

Abstract
Activation of β-Catenin, the central effector of canonical Wnt pathway and a recognized oncogene, is implicated in hepatocellular carcinoma (HCC). Here, we examine N-nitrosodiethylamine (DEN)-induced tumorigenesis in hepatic β-catenin conditional knockout mice (β-cat KO). Only, male β-cat KO and age- and sex-matched littermate controls were given a single intraperitoneal DEN injection and followed for 6–12 months for hepatic tumors. Hepatic tumors were characterized for histology, proliferation, apoptosis, oxidative stress, and specific proteins by western blots, immunohistochemistry and coprecipitation studies. For in vivo tumor intervention studies, specific inhibitors were administered intraperitoneally or through drinking water. Intriguingly, β-cat KO mice show a paradoxical increase in the susceptibility to DEN-induced tumorigenesis. The accelerated tumorigenesis is due to increased injury and inflammation, unrestricted oxidative stress, fibrosis and compensatory increase in hepatocyte proliferation secondary to PDGFRα/phosphoinositide 3-kinase (PIK3CA)/Akt activation and c-Myc overexpression. In vitro suppression of β-catenin expression in hepatoma cells led to enhanced PDGFRα expression, which was abrogated in the presence of NF-κB inhibitor. Daily treatment of 6 months old DEN-exposed β-cat KO with PDGFRα inhibitor dramatically reduced tumor numbers and size. Inclusion of N-acetyl-L-cysteine (NAC), a known antioxidant and NF-κB-inhibitor, in the drinking water led to complete abolition of tumorigenesis in DEN-exposed β-cat KO. In conclusion, loss of β-catenin impairs the ability of liver to counteract DEN-induced oxidative stress and enhances tumorigenesis through PDGFRα/PIK3CA/Akt signaling. Blockade of PDGFRα or oxidative stress dramatically impacts β-catenin-deficient tumorigenesis. Also, hepatoma cells utilize PDGFRα/PIK3CA signaling as an escape mechanism following β-catenin suppression and their sequential suppression profoundly impedes tumor proliferation.
Keywords: Wnt signaling, HCC, PI3kinase, injury, fibrosis, regeneration
β-Catenin, the key mediator of canonical Wnt pathway, was originally identified as a binding partner of E-cadherin and actin cytoskeleton, thereby mediating intercellular adhesion (1, 2). β-Catenin has a core domain of twelve armadillo-repeats responsible for pertinent nuclear interactions and target gene expression (3). In a normal steady state, in the absence of Wnt, β-catenin is phosphorylated at amino-terminal serine and threonine residues and targeted for ubiquitination (4). The binding of Wnt proteins to its cell surface receptor Frizzled and co-receptor low-density lipoprotein-related protein-5/6 leads eventually to glycogen synthase kinase 3β (GSK3β) inactivation. β-Catenin dissociates from the degradation complex composed of adenomatous polyposis coli gene product (APC), axin, GSK3β and casein kinase α to translocate to nucleus to bind to lymphoid enhancer-binding factor/T-cell factor and transactivate target genes.
Presently, the significance of Wnt/β-catenin pathway is well-documented in cellular development, growth, survival, regeneration and self-renewal (5–7). β-Catenin is implicated in a variety of cancers, including hepatocellular carcinoma (HCC). Activating mutations in β-catenin gene (CTNNB1) were found in 70% of hepatoblastomas and 20–40% of HCC (8, 9). Preclinical models such as conditional APC deletion in mouse liver also exhibit hepatic tumors through β-catenin activation (10). Therefore, β-catenin is a recognized oncogene. With this background, we hypothesized that lack of β-catenin in hepatocytes might protect against chemical-induced carcinogenesis in liver.
While, global genetic knockout of β-catenin results in embryonic lethality caused by gastrulation defect (11), the field has had a major boost with the generation of the floxed β-catenin mice, which enables tissue specific deletion (12). To address the role of β-catenin in hepatocarcinogenesis, we used the hepatocyte-specific knockout mice (13), generated by interbreeding mice homozygous for floxed-β-catenin allele (Ex2-6) and mice expressing Cre recombinase under albumin promoter and enhancer (Alb-Cre mice) (14). Tumor development was studied in the Ctnnb1 loxp/loxp; Alb-Cre+/− or β-cat KO mice in response to N-nitrosodiethylamine (DEN), a well-known hepatocarcinogen (15). Intriguingly, we observed a paradoxical increase in susceptibility to DEN-induced tumors role in β-cat KO mice. The tumor promoting effect was associated with increased injury, inflammation and associated oxidative stress, along with compensatory regeneration that occurred through platelet-derived growth factor receptor-α (PDGFRα)-induced activation of Phosphoinositide 3-kinase (PIK3CA)/Akt pathway. Inhibition of PDGFRα or abrogation of oxidative stress led to dramatically reduced tumorigenesis. We also identify PDGFRα/PIK3CA as the ‘escape’ pathway following β-catenin inhibition and successive suppression of these pathways diminished hepatoma cell growth. These observations thus suggest an important role of β-catenin in maintaining redox homeostasis in the liver.
EXPERIMENTAL PROCEDURES
Animals
β-Cat KO (Ctnnb1loxp/loxp; Alb-Cre+/−) mice were described previously (13). Cre-Ctrl (Ctnnb1loxp/loxp; Alb-Cre−/−, Ctnnb1loxp/Wt; Alb-Cre+/−, and Ctnnb1loxp/Wt; Alb-Cre−/−) mice are referred to as controls (Cre-Ctrl). All experiments were performed under strict guidelines of the NIH and the Institutional Animal Use and Care Committee at the University of Pittsburgh.
Tumor induction and treatments
β-Cat KO (n=24) and Cre-Ctrl (n=24) male mice were injected i.p. with N-Nitrosodiethylamine (DEN; Sigma-Aldrich, Inc) at a dose of 5 μg/g body weight at postnatal day 14 (P14). Specifically, Cre-Ctrl mice used were of the following genotypes: Ctnnb1loxp/loxp; Alb-Cre−/− (n=6); Ctnnb1loxp/Wt; Alb-Cre+/− (n=7); and Ctnnb1loxp/Wt; Alb-Cre−/− (n=11). Mice were sacrificed when they showed signs of morbidity, which occurred inβ-cat KO mice only. At this time, DEN-exposed littermate Cre-Ctrl mice were also sacrificed for direct comparison of disease between the two genotypes. Liver tissues were collected for histology and protein analysis. In the N-acetyl-L-cysteine (NAC) treatment group, after DEN injection at P14, both β-cat KO and Cre-Ctrl male mice were given NAC (Sigma) water at a dose of 2 mg/g body weight from day 25 to 7–8 months, and sacrificed. In the Gleevec (STI-571, Imatinib; Cayman Chemicals) treatment group, 6-month-old DEN-injected β-cat KO male mice were given Gleevec (dissolved in dH2O) at daily dose of 50 mg/kg by i.p. injection for 15 days, and sacrificed for liver analysis.
