

Abstract
Skeletal muscle myosin light chain kinase (skMLCK) is a dedicated Ca2+/calmodulin-dependent serine-threonine protein kinase that phosphorylates the regulatory light chain (RLC) of sarcomeric myosin. It is expressed from the MYLK2 gene specifically in skeletal muscle fibers with most abundance in fast contracting muscles. Biochemically, activation occurs with Ca2+ binding to calmodulin forming a (Ca2+)4•calmodulin complex sufficient for activation with a diffusion limited, stoichiometic binding and displacement of a regulatory segment from skMLCK catalytic core. The N-terminal sequence of RLC then extends through the exposed catalytic cleft for Ser15 phosphorylation. Removal of Ca2+ results in the slow dissociation of calmodulin and inactivation of skMLCK. Combined biochemical properties provide unique features for the physiological responsiveness of RLC phosphorylation, including (1) rapid activation of MLCK by Ca2+/calmodulin, (2) limiting kinase activity so phosphorylation is slower than contraction, (3) slow MLCK inactivation after relaxation and (4) much greater kinase activity relative to myosin light chain phosphatase (MLCP). SkMLCK phosphorylation of myosin RLC modulates mechanical aspects of vertebrate skeletal muscle function. In permeabilized skeletal muscle fibers, phosphorylation-mediated alterations in myosin structure increase the rate of force-generation by myosin cross bridges to increase Ca2+-sensitivity of the contractile apparatus. Stimulation-induced increases in RLC phosphorylation in intact muscle produces isometric and concentric force potentiation to enhance dynamic aspects of muscle work and power in unfatigued or fatigued muscle. Moreover, RLC phosphorylation-mediated enhancements may interact with neural strategies for human skeletal muscle activation to ameliorate either central or peripheral aspects of fatigue.
Keywords: myosin light chain kinase, regulatory light chain, calmodulin, calcium, contraction
Introduction
Skeletal muscle sarcomeres are organized into regular arrays of actin thin filaments and myosin thick filaments of well-defined length. An action potential propagated from the neuromuscular junction spreads across the sarcolemma and into T-tubules to trigger the release of Ca2+ from the sarcoplasmic reticulum. The elevated intracellular Ca2+ rapidly activates skeletal muscle contraction by binding to troponin in thin filaments of the sarcomere, thereby allowing myosin cross bridges in thick filaments to bind actin in thin filaments [1]. Contraction occurs when myosin cross bridges exert force on actin filaments with ATP hydrolysis. This force causes the thin filament to slide past the thick filament, and allows the muscle to shorten and to develop force.
A myosin cross bridge, containing the actin-binding surface and ATP pocket in the head, or motor domain, tapers to an alpha-helical neck that connects to the myosin rod region responsible for self assembly into thick filaments (Fig. 1)[1]. Two small protein subunits, the essential light chain and the regulatory light chain (RLC), wrap around each alpha-helical neck region providing mechanical support [2, 3]. Additionally, different domains in the myosin molecule interact to produce an inactive off-state with head-head and head-rod interactions involving RLC.
Fig. 1.
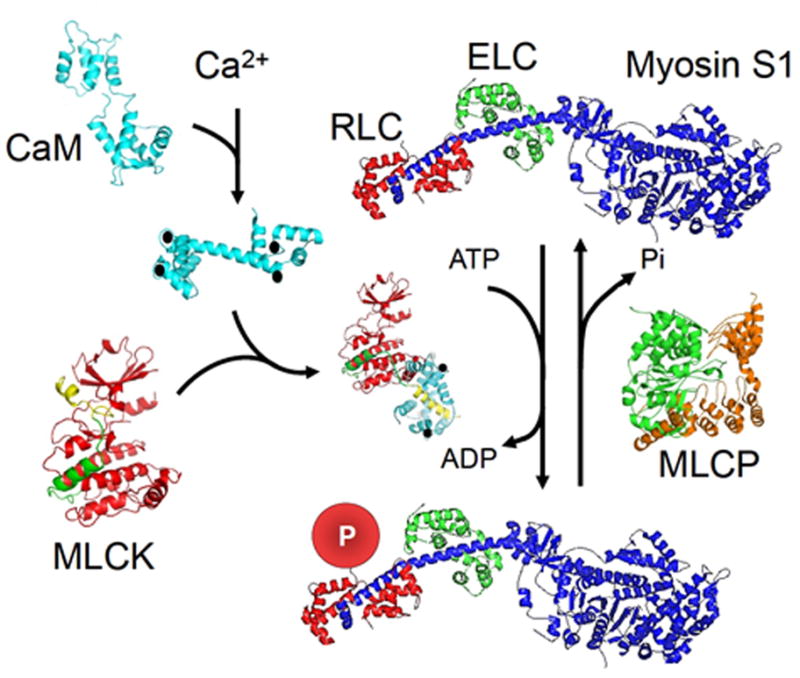
Scheme for myosin RLC phosphorylation in skeletal muscle based on structures of involved proteins. SkMLCK is inactive due to the regulatory segment containing autoinhibitory (green) and calmodulin-binding (yellow) sequences binding to the catalytic core. The phosphorylatable Ser in the N-terminus of RLC is thus prevented from binding in the catalytic cleft between the N-and C-domains of the catalytic core (red). Ca2+ (●) binds to four Ca2+ -binding sites in calmodulin (CaM), and the complex binds to skMLCK to displace the regulatory segment from the catalytic cleft. The subfragment 1 (S1) myosin head and neck domain comprises heavy chain (blue) with essential light chain (green, ECL) and RLC (red, RLC) bound to the α-helical neck region of the heavy chain. The disordered N-terminus of RLC (not shown) extends from RLC bound to the heavy chain for phosphorylation (P) by activated skMLCK. Myosin light chain phosphatase (MLCP) containing the catalytic subunit (green) bound to the regulatory subunit (orange) dephosphorylates phosphorylated RLC. The protein structures are discussed with references in the text.
Ca2+ released to the sarcomeres may also activate Ca2+/calmodulin-dependent skMLCK that phosphorylates RLC [4]. RLC phosphorylation has no significant effect on actin-activated ATPase activity of purified myosin, but promotes movement of the myosin head out of the off-state in sarcomeres, resulting in modulation of Ca2+/troponin-dependent force generation. This review will focus on the biochemical properties of skMLCK important for understanding its role in physiological contractions of skeletal muscle in addition to the role of RLC phosphorylation.
Biochemical Properties of MLCK
Structural properties
MLCKs belong to the family of Ca2+/calmodulin-dependent protein kinases with four distinct MYLK genes expressing tissue and substrate specific kinases [5, 6]. The MYLK2 gene expresses skMLCK specifically in skeletal muscles with greater amounts in fast skeletal muscles compared to slow muscles [4, 7–9]. SkMLCK is a monomer containing an N-terminal sequence with no known function followed by a prototypical protein kinase catalytic core and a regulatory segment containing an autoinhibitory inhibitory sequence and a calmodulin binding sequence (Figs. 1 and 2). SkMLCK is expressed in different sizes in different animal species due to variations in the size of the N-terminus. Although no three-dimensional structure exists for skMLCK, based on x-ray scattering measurements and sequence similarities with other protein kinases, including Ca2+/calmodulin-dependent protein kinases, it shares the same bi-lobed structure for the catalytic core (Fig. 2)[10–13]. Results from protein fragmentation complementation analyses indicate that the principal autoinhibitory motif is contained within the sequence between the catalytic core and the calmodulin-binding sequence consistent with previous results obtained with truncation mutants [14, 15]. The autoinhibitory sequence makes an extensive network of contacts on the surface of the larger C-domain of the catalytic core extending towards the catalytic cleft with the calmodulin-binding sequence to block RLC, but not ATP binding in the catalytic core N-domain (Fig. 2) [16, 17]. In the absence of Ca2+, the kinase is auto inhibited and calmodulin is not bound. The structure of the auto inhibited skMLCK is predicted to be similar in principle with other related kinases [12, 13]. When skMLCK is activated by Ca2+/calmodulin, the regulatory segment is displaced from the catalytic cleft allowing the phosphorylatable Ser15 of RLC to bind to residues within the cleft with the remainder of the N-terminus traversing through to solvent [18]. Thus, skMLCK has an intrasteric regulatory mechanism controlling its activity [19].
Fig. 2.
