

Abstract
Radiation therapy is a first line treatment for prostate cancer patients with localized tumors. Although some patients respond well to the treatment, approximately 10% of low-risk and up to 60% of high-risk prostate cancer patients experience recurrent tumors. However, the molecular mechanisms underlying tumor recurrence remain largely unknown. Here we show that fractionated ionizing radiation (IR) induces differentiation of LNCaP prostate cancer cells into neuroendocrine (NE)-like cells, which are known to be implicated in prostate cancer progression, androgen independent growth and poor prognosis. Further analyses revealed that two CRE-binding transcription factors, CREB and ATF2, function as a transcriptional activator and repressor, respectively, of NE-like differentiation and that IR induces NE-like differentiation by increasing the nuclear content of phospho-CREB and cytoplasmic accumulation of ATF2. Consistent with this notion, stable expression of a non-phosphorylatable CREB or a constitutively nuclear-localized ATF2 in LNCaP cells inhibits IR-induced NE-like differentiation. IR-induced NE-like morphologies are reversible, and three IR-resistant clones isolated from dedifferentiated cells have acquired the ability to proliferate and lost the NE-like cell properties. Also, these three IR-resistant clones exhibit differential responses to IR- and androgen depletion-induced NE-like differentiation. However, they are all resistant to IR-and the chemotherapeutic agent docetaxel-induced cell death, and to androgen depletion-induced growth inhibition. These results suggest that radiation therapy-induced NE-like differentiation may represent a novel pathway by which prostate cancer cells survive the treatment and contribute to tumor recurrence.
Kew words: prostate cancer, neuroendocrine differentiation, ionizing radiation, ATF2, CREB
Introduction
Radiation therapy is a first line treatment for prostate cancer. Although some patients with localized tumors respond well to the treatment (1), approximately 10% of low-risk and up to 60% of high-risk prostate cancer patients experience recurrent tumors (2). However, the molecular mechanisms underlying tumor recurrence remain largely unknown.
Neuroendocrine (NE) cells are one of three types of epithelial cells in the human prostate and are present in 30–100% cases of prostatic adenocarcinoma (3, 4). Although the physiological role of NE cells remains unclear, increased numbers of NE-like cells appear to be associated with prostate cancer progression, androgen independent growth and poor prognosis (5, 6). Interestingly, androgen ablation, cytokines such as IL-6, and agents that elevate the intracellular levels of cAMP can induce NE-like differentiation (NED) in LNCaP prostate cancer cells by activating several distinct signaling pathways (5, 6). Like NE cells, the differentiated NE-like cells also produce a number of neuropeptides that facilitate the growth of surrounding tumor cells in a paracrine manner (5–7). They are generally androgen receptor (AR) negative (8, 9), highly resistant to apoptosis (10, 11), and their differentiation state is reversible (12). Thus, NE-like cells may survive in a dormant state and contribute to prostate cancer recurrence upon dedifferentiation (12).
cAMP response element binding protein (CREB) belongs to the basic region leucine zipper (bZIP) family of transcription factors (13–15). It functions as homodimers or heterodimers to bind a specific DNA sequence, the cAMP responsive element (16), to regulate transcription of target genes responsible for many cellular processes including cell proliferation and differentiation (15). CREB is implicated in prostate cancer growth (17), acquisition of androgen independent growth (18), and transcription of chromogranin A (CgA) (19) and prostate-specific antigen (20). Although it is known that CREB is activated by protein kinase A through the phosphorylation at Ser133 of CREB1B in response to cAMP (14, 21), whether CREB itself can induce NED remains to be determined.
Activating transcription factor 2 (ATF2) also belongs to the bZIP family of transcription factors (22, 23) and is a member of the activator protein 1 (AP–1) (24). AP-1 activity is required for many cellular processes, and deregulated AP-1 activity is implicated in many cancers including prostate cancer (25). Interestingly, ATF2 and CREB share the same CRE sequence, and regulate the transcription of CRE-containing genes. While some CRE-containing target genes are activated by CREB and ATF2 equally or cooperatively (26), differential regulation of other target genes by CREB and ATF2 has also been observed (27–31). Unlike CREB, the role of ATF2 in prostate cancer is little known. A recent study reported that increased cytoplasmic localization of phospho-ATF2 in prostate cancer specimens correlates with the clinical progression of prostate cancer (32), suggesting that alteration of ATF2 subcellular localization may contribute to clinical progression of prostate cancer.
We recently demonstrated that ATF2 is a nucleocytoplasmic shuttling protein and its subcellular localization is regulated by AP-1 dimerization (33). Here we present evidence that ATF2 constantly shuttles between the cytoplasm and nucleus in proliferating LNCaP cells and that fractionated ionizing radiation (IR) induces NED by impairing the nuclear import of ATF2 and increasing the nuclear phospho-CREB at Ser133 (pCREB).
Materials and Methods
Plasmid construction
To construct a constitutively activated form of CREB, cDNA encoding residues 413–490 of VP16 was amplified by PCR from VP16 (Clontech) and subcloned into pHA-CMV. To make VP16-bCREB fusion proteins, cDNA encoding the bZIP domain of CREB1B (residues 285–314) was amplified by PCR from a human cDNA library and subcloned into pHA-VP16. A flexible glycine spacer (GGGGSx4) was inserted between VP16 and bCREB. For the construction of nATF2, the sequence encoding a nuclear localization signal (NLS) (PKKKRKV) from the large T antigen of SV40 (34) was subcloned upstream of ATF2 coding sequences in pFlag-ATF2. pFlag-cATF2 is a deletion mutant of ATF2 in which two NLSs are deleted (33). Both cATF2 and nATF2 were expressed as a fusion protein with the fluorescent protein, Venus, in transient transfection experiments. To knockdown ATF2, sense and antisense oligos (19-mer) were synthesized and subcloned into pSUPER (OligoEngine). Four short interference RNA (siRNA) constructs were made, and their effect on ATF2 expression in LNCaP cells was verified by transient transfection, followed by immunoblotting of ATF2. One ATF2 siRNA construct targeting the 5’ UTR (148–167 of ATF2 mRNA) proved to be the most potent and was used in this work. All plasmids were verified by DNA sequencing.
IR-induced NE-like differentiation
Cells were cultured in 10 cm dishes in RPMI 1640 supplemented with 10% fetal bovine serum (FBS) and antibiotics and were continuously irradiated (2 Gy/day, 5 days/week) in a GC-220 Co-60 for the indicated times. NE-like cells were visualized by morphological changes, and the induction of the NE markers, CgA and neuron-specific enolase (NSE), was determined using immunoblotting with anti-CgA and anti-NSE antibodies (Abcam). To determine the effect of CREB-S133A and nATF2 on IR-induced NED, we used a tetracycline-on system (Invitrogen) to establish stable cell lines that inducibly expressed HA-CREB-S133A or Flag-nATF2. The established cell lines were maintained in the presence of selectable markers (zeocin and blasticidin), and 5 μg/ml of tetracycline was applied while cells were irradiated as described above. Media were changed twice a week, and antibiotics and tetracycline were added accordingly. Cells that extended neurites longer than 2 cell bodies were scored as differentiated, and the induction of CgA and NSE was analyzed by immunoblotting and quantified using ImageJ software. Values were normalized to β-actin.