Oxidative stress analysis
Oxidized (GSSG) and total (GSH) glutathione content were measured using an assay kit (Cayman chemicals) and ratio presented. Lipid peroxidation was determined by measuring malondialdehyde (MDA) level using an assay kit (OxisResearch). 8-hydroxy-2’-deoxyguanosine (8-OHdG) levels in genomic DNA isolated from liver tissues were analyzed as described previously (16). The ratio of 8-OHdG/2-dG was presented.
Statistical analysis
Data are presented as the mean ± SE. Cultures were done in triplicates and repeated twice. Data were analyzed by the student t test or ANOVA, and p<0.05 were considered statistically significant.
Additional methods are included as an online supplement.
RESULTS
β-Catenin loss in hepatocytes promotes DEN-induced hepatocarcinogenesis
A single injection of DEN was administered at postnatal day 14 (P14) to β-cat KO and Cre-Ctrl male mice at a dose of 5 mg/kg. Comparable levels of CYP2E1, an enzyme essential for bioactivation of DEN, are observed in both genotypes at this stage (data not shown), while its levels are downregulated in 4–6-week old β-cat KO mice as reported previously and based on the DNA excision kinetics of albumin-cre transgene (13, 14). In response to DEN and absence of β-catenin in hepatocytes, mice showed early signs of morbidity with tumors appearing and progressing more rapidly (Fig.1). The β-cat KO mice often showed hunched posture and limited physical activity. Upon autopsy, the β-cat KO mice show hepatic tumors and signs of upper gastrointestinal obstruction observed as gross distention of stomach possibly due to lymph node involvement at porta hepatis. Several mice also showed development of pulmonary metastasis. Higher percentage of β-cat KO mice showed macroscopic tumors as compared to Cre-Ctrl mice (Fig. 1A). Cumulative incidence rate for β-cat KO and Cre-Ctrl was calculated as percentage of the total numbers of mice displaying tumors at or before a specific time point over the total number of mice in each experimental group. Cumulative incidence rate of tumors in β-cat KO was significantly greater than Cre-Ctrl at six months and all later time points (33% versus 8%, 42% versus 8%, 50% versus 17%, 71% versus 38%, 83% versus 46%, and 92% versus 46%, all p<0.05). By 13 months, most β-cat KO mice developed tumors (22/24, >90%) as compared to around <50% (11/24) showed disease in the controls (Fig.1B). There were no differences in appearance of tumors among various control genotypes, which exhibited lower cumulative incidence rates than β-cat KO at all times (Fig. 1C). The tumors were significantly larger in β-cat KO livers than Cre-Ctrl livers at 6–8 and 12 month, respectively (p<0.05 and p<0.01, Fig.1D). β-Cat KO livers also showed 3- and 2-fold higher numbers of tumor nodules at 6–8 and 12 month respectively, than time-matched Cre-Ctrl (p<0.05, Fig.1E). Notably, by immunohistochemistry (IHC) we found most tumors to be α-fetoprotein (AFP) positive in β-cat KO livers, and negative in Cre-Ctrl livers (Fig. 1F). This suggests an earlier progression of substantial numbers of tumors in β-cat KO livers to HCC, than the age-matched Cre-Ctrls that showed AFP-negative hepatic adenomas or dysplastic foci (Fig.1D).
Figure 1. Enhanced tumor development in the β-cat KO mice.

Greater percentages of β-cat KO mice show evidence of tumorigenesis than cre-ctrl at all time points (A). Higher cumulative incidence rate of tumors is observed in β-cat KO mice (n=24) than Cre-Ctrl mice (n=24) after DEN (B). All cre-ctrl genotypes showed lower rate of tumor development than β-cat KO (C). Higher average area of tumors per square centimeter of the section (D) and higher numbers of tumors (E) in β-cat KO (n≥3/genotype/time-point). H&E and IHC for AFP in β-cat KO and Cre-Ctrl livers (F). Results in (A–C) are average ± S.E. *, p<0.05, **, p<0.01.
β-Catenin knockout livers after DEN display higher genotoxic injury, inflammation and oxidative stress leading to greater apoptosis, fibrosis and compensatory hepatocyte proliferation
DEN is known to induce DNA damage, injury and inflammation, which lead to excessive oxidative damage and HCC (17). We tested oxidative stress through multiple modalities in the β-cat KO and Cre-Ctrl livers at 6–8 months after DEN-exposure. The ratio of total to oxidized glutathione (GSH/GSSG) was significantly decreased in β-cat KO livers compared to Cre-Ctrl after DEN treatment (Fig. 2A). Lipid peroxidation, as assessed by MDA content, was also significantly higher in β-cat KO livers after DEN (Fig. 2A). 8-OHdG, an oxidized derivative of deoxyguanosine, is a major product of DNA oxidation (18) was also augmented in β-cat KO livers after DEN exposure (Fig 2A). It is important to note that no appreciable differences in oxidative stress in non-DEN exposed age-matched β-cat KO and Cre-ctrl livers were observed as indicated by insignificant baseline differences in 8-OHdG (Fig. 2A).
Figure 2. Loss of β-catenin enhances oxidative stress, fibrosis, inflammation, cell apoptosis, and compensatory proliferation after DEN exposure.

A. Increased ratio of GSSG/GSH (left), increased malondialdehyde adducts (MDA) (middle) and high 8-hydroxy-2’-deoxyguanosine/2’-deoxyguanosine ratio (right) in 6- to 8-month-old mice after DEN injection (n≥5/group). B. Increased numbers of CD14- and CD45-positive cells in DEN-injected mice β-cat KO livers at 6 months. C. IHC for TUNEL. D. TUNEL positive cells were counted in 10 random fields at 50X magnification in 3 livers from each group. E. Fibrosis and cell proliferation was assessed by Masson’s Trichrome staining and IHC for PCNA, respectively. *p<0.05, **p<0.01
To address the mechanism of sustained oxidative stress, we examined β-cat KO livers for any evidence of ongoing inflammation. As seen at 6 months, notable leukocyte infiltration and Kupffer cell activation is observed by IHC for CD45- and CD14 in DEN-exposed β-cat KO as compared to controls (Fig. 2B). Also evident were higher numbers of apoptotic nuclei as seen by IHC for TUNEL in β-cat KO at 6 and 8 months only (Fig. 2C), which were significant (Fig. 2D). The ongoing injury was also reflected by aberrant wound healing response in the form of fibrosis, which was observed by Masson’s trichrome staining (Fig 2E).
