

Abstract
In Alzheimer's disease (AD), dementia severity correlates strongly with decreased synapse density in hippocampus and cortex. Numerous studies report that hippocampal long-term potentiation (LTP) can be inhibited by soluble oligomers of amyloid β-protein (Aβ), but the synaptic elements that mediate this effect remain unclear. We examined field EPSPs and whole-cell recordings in wild-type mouse hippocampal slices. Soluble Aβ oligomers from three distinct sources (cultured cells, AD cortex, or synthetic peptide) inhibited LTP, and this was prevented by the selective NR2B inhibitors ifenprodil and Ro 25-6981. Soluble Aβ enhanced NR2B-mediated NMDA currents and extrasynaptic responses; these effects were mimicked by the glutamate reuptake inhibitor dl-threo-β-benzyloxyaspartic acid. Downstream, an Aβ-mediated rise in p38 mitogen-activated protein kinase (MAPK) activation was followed by downregulation of cAMP response element-binding protein, and LTP impairment was prevented by inhibitors of p38 MAPK or calpain. Thus, soluble Aβ oligomers at low nanomolar levels present in AD brain increase activation of extrasynaptic NR2B-containing receptors, thereby impairing synaptic plasticity.
Introduction
Evidence from many laboratories suggests that the accumulation of amyloid-β protein (Aβ) plays a central role in the pathogenesis of Alzheimer's disease (AD). Aβ peptides are normally generated from β-amyloid precursor protein (APP) via proteolytic cleavages by the β- and γ-secretases (Selkoe, 1998; Marks and Berg, 2008). The soluble monomers can assemble sequentially into soluble oligomers, protofibrils, and fibrils that accumulate in the brains of AD subjects, forming insoluble amyloid plaques. Among these Aβ assembly forms, the levels of soluble Aβ (which includes monomers and soluble oligomers) correlate most strongly with the severity of dementia in AD patients (Lue et al., 1999; McLean et al., 1999). Experimentally, soluble Aβ oligomers (but not monomers) have been found to block hippocampal long-term potentiation (LTP), a synaptic correlate of memory and learning (Lambert et al., 1998; Walsh et al., 2002; Wang et al., 2002; Klyubin et al., 2005; Townsend et al., 2006; Shankar et al., 2007, 2008). Importantly, Aβ immunotherapy can protect against the neuropathology and cognitive deficits observed in APP transgenic mice (Janus et al., 2000; Morgan et al., 2000) and also prevent the LTP inhibition induced by Aβ oligomers (Klyubin et al., 2005). Synthetic Aβ assemblies can inhibit N-methyl-d-aspartate receptor (NMDAR)-dependent but not NMDAR-independent LTP (Chen et al., 2002; Zhao et al., 2004) (but see Raymond et al., 2003). This result is consistent with reports that synthetic Aβ can decrease surface expression of NMDAR (Snyder et al., 2005; Dewachter et al., 2009) and increase (Wu et al., 1995; Molnár et al., 2004) or decrease (Chen et al., 2002; Raymond et al., 2003) NMDAR-mediated synaptic responses. These various findings suggest that the effects of Aβ on NMDAR impair synaptic plasticity, but the detailed mechanism remains unknown.
Synaptic NMDARs are important for LTP, whereas extrasynaptic NMDARs can trigger de novo long-term depression (LTD) under certain conditions (Lu et al., 2001; Massey et al., 2004). Synaptic and extrasynaptic receptors may also have distinct roles in signaling pathways, gene regulation, and cell death (Hardingham et al., 2002). For example, stimulation of synaptic NMDAR is responsible for extracellular signal-regulated kinase (ERK) phosphorylation and activation (Ivanov et al., 2006), cAMP response element binding protein (CREB) activation and neuroprotective effects (Hardingham et al., 2002), whereas activation of extrasynaptic NMDAR contributes to ERK dephosphorylation and inactivation, shuts off the CREB pathway, and induces neuronal death (Hardingham et al., 2002). Increased extrasynaptic NMDA activity has been shown to be deleterious in mouse models of Huntington's disease (Okamoto et al., 2009; Milnerwood et al., 2010).
We recently reported that soluble Aβ oligomers at the pathophysiological levels present in AD brain facilitate hippocampal LTD by increasing the activation of NR2B-containing extrasynaptic NMDARs (Li et al., 2009). We obtained evidence that the enhanced NMDAR responses can be explained, at least in part, by an oligomer-mediated decrease in neuronal glutamate reuptake, thereby inducing glutamate “spillover” to extrasynaptic sites. To further explore the mechanism by which soluble Aβ oligomers inhibit LTP, we have now combined electrophysiological, biochemical, and immunohistological methods to show that the oligomers inhibit LTP by activating extrasynaptic NR2B-containing NMDA receptors.
Materials and Methods
Hippocampal slice preparations.
Mice (C57BL/6 × 129, were killed by isoflurane anesthesia at age 3∼4 wk for patch clamping or at 6∼8 wk for field recordings. Brains were quickly removed and submerged in ice-cold, oxygenated, sucrose-replaced, artificial CSF (ACSF) cutting solution containing (in mm): 206 sucrose, 2 KCl, 2 MgSO4, 1.25 NaH2PO4, 1 CaCl2, 1 MgCl2, 26 NaHCO3, 10 d-glucose, pH 7.4, 315 mOsm). Transverse slices (350 μm thick) were cut with a vibroslicer from the middle portion of each hippocampus. After dissection, slices were incubated in ACSF containing the following (in mm): 124 NaCl, 2 KCl, 2 MgSO4, 1.25 NaH2PO4, 2.5 CaCl2, 26 NaHCO3, 10 d-glucose, pH 7.4, 310 mOsm, in which they were allowed to recover for at least 90 min before recording. A single slice was then transferred to the recording chamber and submerged beneath continuously perfused ACSF saturated with 95% O2 and 5% CO2. Slices were incubated in this chamber for 20 min before stimulation at room temperature (RT) (∼24°C).
Electrophysiology.
Standard field EPSPs (fEPSPs) were recorded in the CA1 region of hippocampus. A unipolar stimulating electrode (World Precision Instruments) was placed in the Schaffer collaterals to deliver test and conditioning stimuli. A borosilicate glass recording electrode filled with ACSF was positioned in stratum radiatum of CA1, 200∼300 μm from the stimulating electrode. fEPSPs in CA1 were induced by test stimuli at 0.05 Hz with an intensity that elicited a fEPSP amplitude of 40∼50% of maximum. Test responses were recorded for 30–60 min before beginning the experiment to ensure stability of the response. NMDAR-mediated fEPSPs were recorded in 0.1 mm Mg2+ ACSF containing 10 μm bicuculline and 10 μm 2,3-dihyro-6-nitro-7-sulfamoyl-benzo(f)quinoxaline (NBQX) to block GABA and AMPA receptors, respectively. To induce LTP, two consecutive trains (1 s) of stimuli at 100 Hz separated by 20 s, a protocol that induces LTP lasting ∼1.5 h in wild-type mice of this genetic background, were applied to the slices. The field potentials were amplified 100× using Molecular Devices 200B amplifier and digitized with Digidata 1322A. The data were sampled at 10 kHz and filtered at 2 kHz. Traces were obtained by pClamp 9.2 and analyzed using the Clampfit 9.2.
Whole-cell recordings were made from the somata of visually identified CA1 pyramidal neurons. Patch pipettes (5∼7 MΩ) were filled with an internal solution containing the following (in mm): 110 Cs-gluconate, 20 CsCl, 10 HEPES, 4 NaCl, 0.5 EGTA, 2 MgCl2, 2 Na2ATP, and 0.25 NaGTP, titrated with KOH to pH 7.4. EPSCs were recorded following stimulation of Schaffer collaterals ∼150 μm from the CA1 cell body with a bipolar electrode. NMDA-mediated EPSCs were recorded at +45 mV in ACSF containing 10 μm bicuculline and 10 μm NBQX, or else at −70 mV in 0.1 mm Mg2+ ACSF containing bicuculline and NBQX. In some experiments, we blocked active synaptic NMDAR by applying MK-801 (20 μm) in the above buffer and changed the stimulation protocol to 0.125 Hz using paired-pulse stimulation (100 ms interpulse interval) because this effects presynaptic release of glutamate, which in turn facilitates the activation of synaptic NMDAR currents. After washout of the MK801, the remaining EPSC can be considered as extrasynaptic NMDA currents. Series resistance was kept at 15∼30 MΩ and monitored throughout each recording. Cells were excluded from data analysis if the series resistance changed by >20% during the course of the experiment. All patch-clamp experiments were performed at 24°C.
Hippocampal neuronal cultures.
Primary hippocampal cultures were generated from embryonic day 18 Sprague Dawley rat embryos. The hippocampus was dissected out in HBSS solution buffered with HEPES and dissociated with 0.125% trypsin (Invitrogen) for 15 min at 37°C, followed by trituration. Dissociated cells were plated at 1.5 × 106 cells on 6-well plates precoated with poly-dl-lysine and laminin. After culturing 4 d in Neurobasal medium with B-27 supplement and GlutaMAX (Invitrogen), cytosine arabinofuranoside was added to reduce glial proliferation. Half the medium was exchanged every 4 d.
Calcium imaging.
Primary hippocampal neurons were prepared as described previously (Townsend et al., 2006). Briefly, hippocampal neurons were prepared from prenatal day 18 embryonic CD1 mice (Charles River). Pregnant mice were killed with CO2, the uterus quickly dissected, and the pups removed. After decapitation, the brains were removed and immediately placed in cold HBSS buffer containing the following (in mm): 145 NaCl, 5 KCl, 2 CaCl, 1 MgCl, 10 glucose, and 10 HEPES. After the hippocampi were dissected out, they were triturated using the Neuronal Tissue Dissociation kit (Miltenyi Biotec). Cells were then counted and initially plated in DMEM plus 10% fetal calf serum on poly-l-lysine-coated (Sigma) 24-well plate coverslips (15,000 cells/cm2 for microscopy and 50,000–100,000 cells/cm2 for calcium imaging and excitotoxicity assays). One hour after plating, the medium was changed to Neurobasal containing B27 and GlutaMAX. At days in vitro (DIV) 14, cells were loaded with Fluo-4 (Invitrogen) according to the manufacturer's recommendations. Briefly, cells were incubated 30 min in complete Fluo-4 loading solution (containing loading buffer, Fluo-4 dye reagent, probenecid, and 10 μm glycine) at 37°C followed by a 30 min incubation at RT. Cells were exposed to conditioned medium (CM) from either 7PA2 cells [Chinese hamster ovary (CHO) cells stably expressing V717F hAPP751] or untransfected CHO− cells for 20 min in the same buffer, and extrasynaptic NMDARs were isolated as described (50 μm bicuculine and 10 μm CNQX for 10 min and 50 μm MK801 for 20 min; Tocris Bioscience). The injectors of the multiwell plate reader were loaded with NMDA (Sigma) in PBS, and after injection of NMDA to a final concentration of 75 μm, fluorescence emission was measured continuously for 2.5 min (excitation at 285 nm, emission at 535 nm). Raw data were analyzed using Prism data analysis software (GraphPad) and expressed as the mean ΔRFU [(F − Fmin)/Fmin] (where RFU is relative fluorescence unit, F is fluorescence, and min is minimum).