Analysis of ATF2 and CREB subcellular localization
LNCaP cells were fixed in ice-cold 3.7% formaldehyde for 20 min, followed by permeabilization in ice-cold 0.2% Triton X-100 for 5 min. Cells were incubated with anti-ATF2 (c-19) (Santa Cruz Biotechnology) overnight, followed by three washes and incubation with the secondary antibody conjugated with Texas Red (Jackson ImmunoResearch Laboratories) for 1 h. To stain DNA, 4',6-diamidino-2-phenylindole (DAPI) was added to the secondary antibody staining reaction at the final concentration of 0.5 μg/ml. Subcelluar localization of ATF2 was examined by microscopic analysis, and fluorescent images were captured using a charge-coupled device camera mounted on a Nikon TE2000 inverted fluorescence microscope with the DAPI and Texas Red filters.
For biochemical subcellular fractionation analysis, cytosolic and nuclear fractions were prepared as described before (33). Cytosolic and nuclear fractions were verified by anti-β-tubulin (Sigma) or anti-histone 3 (Abcam), respectively, in immunoblotting assays. The amount of ATF2, pCREB and CREB was determined with anti-ATF2 and anti-pCREB or anti-CREB (Cell Signaling) antibodies. The amount of ATF2 and pCREB in the cytoplasm or nucleus, respectively, relative to total protein was quantified using ImageJ software.
Transient Transfection
To evaluate the effect of ATF2 knockdown, mutant ATF2 or mutant CREB on NED, 60-80% confluent LNCaP cells cultured in 10 cm dishes were transfected with the indicated plasmids using FuGENE HD (Roche). Transfected cells were examined for morphological changes and harvested for determination of expression of NE markers CgA and NSE using immunoblotting six days after transfection. The induction of CgA and NSE was quantified using ImageJ software and normalized to β-actin.
IR- and androgen depletion-induced NE-like differentiation in IR-resistant clones
To study IR-induced NED in IR-resistant clones, cells were similarly treated as described above for wild-type LNCaP cells. NE-like cells were visualized by morphological changes, and the induction of NE markers CgA and NSE and the expression of AR were determined using immunoblotting with anti-CgA, anti-NSE and anti-AR (Santa Cruz Biotechnology) antibodies. To determine the response of IR-resistant clones to androgen depletion treatment, cells were cultured in phenol-free RPMI 1640 supplemented with 10% Charcoal/Dextran-treated FBS (C/D-FBS) for three weeks and similarly assayed for morphological changes and the induction of NE markers CgA and NSE. Note that although androgen depletion treatment for one week was sufficient to induce neurite outgrowth, the induction of CgA and NSE expression was barely detectable by immunoblotting even for wild-type LNCaP cells.
Cell viability and growth inhibition assay
Wild-type or IR-resistant clones were cultured in 48-well plates and irradiated with fractionated IR (2 Gy/day), or treated with docetaxel (5 nM) or cultured in phenol-free RPMI 1640 supplemented with 10% C/D-FBS for the indicated times. Cell viability for IR- and docetaxel-treated cells was determined using an MTT assay as described previously (33). Because irradiated cells only showed cell death after the second week of irradiation, cell viability of wild-type LNCaP or IR-resistant clones was determined by comparing with cells that had received 10-Gy irradiation. Because wild-type and IR-resistant clones showed different growth rates and because C/D-FBS treatment only inhibited cell growth without inducing cell death, cells cultured in normal FBS were used as controls to first calculate the percentage of growth inhibition (percentage of viable cells in C/D-FBS over those in normal FBS), which was subsequently used to calculate the percentage of growth inhibition at different times when compared with cells immediately after treatment (Day 0). A Student’s t-test was applied for statistical analysis.
Results
IR induces NE-like differentiation in LNCaP cells
In an attempt to isolate radiation resistant clones by following a clinical protocol (70 Gy) (1), we surprisingly found that upon 40-Gy irradiation (2 Gy/day, 5 days/week), the majority of cells (~80%) died while cells that survived the treatment displayed the growth of extended neurites (Fig. 1A), an NE-like phenotype. Expression of two NE cell markers, CgA and NSE, was significantly induced (Fig. 1B, 1C). Similar treatments failed to induce NED in DU145 and PC-3 prostate cancer cells. Consistent with previous reports that NE cells are apoptosis resistant (10, 11), IR-induced NE-like cells were resistant to IR and survived another three week-irradiation until the completion of the entire radiation protocol (70 Gy). Addition of the chemotherapeutic agent docetaxel into the IR-induced NE-like cells did not cause any change in cell viability either.
Fig. 1. IR induces NE-like differentiation in LNCaP prostate cancer cells.

(A) Shown are representative images of cells that received the indicated times of exposures (2 Gy/day, 5 days/week). Note that cells irradiated 20 times display significant neurite outgrowth and branching. (B) and (C) Immunoblotting of CgA and NSE. Cells that received the indicated dose of radiation were harvested and 20 μg of total protein was used for immunoblotting of CgA and NSE.
IR induces cytoplasmic accumulation of ATF2 and an increase in nuclear pCREB
To determine the subcellular localization of ATF2 in IR-induced NE-like cells, we performed immunostaining and found that ATF2 localization in the cytoplasm was increased compared to non-treated cells (Supplementary SFig. 1A). No significant changes in expression and nuclear localization of c-Jun, JunB and JunD were observed (data not shown), suggesting that increased cytoplasmic localization of ATF2 is not due to an decrease in Jun proteins to anchor ATF2 in the nucleus (33). In contrast, ATF2 was predominantly localized in the nucleus with some cytoplasmic localization in proliferating LNCaP cells, and treatment with the nuclear export inhibitor leptomycin B (LMB) (33, 35) increased nuclear localization of ATF2 (Fig 2A). The nuclear sequestration of ATF2 in proliferating LNCaP cells by LMB was also confirmed by subcellular fractionation analysis (data not shown). These results demonstrate that ATF2 constantly shuttles between the cytoplasm and nucleus in proliferating LNCaP cells. Consistent with our previous observation that phosphorylation at residues T69 and T71 does not regulate ATF2 subcellular localization (33), the subcellular localization of phospho-ATF2 was similar to that of ATF2 in proliferating and the NE-like cells (data not shown).
Fig. 2. IR induces cytoplasmic accumulation of ATF2 and an increase in nuclear pCREB in LNCaP cells.