Based on the potential of liver to regenerate, we speculated compensatory increase in hepatocyte proliferation as a consequence of enhanced genotoxic injury in β-cat KO in response to DEN-exposure. Previously, others and we reported liver regeneration in β-cat KO mice albeit after a delay of 24 hours (13, 19). Indeed, 6-month-old β-cat KO mice after DEN show considerable numbers of PCNA-positive hepatocytes (Fig. 2F).
Thus, β-catenin-deficient livers in response to DEN show inability to resolve the genotoxic injury and oxidative stress as a result of ongoing inflammation, apoptosis and fibrosis, which is followed by compensatory regeneration and tumorigenesis.
DEN induces hepatocarcinogenesis in absence of β-catenin through PIK3CA/Akt pathway
We next explored the molecular basis for increased tumor cell proliferation in β-cat KO mice in response to DEN. We verified that the tumors in β-cat KO livers were β-catenin-negative and negative for its target glutamine synthetase (GS) by both IHC and western blot (WB). This was in contrast to tumors in Cre-Ctrl, which mostly expressed membranous β-catenin and centrizonal GS (Fig. 3B) and only rarely showed cytoplasmic and nuclear β-catenin and a more widespread GS localization (data not shown). While cyclin-D1 levels were modestly lower in β-cat KO, DEN-exposure led to increases in proto-oncogene c-Myc and cyclooxygenase-2 (cox-2) (Fig. 3B), suggesting a β-catenin-independent mechanism of c-Myc and cox-2 regulation.
Figure 3. Loss of β-catenin triggers activation of PIK3CA/Akt pathway after DEN exposure.

IHC for β-catenin and GS in liver tumors (Tu) in DEN-treated β-cat KO and Cre-Ctrl animals (A). Representative WB of whole-cell lysates from β-cat KO and Cre-Ctrl livers at 4 or 5, 6 and 8 months with or without DEN treatment for various proteins as indicated (B, C, D). IHC for phospho-Akt (Thr 308) in β-cat KO and Cre-Ctrl tumors (Tu) at 6 months after DEN treatment (E).
As identified in untreated β-cat KO mice (13), we continued to observe an increase in phospho-GSK3β-Ser9 after DEN-exposure (Fig. 3B). Given that Akt is a well-known upstream modulator of GSK3β, we examined total and phospho-Akt-Thr308 levels, which were increased in β-cat KO livers in the presence or absence of DEN as compared to Cre-Ctrl (Fig. 3C–D), albeit the increase was more dramatic in DEN-samples. Also, phospho-Akt-Thr308 was evident in βcat KO tumors by IHC (Fig 3E). While total or phosphorylated levels of PDK1 and PIK3CA (p85 and p110), known upstream effectors of Akt, did not show any baseline differences in their protein expression between the β-cat KO and Cre-Ctrl, following DEN-exposure, noteworthy increases in phospho-PDK1-Ser241 and phosho-PIK3CA-p85-Tyr458/Tyr199 were evident in βcat KO livers (Fig 3C–D). Thus, β-catenin loss in liver leads to enhanced activation of PIK3CA/Akt pathway in response to DEN.
PDGFRα is the upstream effector of PIK3CA/Akt pathway in the absence of β-catenin in DEN-induced hepatocarcinogenesis
To address the mechanism of PIK3CA/Akt activation inβ-cat KO especially after DEN-exposure, we interrogated several receptor tyrosine kinases (RTK) (20), especially epidermal growth factor receptor (EGFR), hepatocyte growth factor (HGF) receptor-c-Met and PDGFRα, for their role in HCC (21). Intriguingly, decreased levels of HGFα, HGF precursor, EGFR and Met were evident in β-cat KO livers (Fig. 4A). However, we identified a dramatic increase in total PDGFRα protein and not PDGFRβ in DEN-exposed β-cat KO livers as compared to controls (Fig. 4B). Immunoprecipitation (IP) studies also revealed enhanced tyrosine-phosphorylated-PDGFRα in DEN-exposed β-cat KO livers (Fig. 4B). Also, IHC detected a noteworthy increase in cytoplasmic and membranous PDGFRα in tumor nodules in β-cat KO livers and not controls (Fig 4C). Similar analysis of samples from tumors in older βcat KO and cre-ctrl mice also showed greater PDGFRα and phospho-Akt (Thr-308) levels in βcat KO livers only (Fig. 4D). These results suggested PDGFRα to be the candidate upstream effector of PIK3CA/Akt pathway activation in DEN-induced carcinogenesis in β-cat KO livers.
Figure 4. PDGFRα is activated in DEN-treated β-cat KO mice & β-catenin deficient hepatoma cells secondary to NF-κB activation.
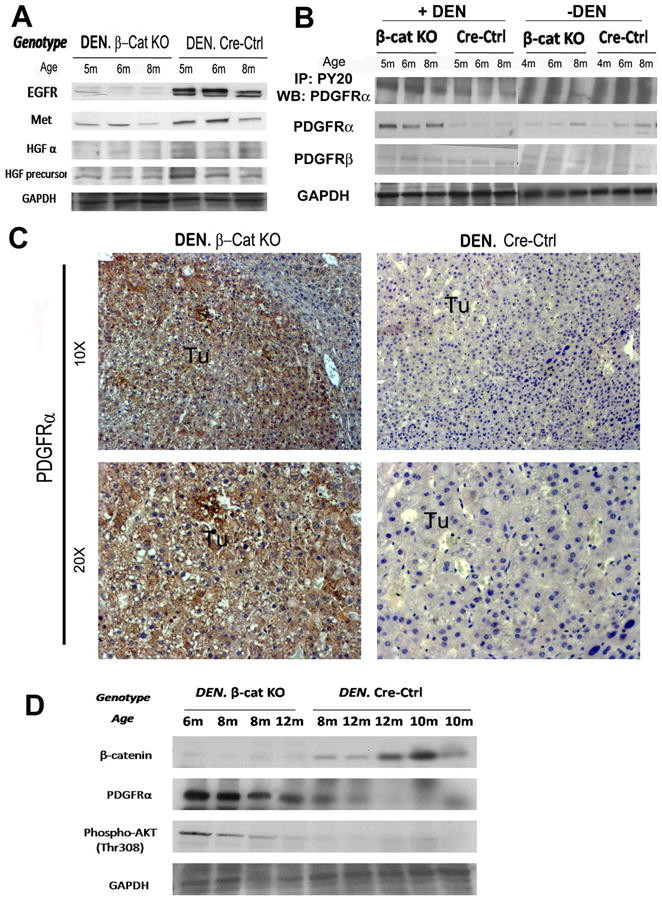
Representative WB of whole-cell lysates from β-cat KO and Cre-Ctrl livers with or without DEN exposure at 4 or 5, 6 and 8 months with antibodies as indicated (A, B). IHC for PDGFRα in β-cat KO and Cre-Ctrl tumors (Tu) at 6 month after DEN (C). Representative WB from whole cell lysates from β-cat KO and Cre-Ctrl livers with tumors at different ages with antibodies as indicated (D).