Enrichment of synaptic and extrasynaptic membranes.
Separation of synaptic and extrasynaptic fractions was performed as described by Goebel-Goody et al. (2009) with minor modifications. In brief, the hippocampal slices were prepared as for electrophysiology (see above, Electrophysiology), treated with either CHO− CM or 7PA2 CM after recovery, and then the slices were harvested in ice-cold sucrose homogenization buffer (50 μl per slice) containing the following (in mm): 320 sucrose, 10 Tris, pH 7.4, 1 Na3VO4, 5 NaF, 1 EDTA, and 1 EGTA. Slices were then homogenized in a glass grinding vessel using a rotating Teflon pestle (2000 rpm) with at least 20 passes to create a Dounce homogenate. The homogenate was centrifuged at 1000 × g for 10 min to remove nuclei and incompletely homogenized material (P1). The resulting supernatant (S1) was spun at 10,000 × g for 15 min to obtain a P2. The P2 was subsequently resuspended in 120 μl sucrose buffer using a motorized pestle mixing/grinding rod (Kontes) directly in the microfuge tube with 30 pulses. The P2 was then subjected to detergent extraction by adding 8 vol of Triton X-100 buffer (final = 0.5% v/v) containing the following reagents (in mm): 10 Tris, pH 7.4, 1 Na3VO4, 5 NaF, 1 mm EDTA, and 1 EGTA. This suspension was incubated at 4°C for 20 min with gentle rotation and then centrifuged at 32,000 × g for 20 min. We operationally defined the pellet as the postsynaptic density (PSD)-associated (or synaptic) fraction and the supernatant as the extrasynaptic fraction. Protein concentrations were determined using the bicinchoninic acid (BCA) assay.
Western blotting.
Different cultures in 6-well plates were lysed in 0.2 ml of lysis buffer (0.1% SDS, 1% Nonidet P-40, 50 mm HEPES, pH 7.4, 2 mm EDTA, 100 mm NaCl, 5 mm Na3VO4, 40 μm p-nitrophenyl phosphate, and 1% protease inhibitor mixture set I (Calbiochem). The lysates were centrifuged at 12,000 rpm for 25 min. The supernatant was collected and denatured. The concentration of protein was determined by BCA assay. We loaded 20 μg of total protein in each lane, separated by 4–12% SDS-PAGE and blotted onto nitrocellulose membrane. The blot was blocked for 1 h at RT, followed by incubation overnight at 4°C with the rabbit polyclonal antibodies NR2B (Invitrogen), p-CREB (Millipore Bioscience Research Reagents), p-ERK1/2, p-p38, and p38 (Cell Signaling Technology) or the mouse monoclonal antibodies PSD95, synaptophysin (Millipore Bioscience Research Reagents), and ERK1/2 (Thermo). Membranes were rinsed and incubated for 1 h with fluorescence-conjugated goat anti-rabbit or mouse immunoglobulin G (IgG) (1:5000; Invitrogen). Blots were scanned using a LI-COR Odyssey system.
Immunocytochemistry and confocal microscopy.
Hippocampal neurons cultured for 14 d were treated with CHO− CM or 7PA2 CM for different times. After treatment, cells were fixed by 4% paraformaldehyde with 4% sucrose for 30 min at RT. Neurons were rinsed three times with PBS and then penetrated with 0.5% Triton X-100 in PBS for 15 min. Following blocking with 5% BSA in PBS, neurons were incubated with rabbit antibody against NR2B (Invitrogen, 1:500) and mouse antibody against synapsin (BD Bioscience, 1:500) at 4°C overnight. After rinsing with PBS three times, cells were incubated with Alexa Fluor 488 goat anti-rabbit IgG and Alexa Fluor 546 goat anti-mouse IgG (Invitrogen, 1:1000) at 4°C overnight. After rinsing three times with PBS, cells were mounted with mounting medium (Southern Biotech). Confocal microscopy was performed on a Zeiss LSM510 confocal microscope. We used a 100×/1.4 objective and scanned the samples in a Z-stack manner (three stacks, interval 0.5 μm). Neurons under different conditions were selected randomly, and the secondary branches were captured.
Drug treatments.
Paired control and experimental slices from a single mouse were maintained together in a single chamber, except during drug treatments. dl-2-Amino-5-phosphonopentanoic acid (AP-5), MK-801, SB203580, calpeptin, ifenprodil, (αR,βS)-α-(4-hydroxyphenyl)-β-methyl-4-(phenylmethyl)-1-piperidinepropanol maleate (Ro 25-6981), (2R*,4S*)-4-(3-phosphonopropyl)-2-piperidinecarboxylic acid (PPPA), (2S*, 3R*)-1-(phenanthren-2-carbonyl)piperazine-2,3-dicarboxylic acid (PPDA), NBQX, and bicuculline were from Tocris Bioscience. dl-Threo-β-benzyloxyaspartic acid (TBOA), glutamic-pyruvic transaminase (GPT), and pyruvic acid were from Sigma. Among these, NBQX, SB203580, calpeptin, Ro 25-6981, PPDA, and TBOA were each dissolved in dimethylsulfoxide (DMSO). The DMSO concentration used in experiments was <0.01%.
Statistical analysis.
LTP values expressed here are those at 60 min after the conditioning stimulus, unless stated otherwise. Results are expressed as means ± SEM. from at least four independent biological samples. Two-tailed Student's t test and one-way ANOVA were used to determine statistical significance throughout.
Results
Soluble Aβ oligomers inhibit hippocampal LTP
We reported previously (Shankar et al., 2007, 2008) that naturally occurring soluble Aβ dimers isolated by size-exclusion chromatography (SEC) from postmortem AD cortex or from the medium of a cultured cell line inhibit LTP in mouse hippocampal slices. Here, we initially examined soluble Aβ derived from a CHO cell line (7PA2) that stably expresses APP carrying the V717F AD mutation. The CM of this cell line is enriched in soluble human Aβ, including monomeric and oligomeric Aβ, without larger insoluble aggregates (Walsh et al., 2002; Shankar et al., 2007). As expected, LTP was inhibited in acute hippocampal slices exposed to the soluble Aβ-containing 7PA2 CM, but not by CM from wild-type CHO cells (105 ± 4% vs 155 ± 8%, n = 8, p < 0.001). Upon fractionation of the CM by SEC (Walsh et al., 2005), we observed that the inhibition of LTP was mediated primarily by low-n Aβ oligomers (150 ± 9% with monomers vs108 ± 3% with oligomers, n = 8, p < 0.01). To further verify this apparent oligomer-mediated inhibition of LTP, pure synthetic Aβ40 S26C cross-linked dimers (Hu et al., 2008; Shankar et al., 2008) were applied to the perfusion bath. Again, hippocampal LTP was significantly reduced versus vehicle alone (116 ± 4%, n = 8, vs 156 ± 11%, n = 5, p < 0.01), and this effect occurred in a dose-dependent manner (Shankar et al., 2008). These results further establish that different sources of small, diffusible Aβ oligomers can perturb synapse strengthening in hippocampus.
LTP inhibition by soluble Aβ depends on activation of NR2B-containing NMDA receptors
The 100 Hz high-frequency stimulation protocol we use is well known for its NMDAR dependence in the CA1 region. In support, we found that an NMDAR antagonist, AP-5 (50 μm; 108 ± 3%, n = 8, p < 0.001), and the use-dependent channel blocker MK-801 (10 μm; 108 ± 2%, n = 6, vs ACSF vehicle 150 ± 6%, n = 7, p < 0.001) each fully blocked LTP in our experimental system. We proceeded to apply selective antagonists to test whether the inhibitory effect of Aβ on hippocampal LTP was attributable to a specific NMDA receptor subtype. The selective NR2B antagonists ifenprodil (3 μm) (Fig. 1A) or Ro 25-6981 (1 μm) (Fig. 1B), which have been reported to have no (Massey et al., 2004; Hu et al., 2009) or only partial (Berberich et al., 2007) effects on LTP, were added to the perfusion solution before 7PA2 or CHO− CM application. Each of these compounds produced a partial rescue of the LTP inhibition caused by 7PA2 CM (ifenprodil, 140 ± 8%, n = 7, p < 0.01; Ro 25-6981, 132 ± 10%, n = 7, p < 0.01 vs 7PA2 CM alone, 105 ± 4%, n = 8). Importantly, similar results were obtained for ifenprodil rescue of LTP inhibition by soluble (TBS) extracts of AD cortex (AD TBS, 111 ± 3%, n = 9 vs ifenprodil plus AD TBS, 131 ± 3%, n = 9, p < 0.01) (Fig. 1C) and by synthetic S26C Aβ dimers (dimer, 107 ± 3%, n = 6 vs ifenprodil plus dimer, 131 ± 5%, n = 7, p < 0.01) (Fig. 1D). In contrast, neither the selective antagonist of NR2A subunits, PPPA (0.5 μm) nor an antagonist of NR2D subunits, PPDA (1 μm), could rescue the soluble Aβ-mediated inhibition of LTP [7PA2, CM plus PPPA, 124 ± 5%, n = 6 vs CHO CM plus PPPA, 150 ± 5%, n = 6, p < 0.01 (Fig. 1E); 7PA2 CM plus PPDA, 125 ± 7%, n = 6 vs CHO CM plus PPDA, 143 ± 6%, n = 5, p < 0.05 (Fig. 1F)], similar to a prior report (Hu et al., 2009). These findings suggest that activation of NR2B-containing NMDA receptors mediates the effect of soluble Aβ on LTP.
Figure 1.