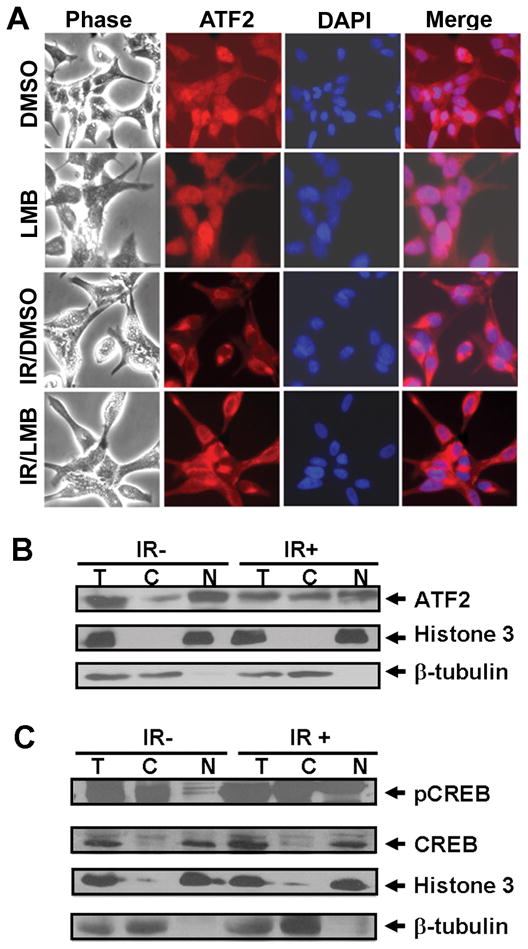
(A) LNCaP Prostate cancer cells cultured in 12-well plates were treated with DMSO or LMB (40 ng/ml) overnight or irradiated (2 Gy/day) for 5 days followed by treatments with DMSO or LMB overnight. Subcellular localization of ATF2 was determined by immunostaining with anti-ATF2 antibody, and DNA in the nucleus was stained with DAPI. (B) and (C) Non-irradiated (IR-) or irradiated LNCaP cells (2 Gy x 5) (IR+) were harvested, and cytosolic, nuclear and total cellular extracts were prepared. Approximately 20 μg of total cellular extracts (T) and an equal portion of cytosolic (C) and nuclear (N) extracts were used for immunoblotting of ATF2, pCREB and CREB.
To determine whether ATF2 cytoplasmic localization is a consequence or a potential cause of NED, we examined ATF2 subcellular localization at different time points before cells underwent morphological changes. Irradiation of cells up to five times increased cytoplasmic ATF2 without inducing striking morphological alterations (Fig. 2A). However, treatment of the irradiated cells with LMB failed to induce nuclear accumulation of ATF2 in irradiated cells (Fig. 2A), indicating that IR impairs the nuclear import of ATF2. No significant change in ATF2 subcellular localization was observed when irradiated less than five times. Subcellular fractionation analysis showed that IR treatment increased cytoplasmic ATF2 from 24% to 45% of total ATF2 (Fig. 2B)
Because CREB regulates transcription of CgA (19), we examined expression and subcellular localization of CREB and pCREB in proliferating and IR-irradiated cells, as we did for ATF2. Because all available pCREB antibodies we tested cross-reacted with phospho-ATF1 and another ~80 kDa cytoplasmic protein (data not shown), we performed subcellular fractionation analysis and determined that IR treatment increased nuclear pCREB from 25% to 49% of the total pCREB (Fig. 2C). Unlike pCREB, the nuclear content of CREB was not altered by IR treatment (Fig. 2C). Interestingly, pCREB was also detected in the cytoplasm in proliferating LNCaP cells and IR treatment did not seem to alter the phosphorylation extent of cytoplasmic CREB. IR-induced NE-like cells maintained a high level of pCREB in the nucleus (Supplementary SFig. 1B). Taken together, these results demonstrate that IR-induced cytoplasmic accumulation of ATF2 and nuclear pCREB occur before cells undergo differentiation.
CREB and ATF2 play an opposing role in NE-like differentiation
The IR-induced cytoplasmic accumulation of ATF2 and nuclear pCREB prompted us to test the hypothesis that nuclear CREB and ATF2 may play an opposing role in NED. Indeed, 50% knockdown of ATF2 resulted in an NE-like morphological change (Supplementary SFig. 2A) and a 1.6-fold induction of NSE (Fig. 3A). No induction of CgA was observed (data not shown). In contrast, transient expression of VP16-bCREB, a constitutively activated and nuclear-localized mutant of CREB (36, 37), induced an NE-like morphological change (Supplementary SFig. 2B) and increased CgA and NSE expression by 2-3 fold (Fig. 3B). However, overexpression of a constitutively nuclear-localized ATF2, nATF2, that has an NLS from the large T antigen of SV40 fused to the N-terminus of ATF2 as others did (34), inhibited VP16-bCREB-mediated morphological changes and the induction of NSE. To determine whether increased cytoplasmic accumulation of endogenous ATF2 can induce NED, we overexpressed a constitutively cytoplasmic-localized ATF2 (cATF2), which lacks the two NLSs (33), in LNCaP cells. Because ATF2 homodimerization impairs ATF2 nuclear import (33), overexpression of cATF2 increased cytoplasmic localization of ATF2 to approximately 50% of total ATF2 (data not shown). Indeed, cATF2, but not nATF2, induced neurite outgrowth (Supplementary SFig. 2C) and a 5.4-fold increase in NSE expression (Fig. 3C). No induction of CgA by cATF2 or nATF2 was observed. Transiently expressed cATF2-Venus and nATF2-Venus were predominantly localized to the cytoplasm and nucleus, respectively (Supplementary SFig. 2D). Immunoblotting analysis confirmed the exogenous expression of VP16-bCREB, cATF2-Venus and nATF2-Venus in these experiments (data not shown). Knockdown of ATF2 or expression of cATF2 had no effect on the localization and amount of pCREB, and overexpression of VP16-bCREB did not alter subcellular localization of ATF2 (data not shown). Taken together, these results support the hypothesis that CREB and ATF2 play an opposing role in NED.
Fig. 3. ATF2 and CREB play opposing roles in NE-like differentiation.
