

Abstract
Changes in the expression of ion channels, contributing to altered neuronal excitability, are emerging as possible mechanisms in the development of certain human epilepsies. In previous immature rodent studies of experimental prolonged febrile seizures, isoform-specific changes in the expression of hyperpolarization-activated cyclic nucleotide-gated cation channels (HCNs) correlated with long-lasting hippocampal hyperexcitability and enhanced seizure susceptibility. Prolonged early-life seizures commonly precede human temporal lobe epilepsy (TLE), suggesting that transcriptional dysregulation of HCNs might contribute to the epileptogenic process. Therefore, we determined whether HCN isoform expression was modified in hippocampi of individuals with TLE. HCN1 and HCN2 expression were measured using in situ hybridization and immunocytochemistry in hippocampi from three groups: TLE with hippocampal sclerosis (HS; n = 17), epileptic hippocampi without HS, or non-HS (NHS; n = 10), and autopsy material (n = 10). The results obtained in chronic human epilepsy were validated by examining hippocampi from the pilocarpine model of chronic TLE.
In autopsy and most NHS hippocampi, HCN1 mRNA expression was substantial in pyramidal cell layers and lower in dentate gyrus granule cells (GCs). In contrast, HCN1 mRNA expression over the GC layer and in individual GCs from epileptic hippocampus was markedly increased once GC neuronal density was reduced by >50%. HCN1 mRNA changes were accompanied by enhanced immunoreactivity in the GC dendritic fields and more modest changes in HCN2 mRNA expression. Furthermore, similar robust and isoform-selective augmentation of HCN1 mRNA expression was evident also in the pilocarpine animal model of TLE. These findings indicate that the expression of HCN isoforms is dynamically regulated in human as well as in experimental hippocampal epilepsy. After experimental febrile seizures (i.e., early in the epileptogenic process), the preserved and augmented inhibition onto principal cells may lead to reduced HCN1 expression. In contrast, in chronic epileptic HS hippocampus studied here, the profound loss of interneuronal and principal cell populations and consequent reduced inhibition, coupled with increased dendritic excitation of surviving GCs, might provoke a “compensatory” enhancement of HCN1 mRNA and protein expression.
Keywords: epilepsy, h-channels, Ih, dentate gyrus, hippocampus, ion channels, hyperpolarization-activated cation channels, pilocarpine, human, experimental epilepsy, temporal lobe, sclerosis, epileptogenesis
Introduction
Temporal lobe epilepsy (TLE) (i.e., spontaneous seizures involving the hippocampal formation) is the most prevalent of refractory epilepsies (Engel, 1993), but the mechanisms converting the normal hippocampus into an epileptic one are not fully understood. The hippocampus in many individuals with TLE is characterized by cell loss in specific regions, most prominently CA1 and CA3/4, a pattern termed hippocampal (or mesial temporal) sclerosis (HS). Whether loss of specific neuronal populations in the “HS pattern” precedes and provokes epilepsy, or is a result of recurrent seizures, has remained a focus of debate (Sutula and Pitkänen, 2002).
The very frequent (30-70%) history of prolonged febrile seizures early in life in individuals with TLE and HS has led to the notion that these seizures might contribute to the pathogenesis of HS and TLE (Cendes et al., 1993; French et al., 1993) (for review, see Baram et al., 2002). It has been suggested that these seizures, or similar early-life “initial precipitating events,” provoke the death of vulnerable hippocampal neurons leading to HS, which in turn results in TLE. Alternative mechanisms include seizure-induced epileptogenesis with secondary cell loss, or a combination of these two processes (Dubé et al., 2000; Mathern et al., 2002). To probe directly the mechanisms by which early-life seizures might contribute to HS and TLE, an animal model of prolonged febrile seizures was created and characterized. In this model, prolonged febrile seizures did not kill neurons but led to transient neuronal injury in regions closely overlapping those involved in human HS (Toth et al., 1998). Importantly, this animal model implicated long-lasting functional (K. Chen et al., 2001) and molecular (Brewster et al., 2002) changes in the hyperpolarization-activated cyclic nucleotide-gated cation channels (HCNs) as potential mediators in the pathogenesis of TLE. These data suggested that the normal or modified HCN channels might be involved in epileptogenic or “compensatory” protective mechanisms in the epileptic hippocampus (Walker and Kullmann, 1999). Therefore, the current study aimed to evaluate the presence and potential alterations of HCN expression in epileptic human hippocampus at the mRNA and protein levels. Because hippocampi from individuals with long-term TLE and HS might also reflect chronic or compensatory changes that are not related to the epileptic process, HCN mRNA expression was also determined in an experimental model of chronic hippocampal epilepsy.
Materials and Methods
Diagnosis assignment and tissue handling. Hippocampi were resected from patients with intractable TLE who were evaluated extensively before surgery, including history and neurological examinations, interictal and ictal EEGs, and neuropsychological and intracarotid amobarbital tests for memory and speech localization. Neuroimaging studies included high-resolution magnetic resonance imaging with visualization of the hippocampal formation and 18fluoro-2-deoxyglucose positron emission tomography (Engel, 1993). If required clinically, intracerebral electrodes determined the epileptogenic zone. Patients were assigned to the HS or non-HS (NHS) groups on the basis of clinical, EEG, neuroimaging, and pathological analyses (Engel, 1993; Mathern et al., 1997a). The HS group (n = 17) consisted of patients without mass lesions but with a severely damaged hippocampus on imaging and neuropathological assessment including cell counts. These individuals generally had onset of seizures during childhood. NHS patients (n = 10) had a macroscopic mass lesion, such as a tumor (n = 4), traumatic scar (n = 1), or cavernous malformation (n = 1), or had nonlesional cryptogenic TLE (n = 4). Cell loss in NHS hippocampi was generally less severe than that in HS tissue. Autopsy hippocampi (n = 10) from individuals without known neurologic disease were obtained 3-11 hr postmortem (6.6 ± 0.75). None showed macroscopic signs of cerebral pathology.
Hippocampal specimens that were cut transverse to the anterior-posterior axis were immersion-fixed in fresh phosphate-buffered 4% paraformaldehyde (PFA) for 24-48 hr, cryoprotected, and then cryostat-cut (30 μm) and mounted on Superfrost-Plus slides (Fisher Scientific, Pittsburgh, PA) for cell counts, in situ hybridization (ISH), and immunocytochemistry (ICC). Tissue was protected from RNase contamination, and all procedures were approved by the appropriate institutional committees.
Induction of experimental chronic TLE and tissue handling. Established protocols, approved by the local Animal Care and Use Committee, were used to induce status epilepticus (SE) in rats (Cavalheiro et al., 1991). Briefly, male Wistar rats (150-200 gm) were injected with a single dose of pilocarpine (340 mg/kg i.p.). Peripheral muscarinic effects were reduced by previous administration of methyl-scopolamine (1 mg/kg s.c.; 30 min before injecting pilocarpine). Approximately 80% of the injected rats experienced SE. Diazepam (0.1 mg/kg s.c.) was administered to all animals 40 min after onset of SE, which terminated the convulsions and sedated all animals. The animals required special care until they recovered from the acute insult and were closely tended, hydrated, and fed in the laboratory. A group of age-matched rats was treated identically but was substituted saline injection for pilocarpine injections. This group was used as the control group. After treatment, animals were monitored daily for the development of spontaneous seizures. Within 2-4 weeks after this treatment, all rats that experienced SE developed a chronic epileptic condition expressed as 2-4 spontaneous “limbic” seizures (characterized by chewing, head nodding, forelimb clonus, rearing, and falling) per week. Animals were killed 30 d after treatment and their brains were rapidly removed and immediately frozen over liquid nitrogen and stored at -80°C until additional use.
In situ hybridization: nonradioactive and quantitative (radioactive). Both nonradioactive and semiquantitative radioactive ISH were used. The former provides clear visualization of mRNA expression in single neurons but does not provide quantitative measures (Bender et al., 2001). The latter has been extensively standardized in our hands to permit relative quantitative analyses (Brunson et al., 2001; Brewster et al., 2002).