NF-κB signaling in the absence of β-catenin contributes to PDGFRα activation
To determine the mechanism of PDGFRα upregulation in the absence of β-catenin, we explored the possibility of establishing an in vitro model utilizing two human hepatoma cell lines- Hep3B and HepG2 cell. HepG2 cells, which harbor a truncated and constitutively active β-catenin exhibited low PDGFRα levels, and Hep3B cells, which harbors full-length, non-mutated and non-active β-catenin showed high endogenous PDGFRα expression by western blots (Fig. 5A). Next, we determined the impact of β-catenin suppression on PDGFRα levels. Hep3B and HepG2 cells transfected with either CTNNB1 siRNA (shown-Hep3B) or antisense (shown-HepG2) dramatically decreased β-catenin protein and led to concomitant increase in PDGFRα levels (Fig. 5B). These results were analogous to the in vivo observations and thus provided us a model to interrogate the mechanism of PDGFRα expression secondary to β-catenin loss.
Figure 5. β-Catenin suppression induced PDGFRα expression through NF-κB, and sequential inhibition of β-catenin and PDGFRα compromises hepatoma cell proliferation.

WB using lysates from untreated Hep3B and HepG2 cells (A), siRNA transfected-Hep3B (left) & β-catenin antisense-transfected HepG2 cells (right) (B). WB for NF-κB and its targets in β-cat KO and Cre-Ctrl livers with or without DEN treatment. (C). NF-κB inhibitor abolishes PDGFRα upregulation after β-catenin knockdown in Hep3B cells (D). A significant decrease in thymidine uptake by Hep3B cells after β-catenin siRNA (left) or antisense (right) transfection (p<0.05), which is further accentuated upon inclusion of STI-571 (p=0.01), when compared to respective controls (E).
Based on previously reported increase in PDGFRα gene expression in β-Cat KO livers especially after hepatectomy (13), and presence of NF-κB binding sites in PDGFRA promoter (22), we examined NF-κB status in β-cat KO and cre-ctrl livers with and without DEN. Representative data from 5–8-month old animals shows a mild increase in total levels of NF-κB subunit p65, and its target gene IκB-α and Traf1 in β-cat KO, however these differences along with changes in levels of NF-κB subunit p50 or NF-κB target Fas were statistically insignificant between the β-cat KO and Cre-Ctrl livers at baseline (Fig. 5C). However, protein levels of both p65 and p50, and targets IκB-α, Fas and Traf1 were induced in β-cat KO livers indicating NF-κB activation following DEN-exposure at the examined time-points (Fig. 5C).
We next determined the relevance of NF-κB activation in PDGFRα overexpression uponβ-catenin suppression in vitro. We simultaneously silenced β-catenin expression and NF-κB activity, the latter being achieved by an inhibitor that blocks translocation of NF-κB subunits (p65 and p50) into the nucleus (23). NF-κB blockade prevented PDGFRα increase following β-catenin suppression (Fig. 5D). Thus, NF-κB activation may be contributing to PDGFRα overexpression and enhanced DEN-induced hepatocarcinogenesis in β-cat KO mice.
PDGFRα inhibition affects tumor growth after β-catenin knockdown in hepatoma cells
Since PDGFRα activation is evident in vivo and in vitro following β-catenin loss or knockdown, respectively, we next asked if there are any biological consequences associated with such compensation. To address this we investigated the potential role of PDGFRα in cell proliferation following CTNNB1 knockdown in vitro. A single dose of STI-571 was used to inhibit PDGFRα signaling 24 hours after CTNNB1 siRNA (Fig. 5E) or antisense (Fig. 5F) treatment. While β-catenin suppression, or treatment with STI-571, independently decreased DNA synthesis, their sequential inhibition led to a greater reduction in thymidine incorporation (Fig. 5E–F). Thus PDGFRα promotes hepatoma growth especially in the event of β-catenin suppression,
PDGFRα inhibition affects in vivo tumor growth in DEN-exposed β-cat KO mice
To further address if PDGFRα activation accounts for enhanced susceptibility to DEN-induced HCC, DEN-injected 6 months old β-cat KO mice (n=4; 2 showing signs of morbidity) were administered 50 mg/kg/day of STI-571 for 15 days and sacrificed to determine tumor burden. Compared to the historic controls (n=5 at 6 months), decrease in tumor area by 2.5-fold, and tumor numbers by 3.5-fold, was evident in the treatment group (Fig. 6A & B). The STI-571 treated β-cat KO group showed tumor size and numbers comparable to DEN-exposed Cre-Ctrl mice at 6 months (n=4) (Fig. 6A & B). Liver tissue from the treatment and non-treated group were examined for expression of phospho-tyrosine and total PDGFRα, and c-Myc. The ratio of phospho- to total-PDGFRα, and total c-Myc levels were significantly decreased in the treatment group as untreated controls (Fig. 6C). Thus, PDGFRα activation plays an important role in DEN-induced tumorigenesis in β-cat KO mice.
Figure 6. Treatment with PDGFRα inhibitor and long-term feeding with NAC dramatically affects DEN-induced tumorigenesis in β-cat KO mice.

Percentage of area occupied by tumor (A) and tumor number per square centimeter (B) in DEN (n=5), DEN+STI-571-treated β-cat KO livers (n=4) and DEN-treated Cre-Ctrl mice (n=4). WB and IP studies identify protein changes (C) in DEN and DEN + STI-571- treated groups (left) and densitometric analysis shows a significant decrease in phospho-PDGFRα in experimental group (right). Tumor incidence (D) in β-cat KO & Cre-Ctrl mice with and without NAC at 8 month after DEN exposure. WB with lysates from livers of regular, DEN-injected and DEN + NAC treated β-cat KO mice at 8 month (E). A significant decrease in MDA content in DEN + NAC treated β-cat KO mice at 7–8 months as compared to untreated controls (F). *p<0.05
NAC, an antioxidant and NF-κB inhibitor, protects β-cat KO mice from DEN-induced hepatocarcinogenesis
We next tested if sustained oxidative stress may be an important contributor towards NF-κB–mediated PDGFRα/PIK3CA/Akt signaling and c-Myc overexpression leading to accelerated DEN-induced HCC in β-cat KO mice. We utilized N-acetyl-L-cysteine (NAC), a well-known ROS scavenger, which is also known to inhibit NF-κB activation (24, 25). Long-term feeding with NAC water from 25 days (P25) after birth to 7–8 month of age had a profound protective effect against tumorigenesis in β-cat KO mice, as seen by lack of any gross or microscopic disease (Fig. 6D). This was in stark contrast to untreated age-matched DEN-exposedβ-cat KO animals at 7–8 months, where 80% these animals showed tumors. A modest protective effect of NAC was also observed in DEN-induced carcinogenesis in the Cre-Ctrl mice group supporting global role of oxidative injury in this tumorigenesis model (Fig. 6D).