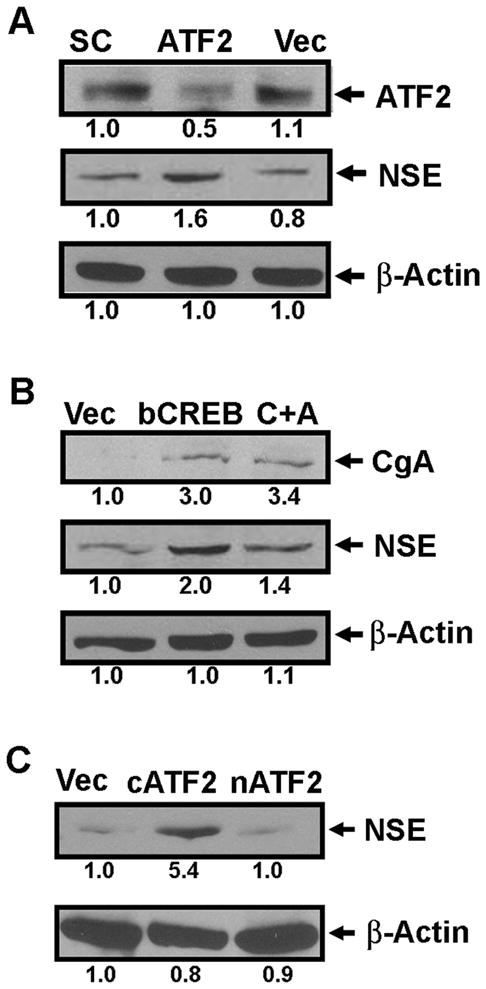
(A) Immunoblotting analysis of ATF2 and NSE expression from LNCaP cells transfected with siRNA constructs for scrambled sequences (SC), ATF2 siRNA (ATF2) or pSUPER vector only (Vec). (B) Immunoblotting analysis of CgA and NSE from LNCaP cells transfected with the vector control (Vec) , the plasmid encoding VP16-bCREB (bCREB), or co-transfected with plasmids encoding VP16-bCREB and nATF2 (C+A). (C) Immunoblotting analysis of NSE from LNCaP cells transfected with the vector control (Vec), or the plasmid encoding cATF2 (cATF2) or nATF2 (nATF2). The number below each lane is the quantified fold change when compared with the first lane.
Stable expression of a non-phosphorylatable CREB or nATF2 inhibits IR-induced NE-like differentiation
To further determine the role of CREB and ATF2 in IR-induced NED, we established tetracycline-inducible stable cell lines that express nATF2 or a non-phosphorylatable CREB (CREB-S133A), which has been used as a dominant negative mutant form of CREB (13, 15). In the absence of tetracycline, these stable cell lines exhibited normal morphology like vector-only cells (Fig. 4A). However, addition of tetracycline significantly induced expression of CREB-S133A or nATF2 (Fig. 4B) and reduced the percentage of cells displaying extended neurites in response to irradiation (Fig. 4C). Interestingly, induction of CgA and NSE by IR was inhibited by nATF2, but not by CREB-S133A (Fig. 4D). These results further support the conclusion that nuclear ATF2 and pCREB play different roles in IR-induced neurite outgrowth.
Fig. 4. Inhibition of IR-induced NE-like differentiation by dominant negative CREB and nATF2.
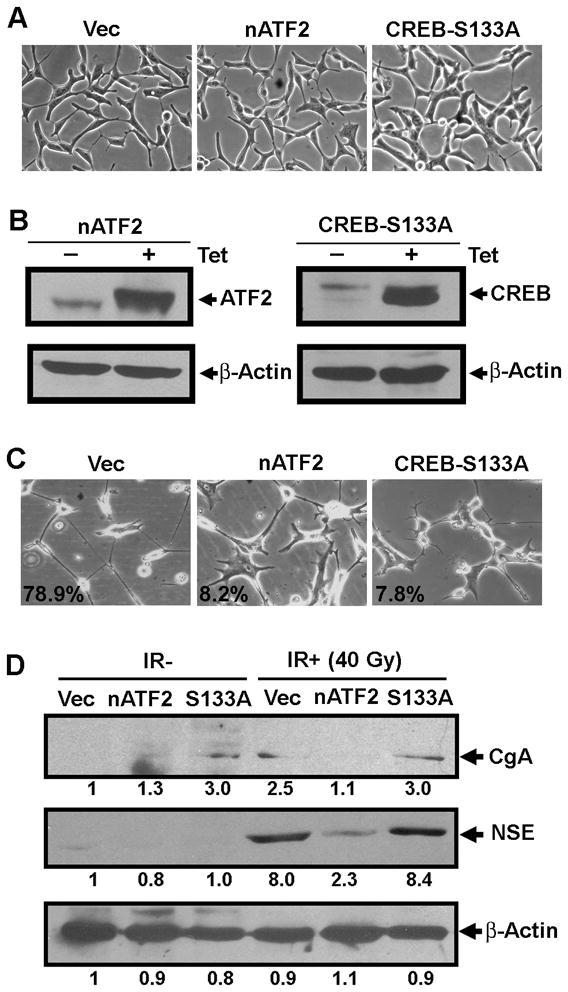
(A) Shown are representative images of stable cell lines that have pcDNA4/TO (Vec), pcDNA4-TO-Flag-nATF2 (nATF2), or pcDNA4/TO-HA-CREB-S133A (CREB-S133A) integrated. (B) Immunoblotting analysis of induced nATF2 and CREB-S133A by tetracycline. Total cell lysates were prepared three days after the induction and Flag-nATF2 and HA-CREB-S133A were detected using anti-ATF2 and anti-HA antibodies, respectively. (C) Shown are representative images acquired from stable cell lines that received 40 Gy-irradiation in the presence of tetracycline. The number indicates the percentage of cells showing extended neurites. (D) Immunoblotting analysis of CgA and NSE from experiments in C. The number below each lane is the quantified fold change when compared with the first lane.
To determine the relationship between the expression of nATF2 and IR-induced phosphorylation of CREB and the relatioship between the expression of CREB-S133A and the subcellular localization of ATF2, we irradiated cells for five days while constantly inducing expression of nATF2 or CREB-S133A. Expression of CREB-S133A did not affect IR-induced cytoplasmic localization of ATF2 (data not shown), whereas expression of nATF2 significantly inhibited IR-induced phosphorylation of CREB (Supplementary SFig. 3). However, expression of nATF2 only did not affect phosphorylation of CREB in the absence of IR (data not shown). These results suggest that IR-induced cytoplasmic sequestration of ATF2 may be a prerequisite for IR-induced phosphorylation of CREB and the subsequent NE-like differentiation.
IR-induced NE-like differentiation is reversible, and dedifferentiated cells lose NE- like properties
Because cAMP-induced NE-like cells are reversible (12), we sought to determine whether IR-induced NE-like cells are also reversible. We irradiated cells for four weeks (40 Gy) to allow all surviving cells to differentiate into NE-like cells and then waited for the growth of any cells that were reversible. Although differentiated NE-like cells were maintained without obvious cell death or growth for the first two months, we isolated three independent clones three months after the completion of the irradiation. We named these clones LNCaP-IRR1 (IRR refers to ionizing radiation resistant), LNCaP-IRR2 and LNCaP-IRR3. These IR-resistant cells showed similar morphology to wild-type LNCaP cells (Supplementary SFig. 4). All three clones lost CgA and NSE expression but retained levels of AR comparable to wild-type LNCaP cells (Fig. 5A), suggesting that these clones have lost their NE-like cell properties.