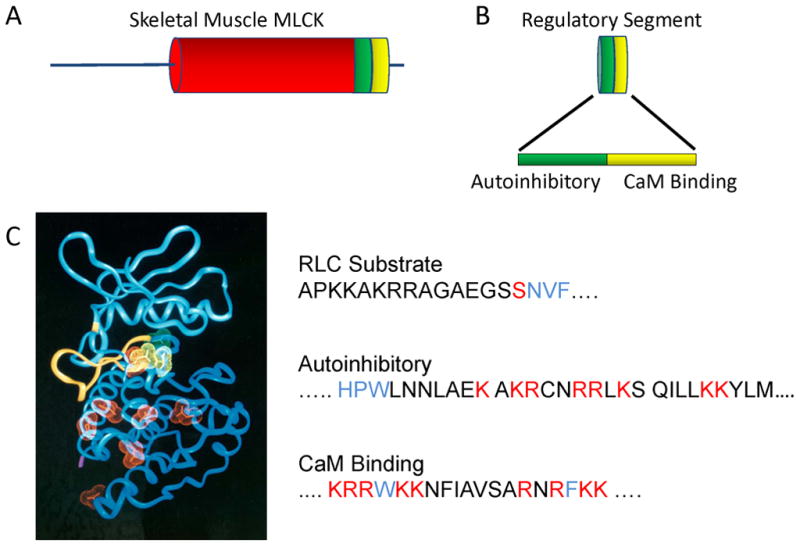
Structural elements important for skMLCK. (A) SkMLCK has an N-terminal sequence followed by a catalytic core (red) and regulatory segment (green, yellow). (B) The regulatory segment has an autoinhibitory sequence immediately C-terminal of the catalytic core followed by the calmodulin-binding sequence. (C) Left panel: Acidic residues (red and yellow) predicted to be on the surface of the modeled catalytic core that may bind to the autoinhibitory sequence with yellow residues important to position RLC for phosphorylation. The N-terminus of RLC extends through the catalytic cleft binding to sites in the N-domain of the catalytic core (yellow ribbon). Right panel: The primary substrate determinants in the N-terminus of skeletal muscle RLC are shown in blue relative to the phosphorylatable Ser (red). The autoinhibitory sequence of skMLCK extends from the catalytic core (blue residues) with basic residues (red) binding to acidic residues on the surface of the catalytic core. Ca2+/calmodulin collapses around the calmodulin-binding sequence containing two hydrophobic residues (blue) with additional ionic interactions with basic residues (red).
Activation by Ca2+/calmodulin
Calmodulin is a highly conserved, ubiquitous Ca2+-binding protein that is composed of N- and C-terminal domains tethered by a highly flexible helical linker sequence [20]. Each domain contains a pair of EF hand motifs that bind Ca2+ (Fig. 1). When saturated with four Ca2+, calmodulin obtains a conformation that permits it to bind to a markedly diverse set of target enzymes, including skMLCK [21]. Binding to target proteins increases its affinity for Ca2+ due to changes in energy coupling [22, 23]. The association of Ca2+/calmodulin with skMLCK is diffusion limited and rapid (10−7M−1 s−1) resulting in activation, while the rate of dissociation of Ca2+/calmodulin is slow (0.03 s−1) resulting in high affinity binding (KCaM = 1 nM) [24, 25]. However, EGTA decreases bound Ca2+ and increases the rate of calmodulin dissociation. These rates are particularly relevant in skeletal muscle physiological responses (see below).
The high-resolution structure of calmodulin bound to a peptide representing the calmodulin-binding sequence of skMLCK illustrates the structural roles of basic and hydrophobic residues in the peptide for high-affinity binding to calmodulin [26]. When calmodulin binds to the calmodulin-binding peptide, it undergoes a large conformational change in which the central helix of calmodulin is bent and twisted bringing the two domains of calmodulin together. The N- and C-terminal domains of calmodulin are brought into close apposition forming a tunnel that engulfs the calmodulin-binding peptide as a single, long α-helix in an antiparallel orientation, i.e., the N-terminal domain of calmodulin binds to the C-terminal end of the calmodulin-binding peptide and the C-terminal domain binds to the peptide N-terminus (Fig. 1). Two hydrophobic residues in the calmodulin-binding sequence (Trp and Phe) separated by 12 intervening residues form important anchor points of interaction (Fig. 2C). One side of the tunnel is comprised of a hydrophobic surface that interfaces with the hydrophobic side of the α-helical peptide. The basic residues within the calmodulin-binding peptide form salt bridges with calmodulin. Additionally, the boundaries at the ends of the peptide are flanked by electrostatic and hydrogen bonds.
Kinetic studies with separated N- and C-terminal domains of calmodulin suggest calmodulin binding to skMLCK is an ordered event with the binding of the C-terminal domain of calmodulin preceding binding of the N-terminal domain [27]. The function of the central helix of calmodulin is to act as a flexible tether that increases the effective concentration of the N-terminal domain, which favors its binding to the C-terminal half of the calmodulin binding sequence and activation of the kinase. These results are consistent with a proposed order of binding for the calmodulin domains based upon the three-dimensional structure of Ca2+/calmodulin-dependent protein kinase I [28]. The N-terminal Trp in the calmodulin-binding sequence protrudes from the kinase with its side chain pointing into solvent, putting it in a position that is readily accessible for binding the C-terminal domain of calmodulin. In contrast, the C-terminus of the calmodulin-binding sequence is buried near the cleft of the two domains of the catalytic core and may not be available initially for calmodulin binding. However, ensuing calmodulin interactions with the skMLCK catalytic core per se appear to contribute to activation [29]. The regulatory segment is subsequently displaced from the catalytic site with calmodulin collapsed at a position near the base but adjacent to the catalytic core (Fig. 1) [10, 30, 31]. The exposed catalytic site of the kinase allows the N-terminus of RLC to enter, followed by closure of the cleft and transfer of phosphate from ATP to RLC.
Catalysis
The folded core structure of the RLC on a myosin heavy chain fragment is observed in the crystal structure of skeletal muscle myosin, but the first 20 residues of the N-terminal domain are too disordered to be visible [2]. The phosphorylatable Ser15 in this disordered N-terminus of skeletal muscle RLC is accessible to skMLCK. Mutational studies in smooth muscle RLC showed that the region encompassing Lys11–Arg16 was important in stabilizing ionic interactions between the myosin heads in the off state and phosphorylation of Ser19 disrupts these multiple weak and predominantly ionic interactions [32]. Presumably, similar but weaker interactions occur for skeletal muscle myosin [3].
Synthetic peptides and mutant light chains have been used to examine the residues that contribute to substrate specificity. The skMLCK phosphorylates RLCs from skeletal, cardiac and smooth muscles with similar catalytic properties (Km, Vmax)[9]. Although not as much is known regarding the details of substrate recognition by skMLCK compared to smooth muscle MLCK, their catalytic properties are distinct [4, 9, 33, 34]. The sequences around the phosphorylatable Ser for different RLCs include: …KKRPQRAT(P)SNVF…, smooth muscle; …KRRTVEGGS(P)SNVF…, fast skeletal muscle; … KKRAGG-AN(P)SNVF…slow skeletal/ventricular muscle[35]. The smMLCK phosphorylates smooth muscle RLC biochemically with favorable substrate properties, but skeletal and cardiac RLCs are poor substrates[9]. Arg16 in smooth muscle RLC is a primary determinant for phosphorylation of Ser19 where Arg16 is proposed to bind to two specific Glu residues at the cleft in the kinase catalytic core [36, 37]. This recognition determinant in smooth muscle RLC is not retained in fast skeletal or slow skeletal/ventricular RLCs which may account for their poor substrate properties with smMLCK. However skMLCK phosphorylates all three RLCs with similar kinetic properties, indicating its substrate recognition determinants are distinct. The sequence GlnValPhe immediately C-terminal of both skeletal and smooth muscle RLCs and additional unidentified structures in the N-terminal half of RLCs are important for substrate recognition by both smooth and skeletal muscle MLCKs. Although skMLCK is a dedicated kinase that appears to only phosphorylate RLCs, it is not selective in regards to phosphorylating fast or slow skeletal muscle RLCs.
Under steady state conditions skMLCK follows a rapid-equilibrium random bi-bi reaction (two substrates converted to two products) whereas the smooth muscle MLCK proceeds by an ordered sequential mechanism in which MgATP binds first, followed by the RLC substrate [24, 38, 39]. Stopped-flow, rapid kinetic experiments show activation occurs at a rate indistinguishable from the binding of the Ca2+/calmodulin to skMLCK [24].
RLC Phosphorylation in vivo
Activation of skMLCK by Ca2+/calmodulin in skeletal muscle fibers
Early physiological observations showed that the extent of RLC phosphorylation depended on the frequency of muscle stimulation with phosphorylation increasing from 0–10% to 50–60% [7, 40–42]. When previously quiescent fast-twitch skeletal muscle is stimulated at a high frequency for 1 s to produce a single, sustained contraction (tetanus), RLC phosphorylation increases at an initial rate consistent with maximal activation of all skMLCK in muscle fibers [40, 42]. However, the rate of phosphorylation is slower than the contraction time, with little phosphorylation occurring during the initial second of stimulation. A transgene for a biosensor skMLCK consisting of skMLCK with an additional calmodulin-binding sequence flanked by enhanced yellow fluorescent protein and enhanced cyan fluorescent protein was expressed specifically in mouse skeletal muscles [43]. Maximal activation and inactivation rates were determined for the biosensor skMLCK in relation to tetanic force development and relaxation (Fig. 3). SkMLCK activation occurred rapidly with no apparent latency relative to force development, consistent with biochemical rapid kinetic measurements showing a diffusion controlled, bimolecular association without an apparent conformational latency [24]. The activation rate was 11 s−1 which was only four-fold slower than the contraction rate of 38 s−1. Computer modeling based on biochemical Ca2+ binding properties suggests that Ca2+ binds to troponin in the actin thin filament and calmodulin at a similar rate [44]. Thus, skeletal muscle force development increases at a faster rate than calmodulin binding to skMLCK because excitation-contraction coupling events distal to Ca2+ binding to troponin occur more rapidly than the formation of the Ca2+/calmodulin/skMLCK complex. Studies with the biosensor skMLCK indicate that Ca2+, not calmodulin availability is a limiting factor for kinase activation, in contrast to conclusions obtained in smooth muscle where calmodulin is limiting [45, 46].