Whole cell liver homogenates from age-matched β-cat KO mice (with and without DEN injection) and DEN-treated-NAC-fed mice were subjected to molecular analysis. DEN injected β-cat KO mice show PDGFRα, Akt and c-Myc upregulation in response to DEN treatment along with evidence of NF-κB activation as evident by Fas and TRAF1 overexpression (Fig 6E). Following NAC-treatment, these mice show a noteworthy decrease in Fas and TRAF1, along with decreased levels of PDGFRα, Akt and c-Myc (Fig. 6E).
Since, we hypothesized that β-cat KO livers were incapable of alleviating increased oxidative stress brought about by DEN and formed the basis of NF-κB driven PDGFRα/PIK3CA activation, we next asked if NAC protected against tumorigenesis through its anti-oxidant properties. Ten-fold reduction in MDA adducts in NAC-fed, DEN-exposed β-cat KO livers demonstrates a robust decrease in oxidative stress (Fig. 6F). Taken together, these data demonstrate the protective effect of NAC on DEN-induced hepatocarcinogenesis especially in the absence of β-catenin, maybe through its antioxidant and NF-κB-inhibitory effect in addition to other mechanisms leading eventually to diminished PDGFRα-PIK3CA/Akt signaling.
DISCUSSION
Around 80% of HCC in patients is frequently observed in the backdrop of chronic liver injury due to etiologies such as viral hepatitis and alcoholic liver disease (26). Chronic insult to the liver leads to inflammation, oxidative stress, hepatocyte death, hepatic fibrosis and regeneration. And due to reasons still unclear some of these responses although meant to be protective and essential for maintenance of liver functions, are also the basis of neoplastic transformation. The dysplasia in regenerating nodules leads eventually to HCC in cirrhotic liver as the cells accumulate chromosomal aberrations and genetic and epigenetic mutations, eventually leading to disproportionate activation of cytoprotective and proliferative signaling. Many pathways, broadly categorized into Ras/MAPK, PIK3CA/AKT and Wnt/β-catenin signaling have been shown to be of relevance in HCC (27). β-Catenin, the chief effector of the Wnt signaling is a widely accepted oncogene due to its broad implications in variety of cancers including around 20–40% of all HCC (28). Interestingly, we report a paradoxical rise in the susceptibility of β-catenin conditional knockout mice to DEN-induced carcinogenesis. Incidentally, hepatocarcinogenesis in this scenario was reminiscent of clinical HCC with evidence of inflammation, injury, fibrosis and regeneration.
DEN is a well-known carcinogen to study HCC in rodent models. It is known to induce DNA damage, mutations, oxidative stress and hepatocyte apoptosis (Fig. 7A) (15, 17). Presence of DEN in conjunction with β-catenin loss led to severe liver injury in the form of inflammation, apoptosis and fibrosis, which were chiefly due to sustained oxidative stress in the absence of β-catenin. The mechanism of unimpeded oxidative stress in the absence of β-catenin maybe multifactorial and is currently being investigated. β-Catenin is known to regulate expression of several glutathione S-transferases involved in counteracting oxidative stress (29). In addition, several cytochrome P540s are misregulated in the absence of β-catenin (13, 30, 31).β-Catenin is also known to interact with FOXO during altered redox state of the cell to dictate expression of genes critical for counteracting oxidative stress signaling, which might be compromised in the absence of β-catenin (32). Thus, β-catenin-deficient hepatocytes are incapable of adequately adapting to oxidative stress initiated by DEN-induced genotoxic injury and sustained by chronic inflammation, activation of Kupffer and stellate cells resulting in apoptosis and fibrosis. These results were further substantiated when NAC, a known antioxidant, completely prevented DEN-induced HCC in β-Cat KO.
Figure 7. Cellular and molecular basis of HCC in β-catenin sufficient and insufficient state.

(A) DEN induces DNA damage and HCC through various signaling mechanisms including β-catenin. In absence of β-catenin, enhanced HCC is secondary to sustained oxidative stress, inflammation, apoptosis and injury that in turn promote cell proliferation in the anomalous microenvironment.
(B) In the absence of β-catenin, oxidative stress initiated by DEN-induced genotoxic injury and sustained by hepatic inflammation, goes unchecked and escalates. The inability of β-catenin-deficient hepatocytes to counteract oxidative stress might be through failure of FOXO-β-catenin interactions and through misexpression of various GSTs and P450s, traditionally regulated by β-catenin signaling in the liver. The increased injury induces cytoprotective NF-κB signaling, which might also be complemented by lack of β-catenin-p65 interactions that are known to sequester NF-κB to prevent its activation. This might in turn play a role in PDGFRα overexpression. In the backdrop of injury, inflammation and activation of nonparenchymal cells, PDGF production induces activation of PDGFRα and downstream PIK3CA signaling, which through increased expression of targets such as c-Myc and cox-2, leads to tumorigenesis.
To understand the molecular basis pertinent to the aforementioned cellular changes, we were interested in signaling pathways specifically implicated in cell proliferation in the absence of β-catenin. Previously, we reported a 10-fold upregulation of PDGFRα in β-cat KO mice compared to Cre-Ctrl after partial hepatectomy (13). Concordant with these findings, DEN-treated β-cat KO livers mice exhibited increased PDGFRα expression and activation (Fig. 7B). The mechanism of increased PDGFRα gene expression in β-cat KO livers is not fully clear but appears to be at least in part be regulated by NF-κB activation. NF-κB binding sites have been previously identified in PDGFRA promoter (22). NF-κB has also been shown to mediate PDGFRα upregulation in response to IL-1β (33). Of relevance in our model is also a previously reported physical interaction between β-catenin and NF-κB (p65), which might be responsible for a low threshold of NF-κB activation in β-cat KO livers after DEN-exposure leading to PDGFRA overexpression (34, 35). It is also relevant to point out that we were unable to demonstrate unequivocal NK-κB activation in β-cat KO at baseline, which might be due existence of feedback inhibition through its targets such as IκB-α and other regulatory mechanisms.