For all ISH procedures, HCN antisense- and sense-riboprobes were generated from transcription vectors containing cDNA of mouse HCN1 (generous gift from Dr. M. Biel, Technische Universität, Munich, Germany) (Ludwig et al., 1998) or HCN2 (courtesy of Dr. Bina Santoro, Columbia University, Columbia, NY) (Santoro et al., 2000). These are highly homologous to the respective cDNA regions of human HCNs (91-96% for HCN1; 90% for HCN2; GenBank accession numbers AF488549.1 and AF064877, respectively). For nonradioactive ISH, probes were labeled with digoxigenin (DIG; 3.5:6.5 DIG-UTP/UTP), and ISH was performed according to protocols published previously (Bender et al., 2001; Brewster et al., 2002) with minor modifications: slide-mounted frozen sections were brought to room temperature, air-dried, and refixed in 4% PFA for 20 min. After fixation, slides were dehydrated and rehydrated through graded ethanols, transferred to 2× SSC, and then hybridized. Briefly, after prehybridization, sections were exposed to a solution containing the DIG-labeled RNA probes for ∼12 hr at 55°C in a humid chamber. After hybridization, sections were subjected to washes of increasing stringency, including 2× SSC at room temperature for 15 min and 0.2× SSC at 55°C for 60 min. Hybrid molecules were detected with an anti-DIG serum tagged with alkaline phosphatase (Bender et al., 2001; Y. Chen et al., 2001).
Radioactive ISH was performed as published previously (Brewster et al., 2002). Briefly, probes were labeled with [35S]-CTP to specific activities of 3.2-5.9 × 107 cpm/μg. Slides were treated as described above, except that incubation in 0.25% acetic anhydride in 0.1 m triethanolamine, pH 8.0, for 8 min followed the last rehydration step. Sections were then dehydrated again, air-dried, and prehybridized for 60 min at 55°C (Brewster et al., 2002). After hybridization (at 55°C) and RNase digestion, sections underwent successive washes (at 55°C), the most stringent in 0.1× and 0.03× SSC for 1 hr each. Sections were dehydrated through 100% ethanol and apposed to film (Biomax MR; Kodak, Rochester, NY) for up to 2 (HCN1) or 3 (HCN2) weeks. For analysis at the single cell level, sections were also dipped in NTB2 nuclear emulsion (Kodak) and exposed for 4-6 weeks. The specificity of the HCN mRNA hybridization signal and the isoform selectivity of the HCN probes have been established (Bender et al., 2001; Brewster et al., 2002).
Quantitative analysis of ISH signal. Relative quantitative analysis of HCN mRNA levels was accomplished using radioactively labeled ISH probes and C14 calibration standards under conditions of signal linearity (Brunson et al., 2001). Briefly, digitized images of each section were acquired using a Studio Star scanner (1200 × 1200 dots per inch; Agfa, Ridgefield, NJ) and analyzed using the ImageTool software program (version 1.25; University of Texas Health Science Center, San Antonio, TX). Densities were corrected for background and expressed in nCi/gm. Two sections per individual were analyzed, and the average was used for group analyses (Brewster et al., 2002). Expression of HCN mRNA “per cell” was calculated as: [mRNA, in nCi/gm]/[cell number/mm3], with the assumption that the density of brain tissue is ∼1. The results are conveyed as pCi/103 cells.
Immunocytochemistry. ICC was modified from published procedures (Y. Chen et al., 2001; Brewster et al., 2002). Briefly, slide-mounted frozen sections were thawed, refixed in 4% PFA (20 min), and dehydrated and rehydrated as describe above. For antigen retrieval, sections were incubated in 10 mm Nacitrate, pH 9.0, at 80°C for 30 min (Jiao et al., 1999). After cooling, sections were quenched with 0.3% H2O2 and immersed in 0.01 m PBS containing 0.3% Triton X-100 (PBS-T), 0.2% SDS, 1% bovine serum albumin (BSA), and 2% normal goat serum for 1 hr to block unspecific binding sites. HCN1-antiserum (rabbit anti-HCN1; 1:2000; Chemicon, Temecula, CA) was added to the solution for 4 d at 4°C, and antibody binding was detected using standard avidin-biotin complex methods (Y. Chen et al., 2001). Briefly, after washes with PBS-T, slides were incubated with biotinylated goat-anti-rabbit IgG (1:250; 4 hr; room temperature; Vector Laboratories, Burlingame, CA). Avidin-biotin-peroxidase complex solution was applied (2 hr; Vectastain Elite-Kit, Vector Laboratories), and antibody binding was visualized by incubating the sections in 0.04% 3,3′-diaminobenzidine containing 0.01% H2O2, 0.01% CoCl2, and 0.01% NiCl2. The specificity of the reaction was evaluated by omitting the primary antibody from the solution, leading to complete loss of signal. To confirm and amplify the data, freshly resected human hippocampi (n = 4) were also subjected to ICC. Here, free-floating sections derived from immersion-fixed cryoprotected frozen tissue were processed through 0.3% H2O2 (30 min) and the preincubation solution (PBS-T containing 1% BSA and 2% normal goat serum) for 1 hr. Sections were exposed to anti-HCN1 (1:2000) or anti-neuronal-specific nuclear protein (NeuN) antiserum (mouse anti-NeuN, 1: 2000; Chemicon) for 48 hr at 4°C, and the presence of antigen was visualized as described above.
Hippocampal neuron densities: counts and analysis. Cell count procedures followed published protocols (Mathern et al., 1997b). Briefly, counts were performed without knowledge of diagnosis at 400× using an ocular grid. For stratum pyramidale, 20 boxes in sequential 2 × 2 box segments (104 × 1040 μm) were selected; for the smaller granule cells, a linear 1 × 5 box was used (52 × 260 μm), and counts over four such areas (both top and bottom granule cell blades) were averaged. Neuronal densities were computed as: N(neurons/mm3) = A[M/(L + M)] divided by the volume of the sampling area (10 μm × area of grid), where A is the number of counted nuclei, L is the average length of the nucleus, and M is the section thickness (Mathern et al., 1997b). The results reflect estimates of the number of neurons per unit volume (i.e., packing density) and not “absolute” calculations of total neurons per hippocampus. Because stereological methods for estimating total neurons require availability of the entire hippocampus, they are not feasible for surgical specimens (West, 1999). However, because tissue from all groups was similarly processed and evaluated “blindly,” statistical differences in neuron densities are accepted as valid in surgically collected material (Abercrombie, 1946; Mathern et al., 1997b). For rat hippocampi, similar methods were followed, as described in detail previously (Brunson et al., 2001).
Results
Characteristics of patients and hippocampal specimens
Table 1 summarizes the clinical characteristics of the autopsy and TLE individuals (HS and NHS). For evaluation of the potential relationship between early life seizures (and other predisposing factors) and the development of TLE plus HS, the table illustrates that, among individuals with TLE plus HS, such initial precipitating events were elicited in 13 (76.4%) and consisted of prolonged/febrile seizures in nine (53%). In contrast, individuals with TLE without HS were less likely to have an identifiable initial precipitating event (2 of 10) and specifically prolonged seizures (0 of 10). Other parameters (age, duration of epilepsy) did not distinguish the two groups. As expected, autopsy material was derived from somewhat older individuals, but age did not correlate with HCN mRNA expression.
Table 1.
Clinical characteristics of autopsy and TLE individuals
|
Parameter |
Autopsy (n = 10) |
Temporal lobe epilepsy Non-HS (n = 10) |
Hippocampal sclerosis (n = 17) |
p value |
|---|---|---|---|---|
| Age at tissue harvest | 45 ± 5.2 | 34.9 ± 2.4 | 34.2 ± 2.3 | p > 0.05 for all pair groups |
| (years ± SEM) (range) | (21-70) | (26-49) | (18-49) | pair groups |
| Age of seizure onset | NA | 19.6 ± 2.1 | 14.5 ± 3.1 | p = 0.25 |
| (years ± SEM) (range) | (10-32) | (3-40) | ||
| Seizure durationa | NA | 15.3 ± 3 | 18.8 ± 2.4 | p = 0.38 |
| (years ± SEM) (range) | (2-37) | (4-35) | ||
| Initial precipitating event | NA | 2 | 13 (9 prolonged seizures) | |
| Gender (male:female) | 5:5 | 5:5 | 11:6 | |
| Hippocampus side (right:left)
|
4:6
|
7:3
|
10:7
|
|
NA, Not applicable; p values computed using ANOVA (age at harvest) or Student's t test (age of seizure onset and duration of seizures).
Seizure duration calculated as time (in years) between onset of habitual seizures and hippocampal resection.
Cell loss was found in hippocampi from HS patients, consonant with published reports (for review, see Armstrong, 1993, Mathern et al., 1997a). As shown in Figure 1, neuronal densities in HS hippocampi were reduced, particularly in CA1 (77%), CA4 (60%), and the granule cell (GC) layer (GCL) (42%), compared with the autopsy group. Counts in NHS hippocampi did not differ significantly from those of autopsy-derived tissue.
Figure 1.