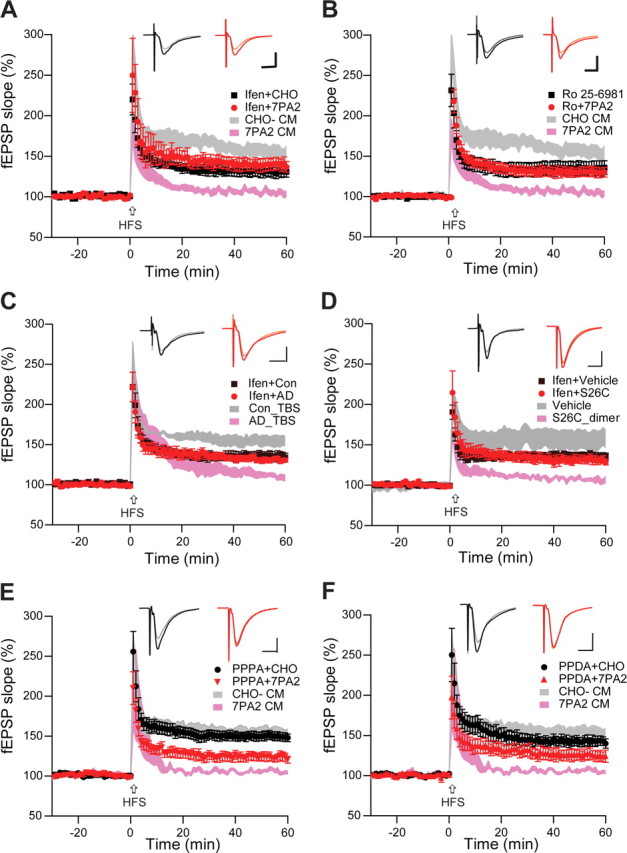
Soluble Aβ impairment of LTP in the CA1 region of hippocampus requires NR2B-containing NMDA receptors. A, 7PA2 CM rich in soluble Aβ inhibited LTP (pink tracing) induced by high-frequency stimulation (HFS, arrow), but this was restored by the NR2B antagonist, ifenprodil (Ifen) (3 μm) (red circles, n = 7) to its control level (black squares, n = 8). CHO− CM had no effect on LTP (gray tracing). Pink and gray tracings are basic data from just 7PA2 CM or CHO− CM. B, Soluble Aβ impaired-LTP was also rescued by another NR2B antagonist, Ro 25-6981 (Ro) 1 μm, n = 7). C, Soluble Aβ-rich TBS extracts of AD brains (AD_TBS) similarly impaired HFS-induced LTP, but this was restored by ifenprodil (3 μm) (red circles, n = 12) to its control level (black squares, n = 8). D, Synthetic human Aβ S26C dimers (10 nm) impaired LTP, but this was restored by ifenprodil (3 μm) (red circles, n = 7) to its control level (black squares, n = 7). E, A low dose of the relative NR2A antagonist PPPA (0.5 μm) alone had no effect on LTP (administered in CHO− CM) (black circles, n = 7) and failed to restore LTP impaired by 7PA2 CM (red triangles, n = 7). Pink and gray tracings are basic data from just 7PA2 CM or CHO− CM. F, A NR2C/NR2D selective antagonist PPDA (1 μm), had no significant effect on the HFS-LTP (black circles, n = 6), and it failed to fully restore Aβ-impaired LTP (red triangles, n = 6). All the NMDA receptor selective antagonists were applied 10 min before Aβ was added. Inset traces are typical field EPSPs recorded before (gray or orange) and after (black or red) HFS for each condition. Horizontal calibration bars, 10 ms; vertical bars, 0.5 mV.
Soluble Aβ activates extrasynaptic NMDA receptors and causes LTP impairment
Consistent with our previous finding that soluble Aβ can activate extrasynaptic NR2B receptors and thereby contribute to the induction of LTD (Li et al., 2009), whole-cell recordings in hippocampal slices showed that 7PA2 CM significantly increased the extrasynaptic NMDA current (isolated by MK801 pretreatment, which blocks active synaptic NMDAR) (charge transfer = 585 ± 165% of pre-7PA2, n = 6) (Fig. 2A1,A2). Similarly, the glutamate reuptake inhibitor TBOA (30 μm) alone markedly increased the NMDAR charge transfer (800 ± 184% of pre-TBOA, n = 5) (Fig. 2B1,B2), and this could be blocked completely by AP-5 (50 μm) (data not shown). As a control for compound specificity, we found by whole-cell recordings that the extrasynaptic NMDA currents were significantly decreased by the NR2B antagonist ifenprodil (3 μm) (57 ± 2%, n = 9) (Fig. 2C), while the synaptic NMDA currents were much less affected by NR2B blockade (Fig. 2C). Importantly, the increase of extrasynaptic NMDA currents by soluble Aβ was specifically attributable to NR2B-containing receptors (7PA2 CM, 448 ± 54%, n = 8 vs Ro 25-6981 plus 7PA2, 148 ± 16%, n = 7, p < 0.001) (Fig. 2D), and a closely similar result was observed for the TBOA-caused enhancement of extrasynaptic NMDA currents (Fig. 2E). To further verify this Aβ oligomer effect on extrasynaptic NMDA receptors, we measured intracellular calcium responses in cultured neurons using Fluo-4 dye. Again, synaptic NMDAR were first blocked by MK-801 to isolate extrasynaptic receptors. The extrasynaptic-mediated cytosolic calcium change in response to NMDA application was significantly greater in 7PA2 CM-treated neurons than in CHO− CM-treated neurons (2.06 ± 0.38%, n = 17 vs 1.02 ± 0.25%, n = 17, p < 0.05) (data not shown).Together, the data presented so far suggest that LTP inhibition by soluble Aβ oligomers involves excessive activation of extrasynaptic NMDA receptors.
Figure 2.
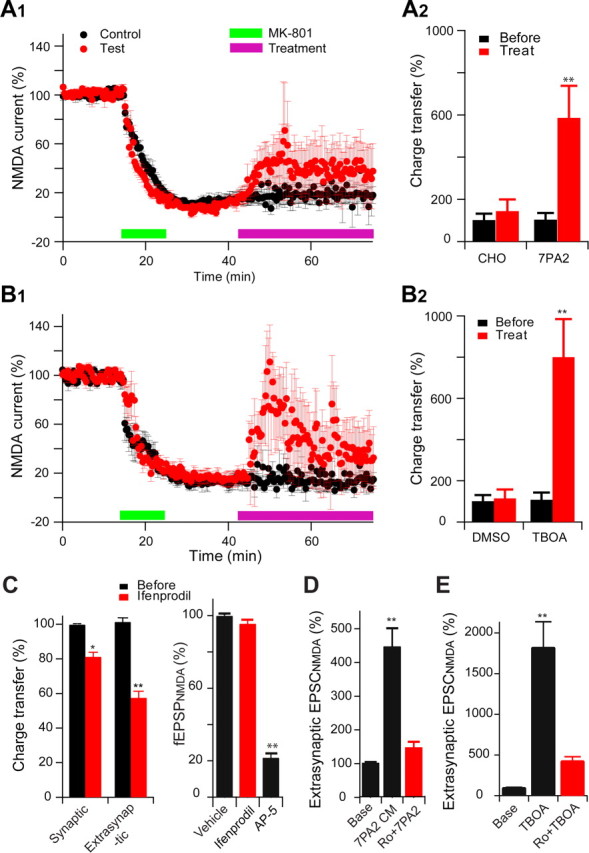
Soluble Aβ oligomers enhance extrasynaptic NMDA currents in the hippocampus. A, Extrasynaptic NMDA currents were recorded when evoked NMDA EPSCs were isolated from synaptic currents by MK801 treatment (20 μm, green bar) followed by wash out. The magenta bars in A1 and B1 represent the application of either 7PA2 CM versus CHO− CM (A1) or TBOA versus DMSO (B1) treatments. A2 and B2 are the summary data for the extrasynaptic responses before (black) and 30 min after (red) treatments (Treat). B, Extrasynaptic NMDA currents were also increased after administering the glutamate reuptake inhibitor TBOA (30 μm, green bar) (B1); summary data shown in B2. C, Effects of NR2B antagonist ifenprodil (3 μm) on isolated synaptic and extrasynaptic NMDAR-mediated EPSCs or on NMDAR synaptic fEPSPs in low Mg2+ (0.1 mm) ACSF plus NBQX (10 μm) and bicuculline (20 μm). D, Isolated extrasynaptic NMDA currents in 7PA2 CM alone or 7PA2 CM plus Ro 25-6981 (Ro). E, TBOA significantly increased extrasynaptic NMDA currents, which were mostly inhibited by the NR2B antagonist Ro 25-6981 (1 μm). Error bars are ± SEM. *p < 0.05, **p < 0.01.
Extrasynaptic NMDARs can be activated by high-frequency stimulation (HFS) resulting in “spillover” of the excess released glutamate, something that theoretically does not occur with low-frequency stimulation (Okubo et al., 2010). Measuring MK801-isolated extrasynaptic NMDA EPSCs, we found that, as expected, conventional HFS caused an increase of NMDA currents (250 ± 60%, n = 4, p < 0.05 vs baseline), but a strong HFS (1.5× test stimulus intensity, which partially blocks LTP induction (Fig. 3A), caused much more enhancement (441 ± 49%, n = 4, p < 0.01 vs baseline; p < 0.05 vs conventional HFS), an effect similar to that of soluble Aβ (636 ± 182%, n = 4, p < 0.01 vs baseline; p > 0.05 vs strong HFS) (Fig. 3C). Interestingly, the soluble Aβ enhancement of extrasynaptic NMDA currents occluded the effect of a subsequent HFS (748 ± 133%, n = 4, p > 0.05 vs 7PA2 CM) (Fig. 3C), suggesting that they share a similar mechanism.
Figure 3.
