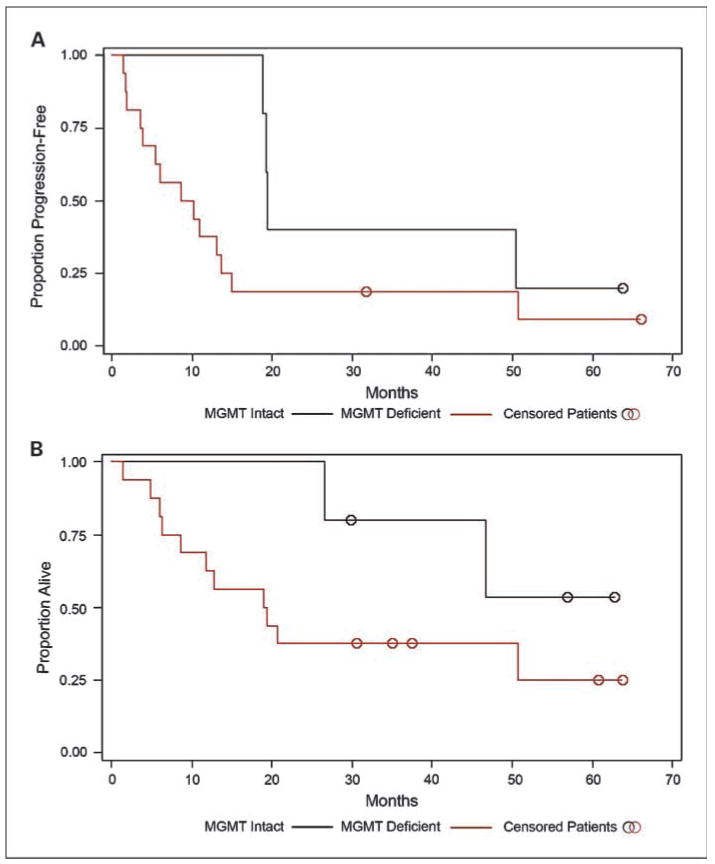
PFS and OS in patients with MGMT-intact or MGMT-deficient neuroendocrine tumors treated with temozolomide-based therapy. A, median PFS was 19.2 mo for MGMT-deficient neuroendocrine tumors and 9.3 mo for MGMT-intact tumors (P = 0.11). B, median OS for patients with MGMT-deficient tumors was not reached; median OS for patients with MGMT-intact tumors was 19.1 mo (P = 0.14).
Discussion
In a large cohort of archival neuroendocrine tumor specimens, we found that MGMT deficiency, as measured by immunohistochemistry, was more common in pancreatic neuroendocrine tumors than in carcinoid tumors. Consistent with this difference, we found that 34% of patients with pancreatic neuroendocrine tumors treated with temozolomide-based regimens experienced a Response Evaluation Criteria in Solid Tumors-defined radiologic tumor regression, whereas responses in carcinoid tumor patients were rare. MGMT deficiency was directly associated with treatment response to temozolomide in the subgroup of 21 treated patients who also had available tumor tissue.
Like temozolomide, streptozocin and dacarbazine induce methylation at the O6 position of guanine (27–30). This common cytotoxic mechanism suggests that the mechanisms of drug resistance for these agents may also be similar and that the ability of MGMT to repair treatment-induced formation of O6 methylguanine may contribute to drug resistance to all three drugs. Our observations that temozolomide-based therapy is more effective in pancreatic neuroendocrine tumors than in carcinoid tumors in fact mirror earlier results with the alkylating agents streptozocin and dacarbazine.
In an initial randomized trial, the combination of streptozocin and doxorubicin was associated with a combined biochemical and radiologic response rate of 69% in patients with pancreatic neuroendocrine tumors (5). In a retrospective analysis of 84 pancreatic neuroendocrine tumor patients treated with streptozocin, 5-fluorouracil, and doxorubicin, using more formal radiologic response criteria, the overall response rate was 39% (4). Dacarbazine was associated with an overall response rate of 33% in patients with pancreatic neuroendocrine tumors in a phase II study (6). The response rate of 34% observed with temozolomide in the current study is similar to that observed in these prior studies.