Neuronal densities in hippocampal fields involved in the neuropathology of hippocampal sclerosis. A, CA1. B, CA4 (corresponding to CA3c in the rodent). C, GCL of the dentate gyrus. Neuronal counts and analyses were performed as described in Materials and Methods. As evident from the figure, whereas specimens from epileptic individuals without hippocampal sclerosis (NHS) had neuronal densities comparable with those of autopsy specimens (AUT), significant reductions were noted in the HS tissue. Values are means ± SEM from 10 AUT, 10 NHS, and 17 HS hippocampi. CA, Cornu ammonis. Asterisks indicate statistical significance.
HCN1 expression patterns in the human hippocampal formation
A novel finding of this study was the demonstration of HCN1 and HCN2 mRNA expression in specific neuronal populations of the human hippocampal formation. In hippocampal tissue derived from autopsies, robust HCN1 mRNA signal was evident throughout the CA pyramidal cell layer (Figs. 2A,F,G, 3A,C), as well as in hilar neurons of the dentate gyrus (Figs. 2A,D, 3E). In contrast, the well preserved GC layer (Figs. 2A,D,3E,4A,D) and individual GCs (Fig. 4F) expressed relatively little HCN1 mRNA. This mRNA expression pattern resembles that described for HCN1 in rodent hippocampus (Moosmang et al., 1999; Santoro et al., 2000; Bender et al., 2001), with the exception that the increased mRNA expression of the channel in CA1 compared with CA3 pyramidal cell layers was not apparent in the human. For HCN2 mRNA, the relatively homogenous modest expression levels in CA1, CA3, and the GC layer of human autopsy hippocampus were rather similar to rodent studies (Fig. 5A) (cf. Moosmang et al., 1999; Bender et al., 2001).
Figure 2.
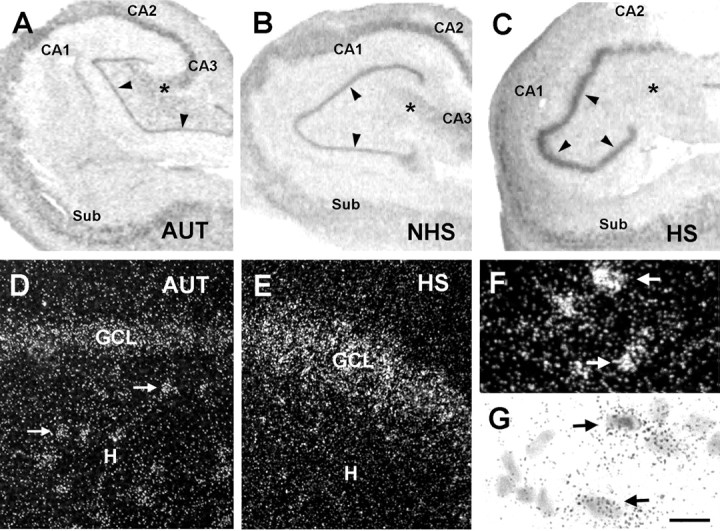
A-C, HCN1 mRNA expression patterns in hippocampal sections from autopsy (AUT) material (A), an individual with TLE NHS (B), and a patient with TLE plus severe HS (>50% cell loss in dentate gyrus) (C). In situ hybridization using radioactively labeled HCN1 cRNA probes was performed and analyzed blindly (see Materials and Methods). The overall HCN1 mRNA expression pattern was similar in AUT and NHS hippocampi, with prominent mRNA signal over neurons in all CA subfields and a low signal over the GCL (arrowheads). HCN1 mRNA expression pattern was strikingly different in hippocampi with severe HS; the signal was strongly increased over the GCL despite substantial GC loss (see Fig. 4). HCN1 mRNA expression was relatively low in CA1 and CA4 (denoted by an asterisk), where cell loss was profound. D, E, Dark-field photomicrographs showing HCN1 mRNA signal in AUT dentate gyrus (D) and the corresponding region from an individual with severe HS (E). In AUT sections, HCN1 mRNA expression was clearly visible over hilar neurons (arrows) and GCL. E, In epileptic HS hippocampus, the hilar neurons were not visible (and are typically lost), whereas robust signal was found over the GCL. F, G, Strongly HCN1 mRNA-expressing neurons in CA1 of an AUT hippocampus (arrows) demonstrated that the low HCN signal in AUT GCL is not attributable to tissue or RNA degradation. Note that, for quantitative analyses, optical density of film autoradiographs, rather than grain counts or size, have been used. Scale bars: A-C, 250 μm; D, E, 30 μm; F, G, 10 μm. CA, Cornu ammonis; Sub, subiculum; H, hilus.
Figure 3.

Differential expression of HCN1 mRNA in specific regions of hippocampal formation from autopsy (AUT) and severe HS epileptic tissue. The distribution of HCN1 mRNA-expressing individual neurons in the CA1-CA2 region (A-D) and the dentate gyrus (E, F) is demonstrated using nonradioactive in situ hybridization (see Materials and Methods). Low-magnification views (A, B) show robust HCN1 mRNA signal in CA1 pyramidal cells (as well as in stratum oriens interneurons; arrows) of autopsy-derived hippocampus (A) compared with sparser HCN1-expressing cells (and drastic reduction of neuronal density; Fig. 1) in epileptic sclerotic hippocampus (B). C, D, Higher magnification demonstrates reduced HCN1 mRNA signal over the CA1 pyramidal cell layer in HS (D) compared with AUT (C) tissue. Signal over individual remaining pyramidal cells is faint, whereas strong HCN1 mRNA expression is observed in presumed interneurons bordering the layer (arrows). E, F, Compared with AUT dentate gyrus (E), GCL from HS hippocampus (F) is depleted, yet HCN1 mRNA expression is clearly apparent in remaining neurons located well within the GCL (arrows). HCN1 mRNA-expressing hilar neurons (E, arrowheads) are virtually absent in the epileptic severe HS material (F). Scale bars: A, B, 150 μm; C-F, 30 μm. CA, Cornu ammonis; H, hilus.
Figure 4.

Expression levels of HCN1 mRNA in human dentate gyrus GC layer correlate highly with reduced neuronal densities. The top panels show HCN1 mRNA expression levels (determined using quantitative in situ hybridization analysis) in individual specimens, plotted against GC layer neuronal density values. Hippocampi from autopsies (AUT) (A), epileptic individuals without hippocampal sclerosis (NHS) (B), and those with the sclerosis pattern (HS) (C) were compared. Autopsy and NHS tissues were characterized by similar neuronal densities and relatively low HCN1 mRNA signal. In contrast, among HS hippocampi, a striking increase of HCN1 mRNA expression correlated with a ∼50% reduction of cell densities to below ∼80,000 per mm3 (C). Therefore, this group was dichotomized into mild (>50% cell densities, compared with AUT) and severe HS. D, Mean ± SEM of HCN1 mRNA levels (right y-axis) in tandem with corresponding mean cell densities in GC layer (left y-axis) of the three hippocampi groups. Cell loss in HS GC layer was significant compared with the autopsy group (*), but HCN1 mRNA signal was not correspondingly reduced in the group as a whole. E, Within the HS group, HCN1 mRNA levels in the depleted GC layer of the severe subgroup [in which cell number was significantly lower than in the mild HS group (*)] were significantly higher compared with tissue with minimal GC cell loss (**p = 0.0065; Student's t test with Welch's correction). F, The basis for the augmented HCN1 mRNA levels in severe HS (absolute levels exceeded those in the AUT and NHS brains; p = 0.03) was the significant (*) upregulation of HCN1 mRNA expression in individual GCs compared with per-cell mRNA expression in AUT, mild HS, or NHS epileptic hippocampus.
Figure 5.

HCN2 mRNA expression in human and rat epileptic hippocampus is lower and less influenced by the epileptic state and the associated cell loss compared with HCN1. A, Autoradiographs of sections subjected to in situ hybridization using probes for HCN2 mRNA revealing that, in contrast to HCN1, expression of this isoform is generally lower and is not visibly increased over the GCL (arrowheads) of the epileptic HS hippocampus. The HS section (bottom) was derived from the same individual shown in Figure 2C. B, Quantitative analysis of HCN2 mRNA expression in autopsy-derived (AUT) and epileptic dentate gyrus with (HS) or without (NHS) hippocampal sclerosis. The HS group was also divided into severe and mild groups on the basis of the extent of neuronal loss (see Fig. 4). Overall HCN2 mRNA levels in the GCL (right y-axis) did not vary significantly with cell numbers or disease state (left y-axis), in contrast to HCN1 mRNA (see Fig. 4 E). Asterisks indicate reduced cells compared with AUT. C demonstrates a modest but significant increase in the overall low HCN2 mRNA per-cell expression in severe HS GCs. D, In the rat, HCN2 mRNA levels were significantly reduced in the GC layer of epileptic hippocampus, but expression per individual GC did not differ among experimental groups. Scale bar, 200 μm. CA, Cornu ammonis; Sub, subiculum.