Fig. 3.
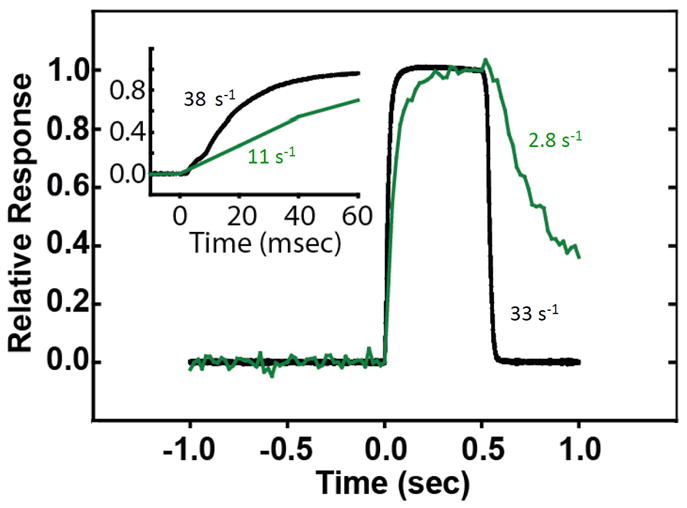
SkMLCK is rapidly activated (green) by Ca2+/calmodulin in skeletal muscle stimulated to develop force (black) but inactivated more slowly relative to relaxation [43]. The slower inactivation rate provides a biochemical memory effect for RLC phosphorylation.
The rate of dissociation of Ca2+ from calmodulin bound to purified skMLCK is slow, resulting in dissociation and inactivation at 3 s−1 [25]. This biochemical dissociation rate is similar to the rate measured in vivo with the biosensor skMLCK [43]. Importantly, this slow rate of CaM dissociation from skMLCK allows for a longer period of skMLCK activity after myoplasmic Ca2+ concentration returns to values low enough for relaxation. Thus, RLC phosphorylation continues for several seconds after relaxation from a brief tetanic contraction, but the rate of phosphorylation slows because the kinase is being inactivated during this time [40–42]. This slow rate of inactivation provides a biochemical memory to enhance RLC phosphorylation after skeletal muscle fibers have relaxed (Fig. 4).
Fig. 4.

Biochemical determinants for physiological phosphorylation of RLC in fast skeletal muscle. A brief tetanic contraction results in RLC phosphorylation related to the amount of fully activated skMLCK in muscle fibers, with slow inactivation due to the slow dissociation of Ca2+/calmodulin from the kinase after relaxation. RLC phosphorylation persists for some time after the skMLCK is inactivated due to low phosphatase activity. The insert shows isometric twitch force responses without (black) and with (green) RLC phosphorylation.
Relative kinase and phosphatase activities
Almost all protein phosphatase activity towards RLC is protein phosphatase type 1[47, 48]. The smooth muscle myosin light chain phosphatase has been extensively characterized biochemically and physiologically and is composed of PP1c in complex with a distinct regulatory subunit, myosin phosphatase target subunit (MYPT1) and another small subunit (M20)[47, 49, 50]. The smooth muscle light chain phosphatase activity is regulated by phosphorylation of MYPT1 as well as a phosphorylated inhibitor protein (CPI-17). Another member of the MYPT family, MYPT2, is expressed preferentially in heart, skeletal muscle and brain where it is in a complex with PP1cδ in striated muscles[47]. There is a transition in expression from MYPT1 to increased expression of MYPT2 with differentiation of C2C12 cells into myotubes in culture[51].
Several biochemical features of MYPT2 and MYPT1 are similar and include: a specific interaction with PP1cδ; interaction of MYPT2 with the small heart-specific myosin phosphatase subunit; interaction of the C-terminal region of MYPT2 with the active form of RhoA; and phosphorylation by Rho-kinase at an inhibitory site that inhibits myosin light chain phosphatase[52]. MYPT2 activates PP1cδ activity, although the extent of activation is less than MYPT1 activation. PP1cδ binds to a RVXF motif in MYPT1 and forms secondary interactions with the N-terminal sequence, some of the ankyrin repeats and with a site within the sequence 304–501[53]. An acidic patch (326–372) may be involved in activation of phosphatase activity towards phosphorylated RLC[54]. The crystal structure of PP1cδ and an N-terminal fragment of MYPT1 shows these hierarchical interactions extend the catalytic groove of PP1c and possibly modify its catalytic properties[55]. Some of these structural features may be similar with MYPT2.
Cardiac myosin is clearly a substrate for the MYPT2-containing myosin light chain phosphatase in vivo [52, 56], but it is not clear that regulation occurs by MYPT2 phosphorylation. RLC is dephosphorylated about 100 times more slowly in cardiac muscle compared to skeletal muscle[41, 42, 57, 58], and skeletal muscle RLC dephosphorylation is about 25 times slower than smooth muscle RLC dephosphorylation[41, 42, 59]. Thus, RLC phosphorylation-dephosphorylation is a more dynamic system in smooth muscles where RLC phosphorylation initiates contraction compared to striated muscles where RLC phosphorylation modulates contraction[8, 56, 59, 60]. In intact fast-twitch skeletal muscle fibers the maximal rate of RLC phosphorylation is 50-fold faster than the rate of dephosphorylation which is an important physiological property related to the function of RLC phosphorylation [41, 42].
Repetitive muscle activity
At low but constant frequencies of stimulation (0.5–10 Hz) that elicit rapid but transient contractions referred to as twitches, RLC is phosphorylated in a time- and frequency-dependent manner even though the contractions are not fused [42, 61]. The rate and extent of biosensor skMLCK activation is proportional to the frequency of muscle contractions from 0.2 to 50 Hz. Rapid increases in Ca2+ concentration during a single twitch are not sufficient to activate skMLCK maximally. In the period between Ca2+ transients at some low frequency, the fraction of activated kinase declines at a slow rate because of the slow rate of calmodulin dissociation from skMLCK. If another twitch is elicited before complete skMLCK inactivation, an additional fraction of the kinase is activated, resulting in an increase in the total active skMLCK. Thus, a maintained high concentration of Ca2+ such as that found with a tetanus produced at a high frequency of stimulation is not necessary for RLC phosphorylation. Equilibrium is established between the rates of activation and inactivation of skMLCK with continuous stimulation at a particular frequency. The biochemical memory related to slow skMLCK inactivation results in a longer period of kinase activity between Ca2+ transients at low contraction frequencies which are sufficient to activate only a small fraction of skMLCK.
In summary, the biochemical properties of skMLCK and MLCP show 1) skMLCK is rapidly activated by Ca2+/calmodulin and the interpulse interval between contractions determines the fraction of skMLCK activated, 2) the activity of activated skMLCK is limiting so that RLC is phosphorylated in secs, not msec with contraction and 3) skMLCK activity is much greater than MLCP activity so a small fractional activation of kinase results in RLC phosphorylation that may be sustained (Fig. 4) [42, 43, 60].
RLC phosphorylation in different skeletal muscle fiber types
Investigations in mouse [62], rabbit [63] and rat [42] skeletal muscle all show a greater effect of repetitive contractions on the extent of RLC phosphorylation in fast-twitch versus slow-twitch muscle. When detectable, stimulation-induced RLC phosphorylation in slow-twitch muscle required higher frequencies and longer stimulation times than did fast-twitch muscle, conditions that typically lead to fatigue [42, 62, 63]. Reduced RLC phosphorylation may be due to lower skMLCK and greater MLCP activities, respectively, in slow-twitch muscle [42]. Calmodulin could also be limiting for skMLCK activation in slow twitch fibers like it is for smooth muscle [45].
Physiological Function of RLC Phosphorylation
Evidence from a variety of muscle models suggests that phosphorylation of myosin RLC by the skMLCK enzyme alters myosin motor function within the myofilaments of striated skeletal muscle. This molecular mechanism increases the Ca2+ sensitivity of the contractile apparatus, an effect that may enhance basic mechanical properties to affect dynamic aspects of muscle force, work and power [60, 64]. Complex interactions between RLC phosphorylation and muscle energetics, as well as to metabolism-related alterations in cross bridge function and/or muscle activation, may modulate muscle function during fatigue.