Response rates associated with these alkylating agents in carcinoid tumors are lower. In a recent trial, 249 patients with advanced carcinoid tumors were randomized to receive either streptozocin/5-fluorouracil or 5-fluorouracil/doxorubicin (7). The response rates associated with these regimens were 16% and 15.9%, respectively. The reported response rates associated with single-agent dacarbazine in carcinoid tumors are 8% to 16% (2, 7). Only a single carcinoid tumor patient (2%) responded to temozolomide-based therapy in our series.
Our results are similar to those of a smaller study of temozolomide monotherapy in 36 patients with neuroendocrine tumors (13). As in our study, 4 of 5 responding patients in the monotherapy study had low MGMT expression; responses were uncommon in patients with high MGMT expression. In contrast to our observations, however, temozolomide monotherapy was associated with an overall response rate of 31% (4 of 13) in patients with bronchial carcinoid tumors. We identified only 8 bronchial carcinoid tumor patients in our series, limiting our ability to more formally evaluate the efficacy of temozolomide in this subpopulation. Interestingly, the single carcinoid patient who responded to temozolomide in our study had a bronchial carcinoid tumor. Although several tumors classified as MGMT “intact” showed heterogeneous staining in our study, we observed a markedly heterogeneous pattern of MGMT expression in three atypical bronchial carcinoid tumors, providing a possible explanation for the sensitivity of some carcinoid tumors to temozolomide.
Streptozocin-based therapy has been associated with improved OS in patients with pancreatic neuroendocrine tumors (5). We observed trends toward improved PFS and OS among temozolomide-treated patients with pancreatic neuroendocrine tumors when compared with treated patients with carcinoid tumors in our study. Survival comparisons in our cohort are limited by both the retrospective nature of our analysis and potential differences in the treated subpopulations. Nevertheless, given the often similar natural history of patients with these malignancies, our observations raise the possibility that the higher observed rate of treatment response may also translate into improved survival in patients with pancreatic neuroendocrine tumors. Although we also observed a trend toward improved PFS and OS in patients with MGMT-deficient compared with MGMT-intact tumors, we cannot rule out the possibility that MGMT status had an independent effect on survival. Prospective, randomized studies will be necessary to confirm these associations.
There remains considerable controversy regarding the optimal method of MGMT analysis in clinical studies. Direct analysis of MGMT enzymatic activity generally requires use of carefully preserved frozen tissue or cell lysates and is not readily applicable to analysis of archival tumor samples from large clinical studies (31–33). Epigenetic silencing of the MGMT gene by CpG island promoter methylation is a common mechanism of MGMT gene regulation, and promoter methylation status, assessed by methylation-specific PCR, has been widely used as a surrogate marker of MGMT activity in clinical specimens (34). In patients with glioblastoma, MGMT promoter methylation has been associated with improved survival and benefit from temozolomide in most, although not all, studies (20, 21, 23, 35–37). Direct measurement of MGMT protein expression using immunohistochemistry, as was done in our study, is the technically easiest and perhaps the most commonly used technique to measure MGMT status in tumor samples. As with MGMT promoter methylation, low levels of immunohistochemical MGMT expression have been associated with improved response to temozolomide in glioblastoma in many studies, although correlations have not always been consistent (19, 22–25, 38).
Our observation that MGMT deficiency is more common in pancreatic neuroendocrine than in carcinoid tumors would suggest that MGMT promoter methylation status may also be more prevalent in pancreatic neuroendocrine tumors. However, previously reported studies of CpG island methylation in neuroendocrine tumors have found either no significant difference in MGMT promoter methylation rates between carcinoid and pancreatic neuroendocrine tumors or higher rates of promoter methylation in carcinoid tumors compared with pancreatic neuroendocrine tumors (39, 40). A poor correlation between MGMT promoter methylation and immunohistochemical expression of MGMT has been reported in several studies directly comparing these two methods (41–43). One study evaluating 31 glioblastoma samples found evidence of MGMT promoter methylation in 61% of samples but low level immunohistochemical MGMT expression (<20% nuclear staining) in only 31% (41). In a second study, substantial numbers of MGMT-positive cells were detected in the majority (73%) of tumor specimens carrying a methylated promoter (42). Tumor heterogeneity, as well as the presence of endothelial cells and other nonneoplastic components expressing MGMT in tumor samples, may have contributed to the discordant results observed in these studies.