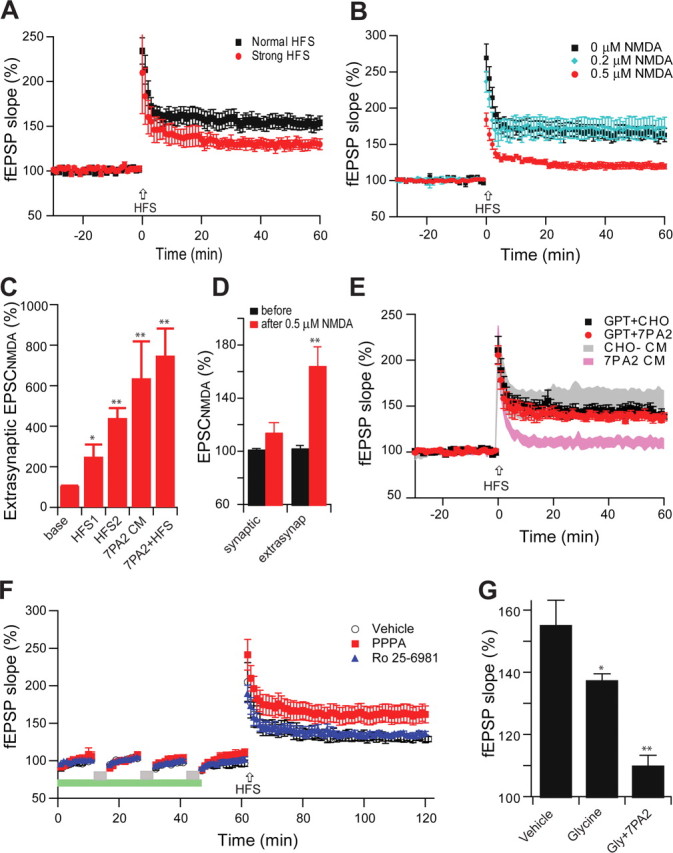
Soluble Aβ impairs LTP via increased extracellular glutamate levels in the Schaffer collateral-CA1 pathway of hippocampus. A, In contrast with the conventional high-frequency stimulation (two trains of 100 Hz, 1 s, separated by 20 s at test stimulus intensity, black squares, n = 6), a strong HFS (at 1.5× test stimulus intensity) significantly reduces the LTP induction (red circles, n = 5). B, A low dose of NMDA (0.5 μm, red circles, n = 7) prevented HFS-induced hippocampal LTP. C, Extrasynaptic NMDA current changes induced by conventional HFS (HFS1), strong HFS (HFS2), 7PA2 CM, or 7PA2 CM plus HFS (7PA2+HFS). D, Responses of isolated NMDAR-mediated synaptic and extrasynaptic (extrasynap) EPSCs to the 0.5 μm NMDA. E, An enzymatic glutamate scavenger system (GPT plus pyruvate) has no significant effect on normal LTP in CHO− CM (black squares, n = 7) but fully prevented the 7PA2 CM impairment of LTP (red circles, n = 8). F, Three repetitive 5 min episodes of subthreshold, low-frequency stimulation (0.5 Hz) (gray bar) desensitize the synaptic NMDAR effect on LTP induction. Green bar represents the time of application of either NR2A-selective (red squares) or NR2B-selective (blue triangles) antagonists given to prevent the desensitization. G, Summary data for the effect of the NMDAR desensitization regulator, glycine (Gly) (100 μm), on Aβ-impaired LTP. Error bars represent ± SEM. *p < 0.05, **p < 0.01.
To mimic these apparent glutamate spillover effects, we applied low-dose (0.5 μm) NMDA exogenously to hippocampal slices for 30 min. This treatment did not affect baseline synaptic transmission, but a subsequent HFS failed to induce LTP (120 ± 4% at 0.5 μm NMDA, n = 6, vs 165 ± 9% at 0 or 0.2 μm NMDA, n = 7, p < 0.001) (Fig. 3B). To explore the mechanism of LTP inhibition by this low-dose NMDA exposure, whole-cell recordings were performed to distinguish extrasynaptic from synaptic components of the NMDAR EPSC. Here, we found that 0.5 μm NMDA significantly enhanced the extrasynaptic NMDA EPSC, while the synaptic component was unaffected (164 ± 14%, n = 5 vs 114 ± 8%, respectively, n = 6) (Fig. 3D). These results are consistent with previous reports that increasing extracellular glutamate levels by very strong tetanic stimulation (Katagiri et al., 2001), exogenous application of low-dose NMDA (Izumi et al., 2008), or inhibition of glutamate reuptake (Katagiri et al., 2001; Li et al., 2009) can each perturb LTP induction.
To further support the hypothesis that inhibition of LTP by soluble Aβ is attributable to increased extracellular glutamate concentration, we used an enzymatic glutamate scavenger system (glutamic-pyruvic transaminase, also known as GPT, plus pyruvate) to reduce extracellular glutamate levels (Overstreet et al., 1997; Min et al., 1998; Li et al., 2009) Exposure of hippocampal slices to GPT alone (5 U/ml) had no effect on baseline synaptic transmission (data not shown). Similarly, GPT plus pyruvate (2 mm) treatment 20 min before the application of CHO− CM affected neither baseline activity nor the magnitude of subsequent LTP induction by HFS (144 ± 5%, n = 6 vs 155 ± 8%, n = 8, p > 0.05) (Fig. 3E). However, this treatment did prevent 7PA2 CM-induced LTP inhibition (138 ± 4%, n = 6 vs 105 ± 4%, n = 8, p < 0.01) (Fig. 3E). The rescue of the LTP impairment by either reducing extracellular glutamate concentrations or blocking NR2B-containing NMDARs suggests that soluble Aβ acts to enhance activation of extrasynaptic NR2B receptors by increasing glutamate spillover.
Synaptic NMDA receptor desensitization is not implicated in LTP inhibition by soluble Aβ
An increase in extracellular glutamate levels by soluble Aβ could persistently activate postsynaptic NMDA receptors and thereby decrease their sensitivity (desensitization) (Mayer et al., 1989) Because there are no commercially available NMDAR desensitization modulators, we applied subthreshold, low-frequency stimulation (0.5 Hz for 5 min applied 3 times 10 min apart) to desensitize the postsynaptic receptors. Subsequent HFS only induced a small LTP (135 ± 3%, n = 6 vs naive LTP 155 ± 8%, n = 7, p < 0.05) (Fig. 3F). If we blocked the synaptic NMDA receptors during the repetitive stimulation using PPPA (1 μm), which does not significantly prevent the Aβ-induced LTP blockade (Fig. 1E), normal LTP occurred (153 ± 7%, n = 6) (Fig. 3F). In contrast, the selective NR2B inhibitor, Ro 25-6981 (1 μm), could not rescue the synaptic desensitization-mediated decrease in LTP (136 ± 3%, n = 5) (Fig. 3F). These results suggest that impaired LTP from synaptic NMDA desensitization is principally mediated by synaptic NR2A receptors, in contrast to impaired LTP from soluble Aβ oligomers. To further support this interpretation, we applied glycine (100 μm), a NMDAR desensitization regulator (Mayer et al., 1989), to the recording solution. Here, HFS-induced LTP was partially blocked, but there was no effect on the 7PA2 CM inhibition of LTP (137 ± 2%, n = 6 vs 111 ± 3%, n = 6, p < 0.01) (Fig. 3G). Thus, blockade of LTP by soluble Aβ does not arise from desensitization of synaptic NMDAR.
Involvement of NR2B-containing receptors in Aβ-induced phosphorylation of p38 mitogen-activated protein kinase and dephosphorylation of ERK1/2 and CREB
Several reports have demonstrated that stimulation of extrasynaptic NR2B-containing NMDA receptors contributes to activation of p38 mitogen-activated protein kinase (MAPK) (Waxman and Lynch, 2005), inactivation of ERK1/2 (Ivanov et al., 2006; Mulholland et al., 2008; Gao et al., 2010), and dephosphorylation of CREB (Hardingham et al., 2002; Papadia et al., 2005). In light of all of our results above, we explored the effects of soluble Aβ oligomers on MAPK and CREB signaling pathways. We exposed cultured hippocampal neurons to 7PA2 CM or CHO− CM for increasing durations and quantified the phosphorylation state of MAPK and CREB by Western blotting. Upon treatment with 7PA2 CM, the phosphorylation level of p38 MAPK was increased at 3 h, reached a peak at 6 h, and dropped by 16 h (Fig. 4A,C). In contrast, the phosphorylation levels of ERK1/2 and CREB were decreased at 6 h and remained low at 16 h. In agreement with our electrophysiology findings, both phosphorylation of p38 MAPK and dephosphorylation of ERK1/2 and CREB were restored by the NR2B-selective NMDAR antagonists ifenprodil (3 μm) and Ro 25-6981 (0.5 μm) (Fig. 4B,D), indicating that the alterations of MAPK and CREB signaling were principally mediated by NR2B-containing NMDA receptors. The phosphorylation time course of p38 MAPK, ERK1/2, and CREB suggests that extrasynaptic NR2B activation precedes altered downstream synaptic signaling.
Figure 4.

Involvement of NR2B-containing NMDA receptors in Aβ-induced phosphorylation of p38 MAPK and dephosphorylation of p42/44 MAPK and CREB. A, B, Western blots of the indicated phosphoproteins (with p- prefix) in cultured hippocampal neurons. C, D, Summary histograms quantifying relative immunoreactivity. A, C, Neurons were untreated or treated with 7PA2 CM for 1, 3, 6, or 16 h or with CHO− CM for 16 h. B, D, Hippocampal neurons were exposed as indicated to 7PA2 CM or CHO− CM without or with the NR2B-selective NMDAR inhibitors ifenprodil (Ifen) and Ro 25-6981 (Ro) for 6 h and then analyzed with the indicated antibodies. Data (means ± SEM) are from at least four independent batches of cultured neurons. Error bars represent ± SEM. *p < 0.05, **p < 0.01.
Longer Aβ exposure decreases synaptic NR2B clusters while preserving extrasynaptic clusters
The electrophysiological data above suggest that soluble Aβ significantly increases extrasynaptic NMDA currents, while synaptic NMDA receptor-mediated responses are apparently unaffected within 30 min. To further examine the apparent selectivity of Aβ effects on extrasynaptic versus synaptic NR2B subunits of NMDA receptors, we used a biochemical approach (Goebel-Goody et al., 2009) to separate the synaptic and extrasynaptic fractions of acute hippocampal slices that had been incubated with ACSF, CHO− CM, or 7PA2 CM. As shown in Figure 5A, there was no significant change of NR2B levels in either the synaptic (Triton X-100 insoluble) or extrasynaptic (Triton X-100 soluble) fractions of the slices after treatment with 7PA2 CM for 30 min, a duration of Aβ exposure sufficient to inhibit HFS-induced LTP (above). When slices were instead treated with 7PA2 CM for 6 h, we observed a significant decrease of synaptic NR2B and PSD95 protein levels compared to treatment with CHO− CM; however, the level of NR2B protein in the extrasynaptic fraction remained unaffected (Fig. 5B). To extend this finding, we exposed cultured hippocampal neurons to 7PA2 CM or CHO− CM for increasing time periods, and NR2B clusters were quantified using immunocytochemistry (Carpenter-Hyland et al., 2004). Dual labeling with antibodies to NR2B and synapsin defined synaptic NR2B clusters colocalized with synapsin (a well-known presynaptic marker) versus extrasynaptic NR2B clusters not colocalized with synapsin. Compared to neurons without treatment, there was only a slight and insignificant decrease of synaptic NR2B clusters on neurons treated with 7PA2 CM for up to 6 h (Fig. 6), a time point at which 7PA2 CM had already increased phosphorylation of p38 MAPK and decreased phosphorylation of ERK1/2 and CREB (Fig. 4). Likewise, there was no clear change in the number of extrasynaptic NR2B clusters over 6 h of treatment (Fig. 6). When hippocampal neurons were treated with 7PA2 CM for 16 h, we observed a significant decrease of synaptic NR2B clusters, whereas extrasynaptic NR2B clusters were still not decreased. Taken together, these temporal data suggest that the synaptic signaling impairment by soluble Aβ is a much slower process than the rapid activation of extrasynaptic NMDA receptors.