PDGFRα is involved in tumor growth, angiogenesis and maintenance of tumor microenvironment including in HCC (36–38). Liver-specific transgenic mice overexpressing PDGF-CC, a ligand of PDGFRα, develops cirrhosis and HCC (39). β-Cat KO livers after DEN-exposure exhibited higher total and tyrosine-phosphorylated PDGFRα indicating ligand-receptor interaction. It is likely that PDGFs released from various inflammatory and resident nonparenchymal cells in the liver may be responsible for PDGFRα activation (Fig. 7B) (40). Further analysis revealed a robust activation of PIK3CA/Akt pathway secondary in response to PDGFRα activation (Fig. 7B). PDGFRα is a known upstream effector of PIK3CA/Akt pathway (20). Because of a critical role of EGFR in HCC, it was interesting to note its decreased levels inβ-cat KO livers, however this upstream effector of PIK3CA was previously shown to be a target of Wnt/β-catenin signaling (41). Similarly, HGF receptor c-Met is known to activate PIK3CA pathway and is highly relevant in HCC. However it was downregulated in β-cat KO livers and has also been previously reported as a target of Wnt signaling (42). The upregulation of the proto-oncogene c-Myc, is intriguing since it is a known target of β-catenin with a known role in HCC (43). Likewise, cox-2 is an essential enzyme in prostaglandins synthesis, promotes inflammation and liver cancer and is a target of Wnt pathway (44). However, PIK3CA/Akt signaling has been shown to induce expression of c-Myc and cox-2 and in turn to promote cell survival and proliferation (45, 46). An overall caveat in the western blot analysis is a potential for selection bias since isolated proteins utilized were from KO mice that showed advanced disease as compared to control livers, which did show microscopic disease but not to the same extent. However, immunohistochemistry did display localization of certain proteins such as PDGFRα and phosho-Akt in KO tumors only.
The role of oxidative stress and PDGFRα/PIK3CA/Akt signaling in tumorigenesis in βcat KO mice was verified by successful chemoprevention of DEN-induced HCC by the use of NAC, a widely recognized antioxidant and also a known NF-κB inhibitor (24, 25). NAC ameliorated oxidative stress and inhibited NF-κB activation, which led to decreased PDGFRα, Akt and c-Myc levels and prevented tumorigenesis. The dexterity of NAC in counteracting oxidative stress has now been reported in several studies (17, 47–49). The role of PDGFRα in tumorigenesis in β-cat KO livers was also explicit as validated by reduction in tumor number and size after 15-day treatment with STI-571, which inhibits PDGFR signaling, especially PDGFRα in the existing model, which in turn was accompanied by decrease in Akt and c-Myc expression. It is important to point out that one cannot rule out a potential selection bias in the STI-571 study due to the use of historical controls owing to limited availability of the β-cat KO mice. Specifically, all five animals used as historical controls from DEN-exposed β-cat KO group did display signs of morbidity, whereas only two of the four mice in the experimental group showed any signs of morbidity, when treatment was initiated. Hence the decrease tumor burden in the experimental group could be due to selection bias, albeit in vitro studies with PDGFRα inhibitor support an important role of PDGFRα in β-catenin-deficient carcinogenesis. However additional studies will be necessary to strengthen this observation.
Clearly, β-catenin activation due to mutations in CTNNB1 and other mechanisms play a significant role in tumor growth and progression in a subset of HCC (28). However, the current study also identifies a role of β-catenin as a potential ‘tumor suppressor’ through its antioxidant properties, especially in patients advanced hepatic fibrosis. Thus, in future, as therapeutic inhibition of β-catenin becomes a reality, it maybe relevant to carefully select patients through verification of β-catenin activation in tumors, although additional studies will be essential to substantiate this concern. Another relevant outcome of the study was the observation that not only was PDGFRα upregulation evident in β-cat KO livers, but it was apparent anytime β-catenin was pharmacologically suppressed in hepatoma cells. This implies that PDGFRα/PIK3CA signaling maybe the major ‘escape pathway’ following β-catenin inhibition and maybe of significance if and when therapeutic inhibition of Wnt/β-catenin pathway becomes a reality in HCC (50, 51). However, additional studies will be essential to further validate these results in suitable in vivo models.
Supplementary Material
Acknowledgments
Grant Support: XFZ was supported in part by funding from the China Scholarship Council. This study was funded by NIH grants 1R01DK62277 and 1R01CA124414 to SPSM and by Rango’s Fund for the Enhancement of Pathology Research.
Abbreviations
- TG
transgenic
- WT
wild-type
- PHx
partial hepatectomy
- HGF
hepatocyte growth factor
- EGF
epidermal growth factor
- PCNA
proliferating cell nuclear antigen
- IOD
integrated optical density
- GS
glutamine synthetase
- HCC
hepatocellular cancer
- IHC
immunohistochemistry
- WB
western blot
- IP
immunoprecipitation
- PIK3CA
Phosphoinositide 3-kinase
- PDK1
phosphoinositide-dependent protein kinase
References
- 1.Hulsken J, Birchmeier W, Behrens J. E-cadherin and APC compete for the interaction with beta-catenin and the cytoskeleton. J Cell Biol. 1994;127:2061–2069. doi: 10.1083/jcb.127.6.2061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.McCrea PD, Turck CW, Gumbiner B. A homolog of the armadillo protein in Drosophila (plakoglobin) associated with E-cadherin. Science. 1991;254:1359–1361. doi: 10.1126/science.1962194. [DOI] [PubMed] [Google Scholar]
- 3.Peifer M, Berg S, Reynolds AB. A repeating amino acid motif shared by proteins with diverse cellular roles. Cell. 1994;76:789–791. doi: 10.1016/0092-8674(94)90353-0. [DOI] [PubMed] [Google Scholar]
- 4.Nelson WJ, Nusse R. Convergence of Wnt, beta-catenin, and cadherin pathways. Science. 2004;303:1483–1487. doi: 10.1126/science.1094291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Clevers H. Wnt/beta-catenin signaling in development and disease. Cell. 2006;127:469–480. doi: 10.1016/j.cell.2006.10.018. [DOI] [PubMed] [Google Scholar]