Fig. 5. Response of IR-resistant clones to IR- and androgen depletion-induced NE-like redifferentiation.
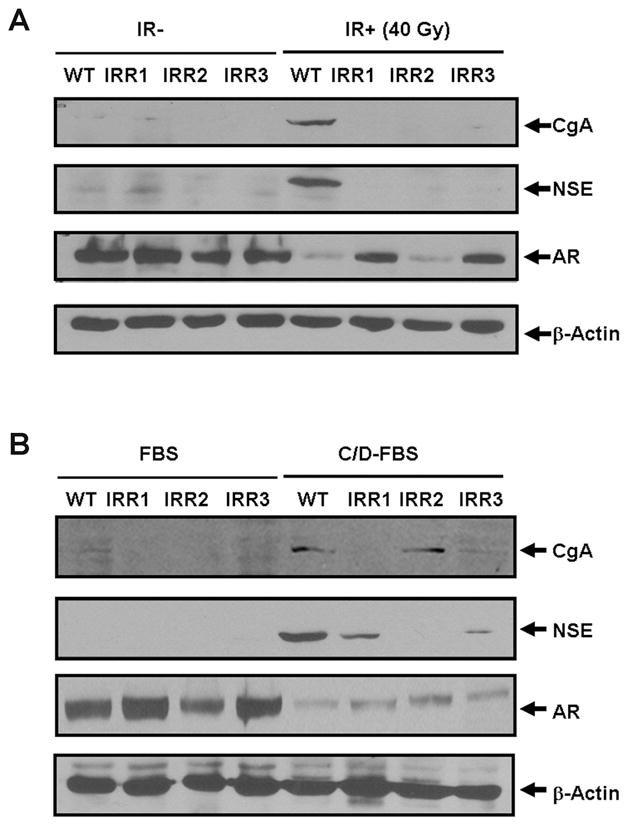
(A) Wild-type LNCaP (WT) and the indicated IR-resistant clones were subjected to fractionated IR (40 Gy), and the induction of CgA and NSE as well as the expression of AR was compared with non-irradiated (IR-) cells. (B) Wild-type LNCaP (WT) and IR-resistant clones were cultured in medium supplemented with 10% FBS (FBS) or C/D-FBS for three weeks, and the induction of CgA and NSE as well as the expression of AR were determined using immunoblotting.
To determine whether these IR-resistant clones can still be induced to redifferentiation, we irradiated them for 40 Gy and examined for morphological changes and the induction of CgA and NSE. While all three clones exhibited extended neurite outgrowth (Supplementary SFig. 5A), the induction of CgA and NSE was completely abrogated (Fig. 5A). Interestingly, AR expression in LNCaP-IRR2 clone was significantly inhibited like parental cells whereas AR expression in LNCaP-IRR1 and LNCaP-IRR3 cells was only slightly attenuated. These distinct responses to IR treatment suggest that these three IR-resistant clones are likely heterogeneous. To determine how these clones respond to androgen depletion treatment, we treated cells in phenol-free medium supplemented with 10% CD-FBS for three weeks. While LNCaP-IRR1 and LNCaP-IRR3 cells exhibited extended neurite outgrowth, LNCaP-IRR2 cells showed only short neurites (Supplementary SFig. 5B). Interestingly, LNCaP-IRR2 showed similar induction of CgA expression by CD-FBS to parental cells, no induction in LNCaP-IRR1 and a significantly attenuated induction in LNCaP-IRR3 cells were observed (Fig. 5B). On the contrary, the induction of NSE in LNCaP-IRR2 was abolished whereas LNCaP-IRR1 and IRR3 responded to the treatment to some extent. Like parental cells, however, the expression of AR in all three clones was significantly down-regulated by the CD-FBS treatment. Taken together, these results suggest that the three IR-resistant clones are heterogeneous and likely have distinct molecular defects in their responses to IR and androgen depletion treatments.
IR-resistant and dedifferentiated cells acquire cross-resistance to therapy
To explore the potential implication of dedifferentiated cells in prostate cancer progression, we examined their response to radiation, the chemotherapeutic agent docetaxel (38) and androgen depletion treatments. Like parental LNCaP cells, all three clones stopped growth during the first-week irradiation (10 Gy) and no cell death was observed (Fig. 6A). During the second-week irradiation, however, all three clones showed significantly reduced cell death when compared with the parental cells. Interestingly, all three IR-resistant cells began to resume growth during the third-week irradiation whereas the parental cells did not show obvious growth or death as all surviving cells differentiated into NE-like cells. Similar to their response to IR treatment, all three IRresistant clones were resistant to cell death induced by the chemotherapeutical agent docetaxel (Fig. 6B), as well as to growth inhibition upon androgen depletion (Fig. 6C). These results suggest that IR-induced NE-like cells have the potential to dedifferentiate back into a proliferating state with the acquisition of cross-resistance to radiotherapy, chemotherapy and hormonal therapy.
Fig. 6. Cross-resistance of IR-resistant clones to therapeutic treatments.
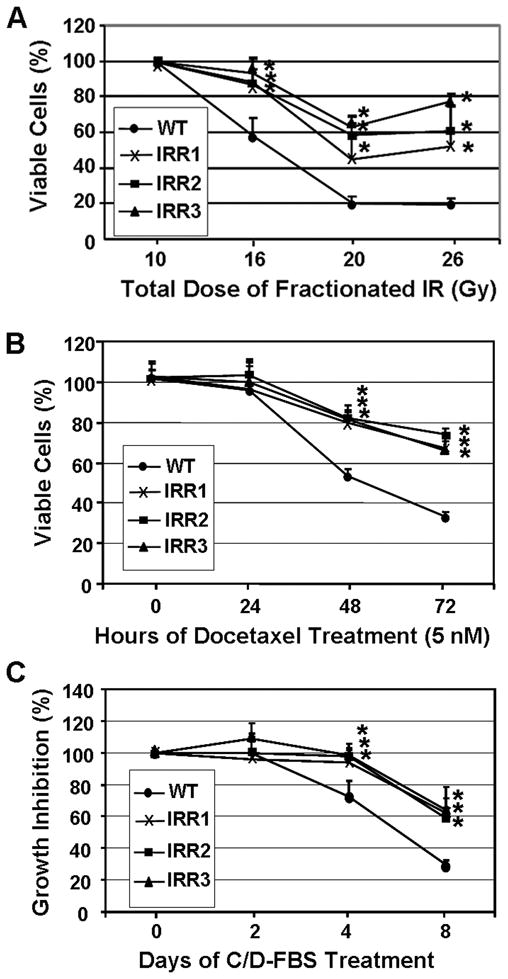
(A) Wild-type LNCaP (WT) and the indicated IR-resistant clones were cultured in 48-well plates and subjected to fractionated IR for the indicated doses. Cell viability was determined one day after the indicated irradiation as the percentage of viable cells that received 10 Gy-irradiation. (B) Cells were treated with docetaxel for the indicated time and cell viability was determined as the percentage of viable cells at 0 hour. (C) Cells were cultured in 10% FBS or C/D-FBS for the indicated time and the inhibition of cell growth by C/D-FBS was determined as described in Materials and Methods. *p<0.01 when compared with wild-type LNCaP cells.