Molecular mechanism: results from permeabilized muscle fibers
Persechini et al. [65]used membrane permeabilized skeletal muscle fibers to show that the addition of skMLCK and corresponding phosphorylation of the RLC increased the sensitivity of the contractile proteins to Ca2+ activation. For example, an increase in phosphorylation from 0.10 to 0.80 mol phosphate per mole RLC increased the steady-state force response of rabbit psoas skeletal muscle fibers at 0.60 μM, but not 10 μM Ca2+. Subsequent studies using a variety of permeabilized skeletal muscle fiber models confirmed that by increasing the force responses at submaximal, but not maximal Ca2+ activations, RLC phosphorylation shifted the force-pCa response to the left [65–69]. Moreover, the increase in Ca2+ sensitivity mediated by RLC phosphorylation in myofilaments was proportional to actomyosin ATPase activity, suggesting that the molecular mechanism for this effect involved an increase in the number of cross-bridges able to cycle against the thin filament, and not to any increase in the force per cycling cross-bridge per se [67]. RLC phosphorylation has no effect on maximum velocity of shortening (Vmax) of skeletal muscle fibers [65, 69].
In structural terms, the ability of RLC phosphorylation to enhance myosin motor function has been described in studies using thick filaments extracted from a variety of vertebrate striated muscle types. In general, the addition of a negatively charged phosphate moiety disorders myosin heads positioned on the thick filament [70–72]. This may be due to charge repulsion between the RLC binding domain and the thick filament, resulting in disruption of an off-state orientation of the myosin heads with displacement of the actin-binding domain axially toward the thin filament [3, 73–76]. During relaxation of smooth muscle, dephosphorylated cross bridges are in an “off” state due to head-head interactions [3]. RLC phosphorylation disrupts the multiple weak, predominantly ionic interactions to release the cross bridges to bind to actin. The heads of striated muscle myosins can undergo similar but much weaker interactions to produce an ordered array in relaxed muscle with unphosphorylated RLC. The head-head interaction does not switch activity off, but may be a resting position where the head-head interaction is weak, and thus, does not inhibit myosin function [77]. Phosphorylation of striated RLC increases the mobility of myosin cross bridges such that they move away from the thick filament surface towards actin thin filaments in skeletal and cardiac fibers. The relationship between RLC phosphorylation-induced alterations in myosin structure and crossbridge function is illustrated in Fig. 5. At the cross bridge level, myosin phosphorylation may modulate the Ca2+-controlled transition from non-force to force-generating states, i.e. the rate constant fapp, that regulates muscle force development whereas the transition of force-generating to non-force generating states, i.e. the rate constant gapp, is unaffected by myosin phosphorylation [67, 78]. In terms of cross bridge models, fapp may correspond to the release of the ATP hydrolysis product inorganic phosphate that triggers the cross bridge power stroke [79, 80]. On the other hand, the gapp term may correspond to ADP release and/or ATP rebinding steps subsequent to the power stroke [81]. Myosin phosphorylation-mediated increases in fapp increases the force output at low levels of Ca2+ activation because the fraction of cross bridges in the force-generating state is low (i.e. αFs = fapp/(fapp + gapp)) [67, 78]. On the other hand, a myosin phosphorylation-mediated increase in fapp has little effect on force at high levels of Ca2+ activation because the fraction of cross bridges in the force-generating state is already high. The inability of myosin phosphorylation to influence Vmax is consistent with the lack of effect this mechanism has on gapp [67, 78, 81]. Thus, results from permeabilized skeletal fiber models have provided important mechanistic information towards understanding how myosin phosphorylation modulates intact skeletal muscle function [60].
Fig. 5.
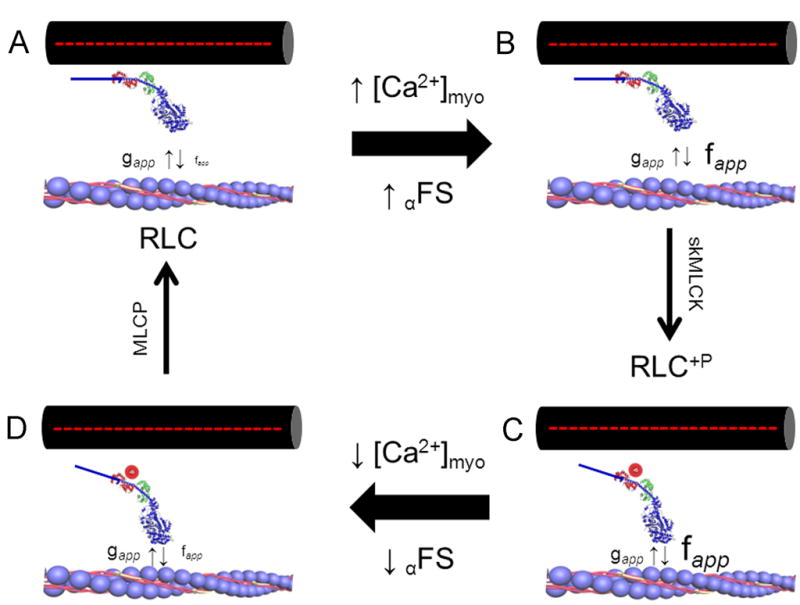
RLC phosphorylation affects myosin cross bridge structure and function during Ca2+ activation of skeletal muscle contraction. In this model, the regulatory influence of Ca2+ on force development is exerted via changes in the rate constant describing the transition of cycling cross bridges from non-force to force-generating states (i.e. fapp). An increase in myoplasmic [Ca2+] ([Ca2+]myo) and concomitant thin filament activation increases force by regulating the fraction of cycling crossbridges able to accumulate in force-generating states (αFS). The addition of a negatively charged phosphate to Ser15 in RLC (red circle) displaces the myosin motor domain laterally from the thick filament towards thin filament binding sites, which affects Ca2+ control of cross bridge kinetics. Clockwise from top left: (A) unphosphorylated cross bridge at rest; when myoplasmic [Ca2+] is low the transition of strong to weak binding states (i.e. gapp) dominates and force is low; (B) unphosphorylated cross bridge during contraction in response to elevated myoplasmic [Ca2+] showing Ca2+ regulated increase in fapp; gapp is not influenced; (C) phosphorylated crossbridge during contraction; the modulation of fapp further increases the fraction of cross bridges able to attain force generating states to increase the Ca2+ sensitivity of force development relative to unphosphorylated state; (D) phosphorylated crossbridge during relaxation; the return of myoplasmic [Ca2+] to low levels may be accompanied by a small residual increase in fapp to slow the rate of relaxation from contraction. Small upward and downward vertical arrows denote the terms fapp and gapp, respectively. MLCP, myosin light chain phosphatase. Based on references [67, 78]
Modulation of intact skeletal muscle contractile properties
The same calcium signal that initiates force development also regulates skMLCK activity, and provides a mechanism that serves as a biochemical memory to facilitate or enhance muscle mechanical function during prolonged or repetitive activity. Since the first demonstration that the RLC of vertebrate striated muscle was phosphorylatable [82], many studies on animal fast-twitch skeletal muscle have shown that this molecular event is temporally correlated with the magnitude of post-tetanic potentiation (PTP), defined as the transient increase in isometric twitch force amplitude following tetanic muscle stimulation [7, 8, 40–43, 61, 83–92]. Direct experimental evidence that skMLCK- mediated phosphorylation of the RLC is mechanistically related to force potentiation comes from studies using muscles devoid of skMLCK that do not display stimulation-induced RLC phosphorylation. For example, Zhi et al. [8]used extensor digitorum longus (EDL) muscles from skMLCK knockout mice to show that tetanic stimulation did not increase RLC phosphorylation and did not potentiate isometric twitch force in contrast to responses in muscles from wild-type mice. Although ablation of skMLCK did not completely inhibit staircase potentiation, i.e. the progressive increase in twitch force during low frequency stimulation, this was attenuated by ~ 50%. Additionally, overexpression of skMLCK enhanced RLC phosphorylation and staircase potentiation [43]. While the primary molecular mechanism for PTP is skMLCK-mediated phosphorylation of RLC, redundant mechanisms, perhaps involving Ca2+ homeostasis, may contribute to staircase potentiation [87, 93].