- 6.Peifer M, Polakis P. Wnt signaling in oncogenesis and embryogenesis--a look outside the nucleus. Science. 2000;287:1606–1609. doi: 10.1126/science.287.5458.1606. [DOI] [PubMed] [Google Scholar]
- 7.Willert K, Brown JD, Danenberg E, Duncan AW, Weissman IL, Reya T, Yates JR, 3rd, et al. Wnt proteins are lipid-modified and can act as stem cell growth factors. Nature. 2003;423:448–452. doi: 10.1038/nature01611. [DOI] [PubMed] [Google Scholar]
- 8.de La Coste A, Romagnolo B, Billuart P, Renard CA, Buendia MA, Soubrane O, Fabre M, et al. Somatic mutations of the beta-catenin gene are frequent in mouse and human hepatocellular carcinomas. Proc Natl Acad Sci U S A. 1998;95:8847–8851. doi: 10.1073/pnas.95.15.8847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wong CM, Fan ST, Ng IO. beta-Catenin mutation and overexpression in hepatocellular carcinoma: clinicopathologic and prognostic significance. Cancer. 2001;92:136–145. doi: 10.1002/1097-0142(20010701)92:1<136::aid-cncr1301>3.0.co;2-r. [DOI] [PubMed] [Google Scholar]
- 10.Colnot S, Decaens T, Niwa-Kawakita M, Godard C, Hamard G, Kahn A, Giovannini M, et al. Liver-targeted disruption of Apc in mice activates beta-catenin signaling and leads to hepatocellular carcinomas. Proc Natl Acad Sci U S A. 2004;101:17216–17221. doi: 10.1073/pnas.0404761101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Haegel H, Larue L, Ohsugi M, Fedorov L, Herrenknecht K, Kemler R. Lack of beta-catenin affects mouse development at gastrulation. Development. 1995;121:3529–3537. doi: 10.1242/dev.121.11.3529. [DOI] [PubMed] [Google Scholar]
- 12.Brault V, Moore R, Kutsch S, Ishibashi M, Rowitch DH, McMahon AP, Sommer L, et al. Inactivation of the beta-catenin gene by Wnt1-Cre-mediated deletion results in dramatic brain malformation and failure of craniofacial development. Development. 2001;128:1253–1264. doi: 10.1242/dev.128.8.1253. [DOI] [PubMed] [Google Scholar]
- 13.Tan X, Behari J, Cieply B, Michalopoulos GK, Monga SP. Conditional deletion of beta-catenin reveals its role in liver growth and regeneration. Gastroenterology. 2006;131:1561–1572. doi: 10.1053/j.gastro.2006.08.042. [DOI] [PubMed] [Google Scholar]
- 14.Postic C, Magnuson MA. DNA excision in liver by an albumin-Cre transgene occurs progressively with age. Genesis. 2000;26:149–150. doi: 10.1002/(sici)1526-968x(200002)26:2<149::aid-gene16>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]
- 15.Verna L, Whysner J, Williams GM. N-nitrosodiethylamine mechanistic data and risk assessment: bioactivation, DNA-adduct formation, mutagenicity, and tumor initiation. Pharmacol Ther. 1996;71:57–81. doi: 10.1016/0163-7258(96)00062-9. [DOI] [PubMed] [Google Scholar]
- 16.Harvilchuck JA, Pu X, Klaunig JE, Carlson GP. Indicators of Oxidative Stress and Apoptosis in Mouse Whole Lung and Clara Cells Following Exposure to Styrene and its Metabolites. Toxicology. 2009 doi: 10.1016/j.tox.2009.08.001. [DOI] [PubMed] [Google Scholar]
- 17.Maeda S, Kamata H, Luo JL, Leffert H, Karin M. IKKbeta couples hepatocyte death to cytokine-driven compensatory proliferation that promotes chemical hepatocarcinogenesis. Cell. 2005;121:977–990. doi: 10.1016/j.cell.2005.04.014. [DOI] [PubMed] [Google Scholar]
- 18.de Souza-Pinto NC, Eide L, Hogue BA, Thybo T, Stevnsner T, Seeberg E, Klungland A, et al. Repair of 8-oxodeoxyguanosine lesions in mitochondrial dna depends on the oxoguanine dna glycosylase (OGG1) gene and 8-oxoguanine accumulates in the mitochondrial dna of OGG1-defective mice. Cancer Res. 2001;61:5378–5381. [PubMed] [Google Scholar]
- 19.Sekine S, Gutierrez PJ, Lan BY, Feng S, Hebrok M. Liver-specific loss of beta-catenin results in delayed hepatocyte proliferation after partial hepatectomy. Hepatology. 2007;45:361–368. doi: 10.1002/hep.21523. [DOI] [PubMed] [Google Scholar]
- 20.Hunter T. Signaling--2000 and beyond. Cell. 2000;100:113–127. doi: 10.1016/s0092-8674(00)81688-8. [DOI] [PubMed] [Google Scholar]
- 21.Minguez B, Tovar V, Chiang D, Villanueva A, Llovet JM. Pathogenesis of hepatocellular carcinoma and molecular therapies. Curr Opin Gastroenterol. 2009;25:186–194. doi: 10.1097/MOG.0b013e32832962a1. [DOI] [PubMed] [Google Scholar]
- 22.Kitami Y, Inui H, Uno S, Inagami T. Molecular structure and transcriptional regulation of the gene for the platelet-derived growth factor alpha receptor in cultured vascular smooth muscle cells. J Clin Invest. 1995;96:558–567. doi: 10.1172/JCI118068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Condello S, Caccamo D, Curro M, Ferlazzo N, Parisi G, Ientile R. Transglutaminase 2 and NF-kappaB interplay during NGF-induced differentiation of neuroblastoma cells. Brain Res. 2008;1207:1–8. doi: 10.1016/j.brainres.2008.02.044. [DOI] [PubMed] [Google Scholar]
- 24.De Flora S, Izzotti A, D'Agostini F, Balansky RM. Mechanisms of N-acetylcysteine in the prevention of DNA damage and cancer, with special reference to smoking-related end-points. Carcinogenesis. 2001;22:999–1013. doi: 10.1093/carcin/22.7.999. [DOI] [PubMed] [Google Scholar]
- 25.Schreck R, Rieber P, Baeuerle PA. Reactive oxygen intermediates as apparently widely used messengers in the activation of the NF-kappa B transcription factor and HIV-1. EMBO J. 1991;10:2247–2258. doi: 10.1002/j.1460-2075.1991.tb07761.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Farazi PA, DePinho RA. Hepatocellular carcinoma pathogenesis: from genes to environment. Nat Rev Cancer. 2006;6:674–687. doi: 10.1038/nrc1934. [DOI] [PubMed] [Google Scholar]
- 27.Villanueva A, Newell P, Chiang DY, Friedman SL, Llovet JM. Genomics and signaling pathways in hepatocellular carcinoma. Semin Liver Dis. 2007;27:55–76. doi: 10.1055/s-2006-960171. [DOI] [PubMed] [Google Scholar]