We sought to minimize these limitations in our study by prospectively using a strict definition for MGMT deficiency, in which specimens were only considered deficient if they showed complete absence of detectable MGMT in tumor cells by immunohistochemistry. We further specifically identified nonneoplastic components of the tumors using these elements as positive internal controls. Nevertheless, technical limitations and interobserver variability remain a concern in the interpretation of MGMT immunohistochemical assays. We also cannot rule out the possibility that mechanisms other than MGMT expression affect neuroendocrine tumor sensitivity to temozolomide. Parallel DNA repair mechanisms, including the base excision repair system, may affect temozolomide sensitivity, resulting in an imperfect correlation between MGMT expression and treatment response (44, 45).
In summary, MGMT deficiency, as measured immunohistochemically, appears to be more common in pancreatic neuroendocrine tumors than in carcinoid tumors. Consistent with this finding, in a retrospective analysis, we observed a 34% response rate to temozolomide-based therapy in pancreatic neuroendocrine tumors compared with 2% in carcinoid tumors. MGMT deficiency was directly associated with temozolomide response in the patient subgroup with available tumor tissue and treatment data. Our findings suggest that MGMT status could be used as a predictive marker to identify neuroendocrine tumor patients who are likely to respond to treatment with alkylating agents. Standardization of techniques to assess MGMT status in tumor tissue, together with prospective trials to confirm a correlation between MGMT status and treatment response in neuroendocrine tumor patients treated with alkylating agents, is warranted.
Acknowledgments
M.H. Kulke thanks the Caring for Carcinoid Foundation, Stephen and Caroline Kaufer Fund for neuroendocrine tumor research, Dr. Raymond and Beverly Sackler, and Saul and Gitta Kurlat.
Grant support: National Cancer Institute grants CA093401 (M.H. Kulke) and P50 CA127003 (Dana-Farber/Harvard Cancer Center SPORE in Gastrointestinal Cancer).
Footnotes
Disclosure of Potential Conflicts of Interest: No potential conflicts of interest were disclosed.
References
- 1.Bukowski R, Johnson K, Peterson R, et al. A phase II trial of combination chemotherapy in patients with metastatic carcinoid tumors. Cancer. 1987;60:2891–5. doi: 10.1002/1097-0142(19871215)60:12<2891::aid-cncr2820601207>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- 2.Bukowski R, Tangen C, Peterson R, et al. Phase II trial of dimethyltriazenoimidazole carboxamide in patients with metastatic carcinoid. A Southwest Oncology Group study. Cancer. 1994;73:1505–8. doi: 10.1002/1097-0142(19940301)73:5<1505::aid-cncr2820730530>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]
- 3.Engstrom P, Lavin P, Moertel C, et al. Streptozocin plus fluorouracil versus doxorubicin therapy for metastatic carcinoid tumor. J Clin Oncol. 1984;2:1255–9. doi: 10.1200/JCO.1984.2.11.1255. [DOI] [PubMed] [Google Scholar]
- 4.Kouvaraki M, Ajani J, Hoff P, et al. Fluorouracil, doxorubicin, and streptozocin in the treatment of patients with locally advanced and metastatic pancreatic endocrine carcinomas. J Clin Oncol. 2004;22:4762–71. doi: 10.1200/JCO.2004.04.024. [DOI] [PubMed] [Google Scholar]