Discussion
NE-like cells are implicated in prostate cancer progression, androgen independent growth and poor prognosis (3–6, 39, 40). Because androgen ablation treatment can induce NED in vitro and in vivo (3–6), it has been proposed that the presence of NE-like cells may contribute to androgen independent growth, a critical factor leading to the failure of current prostate cancer therapy. We present here the first evidence that in addition to androgen ablation, IR also induces NED in the prostate cancer cell line LNCaP. Significantly, IR-induced NED is reversible, and dedifferentiated cells have lost the NE-like properties. However, all isolated three IR-resistant clones derived from dedifferentiated cells are cross-resistant to radiation, docetaxel, and androgen depletion treatments. These findings, along with other reports (41–46), strongly suggest that radiation- or hormonal therapy-induced NED may represent a common pathway by which cancer cells survive treatment and contribute to prostate cancer recurrence.
Although it has been reported that STAT3 (47) and beta-catenin (48) can mediate IL-6- and androgen depletion-induce NED in prostate cancer cells, respectively, it remains largely unexplored how the switch from proliferation to differentiation is turned on at the transcriptional level. Several pieces of evidence presented in this work demonstrate that CREB functions as a transcriptional activator and ATF2 acts as a transcriptional repressor of NED. First, IR induced cytoplasmic accumulation of ATF2 and increased nuclear pCREB. Second, knockdown of ATF2 or overexpression of VP16-bCREB induced NED. Third, overexpression of nATF2 inhibited NED induced by VP16-bCREB, whereas overexpression of cATF2 induced NED. Last, stable expression of CREB-S133A or nATF2 inhibited IR-induced NED.
The transcriptional regulation of CRE-containing genes by ATF2 and CREB is dependent on individual genes. For example, the insulin promoter contains one CRE-binding site, and both ATF2 and CREB can bind it. However, ATF2 activates the transcription of insulin, whereas CREB inhibits it (31). In the present work, we also observed that overexpression of VP16-bCREB increased expression of endogenous CgA and NSE, whereas overexpression of nATF2 inhibited VP16-bCREB-induced expression of NSE, but not CgA. Likewise, knockdown of ATF2 or overexpression of cATF2 increased expression of NSE, but not CgA. These results support the notion that the effect of CREB and ATF2 on target gene transcription is dependent on gene context. Although VP16-bCREB can induce CgA and NSE expression (Fig. 3B), stable expression of nATF2, but not CREB-S133A, inhibited IR-induced expression of CgA and NSE (Fig. 4D). Despite the fact that the CREB-S133A-expressing stable cell line appears to have a basal level of CgA expression in the absence of tetracycline, which is likely due to leaky expression of CREB-S133A, induction of CREB-S133A by tetracycline did not alter the CgA expression in response to IR (Fig. 4D). Given that overexpression of VP16-bCREB induced expression of both CgA and NSE (Fig. 3B), these observations suggest that CREB is not responsible for IR-induced CgA and NSE expression. Alternatively, phosphorylation of CREB at different sites (21) may contribute to IR-induce CgA and NSE expression. Future studies are needed to distinguish these two possibilities. Interestingly, overexpression of CREB-S133A and nATF2 did not inhibit the growth of shorter neurites, but rather inhibited the elongation of neurites (Fig. 4C). Consistent with a role of CREB in neurite elongation in hippocampal neurons (49), it is likely that CREB and ATF2 may oppose each other in irradiated LNCaP cells to regulate transcription of target genes essential for neurite elongation, one of the phases during neuritogenesis (50). Further identification of the target genes will provide insight into the molecular mechanisms by which CREB and ATF2 play opposing roles in IR-induced NED. Because expression of nATF2 inhibited IR-induced phosphoryaltion of CREB (Supplementary SFig. 3), it is possible that nuclear ATF2 may also antagonize an upstream signaling pathway that contributes to IR-induced phosphorylation of CREB. It will be interesting to determine whether this effect is independent of or dependent on ATF2 transcriptional activity. In addition, identification of cell signaling that regulates cytoplasmic accumulation of ATF2 and phosphorylation of CREB will provide opportunities to develop novel therapeutics for prostate cancer.
The finding that IR can induce NED is clinically important, given that approximately 10-60% of patients treated with radiation therapy experience recurrent tumors (2). Although a detailed and well-controlled examination of NE-like cells in recurrent tumors would shed light on our in vitro findings here, the fact that patients who have biochemical recurrence after radiotherapy normally do not undergo surgery or even biopsy prevents us from performing this type of study. In addition, the transient nature of NE-like cells may also not allow us to find a causative link between radiation therapy and the induction of NED in patients. We are therefore currently performing longitudinal analyses to evaluate the effect of radiation therapy on NED and its contribution to tumor recurrence in xenograft nude mice prostate cancer models.
Supplementary Material
Acknowledgments
We would like to thank Drs. Robert Geahlen, David Riese, Jian Jian Li, and Timothy Ratliff for their support and consultation during the course of this work. We are grateful to the members of the Hu laboratory for helpful discussions. This work was supported by the Purdue Cancer Center Small Grants Program and the USAMRAA PCRP (PC073098). DNA sequencing was performed in Purdue Genomic Core Facility supported by NCI CCSG CA23168 to the Purdue Cancer Center.