The functional relationship between RLC phosphorylation and isometric twitch force potentiation of isolated rodent muscle is variable, depending critically upon experimental factors of muscle temperature, length and contraction type. For example, the force potentiation observed in mouse EDL muscle is extremely sensitive to temperature, with the magnitude of PTP increasing by a factor of ~ 2 as muscle temperature is increased in the range 25 – 35° C [88]. Indeed, this temperature effect also accounts for why the magnitude of PTP reported for rat hind limb muscle studied in situ is greater than that reported for mouse muscle studied at lower temperatures in vitro [7, 8, 40–43, 61, 83–92]. Moreover, this temperature response is consistent with permeabilized muscle fiber models showing that the myosin phosphorylation influence on the Ca2+ sensitivity of force development is inversely related to thin filament activation [65, 67–69, 94]. In addition, the magnitude of PTP is length dependent, being decreased and increased at lengths above and below optimal length (Lo), respectively [89–91]. Indeed, this result accords with those from permeabilized skeletal muscle fibers showing that charge replacement on RLC that mimics myosin phosphorylation enhances steady-state force at short, but not long, sarcomere lengths [75, 76]. Because rodent slow-twitch skeletal muscles have a lower amount of skMLCK, they generally have small RLC phosphorylation and PTP responses [40–43]. However, the relationship between RLC phosphorylation and force potentiation phenomena are complex in different muscle fibers. For example, Ryder et al. [43] showed that stimulation-induced increases in RLC phosphorylation (to ~ 0.40 mol phosphate per mol RLC) of soleus muscles isolated from mice overexpressing skMLCK did not display PTP. Because the soleus muscle contains ~ 60 % type IIa skeletal muscle fibers, these data suggest that RLC phosphorylation may selectively enhance contractile properties of type IIb muscle fiber only. The reason for this apparent fiber-type dependence for RLC phosphorylation-mediated force potentiation is presently unclear but may involve actomyosin filament structure or excitation-contraction coupling differences.
Modulation of basic contractile properties by RLC phosphorylation may enhance mechanical function of working skeletal muscle in a number of ways. For example, in addition to an increase in maximal twitch force, an increase in Ca2+ sensitivity by RLC phosphorylation increases the rate of force development. As an example, the effect of stimulation-induced elevations in myosin phosphorylation on isometric twitch force is shown in Figure 6A. This force record reveals that, in addition to increased amplitude, the rate of isometric force development is also enhanced. In contrast, stimulation of muscles lacking skMLCK does not enhance either the rate or extent of isometric twitch force development (Figure 6B). At higher frequencies of stimulation, RLC phosphorylation increases the rate of tetanic force development without increasing maximal tetanic force [84, 85]. Thus, although the potentiation of maximal isometric force is limited to low frequencies of stimulation [92], RLC phosphorylation mediated increases in the rate of force development may enhance peak force levels attained during very brief (i.e. < 200 ms) tetanic activations as may occur during locomotion. Indeed, more recent permeabilized skeletal muscle fiber work indicates that the effect of RLC phosphorylation on the rate of force development may be enhanced at higher Ca2+ activation levels, as occurs during tetanic stimulation [95]. The influence of RLC phosphorylation on muscle force kinetics is not limited to force development, however. For example, in addition to altering the rate of rise of force, RLC phosphorylation slows the rate of relaxation of skinned skeletal muscle fibers [96]. In terms of whole skeletal muscle function, feline caudofemoralis muscle displays a delayed tetanic relaxation in the potentiated versus unpotentiated state (at 37° C) [97]. Indeed, consistent with this, EDL muscles from skMLCK KO mice displayed accelerated relaxation from fully fused tetani compared to EDL muscle from WT mice (at 25° C) [98]. These results suggest that, in addition to enhancing peak force in response to suboptimal Ca2+ activation, the RLC phosphorylation mechanism enhances the kinetics of force development at contraction onset and, in addition, slows the rate of force relaxation following high Ca2+ activation.
Fig. 6.
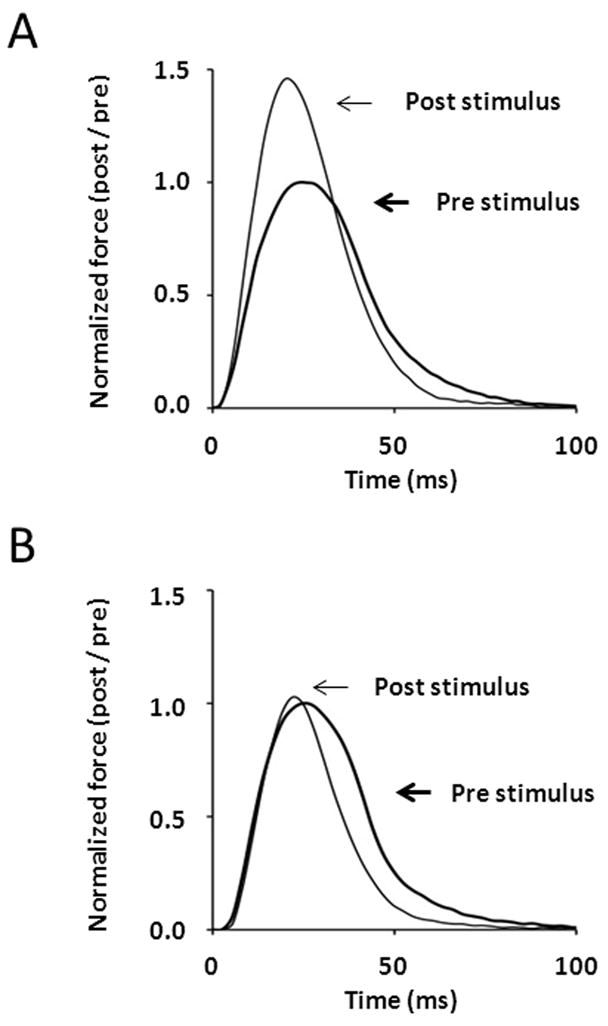
Comparison of isometric twitch responses in EDL muscles from wild-type (A) and skMLCK knockout (B) mice obtained 1 minute before and 15 sec after a potentiating stimulus consisting of four brief trains of 150-Hz stimulation (within 10 sec) that elevated myosin RLC phosphorylation from 0.15 to 0.55 mol phosphate per mol RLC. Note the large (~ 50% post/pre) increase in twitch amplitude of the wild-type muscle relative to the skMLCK knockout muscle. Experiments performed at 25° C with muscles set to 0.90 of optimal length (Lo).
Modulation of dynamic muscle function
The hallmark of skeletal muscle function in vivo is the performance of dynamic mechanical work and power. Although RLC phosphorylation does not appear to enhance Vmax [65, 88, 99], it is associated with enhancements to dynamic aspects of both mouse and rat hindlimb muscle at both low and high-stimulation frequencies [100–105]. The mechanistic explanation for this outcome is an increase in either loaded shortening velocity or shortening displacement following RLC phosphorylation. As an example, the relationship between stimulation-induced increases in myosin RLC phosphorylation and both isometric and concentric twitch force potentiation is shown in Figure 7. Indeed, the apparent sensitizing influence of muscle shortening on concentric twitch force potentiation is consistent with regulatory models incorporating cooperative influences of cycling cross-bridges on thin filament activation [106, 107]. Consistent with previous work from permeabilized and intact muscle models, thin filament deactivation secondary to cross-bridge detachment during shortening could in fact sensitize the contractile proteins to the RLC phosphorylation mechanism. Given this outcome, it seems appealing to believe that activity-dependent increases in RLC phosphorylation could enhance concentric force, work and power during submaximal contractions in vivo.
Fig. 7.
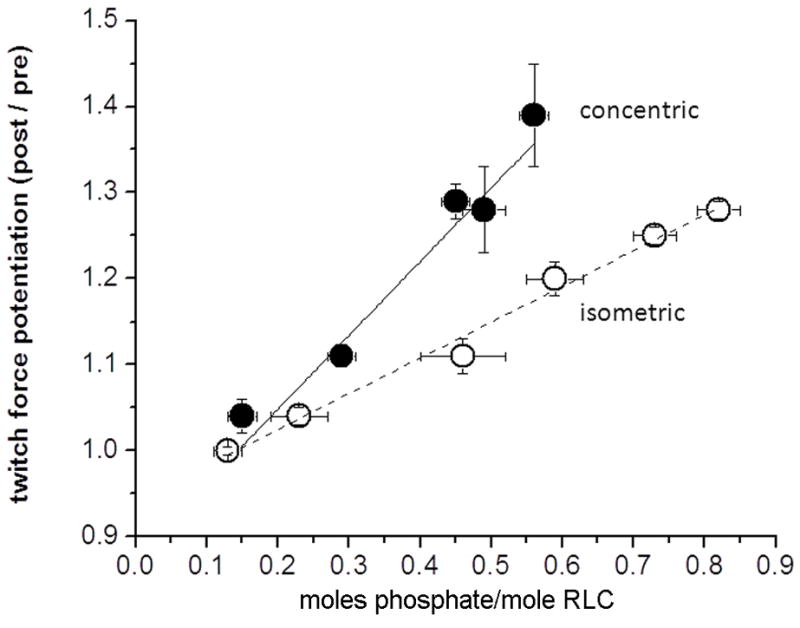
Relation of concentric and isometric twitch force potentiation versus RLC phosphorylation obtained following stimulation at different frequencies (2.5 to 100 Hz). Each data set fitted with linear regression. Concentric (closed circles) data are unpublished; isometric (open circles) data re-plotted from Vandenboom et al.[85]. Note that despite similar y-intercepts, the concentric force – phosphorylation relationship is steeper than is the isometric force - phosphorylation relationship. Data are means ± SEM (n=4–8 muscles each time point).