- 5.Moertel C, Lefkopoulo M, Lipsitz S, et al. Streptozocin-doxorubicin, streptozocin-fluorouracil, or chlorozotocin in the treatment of advanced islet-cell carcinoma. N Engl J Med. 1992;326:519–23. doi: 10.1056/NEJM199202203260804. [DOI] [PubMed] [Google Scholar]
- 6.Ramanathan RK, Cnaan A, Hahn RG, et al. Phase II trial of dacarbazine (DTIC) in advanced pancreatic islet cell carcinoma. Study of the Eastern Cooperative Oncology Group-E6282. Ann Oncol. 2001;12:1139–43. doi: 10.1023/a:1011632713360. [DOI] [PubMed] [Google Scholar]
- 7.Sun W, Lipsitz S, Catalano P, et al. Phase II/III study of doxorubicin with fluorouracil compared with streptozocin with fluorouracil or dacarbazine in the treatment of advanced carcinoid tumors: Eastern Cooperative Oncology Group Study E1281. J Clin Oncol. 2005;23:4897–904. doi: 10.1200/JCO.2005.03.616. [DOI] [PubMed] [Google Scholar]
- 8.Stevens MF, Hickman JA, Langdon SP, et al. Antitumor activity and pharmacokinetics in mice of 8-carbamoyl-3-methyl-imidazo[5,1-d]-1,2,3,5-tetrazin-4(3H)-one (CCRG 81045; M&B 39831), a novel drug with potential as an alternative to dacarbazine. Cancer Res. 1987;47:5846–52. [PubMed] [Google Scholar]
- 9.Middleton MR, Grob JJ, Aaronson N, et al. Randomized phase III study of temozolomide versus dacarbazine in the treatment of patients with advanced metastatic malignant melanoma. J Clin Oncol. 2000;18:158–66. doi: 10.1200/JCO.2000.18.1.158. [DOI] [PubMed] [Google Scholar]
- 10.Stupp R, Mason WP, van den Bent MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352:987–96. doi: 10.1056/NEJMoa043330. [DOI] [PubMed] [Google Scholar]
- 11.Kulke MH, Stuart K, Enzinger PC, et al. Phase II study of temozolomide and thalidomide in patients with metastatic neuroendocrine tumors. J Clin Oncol. 2006;24:401–6. doi: 10.1200/JCO.2005.03.6046. [DOI] [PubMed] [Google Scholar]
- 12.Kulke MH, Stuart K, Earle C, et al. A phase II study of temozolomide and bevacizumab in patients with advanced neuroendocrine tumors. Proc ASCO. 2006;12:A4044. [Google Scholar]
- 13.Ekeblad S, Sundin A, Janson ET, et al. Temozolomide as monotherapy is effective in treatment of advanced malignant neuroendocrine tumors. Clin Cancer Res. 2007;13:2986–91. doi: 10.1158/1078-0432.CCR-06-2053. [DOI] [PubMed] [Google Scholar]
- 14.Isacoff W, Moss R, Pecora A, et al. Temozolomide/ capecitabine therapy for metastatic neuroendocrine tumors of the pancreas. A retrospective review. J Clin Oncol 2006 ASCO Annu Meet Proc. 2006;18S:A14023. [Google Scholar]
- 15.Strosberg J, Choi J, Gardner N, et al. First-line treatment of metastatic pancreatic endocrine carcinomas with capecitabine and temozolomide. J Clin Oncol. 2008;26:A4612. [Google Scholar]
- 16.Liu L, Gerson SL. Targeted modulation of MGMT: clinical implications. Clin Cancer Res. 2006;12:328–31. doi: 10.1158/1078-0432.CCR-05-2543. [DOI] [PubMed] [Google Scholar]
- 17.Gerson SL. Clinical relevance of MGMT in the treatment of cancer. J Clin Oncol. 2002;20:2388–99. doi: 10.1200/JCO.2002.06.110. [DOI] [PubMed] [Google Scholar]
- 18.Middleton MR, Lunn JM, Morris C, et al. O6-methylguanine-DNA methyltransferase in pretreatment tumour biopsies as a predictor of response to temozolomide in melanoma. Br J Cancer. 1998;78:1199–202. doi: 10.1038/bjc.1998.654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chinot OL, Barrie M, Fuentes S, et al. Correlation between O6-methylguanine-DNA methyltransferase and survival in inoperable newly diagnosed glioblastoma patients treated with neoadjuvant temozolomide. J Clin Oncol. 2007;25:1470–5. doi: 10.1200/JCO.2006.07.4807. [DOI] [PubMed] [Google Scholar]