Figure 5.

Soluble Aβ oligomers do not cause detectable synaptic and extrasynaptic fraction changes upon brief exposure, while the synaptic proteins significant decrease after long treatment. A, B, Western blots of biochemically separated synaptic (triton insoluble) versus extrasynaptic (Triton X-100 soluble) fractions from acute hippocampal slices after no treatment (Non) or treatment with CHO− CM or 7PA2 CM for 30 min (A) or 6 h (B). The graphs show quantification of Western blot signals obtained after the three treatments. Error bars represent ± SEM. *p < 0.05, **p < 0.01.
Figure 6.

Soluble Aβ oligomers do not cause detectable synaptic and extrasynaptic morphological changes upon brief exposure. A, Representative photomicrographs of neuronal cultures labeled with antibodies to NR2B (green) or synapsin (Syn) (red). NR2B-positive clusters on dendrites were classified as synaptic or extrasynaptic based on whether they colocalized (yellow in Merge) or not (green in Merge) with synapsin. B, Bar graphs: quantification after 0, 1, 3, 6, or 16 h of exposure to 7PA2 CM or CHO− CM. Scale bar, 10 μm.
Activation of p38 MAPK and calpain are required for Aβ-mediated impairment of hippocampal LTP
Two extrasynaptic related enzymes, p38 MAPK and calpain (Xu et al., 2009), are believed to be involved in Aβ-mediated neurotoxicity (Hsieh et al., 2006; Origlia et al., 2008). In particular, p38 MAPK has been implicated in Aβ-mediated LTP inhibition in the dentate gyrus (Wang et al., 2004). To further examine the role of p38 MAPK, we treated hippocampal slices with SB203580 (5 μm), an inhibitor of this kinase, before CHO− or 7PA2 CM application. Consistent with previous reports, SB203580 alone had no significant effect on LTP, but it prevented the inhibition of LTP by soluble Aβ (141 ± 5%, n = 10 vs 139 ± 6%, n = 8) (Fig. 7A). In addition to p38 MAPK, calpain, a protease, is also significantly activated during extrasynaptic NMDAR stimulation (Xu et al., 2009). The calpain inhibitor calpeptin (100 μm) only partially blocked LTP by itself (155 ± 8% without, n = 8 vs 132 ± 4% with, n = 6, p < 0.05), but it partially and significantly rescued the soluble Aβ effect on LTP (129 ± 5%, n = 6, p < 0.05) (Fig. 7B). Consistent with the electrophysiological findings, the increase of phosphorylation of p38 MAPK and the dephosphorylation of ERK1/2 and CREB by 6 h of treatment with 7PA2 CM were both prevented by calpeptin (Fig. 7C).
Figure 7.

LTP impairment by soluble Aβ involves the p38 MAPK and calpain signaling pathway. A, p38 MAPK inhibitor SB203580 (5 μm) had no effect on normal LTP (black squares, n = 8) but significantly prevented the inhibition of LTP by 7PA2 CM (red circles, n = 7). B, Calpain selective inhibitor calpeptin (100 μm) slightly depressed LTP (black squares, n = 7), but prevented LTP inhibition by 7PA2 CM (red circles, n = 6). C, Increased phosphorylation (p- prefix) of p38 MAPK and dephosphorylation of ERK1/2 and CREB by 6 h treatment with 7PA2 CM were blocked by calpeptin. SB203580 and calpeptin were applied 20 min before soluble Aβ was added. Error bars represent ± SEM. *p < 0.05, **p < 0.01.
Soluble Aβ oligomer effects are mimicked by TBOA
The glutamate reuptake inhibitor TBOA was used to further support the concept that extrasynaptic activation is a basis for the Aβ-mediated LTP inhibition. By itself, 15 μm TBOA was sufficient to inhibit LTP (112 ± 3%, n = 6, p < 0.05) as expected. Similar to the findings with 7PA2 CM treatment, SB203580 and calpeptin partially prevented the TBOA-mediated inhibition of LTP (132 ± 4%, n = 6 and 128 ± 6%, n = 6, respectively, p < 0.05) (Fig. 8A,B). Consistent with the 7PA2 CM-induced signaling changes, TBOA also increase p38 MAPK phosphorylation and Erk1/2/CREB dephosphorylation (Fig. 8C,E), all effects that could be rescued by NR2B antagonists and by calpeptin (Fig. 8D,F). These data further support the conclusion that soluble Aβ, by enhancing extrasynaptic NMDAR activation, can increase extrasynaptic p38 MAPK phosphorylation, an effect that is dependent on calpain activation, and this contributes to its inhibition of synaptic plasticity.
Figure 8.
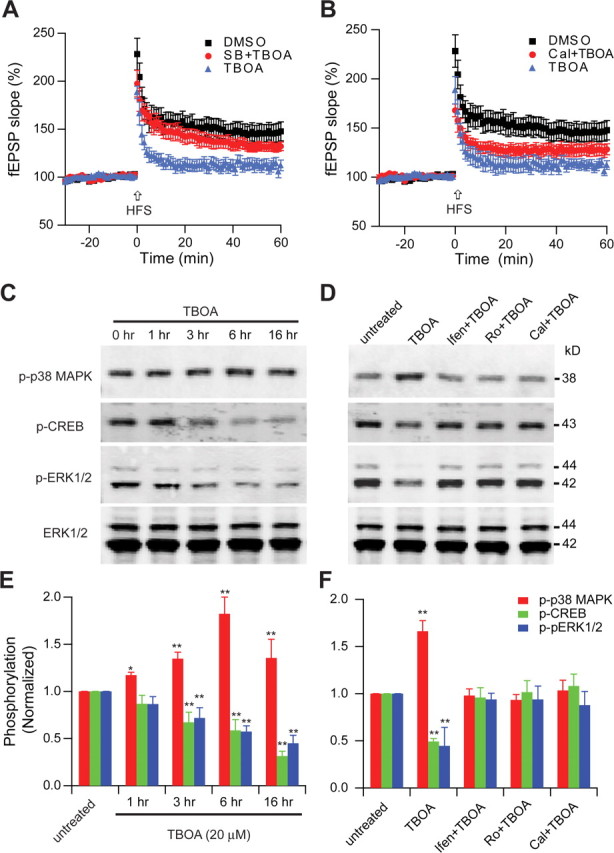
Involvement of NR2B-containing NMDA receptors in TBOA-induced phosphorylation of p38 MAPK and dephosphorylation of ERK1/2 and CREB. A, p38 MAPK inhibitor, SB 203580 (SB) (5 μm), significantly decreased LTP inhibition by the glutamate reuptake inhibitor TBOA (15 μm). B, Calpeptin (Cal) (100 μm), significantly decreased LTP inhibition by TBOA (15 μm). C, D, Western blots of the indicated phosphoproteins (p- prefix) in cultured hippocampal neurons. E, F, Summary histograms quantifying relative immunoreactivity. C, E, Neurons were untreated or treated with TBOA (20 μm) for 1, 3, 6 or 16 h or without TBOA for 16 h. D, F, Hippocampal neurons were exposed as indicated to TBOA without or with the NR2B-selective NMDAR inhibitors ifenprodil (Ifen) and Ro-6981 (Ro) (0.5 μm) or calpain inhibitor calpeptin (100 μm) for 6 h and then analyzed with the indicated antibodies. Data (means ± SEM) are from at least four independent batches of cultured neurons.
Discussion
Elucidating the detailed mechanisms of hippocampal synaptic impairment by soluble amyloid β-protein is important for understanding the basis of the amnestic symptoms that are the initial feature of Alzheimer's disease, with attendant implications for treatment and prevention. Here, we provide multiple lines of evidence that soluble Aβ oligomers from three distinct sources (cultured cells, human brain, and pure synthetic peptide) inhibit NMDAR-dependent hippocampal LTP principally as a result of overactivation of extrasynaptic NR2B-containing receptors. Aβ-inhibited LTP could be rescued by reducing extracellular glutamate levels, by NR2B antagonists, and by inhibitors of the extrasynaptic signaling kinase p38 MAPK or calpain. Whole-cell patch-clamp recordings showed that soluble Aβ significantly increased extrasynaptic NR2B receptor activation in a manner similar to that of a glutamate reuptake inhibitor, TBOA. Soluble Aβ increased p38 MAPK phosphorylation and then impaired the ERK and CREB signaling pathway, and this could be reversed by NR2B-selective antagonists. Measuring synaptic and extrasynaptic NMDAR responses showed that soluble Aβ rapidly increased the extrasynaptic response, preceding the interruption of the synaptic response.
Inhibition of LTP by soluble Aβ involves the overactivation of NR2B-containing NMDA receptors
Our study shows that soluble Aβ oligomers at low to subnanomolar concentrations significantly increase an extrasynaptic NMDA response that is principally mediated by extrasynaptic NR2B subunits, further defining how soluble Aβ can activate certain types of NMDAR-mediated activity (Wu et al., 1995; Molnár et al., 2004; Kelly and Ferreira, 2006; Domingues et al., 2007). NR2B antagonists, but not NR2A and NR2D antagonists, effectively prevented LTP inhibition by Aβ. The partial rescue of LTP by NR2A and NR2D antagonists (Fig. 1E,F) suggests that this is due to their nonselective nature but also that Aβ-mediated inhibition of synaptic plasticity does not depend solely on the NR2B subunit. Although the segregation of NR2A and NR2B to synaptic and extrasynaptic sites, respectively, is not absolute, the activity of synaptic and extrasynaptic NMDARs may be differentially regulated by region-specific signal transduction mechanisms (Hardingham et al., 2002; Ivanov et al., 2006), thus determining NMDAR downstream signaling effects.
Besides altering synaptic plasticity, glutamate excitotoxicity is believed to be a mechanism contributing to progressive neuronal loss in AD (Pomara et al., 1992; Harkany et al., 2000) as it does to the neurodegeneration that follows ischemia or hypoxia (Paschen, 1996; Hardingham et al., 2002; Liu et al., 2007). Considering the small portion of the entire neuronal plasma membrane that is synaptic membrane, the much larger area of the extrasynaptic surface would suggest that NR2B subunits have a much more abundant distribution than the principally synapse-targeted NR2A subunits. It has also been reported that NR2B-containing NMDARs are more sensitive to glutamate than NR2A-containing NMDARs (Kutsuwada et al., 1992). In this context, our prior evidence that extracellular glutamate levels increase because soluble Aβ oligomers impair glutamate reuptake (Li et al., 2009) suggests that an early response to rising levels of soluble Aβ may be an overactivation of NR2B-positive NMDA receptors. This concept could also explain observations that excitotoxicity is triggered by the selective activation of NMDARs that contain NR2B subunits (Sinor et al., 2000; Zhou and Baudry, 2006; Liu et al., 2007; von Engelhardt et al., 2007). Accordingly, NR2B-selective antagonists effectively prevent glutamate-induced excitotoxicity in ischemia (Hardingham et al., 2002; Liu et al., 2007) and rescue Aβ-inhibited LTP (Hu et al., 2009; Rönicke et al., 2010; present study). The depressed synaptic NR2B proteins we observed at 6 h could be explained by endocytosis of surface NMDAR (Snyder et al., 2005) associated with Aβ-mediated effects of excess glutamate levels (Vaslin et al., 2007).