Interaction with fatigue
Muscle fatigue, defined as the reversible decline in muscle force, work and power, is the inevitable outcome of prolonged muscle activity [108]. A well known characteristic of fast-twitch skeletal muscle is, however, the coexistence of twitch force potentiation with declines in tetanic force during fatigue [109]. Because fatigue does not interfere with the ability to phosphorylate the RLC [110], it seems feasible that this mechanism enhances low frequency muscle force, work and/or power to help preserve muscle function under these circumstances. Indeed, when results from muscles from wild-type and skMLCK knockout mice are compared, the absence of RLC phosphorylation is associated with an increased fatigability at low, but not high stimulation frequencies, an effect that is evident until peak tetanic force is depressed by ~ 50% (Figure 8). Similar to previous results, however, muscles lacking skMLCK do display an attenuated degree of twitch potentiation during fatigue [8] to suggest that although RLC phosphorylation may be the primary mechanism, additional processes such as changes in Ca2+ homeostasis may contribute to force potentiation observed during fatigue. It is also possible that low frequency fatigue, i.e. the reduction in muscle force at low, but not high frequencies of stimulation with prolonged impairment of Ca2+ release [108], interacts with the RLC phosphorylation mechanism to enhance the magnitude of twitch force potentiation relative to the unfatigued state [110–113].
Fig. 8.
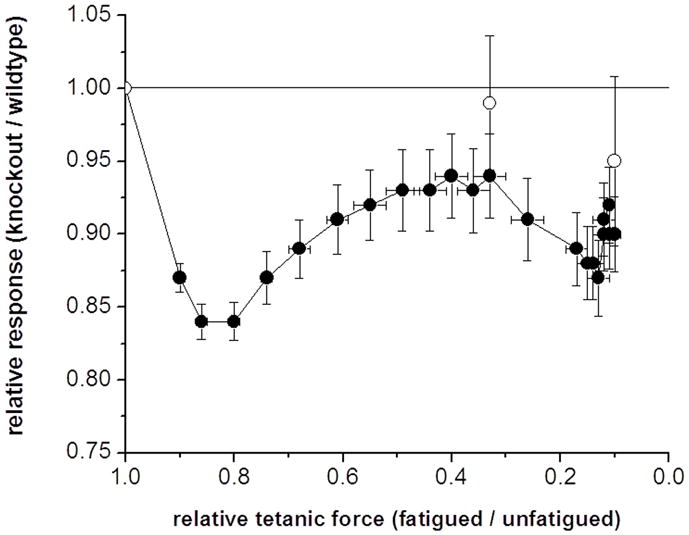
Plot of relative change in low frequency force (Pt) (closed symbols) and unloaded shortening velocity (Vo) (open symbols) measured via the slack test method versus tetanic force decline in muscles from wild-type and skMLCK knockout mice. Muscles were stimulated for 5 minutes during which time Po was reduced to ~ 10% of starting levels in each muscle type. Vo was determined before, during (after 1 minute of stimulation) and immediately after the fatigue run. All data obtained by dividing responses from muscles from skMLCK knockout to wild-type mice and plotted against the corresponding change in tetanic force (the same for muscles from skMLCK knockout and wild-type mice). The Pt of skMLCK KO muscles was reduced more than was the Pt of WT muscles (= 1.00) for all fatigue levels. On the other hand, the Vo of skMLCK KO muscles was similar to the Vo of WT muscles (=1.00) for all levels of fatigue. Data is mean ± SEM (n=12 for each genotype). Muscles were set to 0.90 Lo and subjected to 5 minutes of repetitive high frequency stimulation (150 Hz) (redrawn with permission [98].
Alterations to skeletal muscle function during fatigue are driven by metabolic changes in muscle energetics, substrate depletion and/or metabolic accumulation that singly or in combination, directly influence cross-bridge function [114]. Recent work by Cooke and co-workers suggests that RLC phosphorylation-mediated alterations to the structure and function of the myosin cross-bridge may interact with the metabolic environment of the muscle during fatigue. For example, although shortening velocity of unfatigued muscle is not affected, an RLC phosphorylation mediated increase in the affinity of the myosin motor domain for thin filament binding sites may contribute to the slowing of velocity noted for severely fatigued muscle [115–117]. When results from muscles from wild-type and skMLCK knockout mice are compared, however, the absence of RLC phosphorylation did not alter unloaded shortening velocity of mouse EDL in vitro (25° C) [98]. Moreover, the contraction economy (muscle force/muscle energetic cost) of rat skeletal muscle is reduced in the potentiated compared to non-potentiated state [118]. This outcome may suggest that rather than improve muscle function during prolonged muscle activity, perhaps the energetic cost of RLC-induced increased force (in terms of ATPase) contributes to changes in metabolism that eventually erode muscle performance.
Modulation of human skeletal muscle function
Maximal or submaximal voluntary contractions of human skeletal muscle readily induce myosin RLC phosphorylation and potentiate isometric twitch force amplitude, a response known as post activation potentiation (PAP). Houston and coworkers [119–124] were the first to show RLC phosphorylation in human skeletal muscle was associated with increased low frequency force or torque output. In addition, these studies showed that the magnitude and time course for the appearance of PAP was critically dependent upon contraction duration. For example, a non-fatiguing 10-second maximal voluntary contraction (MVC) that elevated myosin phosphorylation to ~ 0.50 mol phosphate per mol RLC produced an immediate and long lasting PAP that decayed slowly with a time course paralleling myosin dephosphorylation. In contrast, a fatiguing 60-second long MVC delayed the appearance of PAP and blunted its magnitude. Thus, RLC phosphorylation-induced PAP may coexist with fatigue to modulate skeletal muscle performance following muscle activity. Although clear evidence for an effect of PAP on the basic contractile properties of human skeletal muscle has been documented, the functional influence of this modulation on human athletic performance remains equivocal despite extensive study [125, 126]. Reasons for this lack of accord may include large inter-individual differences in fiber type composition and/or fatigue-related complications that mask the influence of PAP on functional performance in vivo.
In addition to interacting with fatigue-processes to modulate mechanical force, it appears that RLC phosphorylation-mediated alterations to basic contractile properties may influence neural strategies for skeletal muscle activation. In principle, PAP-related increases in muscle fiber force could reduce the requirement for fusion/summation processes in recruited motor units. For example, the classic triphasic pattern for motor unit firing rate observed during fatiguing contractions has been associated with the concurrent development of PAP [127]. Moreover, motor unit activities during submaximal contractions have been obtained from human muscle before and after a conditioning contraction that induces PAP. In these experiments, the motor unit firing rates required to achieve the same target force (as % of unfatigued maximum) were reduced by ~ 10% in the potentiated versus unpotentiated muscle (twitch forces increased ~ 200%) [128, 129]. Although the neural feedback mechanism for this putative effect remains to be elucidated, these studies suggest the possibility that the presence of PAP down regulates activation rate perhaps to mitigate the development of either central or peripheral aspects of neuromuscular fatigue.
PAP responses from human skeletal muscle display similarities to PTP response from animal skeletal muscle studies. For example, the magnitude of PAP has been observed to be inversely correlated with the time course of the twitch [130, 131]. PAP has also been correlated with the percent composition and total cross-sectional area of type II muscle fibers [130]. In addition, the magnitude of PAP has been shown to be length-dependent, being greater at 90° (short) than at 135° (long) for dorsiflexion in the tibialis anterior muscle [132]. The magnitude of PAP is also temperature dependent as artificial cooling or heating decreased and increased, respectively, the magnitude of PAP in human dorsiflexor muscles [133]. Thus, although these studies in vivo do not directly associate RLC phosphorylation with downstream changes in muscle function, including PAP, they do provide evidence for an effect of conditioning activity on muscle function consistent with experimental results indicating RLC phosphorylation-mediated increases in Ca2+ sensitivity modulate skeletal muscle function.
In summary, RLC phosphorylation alters myosin motor structure and function to enhance the Ca2+-sensitivity of the contractile apparatus to modulate basic mechanical properties of vertebrate skeletal muscle and enhance dynamic function of working skeletal muscle in the unfatigued or fatigued state. RLC phosphorylation may induce PAP in human skeletal muscle where there are interactions with neural strategies for muscle activation rate to minimize central or peripheral aspects of neuromuscular fatigue.