HCN1 mRNA levels in 9 of 10 NHS hippocampi generally resembled autopsy material; HCN1 mRNA was expressed throughout the CA subfields (Fig. 2B), and quantitative analyses indicated that the mRNA expression in CA1 and the GC layer (Figs. 4B,D, 6) as well as in individual GCs (Fig. 4F) did not differ significantly from those of autopsy material (analysis in CA3 was incomplete because the region was absent or only partially included in many specimens). As in the autopsy material (Figs. 2D, 3E), HCN1 mRNA signal was relatively robust over the preserved, abundant hilar neurons (data not shown).
Figure 6.

HCN1 mRNA expression in the CA1 pyramidal cell layer of three groups of human hippocampi. HCN1 mRNA signal (hatched bars, right y-axis) is coplotted with CA1 pyramidal cell layer neuronal densities (solid bars, left y-axis). The autopsy (AUT) tissue is compared with epileptic hippocampus with (HS) or without (NHS) sclerosis. A drastic (*) reduction of CA1 neuronal densities is found in HS hippocampus and is accompanied by a tendency to lower HCN1 mRNA levels. As shown in Figure 3D, the HCN1 mRNA signal over the depleted HS pyramidal cell layer originates, at least partially, from the preserved interneuronal populations. It should be noted that massive cell loss in CA1 was found in specimens with either mild or severe reduction of GC densities so that the HS group is treated here as a single population. CA, Cornu ammonis.
HCN1 expression pattern, at both mRNA and protein levels, was strikingly different in HS epileptic hippocampus. In CA1 pyramidal cell layer, where cell loss was profound in virtually all specimens (mean, 77%) (Figs. 1, 2C,3B,D), overall mRNA signal tended to be lower. In addition, nonradioactive ISH suggested that signal over the pyramidal cell layer was at least partially attributable to HCN1 mRNA expression in the nondestroyed interneurons (Babb et al., 1989). Particularly striking was the robust mRNA expression of HCN1 in the dentate gyrus GC layer of some HS specimens. Qualitative analysis suggested that HCN1 mRNA expression was most pronounced in specimens demonstrating abnormal contour of the GC layer (Fig. 2C), with visible reduction of neuronal densities (Fig. 3F) (see below). Finally, human hippocampus with the characteristic HS pathology was almost devoid of HCN1 mRNA-expressing hilar neurons (interneurons and mossy cells), likely reflecting depletion of these neuronal populations (Figs. 2E, 3F).
This region and cell-type-specific HCN1 mRNA expression in each of the three groups helped exclude reduced tissue preservation or RNA stability in the autopsy material as potential confounders. This is consistent with previous studies of mRNA stability in simulated postmortem conditions (Johnson et al., 1986) and with our finding of excellent signal preservation in rat hippocampus that was treated to simulate the surgical procedures (data not shown). The region-specific HCN1 mRNA levels in all three experimental groups also excluded differential probe access/penetration among these groups (e.g., attributable to differing lengths of exposure of the tissue to aldehyde fixatives).
Upregulation of HCN1 mRNA expression correlates with substantial cell loss
Quantitative analysis of the HS group of epileptic hippocampi revealed two patterns of GC layer integrity and HCN1 mRNA expression. In the relatively intact GC layer, where neuronal densities were maintained at approximately >50% of those in autopsy material (termed “mild” HS for the purpose of these analyses), HCN1 mRNA signal was weak-to-moderate, as found in most NHS hippocampi. However, when neuronal densities were reduced to <50% of those in autopsies (“severe” HS), a drastic upregulation of HCN1 mRNA expression occurred in the GC layer (Figs. 2C,E, 4C-F).
Despite the profound GC loss, aggregate HCN1 mRNA levels over the severe HS GC layer exceeded those in the autopsy, NHS, and mild HS groups (p < 0.0001; ANOVA; p < 0.01 vs all other groups; Bonferroni's post hoc test). The basis for this strong HCN1 mRNA signal was a dramatic increase of HCN1 mRNA expression in surviving individual GCs (Fig. 4F). As shown in Figures 2E and 3F, the single-cell resolution of radioactive emulsion-dipped ISH, as well as the nonradioactive ISH method-, permitted visualization of increased HCN1 mRNA signal in cells located well within the GC layer (rather than over eccentrically situated GABAergic basket cells).
HCN1 expression changes at the mRNA level were accompanied by enhanced HCN1 protein signal. As shown in Figure 7, little immunoreactive HCN1 was visible in the GC and molecular layers (MLs) of the dentate gyrus with minimal GC loss [mild HS (Fig. 7C); the GC layer is delineated by the neuronal marker NeuN (Fig. 7A)]. In contrast, in epileptic dentate gyrus with severe GC loss, HCN1 immunoreactivity was prominent within the dendritic fields of the GCs in the ML (Fig. 7D; the irregular, depleted GC layer is shown in 7B), and signal was more robust also within the GC layer (Fig. 7, compare C, D). Although the human material did not permit formal quantification of the immunoreactivity, the data strongly suggested that increased HCN1 expression at the mRNA level also resulted in augmented protein expression. Notably, the correlation of GC loss and HCN1 mRNA levels also persisted in the single NHS hippocampus with >50% reduction in GC density (compared with autopsy) (Fig. 4B). This finding suggests that this severe GC loss might be a causal variable in the mechanism of HCN1 mRNA upregulation, or, at a minimum, this severe cell loss might be a surrogate marker of the underlying cause.
Figure 7.
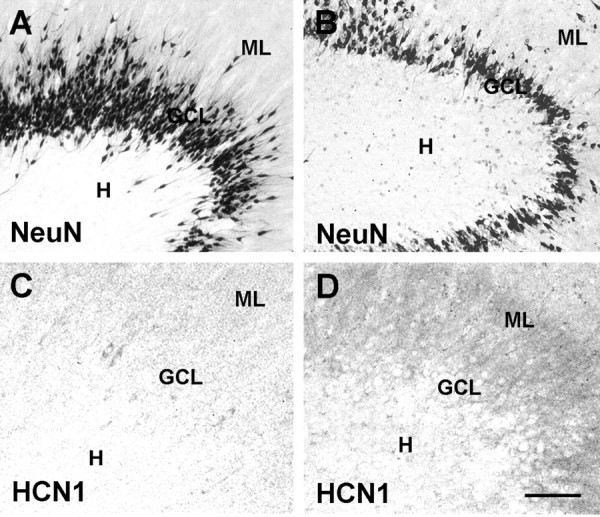
Immunoreactive HCN1 channels located preferentially in the molecular layer are upregulated in dentate gyrus from individuals with TLE and GC loss, indicating that the transcriptional regulation of HCN1 translates to abundance of channel proteins. A and C show tissue derived from epileptic patients with intact GC layer (mild HS). B and D depict epileptic hippocampus with disruption of the GC layer (severe HS) (see Figs. 2, 3). Hippocampal sections shown in A and B underwent immunocytochemistry for NeuN to visualize the presence or loss of neurons in the GCL, and the cell loss in the severe HS (B) is evident. D demonstrates enhanced immunoreactive HCN1 in the layer harboring the GC dendrites, the ML, compared with mild HS-derived material (C). In the human surgical material, it is difficult to resolve with certainty whether the increased signal in the GCL is within GC somata or interneuronal processes. Note that these representative sections were from fresh surgical material, permitting better immunohistochemical analysis. Scale bars: A, B, 100 μm; C, D, 30 μm.
HCN1 mRNA expression in the CA1 pyramidal cell layer
To determine whether increased HCN1 mRNA in the GCL and per individual GC was specific to this neuronal population, we evaluated HCN1 mRNA levels also in the CA1 pyramidal cell layer. Here, HCN1 mRNA levels tended to be lower (43.53 ± 9.54 vs 77 ± 27.49 nCi/gm in HS and NHS, respectively), a difference not reaching statistical significance (Fig. 6). Additional examination of the cells expressing HCN1 mRNA revealed two important facts. First, the cell loss in this layer was profound (Fig. 1), and quantitative per-cell mRNA analysis in the face of profound (∼77%) reduction in cell numbers (the denominator) can lead to artifactual results (Mathern et al., 1999). Furthermore, visual inspection suggested that much of the HCN1 mRNA signal appeared over the intact interneurons rather than over the few remaining pyramidal cells (Fig. 3D). For these two reasons, we relied on qualitative analysis (Fig. 3, compare C, D) that failed to support upregulation of HCN1 mRNA expression in individual surviving CA1 pyramidal cells and suggested that the signal in CA1 pyramidal cell layer emanated from HCN1 mRNA expression in the interneurons. Compared with the findings in the GC layer, these results support the notion of a specific increase of HCN1 mRNA levels in individual GCs in HS hippocampus with severe cell loss.