Soluble Aβ enhances extrasynaptic NMDA receptor responses
Soluble Aβ oligomers were recently found to interrupt glutamate reuptake (Matos et al., 2008; Li et al., 2009) and increase extracellular glutamate levels (O'Shea et al., 2008). Our current work using both whole-cell recording and Fluo-4 calcium measurements confirmed that Aβ rapidly and significantly increased extrasynaptic NMDA responses. To further explore the basis for impairment of LTP by soluble Aβ, TBOA, a glutamate reuptake inhibitor, was analyzed comparatively since it can promote diffusion of glutamate from release sites and activation of NMDARs beyond synaptic sites. We also applied a very low dose of NMDA (0.5 μm) to provide further evidence that the principal effect of Aβ occurs at extrasynaptic receptors. Both this and prior studies suggest that hippocampal LTP can be impaired by extrasynaptic NMDAR overactivation caused by glutamate spillover from TBOA, by increased tetanus stimulation intensity, or by exogenous NMDA (Katagiri et al., 2001; Izumi et al., 2008; Li et al., 2009). Interestingly, we showed that increased glutamate levels cause persistent NMDA receptor activation and therefore desensitization, but this phenomenon is not implicated in the Aβ block of LTP. We further show that the inhibition of synaptic plasticity by Aβ oligomers can be prevented by reducing extracellular glutamate levels via a glutamate scavenger system.
Of potential relevance to our findings with Aβ, several recent studies demonstrate that increased extrasynaptic NMDAR activity plays a key role in the pathogenic mechanisms of Huntington's disease (Okamoto et al., 2009; Milnerwood et al., 2010) and of glutamate excitotoxicity (Stanika et al., 2009). This work further supports a scenario in which synaptic NMDARs activate cellular survival pathways, whereas extrasynaptic NMDARs tend to activate pathways that contribute to cell death (Hardingham et al., 2002; Papadia and Hardingham, 2007). Interestingly, memantine, a NMDAR antagonist used to treat more advanced AD patients, has been shown to prevent Aβ-mediated inhibition of LTP in rodents in vivo (Klyubin et al., 2011) and is mainly targeted to the extrasynaptic NMDAR (Milnerwood et al., 2010; Xia et al., 2010). Our combined electrophysiological and biochemical data show that the soluble Aβ-enhanced extrasynaptic NMDAR response happens much faster than the synaptic depression, suggesting that the extrasynaptic activation triggers downstream signaling pathways, e.g., via PP1 (Knobloch et al., 2007), so that HFS can no longer induce LTP. The current study and the related reports just reviewed suggest that overactivation of extrasynaptic NMDARs in the hippocampus may be an early step in the pathogenesis of the amnestic symptoms of AD.
We have no evidence that the soluble Aβ oligomers bind directly to NR2B, to other NMDAR subunits, or indeed to any specific hydrophilic cell surface receptor, as discussed previously (Shankar et al., 2007, their Fig. 7). Instead, we hypothesize that the cognate targets of Aβ oligomers are likely to be plasma membrane lipids, which would be expected to avidly bind the highly hydrophobic oligomers rather than binding to the hydrophilic ectodomains of protein receptors. Oligomer-mediated perturbation of the fine structure of the lipid bilayer could then lead to secondary biophysical effects on the structure and function of various transmembrane receptors (e.g., the NMDA, AMPA, insulin, and/or α7-nicotinic receptors proposed to date). Pinpointing the binding partners of natural Aβ oligomers will be labor intensive, since one needs to purify them to homogeneity from AD cortex, label them, and expose primary neurons or brain slices to them to identify in an unbiased fashion their molecular targets. Using a synthetic Aβ peptide of a single defined length at supraphysiological concentrations would not be expected to yield the natural binding partner(s).
Soluble Aβ enhances calpain/p38 MAPK signaling pathway and then impairs ERK/CREB phosphorylation
Our result that soluble Aβ significantly inhibits ERK and CREB phosphorylation is consistent with a previous report (Townsend et al., 2007) that the inactivation of ERK/CREB signaling pathway by soluble Aβ species secreted by cultured cells takes a few hours but can be accelerated by chemical stimulation. Although some reports suggest that supraphysiological concentrations of synthetic Aβ can enhance ERK and CREB phosphorylation within minutes (Dineley et al., 2001; Chong et al., 2006), the differences from our time course may relate to our natural sources of soluble Aβ and our low concentrations, as found in AD brain extracts. For example, synthetic fibrillar Aβ, but not small synthetic Aβ oligomers, can directly activate ERK phosphorylation (Sondag et al., 2009).
In Alzheimer's disease brain tissue, abnormally high levels of calpain (Saito et al., 1993) and p38 MAPK (Hensley et al., 1999; Sun et al., 2003) activation can be measured, perhaps even at an early stage in the disease process. Recent work has shown that the concentrations of calpain and p38 MAPK were significantly greater in extrasynaptic membranes than in synaptic membranes in cortical tissue (Xu et al., 2009). It has been reported that Aβ-mediated inhibition of LTP involves in part the p38 MAPK signaling pathway (Wang et al., 2004). p38 MAPK is a part of a complex NMDAR-driven signaling cascade, and increasing its activity contributes to LTP inhibition (Izumi et al., 2008). In this regard, an oligomeric Aβ synthetic peptide, but not its fibrillar form, strongly stimulated p38 MAPK kinase (Origlia et al., 2008; Sondag et al., 2009). Our data confirm that natural Aβ oligomers significantly increase p38 MAPK phosphorylation. That the p38 MAPK and calpain inhibitors also prevented Aβ- and TBOA-mediated LTP inhibition due to glutamate spillover in our study further supports an important role for the p38 MAPK in an extrasynaptic-dominated signaling pathway. Overall, our study suggests that development of effective extrasynaptic NR2B antagonists could help ameliorate the hippocampal amnestic symptoms that occur early in the course of AD and its harbinger, mild cognitive impairment.
Footnotes
This work was supported by National Institutes of Health Grant AG 027443 (D.J.S.). We thank D. Walsh (Brigham and Women's Hospital, Boston, MA) for providing the synthetic S26C dimer and M. Rowan (Trinity College, Dublin, Ireland) and B. Sabatini (Harvard Medical School, Boston, MA) for helpful discussions. T. Yang (Brigham and Women's Hospital, Boston, MA) prepared the AD brain extracts.
The authors declare no competing financial interests.
References
- Berberich S, Jensen V, Hvalby Ø, Seeburg PH, Köhr G. The role of NMDAR subtypes and charge transfer during hippocampal LTP induction. Neuropharmacology. 2007;52:77–86. doi: 10.1016/j.neuropharm.2006.07.016. [DOI] [PubMed] [Google Scholar]
- Carpenter-Hyland EP, Woodward JJ, Chandler LJ. Chronic ethanol induces synaptic but not extrasynaptic targeting of NMDA receptors. J Neurosci. 2004;24:7859–7868. doi: 10.1523/JNEUROSCI.1902-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen QS, Wei WZ, Shimahara T, Xie CW. Alzheimer amyloid beta-peptide inhibits the late phase of long-term potentiation through calcineurin-dependent mechanisms in the hippocampal dentate gyrus. Neurobiol Learn Mem. 2002;77:354–371. doi: 10.1006/nlme.2001.4034. [DOI] [PubMed] [Google Scholar]
- Chong YH, Shin YJ, Lee EO, Kayed R, Glabe CG, Tenner AJ. ERK1/2 activation mediates Aβ oligomer-induced neurotoxicity via caspase-3 activation and tau cleavage in rat organotypic hippocampal slice cultures. J Biol Chem. 2006;281:20315–20325. doi: 10.1074/jbc.M601016200. [DOI] [PubMed] [Google Scholar]
- Dewachter I, Filipkowski RK, Priller C, Ris L, Neyton J, Croes S, Terwel D, Gysemans M, Devijver H, Borghgraef P, Godaux E, Kaczmarek L, Herms J, Van Leuven F. Deregulation of NMDA-receptor function and down-stream signaling in APP[V717I] transgenic mice. Neurobiol Aging. 2009;30:241–256. doi: 10.1016/j.neurobiolaging.2007.06.011. [DOI] [PubMed] [Google Scholar]
- Dineley KT, Westerman M, Bui D, Bell K, Ashe KH, Sweatt JD. Beta-amyloid activates the mitogen-activated protein kinase cascade via hippocampal alpha7 nicotinic acetylcholine receptors: in vitro and in vivo mechanisms related to Alzheimer's disease. J Neurosci. 2001;21:4125–4133. doi: 10.1523/JNEUROSCI.21-12-04125.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domingues A, Almeida S, da Cruz e Silva EF, Oliveira CR, Rego AC. Toxicity of beta-amyloid in HEK293 cells expressing NR1/NR2A or NR1/NR2B N-methyl-d-aspartate receptor subunits. Neurochem Int. 2007;50:872–880. doi: 10.1016/j.neuint.2007.03.001. [DOI] [PubMed] [Google Scholar]
- Gao C, Gill MB, Tronson NC, Guedea AL, Guzmán YF, Huh KH, Corcoran KA, Swanson GT, Radulovic J. Hippocampal NMDA receptor subunits differentially regulate fear memory formation and neuronal signal propagation. Hippocampus. 2010;20:1072–1082. doi: 10.1002/hipo.20705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goebel-Goody SM, Davies KD, Alvestad Linger RM, Freund RK, Browning MD. Phospho-regulation of synaptic and extrasynaptic N-methyl-d-aspartate receptors in adult hippocampal slices. Neuroscience. 2009;158:1446–1459. doi: 10.1016/j.neuroscience.2008.11.006. [DOI] [PubMed] [Google Scholar]