Acknowledgments
This work was supported by grants (NIH HLR01HL080536, JTS; Natural Sciences and Engineering Research Council of Canada, RV), The Fouad A. and Val Imm Bashour Distinguished Chair and the Moss Heart Fund.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Gordon AM, Homsher E, Regnier M. Physiol Rev. 2000;80:853–924. doi: 10.1152/physrev.2000.80.2.853. [DOI] [PubMed] [Google Scholar]
- 2.Rayment I. J Biol Chem. 1996;271:15850–15853. doi: 10.1074/jbc.271.27.15850. [DOI] [PubMed] [Google Scholar]
- 3.Lowey S, Trybus KM. Journal of Biological Chemistry. 2010;285:16403–16407. doi: 10.1074/jbc.R109.025551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kamm KE, Stull JT. J Biol Chem. 2001;276:4527–4530. doi: 10.1074/jbc.R000028200. [DOI] [PubMed] [Google Scholar]
- 5.Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S. Science. 2002;298:1912–1934. doi: 10.1126/science.1075762. [DOI] [PubMed] [Google Scholar]
- 6.Swulius M, Waxham M. Cellular and Molecular Life Sciences. 2008;65:2637–2657. doi: 10.1007/s00018-008-8086-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Moore RL, Houston ME, Iwamoto GA, Stull JT. J Cell Physiol. 1985;125:301–305. doi: 10.1002/jcp.1041250219. [DOI] [PubMed] [Google Scholar]
- 8.Zhi G, Ryder JW, Huang J, Ding P, Chen Y, Zhao Y, Kamm KE, Stull JT. Proc Natl Acad Sci USA. 2005;102:17519–17524. doi: 10.1073/pnas.0506846102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stull JT, Nunnally MH, Michnoff CH. In: The Enzymes. Krebs EG, Boyer PD, editors. Academic Press; Orlando: 1986. pp. 113–166. [Google Scholar]
- 10.Krueger JK, Olah GA, Rokop SE, Zhi G, Stull JT, Trewhella J. Biochemistry. 1997;36:6017–6023. doi: 10.1021/bi9702703. [DOI] [PubMed] [Google Scholar]
- 11.Knighton DR, Zheng J, Ten Eyck LF, Ashford VA, Xuong N-H, Taylor SS, Sowadski JM. Science. 1991;253:407–413. doi: 10.1126/science.1862342. [DOI] [PubMed] [Google Scholar]
- 12.Kobe B, Heierhorst J, Feil SC, Parker MW, Benian GM, Weiss KR, Kemp BE. EMBO J. 1996;15:6810–6821. [PMC free article] [PubMed] [Google Scholar]
- 13.Mayans O, van der Ven PFM, Wilm M, Mues A, Young P, Furst DO, Wilmanns M, Gautel M. Nature. 1998;395:863–869. doi: 10.1038/27603. [DOI] [PubMed] [Google Scholar]
- 14.Padre RC, Stull JT. J Biol Chem. 2000;275:26665–26673. doi: 10.1074/jbc.M001769200. [DOI] [PubMed] [Google Scholar]
- 15.Padre RC, Stull JT. FEBS Lett. 2000;472:148–152. doi: 10.1016/s0014-5793(00)01434-4. [DOI] [PubMed] [Google Scholar]
- 16.Gallagher PJ, Herring BP, Trafny A, Sowadski J, Stull JT. J Biol Chem. 1993;268:26578–26582. [PMC free article] [PubMed] [Google Scholar]
- 17.Stull JT, Lin PJ, Krueger JK, Trewhella J, Zhi G. Acta Physiol Scand. 1998;164:471–482. doi: 10.1111/j.1365-201x.1998.tb10699.x. [DOI] [PubMed] [Google Scholar]
- 18.Gao ZH, Zhi G, Herring BP, Moomaw C, Deogny L, Slaughter CA, Stull JT. J Biol Chem. 1995;270:10125–10135. doi: 10.1074/jbc.270.17.10125. [DOI] [PubMed] [Google Scholar]
- 19.Krueger JK, Padre RC, Stull JT. J Biol Chem. 1995;270:16848–16853. doi: 10.1074/jbc.270.28.16848. [DOI] [PubMed] [Google Scholar]
- 20.Chin D, Means AR. Trends Cell Biol. 2000;10:322–328. doi: 10.1016/s0962-8924(00)01800-6. [DOI] [PubMed] [Google Scholar]
- 21.Blumenthal DK, Stull JT. Biochemistry. 1980;19:5608–5614. doi: 10.1021/bi00565a023. [DOI] [PubMed] [Google Scholar]
- 22.Persechini A, White HD, Gansz KJ. J Biol Chem. 1996;271:62–67. doi: 10.1074/jbc.271.1.62. [DOI] [PubMed] [Google Scholar]
- 23.Olwin BB, Edelman AM, Krebs EG, Storm DR. Journal of Biological Chemistry. 1984;259:10949–10955. [PubMed] [Google Scholar]
- 24.Bowman BF, Peterson JA, Stull JT. J Biol Chem. 1992;267:5346–5354. [PubMed] [Google Scholar]
- 25.Gallagher PJ, Herring BP, Stull JT. J Muscle Res Cell Motil. 1997;18:1–16. doi: 10.1023/a:1018616814417. [DOI] [PubMed] [Google Scholar]
- 26.Ikura M, Clore G, Gronenborn A, Zhu G, Klee C, Bax A. Science. 1992;256:632–638. doi: 10.1126/science.1585175. [DOI] [PubMed] [Google Scholar]
- 27.Persechini A, McMillan K, Leakey P. Journal of Biological Chemistry. 1994;269:16148–16154. [PubMed] [Google Scholar]
- 28.Goldberg J, Nairn AC, Kuriyan J. Cell. 1996;84:875–887. doi: 10.1016/s0092-8674(00)81066-1. [DOI] [PubMed] [Google Scholar]
- 29.Zhi G, Abdullah SM, Stull JT. J Biol Chem. 1998;273:8951–8957. doi: 10.1074/jbc.273.15.8951. [DOI] [PubMed] [Google Scholar]
- 30.Krueger JK, Gallagher SC, Zhi G, Geguchadze R, Persechini A, Stull JT, Trewhella J. J Biol Chem. 2001;276:4535–4538. doi: 10.1074/jbc.C000857200. [DOI] [PubMed] [Google Scholar]
- 31.Heller WT, Krueger JK, Trewhella J. Biochemistry. 2003;42:10579–10588. doi: 10.1021/bi0348664. [DOI] [PubMed] [Google Scholar]
- 32.Ikebe M, Ikebe R, Kamisoyama H, Reardon S, Schwonek JP, Sanders CR, Matsuura M. Journal of Biological Chemistry. 1994;269:28173–28180. [PubMed] [Google Scholar]
- 33.Kemp BE, Pearson RB. J Biol Chem. 1985;260:3355–3359. [PubMed] [Google Scholar]
- 34.Michnoff CH, Kemp BE, Stull JT. J Biol Chem. 1986;261:8320–8326. [PubMed] [Google Scholar]
- 35.Collins J. J Muscle Research & Cell Motility. 2006;27:69–74. doi: 10.1007/s10974-006-9057-6. [DOI] [PubMed] [Google Scholar]
- 36.Herring BP, Gallagher PJ, Stull JT. J Biol Chem. 1992;267:25945–25950. [PubMed] [Google Scholar]
- 37.Zhi G, Herring BP, Stull JT. J Biol Chem. 1994;269:24723–24727. [PubMed] [Google Scholar]
- 38.Geuss U, Mayr GW, Heilmeyer LMG. European Journal of Biochemistry. 1985;153:327–334. doi: 10.1111/j.1432-1033.1985.tb09305.x. [DOI] [PubMed] [Google Scholar]
- 39.Sobieszek A. European Journal of Biochemistry. 1991;199:735–743. doi: 10.1111/j.1432-1033.1991.tb16178.x. [DOI] [PubMed] [Google Scholar]
- 40.Manning DR, Stull JT. Biochem Biophys Res Commun. 1979;90:164–170. doi: 10.1016/0006-291x(79)91604-8. [DOI] [PubMed] [Google Scholar]
- 41.Manning DR, Stull JT. Am J Physiol. 1982;242:C234–241. doi: 10.1152/ajpcell.1982.242.3.C234. [DOI] [PubMed] [Google Scholar]
- 42.Moore RL, Stull JT. Am J Physiol. 1984;247:C462–471. doi: 10.1152/ajpcell.1984.247.5.C462. [DOI] [PubMed] [Google Scholar]
- 43.Ryder JW, Lau KS, Kamm KE, Stull JT. J Biol Chem. 2007;282:20447–20454. doi: 10.1074/jbc.M702927200. [DOI] [PubMed] [Google Scholar]
- 44.Robertson SP, Johnson JD, Potter JD. Biophys J. 1981;34:559–569. doi: 10.1016/S0006-3495(81)84868-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Isotani E, Zhi G, Lau KS, Huang J, Mizuno Y, Persechin A, Geguchadze R, Kamm KE, JTS Proc Natl Acad Sci USA. 2004;101:6279–6284. doi: 10.1073/pnas.0308742101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ding H-L, Ryder JW, Stull JT, Kamm KE. J Biol Chem. 2009;284:15541–15548. doi: 10.1074/jbc.M900888200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ito M, Nakano T, Erdodi F, Hartshorne DJ. Mol Cell Biochem. 2004;259:197–209. doi: 10.1023/b:mcbi.0000021373.14288.00. [DOI] [PubMed] [Google Scholar]