In the human, the inciting cause for these alterations of HCN1 mRNA expression was difficult to determine. Although the severe granule cell loss (perhaps implying concurrent loss of inhibitory interneurons or a more disrupted hippocampal circuit) might be the major precipitant, other variables and particularly the factors that led to the development of the TLE plus HS in the first place could not be excluded. Therefore, we reasoned that if the enhanced HCN1 mRNA expression in surviving GCs resulted from parameters associated with this severe chronic epileptic state, then it should also be evident in experimental models of human TLE plus HS.
HCN1 mRNA expression is upregulated in GCs of experimental TLE and correlates with neuronal loss
Because controlled prospective and mechanistic studies in human material are limited, we chose to study HCN1 mRNA expression in pilocarpine-treated animals, a model that closely reproduces human chronic hippocampal epilepsy (Cavalheiro et al., 1991; Brooks-Kayal et al., 1998). As shown in Figure 8A, strong HCN1 mRNA expression was evident over the GC layer in chronically epileptic (i.e., having spontaneous seizures) animals, compared with the control group (Fig. 8B) (p < 0.05; n = 5 controls, 4 pilocarpine-treated). These findings indicate that increased HCN1 mRNA expression is not unique to human TLE plus HS but represents a consequence of specific factor(s) that are inherent in chronic hippocampal epilepsy with similar cell loss.
Figure 8.

HCN1 mRNA expression is upregulated in GCs of the pilocarpine animal model of severe chronic epilepsy with neuronal loss. A depicts photomicrographs of coronal sections through the hippocampal formation of adult rats 1 month after pilocarpine-induced status epilepticus (when all animals had spontaneous seizures) compared with controls. Significant increase of HCN1 mRNA signal over the GC layer is visible, as quantified in B. C demonstrates the pattern of cell loss in the animal model. Whereas the GC population was significantly reduced (left y-axis; numbers are in thousands of GCs), it was accompanied by a profound loss of hilar neurons (right y-axis; absolute numbers), suggesting that loss of hilar mossy cells and interneurons might be more closely related to the significant upregulation of HCN1 mRNA expression in individual surviving GCs (D). See Materials and Methods for calculations of per-cell mRNA expression. GC-Pilo, GC-Ctl, Hil-Pilo, and Hil-Ctl refer to granule cell and hilar neurons in the pilocarpine-treated and control groups. Asterisks indicate statistical significance.
Cell count analyses indicated that HCN1 mRNA in epileptic GC layer was associated with significant reduction of GC density (Fig. 8C). In agreement, the single pilocarpine-treated rat with blunted increase of HCN1 mRNA levels over the GC layer was found to have less cell loss (data not shown). Interestingly, a striking depletion of neurons in the dentate gyrus hilus (presumed interneurons and mossy cells) was found in the pilocarpine group. Thus, whereas a 20% reduction occurred in the GC layer, a 56% depletion was observed in the hilus. This close association of GC and hilar cell loss is consistent with the notion that profound principal cell loss in the human hippocampus might be a marker of destruction of hilar cell populations, and that the latter might play a key role in altering HCN1 mRNA expression.
Isoform specificity of altered HCN1 mRNA expression in chronically epileptic hippocampal neurons
The subunit composition of HCN channels influences the properties of neuronal Ih currents, including their voltage-dependent activation kinetics and gating by cAMP (Franz et al., 2000; Santoro et al., 2000; Vasilyev and Barish, 2002). Furthermore, differential regulation of HCN1 and HCN2 mRNA expression has been demonstrated (Bender et al., 2001; Bräuer et al., 2001; Brewster et al., 2002). Therefore, having determined altered expression of HCN1 mRNA, we studied the presence and regulation of HCN2 mRNA expression in the three groups of human hippocampi as well as in the pilocarpine experimental model of TLE.
In the human, HCN2 mRNA expression was low throughout the hippocampal formation compared with HCN1 mRNA levels (compare Fig. 5A,B with Figs. 2, 4). Within the GC layer, although overall mRNA signal did not increase even in severe HS, compared with mild HS or NHS and autopsy material (Fig. 5B), a statistically significant (80%) enhancement of HCN2 mRNA expression per cell was noted [Fig. 5C; compare with a 501% increase of HCN1 mRNA in corresponding groups (Fig. 4F)]. In the rat, HCN2 mRNA levels were significantly reduced in the GC layer of epileptic hippocampus; however, expression per individual GC did not differ among experimental groups (Fig. 5D). Together, these data indicate that in both human and rodent chronic TLE hippocampus, changes in HCN2 mRNA expression are relatively modest in the face of severe cell loss, compared with the strong upregulation of HCN1 mRNA levels.
Discussion
The studies reported here indicate the following: (1) HCNs, specifically HCN1 and HCN2 mRNAs, are expressed in human hippocampus; (2) HCN1 mRNA and protein expression is increased in surviving GCs of epileptic HS hippocampi with severe GC cell loss; (3) increased GC HCN1 mRNA levels also occur in an experimental model of chronic hippocampal epilepsy, showing GC as well as profound hilar neuronal loss. These findings suggest that upregulation of HCN1 mRNA expression may be driven by the altered inhibitory and excitatory circuitries of the dentate gyrus, perhaps as a compensatory response.
Structure and function of HCN isforms in the hippocampus
This study demonstrates the expression of two HCN isoforms in human hippocampal formation. HCNs mediate hyperpolarization-activated (Ih) currents in the heart (DiFrancesco, 1993) and brain (Pape, 1996). In the hippocampus, these h-currents contribute to regulating neuronal membrane potential (Lupica et al., 2001) and shaping rhythmic neuronal activity (Maccaferri and McBain, 1996). Short-term modulation of HCN function involves cAMP binding to the channel's C terminal, intracellular domain (Wainger et al., 2001), influencing channel kinetics and voltage-dependent activation curves (DiFrancesco, 1993; Wainger et al., 2001). More recently, long-term modulation of the properties of h-currents has been suggested to result from the regulation of the channels at the transcriptional level (Bräuer et al., 2001; Brewster et al., 2002).
HCN channels are encoded by four characterized genes, two of which (HCN1 and HCN2) are substantially expressed in adult rodent hippocampus (Moosmang et al., 1999; Santoro et al., 2000; Bender et al., 2001). In vivo, four HCN molecules of the same isoform type probably assemble to form homomeric HCN channels. HCN1, encoding fast-kinetics channels with modest cAMP gating, is highly expressed in hippocampal pyramidal cells and CA1 interneurons (Santoro et al., 2000; Bender et al., 2001; Brewster et al., 2002). The Ih currents of these neurons have been implicated in maintenance of resting membrane potential (Lupica et al., 2001), synchronized network activity (Magee, 1999; but see Buhl et al., 2002), and “dampening” the effects of dendritic excitation (depolarization) on somatic output (Magee, 1998; Poolos et al., 2002). In contrast to HCN1, the HCN2 gene encodes a channel with slower kinetics and robust cAMP-evoked shifts in voltage dependence (Ludwig et al., 1998; Santoro et al., 1998, 2000). This channel is highly expressed in pacemaker cells (e.g., in thalamus and heart), where it likely contributes critically to the provocation of repetitive neuronal firing (Pape, 1996; Santoro et al., 2000; Ludwig et al., 2003). The function(s) of the h-currents in GCs, encoded by modest levels of HCN mRNA expression (Santoro et al., 2000; Bender et al., 2001), have not been fully resolved (Chevaleyre and Castillo, 2002; Mellor et al., 2002).
Because the isoform composition of HCN channels determines their physiological responses (S. Chen et al., 2001; Ulens and Tytgat, 2001), the relative abundance of HCN isoforms can be expected to contribute to properties of the h-current of an individual neuron (Franz et al., 2000; Santoro et al., 2000; Vasilyev and Barish, 2002). Therefore, selective or differential regulation of HCN subunit mRNA expression during pathological states, if leading to altered protein levels and membrane insertion of functional channels, might contribute to altered neuronal resting membrane potential and responses to both inhibitory and excitatory network inputs.