References
- 1.Ganswindt U, Paulsen F, Anastasiadis AG, Stenzl A, Bamberg M, Belka C. 70 Gy or more: which dose for which prostate cancer? J Cancer Res Clin Oncol. 2005;131:407–19. doi: 10.1007/s00432-005-0681-0. [DOI] [PubMed] [Google Scholar]
- 2.Allen GW, Howard AR, Jarrard DF, Ritter MA. Management of prostate cancer recurrences after radiation therapy-brachytherapy as a salvage option. Cancer. 2007;110:1405–16. doi: 10.1002/cncr.22940. [DOI] [PubMed] [Google Scholar]
- 3.Daneshmand S, Quek ML, Pinski J. Neuroendocrine differentiation in prostate cancer. Cancer Therapy. 2005;3:383–96. [Google Scholar]
- 4.Nelson EC, Cambio AJ, Yang JC, Ok JH, Lara PN, Jr, Evans CP. Clinical implications of neuroendocrine differentiation in prostate cancer. Prostate Cancer Prostatic Dis. 2007;10:6–14. doi: 10.1038/sj.pcan.4500922. [DOI] [PubMed] [Google Scholar]
- 5.Amorino GP, Parsons SJ. Neuroendocrine cells in prostate cancer. Crit Rev Eukaryot Gene Expr. 2004;14:287–300. doi: 10.1615/critreveukaryotgeneexpr.v14.i4.40. [DOI] [PubMed] [Google Scholar]
- 6.Yuan TC, Veeramani S, Lin MF. Neuroendocrine-like prostate cancer cells: neuroendocrine transdifferentiation of prostate adenocarcinoma cells. Endocr Relat Cancer. 2007;14:531–47. doi: 10.1677/ERC-07-0061. [DOI] [PubMed] [Google Scholar]
- 7.Deeble PD, Cox ME, Frierson HF, Jr, et al. Androgen-independent growth and tumorigenesis of prostate cancer cells are enhanced by the presence of PKA-differentiated neuroendocrine cells. Cancer Res. 2007;67:3663–72. doi: 10.1158/0008-5472.CAN-06-2616. [DOI] [PubMed] [Google Scholar]
- 8.Nakada SY, di Sant'Agnese PA, Moynes RA, et al. The androgen receptor status of neuroendocrine cells in human benign and malignant prostatic tissue. Cancer Res. 1993;53:1967–70. [PubMed] [Google Scholar]
- 9.Bonkhoff H. Neuroendocrine differentiation in human prostate cancer. Morphogenesis, proliferation and androgen receptor status. Ann Oncol. 2001;12 (Suppl 2):S141–4. doi: 10.1093/annonc/12.suppl_2.s141. [DOI] [PubMed] [Google Scholar]
- 10.Fixemer T, Remberger K, Bonkhoff H. Apoptosis resistance of neuroendocrine phenotypes in prostatic adenocarcinoma. Prostate. 2002;53:118–23. doi: 10.1002/pros.10133. [DOI] [PubMed] [Google Scholar]
- 11.Vanoverberghe K, Vanden Abeele F, Mariot P, et al. Ca2+ homeostasis and apoptotic resistance of neuroendocrine-differentiated prostate cancer cells. Cell Death Differ. 2004;11:321–30. doi: 10.1038/sj.cdd.4401375. [DOI] [PubMed] [Google Scholar]
- 12.Cox ME, Deeble PD, Lakhani S, Parsons SJ. Acquisition of neuroendocrine characteristics by prostate tumor cells is reversible: implications for prostate cancer progression. Cancer Res. 1999;59:3821–30. [PubMed] [Google Scholar]
- 13.Mayr B, Montminy M. Transcriptional regulation by the phosphorylation- dependent factor CREB. Nat Rev Mol Cell Biol. 2001;2:599–609. doi: 10.1038/35085068. [DOI] [PubMed] [Google Scholar]
- 14.Brindle PK, Montminy MR. The CREB family of transcription activators. Curr Opin Genet Dev. 1992;2:199–204. doi: 10.1016/s0959-437x(05)80274-6. [DOI] [PubMed] [Google Scholar]
- 15.Shaywitz AJ, Greenberg ME. CREB: a stimulus-induced transcription factor activated by a diverse array of extracellular signals. Annu Rev Biochem. 1999;68:821–61. doi: 10.1146/annurev.biochem.68.1.821. [DOI] [PubMed] [Google Scholar]
- 16.Montminy MR, Bilezikjian LM. Binding of a nuclear protein to the cyclinc-AMP response element of the somatostatin gene. Nature. 1987;328:175–178. doi: 10.1038/328175a0. [DOI] [PubMed] [Google Scholar]
- 17.Garcia GE, Nicole A, Bhaskaran S, Gupta A, Kyprianou N, Kumar AP. Akt-and CREB-mediated prostate cancer cell proliferation inhibition by Nexrutine, a Phellodendron amurense extract. Neoplasia. 2006;8:523–33. doi: 10.1593/neo.05745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Unni E, Sun S, Nan B, et al. Changes in androgen receptor nongenotropic signaling correlate with transition of LNCaP cells to androgen independence. Cancer Res. 2004;64:7156–68. doi: 10.1158/0008-5472.CAN-04-1121. [DOI] [PubMed] [Google Scholar]
- 19.Canaff L, Bevan S, Wheeler DG, et al. Analysis of molecular mechanisms controlling neuroendocrine cell specific transcription of the chromogranin A gene. Endocrinology. 1998;139:1184–96. doi: 10.1210/endo.139.3.5851. [DOI] [PubMed] [Google Scholar]
- 20.Kim J, Jia L, Stallcup MR, Coetzee GA. The role of protein kinase A pathway and cAMP responsive element-binding protein in androgen receptor-mediated transcription at the prostate-specific antigen locus. J Mol Endocrinol. 2005;34:107–18. doi: 10.1677/jme.1.01701. [DOI] [PubMed] [Google Scholar]
- 21.Johannessen M, Moens U. Multisite phosphorylation of the cAMP response element-binding protein (CREB) by a diversity of protein kinases. Front Biosci. 2007;12:1814–32. doi: 10.2741/2190. [DOI] [PubMed] [Google Scholar]
- 22.Hai TW, Liu F, Coukos WJ, Green MR. Transcription factor ATF cDNA clones: an extensive family of leucine zipper proteins able to selectively form DNA-binding heterodimers. Genes Dev. 1989;3:2083–90. doi: 10.1101/gad.3.12b.2083. [DOI] [PubMed] [Google Scholar]
- 23.Maekawa T, Sakura H, Kanei-Ishii C, et al. Leucine zipper structure of the protein CRE-BP1 binding to the cyclic AMP response element in brain. EMBO J. 1989;8:2023–2028. doi: 10.1002/j.1460-2075.1989.tb03610.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wagner EF. AP-1-Introductory remarks. Oncogene. 2001;20:2334–2335. doi: 10.1038/sj.onc.1204416. [DOI] [PubMed] [Google Scholar]
- 25.Eferl R, Wagner EF. AP-1: a double-edged sword in tumorigenesis. Nat Rev Cancer. 2003;3:859–68. doi: 10.1038/nrc1209. [DOI] [PubMed] [Google Scholar]