- Hardingham GE, Fukunaga Y, Bading H. Extrasynaptic NMDARs oppose synaptic NMDARs by triggering CREB shut-off and cell death pathways. Nat Neurosci. 2002;5:405–414. doi: 10.1038/nn835. [DOI] [PubMed] [Google Scholar]
- Harkany T, Abrahám I, Timmerman W, Laskay G, Tóth B, Sasvári M, Kónya C, Sebens JB, Korf J, Nyakas C, Zarándi M, Soós K, Penke B, Luiten PG. β-Amyloid neurotoxicity is mediated by a glutamate-triggered excitotoxic cascade in rat nucleus basalis. Eur J Neurosci. 2000;12:2735–2745. doi: 10.1046/j.1460-9568.2000.00164.x. [DOI] [PubMed] [Google Scholar]
- Hensley K, Floyd RA, Zheng NY, Nael R, Robinson KA, Nguyen X, Pye QN, Stewart CA, Geddes J, Markesbery WR, Patel E, Johnson GV, Bing G. p38 kinase is activated in the Alzheimer's disease brain. J Neurochem. 1999;72:2053–2058. doi: 10.1046/j.1471-4159.1999.0722053.x. [DOI] [PubMed] [Google Scholar]
- Hsieh H, Boehm J, Sato C, Iwatsubo T, Tomita T, Sisodia S, Malinow R. AMPAR removal underlies Aβ-induced synaptic depression and dendritic spine loss. Neuron. 2006;52:831–843. doi: 10.1016/j.neuron.2006.10.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu NW, Smith IM, Walsh DM, Rowan MJ. Soluble amyloid-beta peptides potently disrupt hippocampal synaptic plasticity in the absence of cerebrovascular dysfunction in vivo. Brain. 2008;131:2414–2424. doi: 10.1093/brain/awn174. [DOI] [PubMed] [Google Scholar]
- Hu NW, Klyubin I, Anwyl R, Rowan MJ. GluN2B subunit-containing NMDA receptor antagonists prevent Aβ-mediated synaptic plasticity disruption in vivo. Proc Natl Acad Sci U S A. 2009;106:20504–20509. doi: 10.1073/pnas.0908083106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanov A, Pellegrino C, Rama S, Dumalska I, Salyha Y, Ben-Ari Y, Medina I. Opposing role of synaptic and extrasynaptic NMDA receptors in regulation of the extracellular signal-regulated kinases (ERK) activity in cultured rat hippocampal neurons. J Physiol. 2006;572:789–798. doi: 10.1113/jphysiol.2006.105510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izumi Y, Tokuda K, Zorumski CF. Long-term potentiation inhibition by low-level N-methyl-d-aspartate receptor activation involves calcineurin, nitric oxide, and p38 mitogen-activated protein kinase. Hippocampus. 2008;18:258–265. doi: 10.1002/hipo.20383. [DOI] [PubMed] [Google Scholar]
- Janus C, Pearson J, McLaurin J, Mathews PM, Jiang Y, Schmidt SD, Chishti MA, Horne P, Heslin D, French J, Mount HT, Nixon RA, Mercken M, Bergeron C, Fraser PE, St George-Hyslop P, Westaway D. A beta peptide immunization reduces behavioural impairment and plaques in a model of Alzheimer's disease. Nature. 2000;408:979–982. doi: 10.1038/35050110. [DOI] [PubMed] [Google Scholar]
- Katagiri H, Tanaka K, Manabe T. Requirement of appropriate glutamate concentrations in the synaptic cleft for hippocampal LTP induction. Eur J Neurosci. 2001;14:547–553. doi: 10.1046/j.0953-816x.2001.01664.x. [DOI] [PubMed] [Google Scholar]
- Kelly BL, Ferreira A. beta-Amyloid-induced dynamin 1 degradation is mediated by N-methyl-d-aspartate receptors in hippocampal neurons. J Biol Chem. 2006;281:28079–28089. doi: 10.1074/jbc.M605081200. [DOI] [PubMed] [Google Scholar]
- Klyubin I, Walsh DM, Lemere CA, Cullen WK, Shankar GM, Betts V, Spooner ET, Jiang L, Anwyl R, Selkoe DJ, Rowan MJ. Amyloid β protein immunotherapy neutralizes Aβ oligomers that disrupt synaptic plasticity in vivo. Nat Med. 2005;11:556–561. doi: 10.1038/nm1234. [DOI] [PubMed] [Google Scholar]
- Klyubin I, Wang Q, Reed MN, Irving EA, Upton N, Hofmeister J, Cleary JP, Anwyl R, Rowan MJ. Protection against Aβ-mediated rapid disruption of synaptic plasticity and memory by memantine. Neurobiol Aging. 2011;32:614–623. doi: 10.1016/j.neurobiolaging.2009.04.005. [DOI] [PubMed] [Google Scholar]
- Knobloch M, Farinelli M, Konietzko U, Nitsch RM, Mansuy IM. Aβ oligomer-mediated long-term potentiation impairment involves protein phosphatase 1-dependent mechanisms. J Neurosci. 2007;27:7648–7653. doi: 10.1523/JNEUROSCI.0395-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kutsuwada T, Kashiwabuchi N, Mori H, Sakimura K, Kushiya E, Araki K, Meguro H, Masaki H, Kumanishi T, Arakawa M, Mishina M. Molecular diversity of the NMDA receptor channel. Nature. 1992;358:36–41. doi: 10.1038/358036a0. [DOI] [PubMed] [Google Scholar]
- Lambert MP, Barlow AK, Chromy BA, Edwards C, Freed R, Liosatos M, Morgan TE, Rozovsky I, Trommer B, Viola KL, Wals P, Zhang C, Finch CE, Krafft GA, Klein WL. Diffusible, nonfibrillar ligands derived from Aβ1–42 are potent central nervous system neurotoxins. Proc Natl Acad Sci U S A. 1998;95:6448–6453. doi: 10.1073/pnas.95.11.6448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Hong S, Shepardson NE, Walsh DM, Shankar GM, Selkoe D. Soluble oligomers of amyloid beta protein facilitate hippocampal long-term depression by disrupting neuronal glutamate uptake. Neuron. 2009;62:788–801. doi: 10.1016/j.neuron.2009.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Wong TP, Aarts M, Rooyakkers A, Liu L, Lai TW, Wu DC, Lu J, Tymianski M, Craig AM, Wang YT. NMDA receptor subunits have differential roles in mediating excitotoxic neuronal death both in vitro and in vivo. J Neurosci. 2007;27:2846–2857. doi: 10.1523/JNEUROSCI.0116-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu W, Man H, Ju W, Trimble WS, MacDonald JF, Wang YT. Activation of synaptic NMDA receptors induces membrane insertion of new AMPA receptors and LTP in cultured hippocampal neurons. Neuron. 2001;29:243–254. doi: 10.1016/s0896-6273(01)00194-5. [DOI] [PubMed] [Google Scholar]
- Lue LF, Kuo YM, Roher AE, Brachova L, Shen Y, Sue L, Beach T, Kurth JH, Rydel RE, Rogers J. Soluble amyloid beta peptide concentration as a predictor of synaptic change in Alzheimer's disease. Am J Pathol. 1999;155:853–862. doi: 10.1016/s0002-9440(10)65184-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marks N, Berg MJ. Neurosecretases provide strategies to treat sporadic and familial Alzheimer disorders. Neurochem Int. 2008;52:184–215. doi: 10.1016/j.neuint.2007.06.020. [DOI] [PubMed] [Google Scholar]
- Massey PV, Johnson BE, Moult PR, Auberson YP, Brown MW, Molnar E, Collingridge GL, Bashir ZI. Differential roles of NR2A and NR2B-containing NMDA receptors in cortical long-term potentiation and long-term depression. J Neurosci. 2004;24:7821–7828. doi: 10.1523/JNEUROSCI.1697-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matos M, Augusto E, Oliveira CR, Agostinho P. Amyloid-beta peptide decreases glutamate uptake in cultured astrocytes: involvement of oxidative stress and mitogen-activated protein kinase cascades. Neuroscience. 2008;156:898–910. doi: 10.1016/j.neuroscience.2008.08.022. [DOI] [PubMed] [Google Scholar]
- Mayer ML, Vyklicky L, Jr, Clements J. Regulation of NMDA receptor desensitization in mouse hippocampal neurons by glycine. Nature. 1989;338:425–427. doi: 10.1038/338425a0. [DOI] [PubMed] [Google Scholar]
- McLean CA, Cherny RA, Fraser FW, Fuller SJ, Smith MJ, Beyreuther K, Bush AI, Masters CL. Soluble pool of Aβ amyloid as a determinant of severity of neurodegeneration in Alzheimer's disease. Ann Neurol. 1999;46:860–866. doi: 10.1002/1531-8249(199912)46:6<860::aid-ana8>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- Milnerwood AJ, Gladding CM, Pouladi MA, Kaufman AM, Hines RM, Boyd JD, Ko RW, Vasuta OC, Graham RK, Hayden MR, Murphy TH, Raymond LA. Early increase in extrasynaptic NMDA receptor signaling and expression contributes to phenotype onset in Huntington's disease mice. Neuron. 2010;65:178–190. doi: 10.1016/j.neuron.2010.01.008. [DOI] [PubMed] [Google Scholar]
- Min MY, Rusakov DA, Kullmann DM. Activation of AMPA, kainate, and metabotropic receptors at hippocampal mossy fiber synapses: role of glutamate diffusion. Neuron. 1998;21:561–570. doi: 10.1016/s0896-6273(00)80566-8. [DOI] [PubMed] [Google Scholar]
- Molnár Z, Soós K, Lengyel I, Penke B, Szegedi V, Budai D. Enhancement of NMDA responses by beta-amyloid peptides in the hippocampus in vivo. Neuroreport. 2004;15:1649–1652. doi: 10.1097/01.wnr.0000134471.06244.d2. [DOI] [PubMed] [Google Scholar]
- Morgan D, Diamond DM, Gottschall PE, Ugen KE, Dickey C, Hardy J, Duff K, Jantzen P, DiCarlo G, Wilcock D, Connor K, Hatcher J, Hope C, Gordon M, Arendash GW. A beta peptide vaccination prevents memory loss in an animal model of Alzheimer's disease. Nature. 2000;408:982–985. doi: 10.1038/35050116. [DOI] [PubMed] [Google Scholar]