Seizure-induced HCN channel regulation in the absence of cell loss and with preserved inhibition
We demonstrated previously that prolonged early-life experimental seizures modulate the expression of HCN genes (Brewster et al., 2002). In CA1 neurons expressing multiple HCN isoforms, the seizures induced a coordinated reduction of HCN1 mRNA expression and enhancement of HCN2 mRNA expression (Brewster et al., 2002). This reduction of HCN1 mRNA expression occurred in the presence of augmented presynaptic inhibitory input onto the CA1 pyramidal cells (Chen et al., 1999). Neither principal cells (Toth et al., 1998) nor vulnerable interneuronal populations are lost in this model of early epileptogenesis (Bender et al., 2003). Thus, the observed selective downregulation of HCN1 mRNA expression might occur in response to increased hyperpolarizing input and consequent enhanced activation of this channel.
It should be noted that, in addition to reduced HCN1 mRNA levels in this immature rodent model, concurrent increased HCN2 mRNA expression, accompanied by slowed kinetics and shifted voltage-dependent activation curves of the cellular h-current in the depolarizing direction (the latter perhaps attributable to altered cellular cAMP or channel sensitivity thereto), led to increased bursting behavior in response to hyperpolarizing input and a “hyperexcitable” hippocampus (Dubé et al., 2000; K. Chen et al., 2001; Brewster et al., 2002). Nevertheless, the data suggested that transcriptional dysregulation of one or more HCN subunit isoform expression might be involved in the mechanisms of development of human hippocampal epilepsy (TLE). Therefore, they prompted analysis of HCN expression patterns in hippocampi from human individuals with refractory TLE. These studies were undertaken with the understanding that analysis of changes of HCN mRNA expression early in the epileptogenic process is not feasible in the human, so that the findings in “end-stage” HS hippocampus might reflect “pro-epileptogenic,” compensatory, or even coincidental changes.
Potential mechanisms for altered HCN mRNA expression in epileptic “sclerotic” hippocampus
HCN1 and HCN2 mRNAs were expressed in human hippocampal formation, in distributions generally concordant with those described for rodents. HCN1 mRNA was strongly expressed in pyramidal cells of CA1 and in presumed interneurons in the hilus of the dentate gyrus, whereas signal over the GC layer was low. HCN2 mRNA levels were generally low throughout the hippocampal formation. However, whereas dysregulated HCN mRNA expression occurred in human epileptic hippocampus, the changes differed profoundly from those associated with epileptogenesis in immature rodent models. Thus, in the dentate gyrus, HCN1 mRNA expression was highly augmented in individual GCs of HS specimens with a depleted GC layer. It should be noted that although destruction of the GC layer in HS hippocampi is typically less pronounced compared with CA1 and CA3c (CA4), significant reduction in neuronal densities commonly occurs (Figs. 1, 4) (Houser, 1990; Armstrong, 1993; Haas et al., 2002).
In the present study, a ∼50% reduction of GC density was an apparent threshold for drastic upregulation of HCN1 mRNA expression. What mechanism(s) could underlie this rather abrupt “break point?” It is tempting to speculate that the GC loss might be a marker of profound changes in inhibitory (hyperpolarizing) and excitatory input onto the surviving GCs, and that these changes, in turn, triggered the “neuroplasticity” in HCN mRNA expression. Indeed, both reduced inhibition (Williamson et al., 1999) and increased excitatory input have been documented in human hippocampus with severe HS (Isokawa et al., 1997; but see Swanson et al., 1998). The former might result from the destruction of hilar interneuronal populations, whereas the latter is probably a consequence of “sprouting” of the GC axons, the mossy fibers (Franck et al., 1995).
According to this scenario, in the face of increased hyperpolarizing input from intact interneuronal populations (as in the early-life seizure study) (K. Chen et al., 2001; Brewster et al., 2002), HCN1 mRNA expression might decline. In contrast, loss of interneurons (or their mossy cell afferents) and consequent reduced perisomatic inhibition found in severe human HS hippocampus, perhaps in conjunction with enhanced dendritic excitation, might trigger increased HCN1 mRNA expression. Our findings in the well characterized pilocarpine model of chronic hippocampal epilepsy support this scenario. As in the human, we found strong upregulation of HCN1 mRNA expression in individual GCs; although, compared with the human material, GC cell loss in this animal model was relatively modest (20%). However, the GC loss was accompanied by a striking (>55%) reduction of hilar neurons (including vulnerable mossy cells and interneurons), consistent with the notion that the loss of these populations and consequent loss of inhibition (Kobayashi and Buckmaster, 2003; Sloviter et al., 2003) might be an important determinant of the increased GC HCN1 mRNA levels.
In addition to altered inhibition, the increased excitation in the dentate gyrus circuit as a consequence of GC death and reactive sprouting might also contribute to HCN1 mRNA regulation. As better characterized in animal models, the majority of sprouted mossy fiber contacts are onto neighboring GCs (Okazaki et al., 1995; Wenzel et al., 2000; Buckmaster et al., 2002) and primarily onto their dendrites (Buckmaster et al., 2002), so that the surviving epileptic GC is subjected to enhanced dendritic excitation (Isokawa et al., 1997). Teleologically, increased density of HCN1 channels congregating at the dendritic subcellular compartment (Lörincz et al., 2002), particularly if leading to increased dendritic Ih, should provide a significant dampening effect on dendritic depolarization, “protecting” the soma from excessive dendritic excitation (Magee, 1998; Poolos et al., 2002). Thus, upregulation of HCN1 mRNA expression joins several morphological, physiological, and neurochemical changes that have been described in GCs from human HS hippocampal formation (Isokawa et al., 1997; Jeub et al., 1999; Mathern et al., 1999; Nägerl et al., 2000). Similar neuroplastic and compensatory changes, such as enhanced expression of the GABA-synthesizing enzyme glutamate decarboxylase (Schwarzer and Sperk, 1995), have also been reported in animal models of human TLE.
Potential role of dysregulation of HCN1 mRNA expression in the human epileptic process
Both the mechanisms leading to altered HCN1 mRNA expression in the HS hippocampus and potential consequences of this effect cannot be addressed in human material, but correlations can be made that can provide the foundation for testable hypotheses. For example, the major correlation of GC HCN1 mRNA expression in HS hippocampi was with GC cell loss, and both parameters tended to be higher in individuals with a history of early-life seizures (p = 0.08 for GC density; p = 0.1 for HCN1 mRNA levels; p = 0.07 for HCN1 mRNA expression per individual GC in this group, compared with individuals without this early-life history). Thus, the strong tendency for correlation of early-life prolonged seizures and GC HCN1 mRNA expression was attributable to the severity of pathological changes in these specimens, suggesting potential causal relationships. It should be noted that the age at seizure onset (p = 0.56) duration of epilepsy (p = 0.3), age at tissue harvest (p = 0.9), or other patient characteristics did not influence the HCN1 mRNA signal. This finding supports the biological specificity of the altered HCN1 mRNA expression in GCs of epileptic HS hippocampus, a notion further buttressed by the rat model data.
Finally, these studies, looking at the “end stage” of human intractable epilepsy, do not permit dissecting out causal relationships. Thus, although teleologically logical, whether altered HCN1 mRNA expression is a consequence of the loss of interneurons, mossy cells, and GCs, of the resulting altered balance of excitatory and inhibitory inputs onto surviving GCs, or of combinations of these and other factors, cannot be ascertained (Swanson et al., 1998; Williamson et al., 1999). Similarly, the consequences of selective increase in mRNA and protein expression of a single HCN isoform are impossible to fully predict. In addition to the resulting altered gating properties of the cellular complement of h-channels that might, in itself, lead to different neuronal responses and network excitability, potential changes in subcellular localization, other conductances, and numerous other intrinsic and extrinsic factors are likely to play a role in determining the net impact of a given change in HCN subunit expression on neuronal and network excitability. For example, enhanced somatic HCN1 mRNA expression and the resulting increase of overall h-current might contribute to the hyperexcitability of the epileptic dentate gyrus (Coulter, 2001; Dalby and Mody, 2001). Increased Ih might enhance synchronization of GC action potentials (Magee, 1999), at least partially by converting inhibitory input (at select frequencies) on the GC somata into increased neuronal firing (K. Chen et al., 2001). Therefore, whereas the present study firmly establishes changes of HCN expression as contributory to the human epileptic state, additional work in animal, in vitro, and computational models is required to decipher the mechanistic role of this “transcriptional channelopathy” in the pathogenesis of human hippocampal epilepsy.