- 26.Gueorguiev VD, Cheng SY, Sabban EL. Prolonged activation of cAMP-response element-binding protein and ATF-2 needed for nicotine-triggered elevation of tyrosine hydroxylase gene transcription in PC12 cells. J Biol Chem. 2006;281:10188–95. doi: 10.1074/jbc.M513806200. [DOI] [PubMed] [Google Scholar]
- 27.Ionescu AM, Drissi H, Schwarz EM, et al. CREB Cooperates with BMP-stimulated Smad signaling to enhance transcription of the Smad6 promoter. J Cell Physiol. 2004;198:428–40. doi: 10.1002/jcp.10421. [DOI] [PubMed] [Google Scholar]
- 28.Ionescu AM, Schwarz EM, Zuscik MJ, et al. ATF-2 cooperates with Smad3 to mediate TGF-beta effects on chondrocyte maturation. Exp Cell Res. 2003;288:198–207. doi: 10.1016/s0014-4827(03)00181-2. [DOI] [PubMed] [Google Scholar]
- 29.Niwano K, Arai M, Koitabashi N, et al. Competitive binding of CREB and ATF2 to cAMP/ATF responsive element regulates eNOS gene expression in endothelial cells. Arterioscler Thromb Vasc Biol. 2006;26:1036–42. doi: 10.1161/01.ATV.0000215179.76144.39. [DOI] [PubMed] [Google Scholar]
- 30.Flint KJ, Jones NC. Differential regulation of three members of the ATF/CREB family of DNA-binding proteins. Oncogene. 1991;6:2019–26. [PubMed] [Google Scholar]
- 31.Hay CW, Ferguson LA, Docherty K. ATF-2 stimulates the human insulin promoter through the conserved CRE2 sequence. Biochim Biophys Acta. 2007;1769:79–91. doi: 10.1016/j.bbaexp.2007.01.005. [DOI] [PubMed] [Google Scholar]
- 32.Ricote M, Garcia-Tunon I, Bethencourt F, et al. The p38 transduction pathway in prostatic neoplasia. J Pathol. 2006;208:401–07. doi: 10.1002/path.1910. [DOI] [PubMed] [Google Scholar]
- 33.Liu H, Deng X, Shyu YJ, Li JJ, Taparowsky EJ, Hu CD. Mutual Regulation of c-Jun and ATF2 by Transcriptional Activation and Subcellular Localization. EMBO J. 2006;25:1058–1069. doi: 10.1038/sj.emboj.7601020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Paulmurugan R, Gambhir SS. Firefly luciferase enzyme fragment complementation for imaging in cells and living animals. Anal Chem. 2005;77:1295–302. doi: 10.1021/ac0484777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kudo N, Wolff B, Sekimoto T, et al. Leptomycin B inhibition of signal-mediated nuclear export by direct binding to CRM1. Exp Cell Res. 1998;242:540–7. doi: 10.1006/excr.1998.4136. [DOI] [PubMed] [Google Scholar]
- 36.Reusch JE, Colton LA, Klemm DJ. CREB activation induces adipogenesis in 3T3-L1 cells. Mol Cell Biol. 2000;20:1008–20. doi: 10.1128/mcb.20.3.1008-1020.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Barco A, Alarcon JM, Kandel ER. Expression of constitutively active CREB protein facilitates the late phase of long-term potentiation by enhancing synaptic capture. Cell. 2002;108:689–703. doi: 10.1016/s0092-8674(02)00657-8. [DOI] [PubMed] [Google Scholar]
- 38.de Wit R. Chemotherapy in hormone-refractory prostate cancer. BJU Int. 2008;101 (Suppl 2):11–5. doi: 10.1111/j.1464-410X.2007.07485.x. [DOI] [PubMed] [Google Scholar]
- 39.di Sant'Agnese PA. Neuroendocrine differentiation in prostatic carcinoma: an update on recent developments. Ann Oncol. 2001;12 (Suppl 2):S135–40. [PubMed] [Google Scholar]
- 40.Huang J, Wu C, di Sant'Agnese PA, Yao JL, Cheng L, Na Y. Function and molecular mechanisms of neuroendocrine cells in prostate cancer. Anal Quant Cytol Histol. 2007;29:128–38. [PubMed] [Google Scholar]
- 41.Yuan TC, Veeramani S, Lin FF, et al. Androgen deprivation induces human prostate epithelial neuroendocrine differentiation of androgen-sensitive LNCaP cells. Endocr Relat Cancer. 2006;13:151–67. doi: 10.1677/erc.1.01043. [DOI] [PubMed] [Google Scholar]
- 42.Ismail AH, Landry F, Aprikian AG, Chevalier S. Androgen ablation promotes neuroendocrine cell differentiation in dog and human prostate. Prostate. 2002;51:117–25. doi: 10.1002/pros.10066. [DOI] [PubMed] [Google Scholar]
- 43.Wright ME, Tsai MJ, Aebersold R. Androgen receptor represses the neuroendocrine transdifferentiation process in prostate cancer cells. Mol Endocrinol. 2003;17:1726–37. doi: 10.1210/me.2003-0031. [DOI] [PubMed] [Google Scholar]
- 44.Jin RJ, Wang Y, Masumori N, et al. NE-10 neuroendocrine cancer promotes the LNCaP xenograft growth in castrated mice. Cancer Res. 2004;64:5489–95. doi: 10.1158/0008-5472.CAN-03-3117. [DOI] [PubMed] [Google Scholar]
- 45.Jiborn T, Bjartell A, Abrahamsson PA. Neuroendocrine differentiation in prostatic carcinoma during hormonal treatment. Urology. 1998;51:585–9. doi: 10.1016/s0090-4295(97)00684-5. [DOI] [PubMed] [Google Scholar]
- 46.Zhang XQ, Kondrikov D, Yuan TC, Lin FF, Hansen J, Lin MF. Receptor protein tyrosine phosphatase alpha signaling is involved in androgen depletion-induced neuroendocrine differentiation of androgen-sensitive LNCaP human prostate cancer cells. Oncogene. 2003;22:6704–16. doi: 10.1038/sj.onc.1206764. [DOI] [PubMed] [Google Scholar]
- 47.Spiotto MT, Chung TD. STAT3 mediates IL-6-induced neuroendocrine differentiation in prostate cancer cells. Prostate. 2000;42:186–95. doi: 10.1002/(sici)1097-0045(20000215)42:3<186::aid-pros4>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- 48.Yang X, Chen MW, Terry S, et al. A human- and male-specific protocadherin that acts through the wnt signaling pathway to induce neuroendocrine transdifferentiation of prostate cancer cells. Cancer Res. 2005;65:5263–71. doi: 10.1158/0008-5472.CAN-05-0162. [DOI] [PubMed] [Google Scholar]
- 49.Karasewski L, Ferreira A. MAPK signal transduction pathway mediates agrin effects on neurite elongation in cultured hippocampal neurons. J Neurobiol. 2003;55:14–24. doi: 10.1002/neu.10197. [DOI] [PubMed] [Google Scholar]
- 50.Aletta JM, Greene LA. Growth cone configuration and advance: a time-lapse study using video-enhanced differential interference contrast microscopy. J Neurosci. 1988;8:1425–35. doi: 10.1523/JNEUROSCI.08-04-01425.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.