- 20.Brandes AA, Tosoni A, Cavallo G, et al. Correlations between O6-methylguanine DNA methyltransferase promoter methylation status, 1p and 19q deletions, and response to temozolomide in anaplastic and recurrent oligodendroglioma: a prospective GICNO study. J Clin Oncol. 2006;24:4746–53. doi: 10.1200/JCO.2006.06.3891. [DOI] [PubMed] [Google Scholar]
- 21.Hegi ME, Diserens AC, Gorlia T, et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med. 2005;352:997–1003. doi: 10.1056/NEJMoa043331. [DOI] [PubMed] [Google Scholar]
- 22.Friedman HS, McLendon RE, Kerby T, et al. DNA mismatch repair and O6-alkylguanine-DNA alkyltransferase analysis and response to Temodal in newly diagnosed malignant glioma. J Clin Oncol. 1998;16:3851–7. doi: 10.1200/JCO.1998.16.12.3851. [DOI] [PubMed] [Google Scholar]
- 23.Brell M, Tortosa A, Verger E, et al. Prognostic significance of O6-methylguanine-DNA methyltransferase determined by promoter hypermethylation and immunohistochemical expression in anaplastic gliomas. Clin Cancer Res. 2005;11:5167–74. doi: 10.1158/1078-0432.CCR-05-0230. [DOI] [PubMed] [Google Scholar]
- 24.Levin N, Lavon I, Zelikovitsh B, et al. Progressive low-grade oligodendrogliomas: response to temozolomide and correlation between genetic profile and O6-methylguanine DNA methyltransferase protein expression. Cancer. 2006;106:1759–65. doi: 10.1002/cncr.21809. [DOI] [PubMed] [Google Scholar]
- 25.Pollack IF, Hamilton RL, Sobol RW, et al. O6-methylguanine-DNA methyltransferase expression strongly correlates with outcome in childhood malignant gliomas: results from the CCG-945 cohort. J Clin Oncol. 2006;24:3431–7. doi: 10.1200/JCO.2006.05.7265. [DOI] [PubMed] [Google Scholar]
- 26.Capper D, Mittelbronn M, Meyermann R, et al. Pitfalls in the assessment of MGMT expression and in its correlation with survival in diffuse astrocytomas: proposal of a feasible immunohistochemical approach. Acta Neuropathol. 2008;115:249–59. doi: 10.1007/s00401-007-0310-x. [DOI] [PubMed] [Google Scholar]
- 27.Bennett RA, Pegg AE. Alkylation of DNA in rat tissues following administration of streptozotocin. Cancer Res. 1981;41:2786–90. [PubMed] [Google Scholar]
- 28.Souliotis VL, Boussiotis VA, Pangalis GA, et al. In vivo formation and repair of O6-methylguanine in human leukocyte DNA after intravenous exposure to dacarbazine. Carcinogenesis. 1991;12:285–8. doi: 10.1093/carcin/12.2.285. [DOI] [PubMed] [Google Scholar]
- 29.Newell D, Gescher A, Harland S, et al. N-methyl antitumour agents. A distinct class of anticancer drugs? Cancer Chemother Pharmacol. 1987;19:91–102. doi: 10.1007/BF00254559. [DOI] [PubMed] [Google Scholar]
- 30.Gerson SL. Modulation of human lymphocyte O6-alkylguanine-DNA alkyltransferase by streptozotocin in vivo. Cancer Res. 1989;49:3134–8. [PubMed] [Google Scholar]
- 31.Silber JR, Blank A, Bobola MS, et al. O6-methylguanine-DNA methyltransferase-deficient phenotype in human gliomas: frequency and time to tumor progression after alkylating agent-based chemotherapy. Clin Cancer Res. 1999;5:807–14. [PubMed] [Google Scholar]
- 32.Bobola MS, Berger MS, Ellenbogen RG, et al. O6-methylguanine-DNA methyltransferase in pediatric primary brain tumors: relation to patient and tumor characteristics. Clin Cancer Res. 2001;7:613–9. [PubMed] [Google Scholar]