Footnotes
This work was supported by National Institutes of Health Grants NS35439 (T.Z.B.), NS28912 (A.L.B., T.Z.B.), and NS02808 and NS38992 (G.W.M.) and by an Epilepsy Foundation of America, Milken Foundation postdoctoral research fellowship (R.A.B.). We thank Dr. Y. Chen for technical help.
Correspondence should be addressed to Dr. Gary W. Mathern, Reed Neurological Research Center, Room 2123, 710 Westwood Plaza, Los Angeles, CA 90095-1769. E-mail: gmathern@ucla.edu.
Copyright © 2003 Society for Neuroscience 0270-6474/03/236826-11$15.00/0
References
- Abercrombie M ( 1946) Estimation of nuclear population from microtome sections. Anat Rec 94: 239-247. [DOI] [PubMed] [Google Scholar]
- Armstrong DD ( 1993) The neuropathology of temporal lobe epilepsy. J Neuropath Exp Neurol 52: 433-443. [DOI] [PubMed] [Google Scholar]
- Babb TL, Pretorius JK, Kupfer WR, Crandall PM ( 1989) Glutamate decarboxylase-immunoreactive neurons are preserved in human epileptic hippocampus. J Neurosci 9: 2562-2574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baram TZ, Eghbal-Ahmadi M, Bender RA ( 2002) Is neuronal death required for seizure-induced epileptogenesis in the immature brain? Prog Brain Res 135: 365-375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bender RA, Brewster A, Santoro B, Ludwig A, Hofmann F, Biel M, Baram TZ ( 2001) Differential and age-dependent expression of hyperpolarization-activated, cyclic nucleotide-gated cation channel isoforms 1-4 suggests evolving roles in the developing rat hippocampus. Neuroscience 106: 689-698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bender RA, Dubé C, Gonzalez-Vega R, Mina EW, Baram TZ ( 2003) Mossy fiber plasticity and enhanced hippocampal excitability, without hippocampal cell loss or altered neurogenesis, in an animal model of prolonged febrile seizures. Hippocampus 13: 357-370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bräuer AU, Savaskan NE, Kole MH, Plaschke M, Monteggia LM, Nestler EJ, Simburger E, Deisz RA, Ninnemann O, Nitsch R ( 2001) Molecular and functional analysis of hyperpolarization-activated pacemaker channels in the hippocampus after entorhinal cortex lesion. FASEB J 15: 2689-2701. [DOI] [PubMed] [Google Scholar]
- Brewster A, Bender RA, Chen Y, Dubé C, Eghbal-Ahmadi M, Baram TZ ( 2002) Developmental febrile seizures modulate hippocampal gene expression of hyperpolarization-activated channels in an isoform- and cell-specific manner. J Neurosci 22: 4591-4599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks-Kayal AR, Shumate MD, Jin H, Rikhter TY, Coulter DA ( 1998) Selective changes in single cell GABAA receptor subunit expression and function in temporal lobe epilepsy. Nat Med 4: 1166-1172. [DOI] [PubMed] [Google Scholar]
- Brunson KL, Eghbal-Ahmadi M, Bender RA, Chen Y, Baram TZ ( 2001) Long-term, progressive hippocampal cell loss and dysfunction induced by early-life administration of corticotropin-releasing hormone reproduce the effects of early-life stress. Proc Natl Acad Sci USA 98: 8856-8861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckmaster PS, Zhang GF, Yamawaki R ( 2002) Axon Sprouting in a model of temporal lobe epilepsy creates a predominantly excitatory feedback circuit. J Neurosci 22: 6650-6658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buhl DL, Morozov A, Mongomery SM, Harris K, Kandel E, Buzsáki G ( 2002) Network patterns in the hippocampus of the HCN1 KO mouse. Soc Neurosci Abstr 28: 577.1. [Google Scholar]
- Cavalheiro EA, Leite JP, Bortolotto ZA, Turski WA, Ikonomidou C, Turski L ( 1991) Long-term effects of pilocarpine in rats: structural damage of the brain triggers kindling and spontaneous recurrent seizures. Epilepsia 32: 778-782. [DOI] [PubMed] [Google Scholar]
- Cendes F, Andermann F, Dubeau F, Gloor P, Evans A, Jones-Gotman M, Olivier A, Andermann E, Robitaille Y, Lopes-Cendes I, Peters T, Melanson D ( 1993) Early childhood prolonged febrile convulsions, atrophy and sclerosis of mesial structures and temporal lobe epilepsy: an MRI volumetric study. Neurology 43: 1083-1087. [DOI] [PubMed] [Google Scholar]
- Chen K, Baram TZ, Soltesz I ( 1999) Febrile seizures in the developing brain result in persistent modification of neuronal excitability in limbic circuits. Nat Med 5: 888-894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen K, Aradi I, Thon N, Eghbal-Ahmadi M, Baram TZ, Soltesz I ( 2001) Persistently modified h-channels after complex febrile seizures convert the seizure-induced enhancement of inhibition to hyperexcitability. Nat Med 7: 331-337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Wang J, Siegelbaum SA ( 2001) Properties of hyperpolarization-activated pacemaker current defined by coassembly of HCN1 and HCN2 subunits and basal modulation by cyclic nucleotide. J Gen Physiol 117: 491-504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Bender RA, Frotscher M, Baram TZ ( 2001) Novel and transient populations of corticotropin-releasing hormone-expressing neurons in developing hippocampus suggest unique functional roles: a quantitative spatiotemporal analysis. J Neurosci 21: 7171-7181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chevaleyre V, Castillo PE ( 2002) Assessing the role of Ih channels in synaptic transmission and mossy fiber LTP. Proc Natl Acad Sci USA 99: 9538-9543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coulter DA ( 2001) Epilepsy-associated plasticity in gamma-aminobutyric acid receptor expression, function, and inhibitory synaptic properties. Int Rev Neurobiol 45: 237-252. [DOI] [PubMed] [Google Scholar]
- Dalby NO, Mody I ( 2001) The process of epileptogenesis: a pathophysiological approach. Curr Opin Neurol 14: 187-192. [DOI] [PubMed] [Google Scholar]
- DiFrancesco D ( 1993) Pacemaker mechanisms in cardiac tissue. Annu Rev Physiol 55: 455-472. [DOI] [PubMed] [Google Scholar]
- Dubé C, Chen K, Eghbal-Ahmadi M, Brunson K, Soltesz I, Baram TZ ( 2000) Prolonged febrile seizures in the immature rat model enhance hippocampal excitability long term. Ann Neurol 47: 336-344. [PMC free article] [PubMed] [Google Scholar]
- Engel Jr J ( 1993) Surgical treatment of the epilepsies, Ed 2 (Engel Jr J, ed). New York: Raven.