- Mulholland PJ, Luong NT, Woodward JJ, Chandler LJ. Brain-derived neurotrophic factor activation of extracellular signal-regulated kinase is autonomous from the dominant extrasynaptic NMDA receptor extracellular signal-regulated kinase shutoff pathway. Neuroscience. 2008;151:419–427. doi: 10.1016/j.neuroscience.2007.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamoto S, Pouladi MA, Talantova M, Yao D, Xia P, Ehrnhoefer DE, Zaidi R, Clemente A, Kaul M, Graham RK, Zhang D, Vincent Chen HS, Tong G, Hayden MR, Lipton SA. Balance between synaptic versus extrasynaptic NMDA receptor activity influences inclusions and neurotoxicity of mutant huntingtin. Nat Med. 2009;15:1407–1413. doi: 10.1038/nm.2056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okubo Y, Sekiya H, Namiki S, Sakamoto H, Iinuma S, Yamasaki M, Watanabe M, Hirose K, Iino M. Imaging extrasynaptic glutamate dynamics in the brain. Proc Natl Acad Sci U S A. 2010;107:6526–6531. doi: 10.1073/pnas.0913154107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Origlia N, Righi M, Capsoni S, Cattaneo A, Fang F, Stern DM, Chen JX, Schmidt AM, Arancio O, Yan SD, Domenici L. Receptor for advanced glycation end product-dependent activation of p38 mitogen-activated protein kinase contributes to amyloid-beta-mediated cortical synaptic dysfunction. J Neurosci. 2008;28:3521–3530. doi: 10.1523/JNEUROSCI.0204-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Shea SD, Smith IM, McCabe OM, Cronin MM, Walsh DM, O'Connor WT. Intracerebroventricular administration of amyloid β-protein oligomers selectively increases dorsal hippocampal dialysate glutamate levels in the awake rat. Sensors. 2008;8:7428–7437. doi: 10.3390/s8117428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overstreet LS, Pasternak JF, Colley PA, Slater NT, Trommer BL. Metabotropic glutamate receptor mediated long-term depression in developing hippocampus. Neuropharmacology. 1997;36:831–844. doi: 10.1016/s0028-3908(97)00031-2. [DOI] [PubMed] [Google Scholar]
- Papadia S, Hardingham GE. The dichotomy of NMDA receptor signaling. Neuroscientist. 2007;13:572–579. doi: 10.1177/10738584070130060401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papadia S, Stevenson P, Hardingham NR, Bading H, Hardingham GE. Nuclear Ca2+ and the cAMP response element-binding protein family mediate a late phase of activity-dependent neuroprotection. J Neurosci. 2005;25:4279–4287. doi: 10.1523/JNEUROSCI.5019-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paschen W. Glutamate excitotoxicity in transient global cerebral ischemia. Acta Neurobiol Exp (Wars) 1996;56:313–322. doi: 10.55782/ane-1996-1136. [DOI] [PubMed] [Google Scholar]
- Pomara N, Singh R, Deptula D, Chou JC, Schwartz MB, LeWitt PA. Glutamate and other CSF amino acids in Alzheimer's disease. Am J Psychiatry. 1992;149:251–254. doi: 10.1176/ajp.149.2.251. [DOI] [PubMed] [Google Scholar]
- Raymond CR, Ireland DR, Abraham WC. NMDA receptor regulation by amyloid-β does not account for its inhibition of LTP in rat hippocampus. Brain Res. 2003;968:263–272. doi: 10.1016/s0006-8993(03)02269-8. [DOI] [PubMed] [Google Scholar]
- Rönicke R, Mikhaylova M, Rönicke S, Meinhardt J, Schröder UH, Fändrich M, Reiser G, Kreutz MR, Reymann KG. Early neuronal dysfunction by amyloid beta oligomers depends on activation of NR2B-containing NMDA receptors. Neurobiol Aging. 2010 doi: 10.1016/j.neurobiolaging.2010.01.01. [DOI] [PubMed] [Google Scholar]
- Saito K, Elce JS, Hamos JE, Nixon RA. Widespread activation of calcium-activated neutral proteinase (calpain) in the brain in Alzheimer disease: a potential molecular basis for neuronal degeneration. Proc Natl Acad Sci U S A. 1993;90:2628–2632. doi: 10.1073/pnas.90.7.2628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selkoe DJ. The cell biology of β-amyloid precursor protein and presenilin in Alzheimer's disease. Trends Cell Biol. 1998;8:447–453. doi: 10.1016/s0962-8924(98)01363-4. [DOI] [PubMed] [Google Scholar]
- Shankar GM, Bloodgood BL, Townsend M, Walsh DM, Selkoe DJ, Sabatini BL. Natural oligomers of the Alzheimer amyloid-β protein induce reversible synapse loss by modulating an NMDA-type glutamate receptor-dependent signaling pathway. J Neurosci. 2007;27:2866–2875. doi: 10.1523/JNEUROSCI.4970-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shankar GM, Li S, Mehta TH, Garcia-Munoz A, Shepardson NE, Smith I, Brett FM, Farrell MA, Rowan MJ, Lemere CA, Regan CM, Walsh DM, Sabatini BL, Selkoe DJ. Soluble amyloid β-protein dimers isolated directly from Alzheimer disease patients potently impair synaptic plasticity and memory. Nat Med. 2008;14:837–842. doi: 10.1038/nm1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinor JD, Du S, Venneti S, Blitzblau RC, Leszkiewicz DN, Rosenberg PA, Aizenman E. NMDA and glutamate evoke excitotoxicity at distinct cellular locations in rat cortical neurons in vitro. J Neurosci. 2000;20:8831–8837. doi: 10.1523/JNEUROSCI.20-23-08831.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snyder EM, Nong Y, Almeida CG, Paul S, Moran T, Choi EY, Nairn AC, Salter MW, Lombroso PJ, Gouras GK, Greengard P. Regulation of NMDA receptor trafficking by amyloid-β. Nat Neurosci. 2005;8:1051–1058. doi: 10.1038/nn1503. [DOI] [PubMed] [Google Scholar]
- Sondag CM, Dhawan G, Combs CK. Beta amyloid oligomers and fibrils stimulate differential activation of primary microglia. J Neuroinflammation. 2009;6:1–13. doi: 10.1186/1742-2094-6-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanika RI, Pivovarova NB, Brantner CA, Watts CA, Winters CA, Andrews SB. Coupling diverse routes of calcium entry to mitochondrial dysfunction and glutamate excitotoxicity. Proc Natl Acad Sci U S A. 2009;106:9854–9859. doi: 10.1073/pnas.0903546106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun A, Liu M, Nguyen XV, Bing G. p38 MAP kinase is activated at early stages in Alzheimer's disease brain. Exp Neurol. 2003;183:394–405. doi: 10.1016/s0014-4886(03)00180-8. [DOI] [PubMed] [Google Scholar]
- Townsend M, Shankar GM, Mehta T, Walsh DM, Selkoe DJ. Effects of secreted oligomers of amyloid β-protein on hippocampal synaptic plasticity: a potent role for trimers. J Physiol. 2006;572:477–492. doi: 10.1113/jphysiol.2005.103754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Townsend M, Mehta T, Selkoe DJ. Soluble Aβ inhibits specific signal transduction cascades common to the insulin receptor pathway. J Biol Chem. 2007;282:33305–33312. doi: 10.1074/jbc.M610390200. [DOI] [PubMed] [Google Scholar]
- Vaslin A, Puyal J, Borsello T, Clarke PG. Excitotoxicity-related endocytosis in cortical neurons. J Neurochem. 2007;102:789–800. doi: 10.1111/j.1471-4159.2007.04564.x. [DOI] [PubMed] [Google Scholar]
- von Engelhardt J, Coserea I, Pawlak V, Fuchs EC, Köhr G, Seeburg PH, Monyer H. Excitotoxicity in vitro by NR2A- and NR2B-containing NMDA receptors. Neuropharmacology. 2007;53:10–17. doi: 10.1016/j.neuropharm.2007.04.015. [DOI] [PubMed] [Google Scholar]
- Walsh DM, Klyubin I, Fadeeva JV, Cullen WK, Anwyl R, Wolfe MS, Rowan MJ, Selkoe DJ. Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature. 2002;416:535–539. doi: 10.1038/416535a. [DOI] [PubMed] [Google Scholar]
- Walsh DM, Townsend M, Podlisny MB, Shankar GM, Fadeeva JV, El Agnaf O, Hartley DM, Selkoe DJ. Certain inhibitors of synthetic amyloid β-peptide (Aβ) fibrillogenesis block oligomerization of natural Aβ and thereby rescue long-term potentiation. J Neurosci. 2005;25:2455–2462. doi: 10.1523/JNEUROSCI.4391-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang HW, Pasternak JF, Kuo H, Ristic H, Lambert MP, Chromy B, Viola KL, Klein WL, Stine WB, Krafft GA, Trommer BL. Soluble oligomers of β amyloid (1–42) inhibit long-term potentiation but not long-term depression in rat dentate gyrus. Brain Res. 2002;924:133–140. doi: 10.1016/s0006-8993(01)03058-x. [DOI] [PubMed] [Google Scholar]
- Wang Q, Walsh DM, Rowan MJ, Selkoe DJ, Anwyl R. Block of long-term potentiation by naturally secreted and synthetic amyloid β-peptide in hippocampal slices is mediated via activation of the kinases c-Jun N-terminal kinase, cyclin-dependent kinase 5, and p38 mitogen-activated protein kinase as well as metabotropic glutamate receptor type 5. J Neurosci. 2004;24:3370–3378. doi: 10.1523/JNEUROSCI.1633-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waxman EA, Lynch DR. N-methyl-d-aspartate receptor subtype mediated bidirectional control of p38 mitogen-activated protein kinase. J Biol Chem. 2005;280:29322–29333. doi: 10.1074/jbc.M502080200. [DOI] [PubMed] [Google Scholar]
- Wu J, Anwyl R, Rowan MJ. beta-Amyloid selectively augments NMDA receptor-mediated synaptic transmission in rat hippocampus. Neuroreport. 1995;6:2409–2413. doi: 10.1097/00001756-199511270-00031. [DOI] [PubMed] [Google Scholar]
- Xia P, Chen HS, Zhang D, Lipton SA. Memantine preferentially blocks extrasynaptic over synaptic NMDA receptor currents in hippocampal autapses. J Neurosci. 2010;30:11246–11250. doi: 10.1523/JNEUROSCI.2488-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Kurup P, Zhang Y, Goebel-Goody SM, Wu PH, Hawasli AH, Baum ML, Bibb JA, Lombroso PJ. Extrasynaptic NMDA receptors couple preferentially to excitotoxicity via calpain-mediated cleavage of STEP. J Neurosci. 2009;29:9330–9343. doi: 10.1523/JNEUROSCI.2212-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao D, Watson JB, Xie CW. Amyloid beta prevents activation of calcium/calmodulin-dependent protein kinase II and AMPA receptor phosphorylation during hippocampal long-term potentiation. J Neurophysiol. 2004;92:2853–2858. doi: 10.1152/jn.00485.2004. [DOI] [PubMed] [Google Scholar]
- Zhou M, Baudry M. Developmental changes in NMDA neurotoxicity reflect developmental changes in subunit composition of NMDA receptors. J Neurosci. 2006;26:2956–2963. doi: 10.1523/JNEUROSCI.4299-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]