- 33.Bobola MS, Silber JR, Ellenbogen RG, et al. O6-methylguanine-DNA methyltransferase, O6-benzylguanine, and resistance to clinical alkylators in pediatric primary brain tumor cell lines. Clin Cancer Res. 2005;11:2747–55. doi: 10.1158/1078-0432.CCR-04-2045. [DOI] [PubMed] [Google Scholar]
- 34.Esteller M, Hamilton SR, Burger PC, et al. Inactivation of the DNA repair gene O6-methylguanine-DNA methyltransferase by promoter hypermethylation is a common event in primary human neoplasia. Cancer Res. 1999;59:793–7. [PubMed] [Google Scholar]
- 35.Paz MF, Yaya-Tur R, Rojas-Marcos I, et al. CpG island hypermethylation of the DNA repair enzyme methyltransferase predicts response to temozolomide in primary gliomas. Clin Cancer Res. 2004;10:4933–8. doi: 10.1158/1078-0432.CCR-04-0392. [DOI] [PubMed] [Google Scholar]
- 36.Eoli M, Menghi F, Bruzzone MG, et al. Methylation of O6-methylguanine DNA methyltransferase and loss of heterozygosity on 19q and/or 17p are overlapping features of secondary glioblastomas with prolonged survival. Clin Cancer Res. 2007;13:2606–13. doi: 10.1158/1078-0432.CCR-06-2184. [DOI] [PubMed] [Google Scholar]
- 37.Blanc JL, Wager M, Guilhot J, et al. Correlation of clinical features and methylation status of MGMT gene promoter in glioblastomas. J Neurooncol. 2004;68:275–83. doi: 10.1023/b:neon.0000033385.37098.85. [DOI] [PubMed] [Google Scholar]
- 38.Jaeckle KA, Eyre HJ, Townsend JJ, et al. Correlation of tumor O6 methylguanine-DNA methyltransferase levels with survival of malignant astrocytoma patients treated with bis-chloroethylnitrosourea: a Southwest Oncology Group study. J Clin Oncol. 1998;16:3310–5. doi: 10.1200/JCO.1998.16.10.3310. [DOI] [PubMed] [Google Scholar]
- 39.Arnold CN, Sosnowski A, Schmitt-Graff A, et al. Analysis of molecular pathways in sporadic neuroendocrine tumors of the gastro-entero-pancreatic system. Int J Cancer. 2007;120:2157–64. doi: 10.1002/ijc.22569. [DOI] [PubMed] [Google Scholar]
- 40.Chan AO, Kim SG, Bedeir A, et al. CpG island methylation in carcinoid and pancreatic endocrine tumors. Oncogene. 2003;22:924–34. doi: 10.1038/sj.onc.1206123. [DOI] [PubMed] [Google Scholar]
- 41.Maxwell JA, Johnson SP, Quinn JA, et al. Quantitative analysis of O6-alkylguanine-DNA alkyltransferase in malignant glioma. Mol Cancer Ther. 2006;5:2531–9. doi: 10.1158/1535-7163.MCT-06-0106. [DOI] [PubMed] [Google Scholar]
- 42.Sasai K, Nodagashira M, Nishihara H, et al. Careful exclusion of non-neoplastic brain components is required for an appropriate evaluation of O6-methylguanine-DNA methyltransferase status in glioma. Am J Surg Pathol. 2008 doi: 10.1097/PAS.0b013e318164c3f0. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 43.Grasbon-Frodl EM, Kreth FW, Ruiter M, et al. Intratumoral homogeneity of MGMT promoter hypermethylation as demonstrated in serial stereotactic specimens from anaplastic astrocytomas and glioblastomas. Int J Cancer. 2007;121:2458–64. doi: 10.1002/ijc.23020. [DOI] [PubMed] [Google Scholar]
- 44.Bobola MS, Tseng SH, Blank A, et al. Role of O6-methylguanine-DNA methyltransferase in resistance of human brain tumor cell lines to the clinically relevant methylating agents temozolomide and streptozotocin. Clin Cancer Res. 1996;2:735–41. [PubMed] [Google Scholar]
- 45.Trivedi RN, Almeida KH, Fornsaglio JL, et al. The role of base excision repair in the sensitivity and resistance to temozolomide-mediated cell death. Cancer Res. 2005;65:6394–400. doi: 10.1158/0008-5472.CAN-05-0715. [DOI] [PubMed] [Google Scholar]