- Franck JE, Pokorny J, Kunkel DD, Schwartzkroin PA ( 1995) Physiologic and morphologic characteristics of granule cell circuitry in human epileptic hippocampus. Epilepsia 36: 543-558. [DOI] [PubMed] [Google Scholar]
- Franz O, Liss B, Neu A, Roeper J ( 2000) Single-cell mRNA expression of HCN1 correlates with a fast gating phenotype of hyperpolarization-activated cyclic nucleotide-gated ion channels (IH) in central neurons. Eur J Neurosci 12: 2685-2693. [DOI] [PubMed] [Google Scholar]
- French JA, Williamson PD, Thadani VM, Darcey TM, Mattson RH, Spencer SS, Spencer DD ( 1993) Characteristics of medial temporal lobe epilepsy. I. Results of history and physical examination. Ann Neurol 34: 774-780. [DOI] [PubMed] [Google Scholar]
- Haas CA, Dudeck O, Kirsch M, Huszka C, Kann G, Pollak S, Zentner J, Frotscher M ( 2002) Role for reelin in the development of granule cell dispersion in temporal lobe epilepsy. J Neurosci 22: 5797-5802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houser CR ( 1990) Granule cell dispersion in the dentate gyrus of humans with temporal lobe epilepsy. Brain Res 535: 195-204. [DOI] [PubMed] [Google Scholar]
- Isokawa M, Levesque M, Fried I, Engel Jr J ( 1997) Glutamate currents in morphologically identified human dentate granule cells in temporal lobe epilepsy. J Neurophysiol 77: 3355-3369. [DOI] [PubMed] [Google Scholar]
- Jeub M, Lie A, Blümcke I, Elger CE, Beck H ( 1999) Loss of dynorphin-mediated inhibition of voltage-dependent Ca2+ currents in hippocampal granule cells isolated from epilepsy patients is associated with mossy fiber sprouting. Neuroscience 94: 465-471. [DOI] [PubMed] [Google Scholar]
- Jiao Y, Sun Z, Lee T, Fusco FR, Kimble TD, Meade CA, Cuthbertson S, Reiner A ( 1999) A simple and sensitive antigen retrieval method for free-floating and slide-mounted tissue sections. J Neurosci Methods 93: 149-162. [DOI] [PubMed] [Google Scholar]
- Johnson SA, Morgan DG, Finch CE ( 1986) Extensive postmortem stability of RNA from rat and human brain. J Neurosci Res 16: 267-280. [DOI] [PubMed] [Google Scholar]
- Kobayashi M, Buckmaster PS ( 2003) Reduced inhibition of dentate gyrus granule cells in a model of temporal lobe epilepsy. J Neurosci 23: 2440-2452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lörincz A, Notomi T, Tamás C, Shigemoto R, Nusser Z ( 2002) Polarized and compartment-dependent distribution of HCN1 in pyramidal cell dendrites. Nat Neurosci 5: 1185-1193. [DOI] [PubMed] [Google Scholar]
- Ludwig A, Zong X, Jeglitsch M, Hofmann F, Biel M ( 1998) A family of hyperpolarization-activated mammalian cation channels. Nature 393: 587-591. [DOI] [PubMed] [Google Scholar]
- Ludwig A, Budde T, Stieber J, Moosmang S, Wahl C, Holthoff K, Langebartels A, Wotjak C, Munsch T, Zong X, Feil S, Feil R, Lancel M, Chien KR, Konnerth A, Pape HC, Biel M, Hofmann F ( 2003) Absence epilepsy and sinus dysrhythmia in mice lacking the pacemaker channel HCN2. EMBO J 22: 216-224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lupica CR, Bell JA, Hoffman AF, Watson PL ( 2001) Contribution of the hyperpolarization-activated current (Ih) to membrane potential and GABA release in hippocampal interneurons. J Neurophysiol 86: 261-268. [DOI] [PubMed] [Google Scholar]
- Maccaferri G, McBain CJ ( 1996) The hyperpolarization-activated current (Ih) and its contribution to pacemaker activity in rat CA1 hippocampal stratum oriens-alveus interneurons. J Physiol (Lond) 497: 119-130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magee JC ( 1998) Dendritic hyperpolarization-activated currents modify the integrative properties of hippocampal CA1 pyramidal neurons. J Neurosci 18: 7613-7624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magee JC ( 1999) Dendritic Ih normalizes temporal summation in hippocampal CA1 neurons. Nat Neurosci 2: 508-514. [DOI] [PubMed] [Google Scholar]
- Mathern GW, Babb TL, Armstrong DL ( 1997a). Hippocampal sclerosis. In: Epilepsy: a comprehensive textbook (Engel Jr J, Pedley TA, eds), pp 133-155. Philadelphia: Lippincott-Raven.
- Mathern GW, Kuhlman PA, Mendoza D, Pretorius JK ( 1997b) Human fascia dentata anatomy and hippocampal neuron densities differ depending on the epileptic syndrome and age at first seizures. J Neuropathol Exp Neurol 56: 199-212. [DOI] [PubMed] [Google Scholar]
- Mathern GW, Pretorius JK, Mendoza D, Leite JP, Chimelli L, Born DE, Fried I, Assirati JA, Ojemann GA, Adelson PD, Cahan LD, Kornblum HI ( 1999) Hippocampal N-methyl-aspartate receptor subunit mRNA levels in temporal lobe epilepsy patients. Ann Neurol 46: 343-358. [DOI] [PubMed] [Google Scholar]
- Mathern GW, Adelson PD, Cahan LD, Leite JP ( 2002) Hippocampal neuron damage in epilepsy: Meyer's hypothesis revisited. Prog Brain Res 135: 237-251. [DOI] [PubMed] [Google Scholar]
- Mellor J, Nicoll RA, Schmitz D ( 2002) Mediation of hippocampal mossy fiber long term potentiation by presynaptic Ih channels. Science 295: 143-147. [DOI] [PubMed] [Google Scholar]
- Moosmang S, Biel M, Hofmann F, Ludwig A ( 1999) Differential distribution of four hyperpolarization-activated cation channels in mouse brains. Biol Chem 380: 975-980. [DOI] [PubMed] [Google Scholar]
- Nägerl UV, Mody I, Jeub M, Lie AA, Elger CE, Beck H ( 2000) Surviving granule cells of the sclerotic human hippocampus have reduced Ca2+ influx because of a loss of calbindin-D28k in temporal lobe epilepsy. J Neurosci 20: 1831-1836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okazaki MM, Evenson DA, Nadler JV ( 1995) Hippocampal mossy fiber sprouting and synapse formation after status epilepticus in rats: visualization after retrograde transport of biocytin. J Comp Neurol 352: 515-534. [DOI] [PubMed] [Google Scholar]
- Pape HC ( 1996) Queer current and pacemaker: the hyperpolarization-activated cation current in neurons. Annu Rev Physiol 58: 299-327. [DOI] [PubMed] [Google Scholar]
- Poolos NP, Migliore M, Johnston D ( 2002) Pharmacological upregulation of h-channels reduces the excitability of pyramidal neuron dendrites. Nat Neurosci 5: 767-774. [DOI] [PubMed] [Google Scholar]
- Santoro B, Liu D, Yao H, Bartsch D, Kandel ER, Siegelbaum SA, Tibbs GR ( 1998) Identification of a gene encoding a hyperpolarization-activated pacemaker channel of brain. Cell 93: 717-729. [DOI] [PubMed] [Google Scholar]
- Santoro B, Chen S, Lüthi A, Pavlidis P, Shumyatsky GP, Tibbs GR, Siegelbaum SA ( 2000) Molecular and functional heterogeneity of hyperpolarization-activated pacemaker channels in the mouse CNS. J Neurosci 20: 5264-5275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarzer C, Sperk G ( 1995) Hippocampal granule cells express glutamic acid decarboxylase-67 after limbic seizures in the rat. Neuroscience 69: 705-709. [DOI] [PubMed] [Google Scholar]
- Sloviter RS, Zappone CA, Harvey BD, Bumanglag AV, Bender RA, Frotscher M ( 2003) “Dormant basket cell” hypothesis revisited; relative vulnerabilities of dentate gyrus mossy cells and inhibitory interneurons after hippocampal status epilepticus in the rat. J Comp Neurol 459: 44-76. [DOI] [PubMed] [Google Scholar]
- Sutula TP, Pitkänen A ( 2002) Do seizures damage the brain? In: Progressive brain research. Amsterdam: Elsevier.
- Swanson TH, Sperling MR, O'Connor MJ ( 1998) Strong paired pulse depression of dentate granule cells in slices from patients with temporal lobe epilepsy. J Neural Transm 105: 613-625. [DOI] [PubMed] [Google Scholar]
- Toth Z, Yan XX, Haftoglou S, Ribak CE, Baram TZ ( 1998) Seizure-induced neuronal injury: vulnerability to febrile seizures in an immature rat model. J Neurosci 18: 4285-4294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulens C, Tytgat J ( 2001) Functional heteromerization of HCN1 and HCN2 pacemaker channels. J Biol Chem 276: 6069-6072. [DOI] [PubMed] [Google Scholar]
- Vasilyev DV, Barish ME ( 2002) Postnatal development of the hyperpolarization-activated excitatory current Ih in mouse hippocampal pyramidal neurons. J Neurosci 22: 8992-9004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wainger BJ, DeGennaro M, Santoro B, Siegelbaum SA, Tibbs GR ( 2001) Molecular mechanism of cAMP modulation of HCN pacemaker channels. Nature 411: 805-810. [DOI] [PubMed] [Google Scholar]
- Walker MC, Kullmann DM ( 1999) Febrile convulsions: a “benign” condition? Nat Med 5: 871-872. [DOI] [PubMed] [Google Scholar]
- Wenzel HJ, Woolley CS, Robbins CA, Schwartzkroin PA ( 2000) Kainic acid-induced mossy fiber sprouting and synapse formation in the dentate gyrus of rats. Hippocampus 10: 244-260. [DOI] [PubMed] [Google Scholar]
- West MJ ( 1999) Stereological methods for estimating the total number of neurons and synapses: issues of precision and bias. Trends Neurosci 22: 51-61. [DOI] [PubMed] [Google Scholar]
- Williamson A, Patrylo PR, Spencer DD ( 1999) Decrease in inhibition in dentate granule cells from patients with medial temporal lobe epilepsy. Ann Neurol 45: 92-99. [DOI] [PubMed] [Google Scholar]