- 28.Monga SP. Role of Wnt/beta-catenin signaling in liver metabolism and cancer. Int J Biochem Cell Biol. 2009 doi: 10.1016/j.biocel.2009.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tan X, Yuan Y, Zeng G, Apte U, Thompson MD, Cieply B, Stolz DB, et al. Beta-catenin deletion in hepatoblasts disrupts hepatic morphogenesis and survival during mouse development. Hepatology. 2008;47:1667–1679. doi: 10.1002/hep.22225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Loeppen S, Koehle C, Buchmann A, Schwarz M. A beta-catenin-dependent pathway regulates expression of cytochrome P450 isoforms in mouse liver tumors. Carcinogenesis. 2005;26:239–248. doi: 10.1093/carcin/bgh298. [DOI] [PubMed] [Google Scholar]
- 31.Sekine S, Lan BY, Bedolli M, Feng S, Hebrok M. Liver-specific loss of beta-catenin blocks glutamine synthesis pathway activity and cytochrome p450 expression in mice. Hepatology. 2006;43:817–825. doi: 10.1002/hep.21131. [DOI] [PubMed] [Google Scholar]
- 32.Essers MA, de Vries-Smits LM, Barker N, Polderman PE, Burgering BM, Korswagen HC. Functional interaction between beta-catenin and FOXO in oxidative stress signaling. Science. 2005;308:1181–1184. doi: 10.1126/science.1109083. [DOI] [PubMed] [Google Scholar]
- 33.Lindroos PM, Rice AB, Wang YZ, Bonner JC. Role of nuclear factor-kappa B and mitogen-activated protein kinase signaling pathways in IL-1 beta-mediated induction of alpha-PDGF receptor expression in rat pulmonary myofibroblasts. J Immunol. 1998;161:3464–3468. [PubMed] [Google Scholar]
- 34.Deng J, Miller SA, Wang HY, Xia W, Wen Y, Zhou BP, Li Y, et al. beta-catenin interacts with and inhibits NF-kappa B in human colon and breast cancer. Cancer Cell. 2002;2:323–334. doi: 10.1016/s1535-6108(02)00154-x. [DOI] [PubMed] [Google Scholar]
- 35.Du Q, Zhang X, Cardinal J, Cao Z, Guo Z, Shao L, Geller DA. Wnt/beta-catenin signaling regulates cytokine-induced human inducible nitric oxide synthase expression by inhibiting nuclear factor-kappaB activation in cancer cells. Cancer Res. 2009;69:3764–3771. doi: 10.1158/0008-5472.CAN-09-0014. [DOI] [PubMed] [Google Scholar]
- 36.Oseini AM, Roberts LR. PDGFRalpha: a new therapeutic target in the treatment of hepatocellular carcinoma? Expert Opin Ther Targets. 2009;13:443–454. doi: 10.1517/14728220902719233. [DOI] [PubMed] [Google Scholar]
- 37.Stock P, Monga D, Tan X, Micsenyi A, Loizos N, Monga SP. Platelet-derived growth factor receptor-alpha: a novel therapeutic target in human hepatocellular cancer. Mol Cancer Ther. 2007;6:1932–1941. doi: 10.1158/1535-7163.MCT-06-0720. [DOI] [PubMed] [Google Scholar]
- 38.Zhang T, Sun HC, Xu Y, Zhang KZ, Wang L, Qin LX, Wu WZ, et al. Overexpression of platelet-derived growth factor receptor alpha in endothelial cells of hepatocellular carcinoma associated with high metastatic potential. Clin Cancer Res. 2005;11:8557–8563. doi: 10.1158/1078-0432.CCR-05-0944. [DOI] [PubMed] [Google Scholar]
- 39.Campbell JS, Hughes SD, Gilbertson DG, Palmer TE, Holdren MS, Haran AC, Odell MM, et al. Platelet-derived growth factor C induces liver fibrosis, steatosis, and hepatocellular carcinoma. Proc Natl Acad Sci U S A. 2005;102:3389–3394. doi: 10.1073/pnas.0409722102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wong L, Yamasaki G, Johnson RJ, Friedman SL. Induction of beta-platelet-derived growth factor receptor in rat hepatic lipocytes during cellular activation in vivo and in culture. J Clin Invest. 1994;94:1563–1569. doi: 10.1172/JCI117497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tan X, Apte U, Micsenyi A, Kotsagrelos E, Luo JH, Ranganathan S, Monga DK, et al. Epidermal growth factor receptor: a novel target of the Wnt/beta-catenin pathway in liver. Gastroenterology. 2005;129:285–302. doi: 10.1053/j.gastro.2005.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Boon EM, van der Neut R, van de Wetering M, Clevers H, Pals ST. Wnt signaling regulates expression of the receptor tyrosine kinase met in colorectal cancer. Cancer Res. 2002;62:5126–5128. [PubMed] [Google Scholar]
- 43.Thorgeirsson SS, Factor VM, Snyderwine EG. Transgenic mouse models in carcinogenesis research and testing. Toxicol Lett. 2000;112–113:553–555. doi: 10.1016/s0378-4274(99)00224-6. [DOI] [PubMed] [Google Scholar]
- 44.Breinig M, Schirmacher P, Kern MA. Cyclooxygenase-2 (COX-2)--a therapeutic target in liver cancer? Curr Pharm Des. 2007;13:3305–3315. doi: 10.2174/138161207782360627. [DOI] [PubMed] [Google Scholar]
- 45.Rodriguez-Barbero A, Dorado F, Velasco S, Pandiella A, Banas B, Lopez-Novoa JM. TGF-beta1 induces COX-2 expression and PGE2 synthesis through MAPK and PI3K pathways in human mesangial cells. Kidney Int. 2006;70:901–909. doi: 10.1038/sj.ki.5001626. [DOI] [PubMed] [Google Scholar]
- 46.Wang GL, Iakova P, Wilde M, Awad S, Timchenko NA. Liver tumors escape negative control of proliferation via PI3K/Akt-mediated block of C/EBP alpha growth inhibitory activity. Genes Dev. 2004;18:912–925. doi: 10.1101/gad.1183304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hanczko R, Fernandez DR, Doherty E, Qian Y, Vas G, Niland B, Telarico T, et al. Prevention of hepatocarcinogenesis and increased susceptibility to acetaminophen-induced liver failure in transaldolase-deficient mice by N-acetylcysteine. J Clin Invest. 2009;119:1546–1557. doi: 10.1172/JCI35722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Reliene R, Fischer E, Schiestl RH. Effect of N-acetyl cysteine on oxidative DNA damage and the frequency of DNA deletions in atm-deficient mice. Cancer Res. 2004;64:5148–5153. doi: 10.1158/0008-5472.CAN-04-0442. [DOI] [PubMed] [Google Scholar]
- 49.Takami T, Kaposi-Novak P, Uchida K, Gomez-Quiroz LE, Conner EA, Factor VM, Thorgeirsson SS. Loss of hepatocyte growth factor/c-Met signaling pathway accelerates early stages of N-nitrosodiethylamine induced hepatocarcinogenesis. Cancer Res. 2007;67:9844–9851. doi: 10.1158/0008-5472.CAN-07-1905. [DOI] [PubMed] [Google Scholar]
- 50.Roberts LR, Gores GJ. Hepatocellular carcinoma: molecular pathways and new therapeutic targets. Semin Liver Dis. 2005;25:212–225. doi: 10.1055/s-2005-871200. [DOI] [PubMed] [Google Scholar]
- 51.Villanueva A, Toffanin S, Llovet JM. Linking molecular classification of hepatocellular carcinoma and personalized medicine: preliminary steps. Curr Opin Oncol. 2008;20:444–453. doi: 10.1097/CCO.0b013e328302c9e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.