- 48.Hartshorne DJ, Ito M, Erdodi F. J Biol Chem. 2004;279:37211–37214. doi: 10.1074/jbc.R400018200. [DOI] [PubMed] [Google Scholar]
- 49.Khromov A, Choudhury N, Stevenson AS, Somlyo AV, Eto M. J Biol Chem. 2009;284:21569–21579. doi: 10.1074/jbc.M109.019729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Eto M. J Biol Chem. 2009;284:35273–35277. doi: 10.1074/jbc.R109.059972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wu Y, Erdodi F, Muranyi A, Nullmeyer KD, Lynch R, Hartshorne D. J Muscle Res Cell Motil. 2003;24:499–511. doi: 10.1023/b:jure.0000009810.36038.53. [DOI] [PubMed] [Google Scholar]
- 52.Okamoto R, Kato T, Mizoguchi A, Takahashi N, Nakakuki T, Mizutani H, Isaka N, Imanaka-Yoshida K, Kaibuchi K, Lu Z, Mabuchi K, Tao T, Hartshorne DJ, Nakano T, Ito M. Cellular Signalling. 2006;18:1408–1416. doi: 10.1016/j.cellsig.2005.11.001. [DOI] [PubMed] [Google Scholar]
- 53.Tóth A, Kiss E, Herberg FW, Gergely P, Hartshorne DJ, Erdödi F. European Journal of Biochemistry. 2000;267:1687–1697. doi: 10.1046/j.1432-1327.2000.01158.x. [DOI] [PubMed] [Google Scholar]
- 54.Hirano K, Phan BC, Hartshorne DJ. Journal of Biological Chemistry. 1997;272:3683–3688. doi: 10.1074/jbc.272.6.3683. [DOI] [PubMed] [Google Scholar]
- 55.Terrak M, Kerff F, Langsetmo K, Tao T, Dominguez R. Nature. 2004;429:780–784. doi: 10.1038/nature02582. [DOI] [PubMed] [Google Scholar]
- 56.Mizutani H, Okamoto R, Moriki N, Konishi K, Taniguchi M, Fujita S, Dohi K, Onishi K, Suzuki N, Satoh S, Makino N, Itoh T, Hartshorne DJ, Ito M. Circulation Journal. 2010;74:120–128. doi: 10.1253/circj.cj-09-0462. [DOI] [PubMed] [Google Scholar]
- 57.Herring BP, England PJ. Biochem J. 1986;240:205–214. doi: 10.1042/bj2400205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.High CW, Stull JT. Am J Physiol. 1980;239:H756–764. doi: 10.1152/ajpheart.1980.239.6.H756. [DOI] [PubMed] [Google Scholar]
- 59.Ding P, Huang J, Battiprolu PK, Hill JA, Kamm KE, Stull JT. Journal of Biological Chemistry. 2010;285:40819–40829. doi: 10.1074/jbc.M110.160499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sweeney HL, Bowman BF, Stull JT. Am J Physiol. 1993;264:C1085–1095. doi: 10.1152/ajpcell.1993.264.5.C1085. [DOI] [PubMed] [Google Scholar]
- 61.Klug GA, Botterman BR, Stull JT. J Biol Chem. 1982;257:4688–4690. [PubMed] [Google Scholar]
- 62.Crow MT, Kushmerick MJ. J Biol Chem. 1982;257:2121–2124. [PubMed] [Google Scholar]
- 63.Westwood SA, Hudlicka O, Perry SV. Biochem J. 1984;218:841–847. doi: 10.1042/bj2180841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Grange RW, Vandenboom R, Houston ME. Can J Appl Physiol. 1993;18:229–242. doi: 10.1139/h93-020. [DOI] [PubMed] [Google Scholar]
- 65.Persechini A, Stull JT, Cooke R. J Biol Chem. 1985;260:7951–7954. [PubMed] [Google Scholar]
- 66.Metzger JM, Greaser ML, Moss RL. J Gen Physiol. 1989;93:855–883. doi: 10.1085/jgp.93.5.855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sweeney HL, Stull JT. Proc Natl Acad Sci USA. 1990;87:414–418. doi: 10.1073/pnas.87.1.414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sweeney HL, Kushmerick MJ. AJP - Cell Physiology. 1985;249:C362–365. doi: 10.1152/ajpcell.1985.249.3.C362. [DOI] [PubMed] [Google Scholar]
- 69.Sweeney HL, Stull JT. Am J Physiol. 1986;250:C657–660. doi: 10.1152/ajpcell.1986.250.4.C657. [DOI] [PubMed] [Google Scholar]
- 70.Craig R, Padrón R, Kendrick-Jones J. The Journal of Cell Biology. 1987;105:1319–1327. doi: 10.1083/jcb.105.3.1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Padron R, Pante N, Sosa H, Kendrick-Jones J. J Muscle Res Cell Motil. 1991;12:235–241. doi: 10.1007/BF01745112. [DOI] [PubMed] [Google Scholar]
- 72.Ritz-Gold CJ, Cooke R, Blumenthal DK, Stull JT. Biochem Biophys Res Commun. 1980;93:209–214. doi: 10.1016/s0006-291x(80)80267-1. [DOI] [PubMed] [Google Scholar]
- 73.Levine RJ, Kensler RW, Yang Z, Stull JT, Sweeney HL. Biophys J. 1996;71:898–907. doi: 10.1016/S0006-3495(96)79293-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Sweeney HL, Yang Z, Zhi G, Stull JT, Trybus KM. Proc Natl Acad Sci USA. 1994;91:1490–1494. doi: 10.1073/pnas.91.4.1490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Levine RJ, Yang Z, Epstein ND, Fananapazir L, Stull JT, Sweeney HL. J Struct Biol. 1998;122:149–161. doi: 10.1006/jsbi.1998.3980. [DOI] [PubMed] [Google Scholar]
- 76.Yang Z, Stull JT, Levine RJC, Sweeney HL. J Struc Biol. 1998;122:139–148. doi: 10.1006/jsbi.1998.3979. [DOI] [PubMed] [Google Scholar]
- 77.Zoghbi ME, Woodhead JL, Moss RL, Craig R. Proc Natl Acad Sci U S A. 2008;105:2386–2390. doi: 10.1073/pnas.0708912105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Brenner B. Proceedings of the National Academy of Sciences of the United States of America. 1988;85:3265–3269. doi: 10.1073/pnas.85.9.3265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Metzger JM, Moss RL. Science. 1990;247:1088–1090. doi: 10.1126/science.2309121. [DOI] [PubMed] [Google Scholar]
- 80.Millar NC, Homsher E. Journal of Biological Chemistry. 1990;265:20234–20240. [PubMed] [Google Scholar]
- 81.Siemankowski RF, Wiseman MO, White HD. Proceedings of the National Academy of Sciences of the United States of America. 1985;82:658–662. doi: 10.1073/pnas.82.3.658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Perrie WT, Smillie LB, Perry SV. Biochem J. 1972;128:105P–106P. doi: 10.1042/bj1280105p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Close RI. Physiol Rev. 1972;52:129–197. doi: 10.1152/physrev.1972.52.1.129. [DOI] [PubMed] [Google Scholar]
- 84.Vandenboom R, Grange RW, Houston ME. AJP - Cell Physiology. 1995;268:C596–603. doi: 10.1152/ajpcell.1995.268.3.C596. [DOI] [PubMed] [Google Scholar]
- 85.Vandenboom R, Xeni J, Bestic NM, Houston ME. AJP - Regulatory Integrative and Comparative Physiology. 1997;272:R1980–1984. doi: 10.1152/ajpregu.1997.272.6.R1980. [DOI] [PubMed] [Google Scholar]
- 86.Rassier DE, Tubman LA, MacIntosh BR. Am J Physiol. 1997;273:C198–C204. doi: 10.1152/ajpcell.1997.273.1.C198. [DOI] [PubMed] [Google Scholar]
- 87.MacIntosh BR, Grange RW, Cory CR, Houston ME. Pflugers Arch. 1993;524:9–15. doi: 10.1007/BF00374497. [DOI] [PubMed] [Google Scholar]
- 88.Moore RL, Palmer BM, Williams SL, Tanabe H, Grange RW, Houston ME. AJP - Cell Physiology. 1990;259:C432–438. doi: 10.1152/ajpcell.1990.259.3.C432. [DOI] [PubMed] [Google Scholar]
- 89.Rassier DE, MacIntosh BR. Can J Physiol Pharmacol. 2002;80:993–1000. doi: 10.1139/y02-127. [DOI] [PubMed] [Google Scholar]
- 90.Rassier DE, Macintosh BR. Braz J Med Biol Res. 2000;33:499–508. doi: 10.1590/s0100-879x2000000500003. [DOI] [PubMed] [Google Scholar]
- 91.Rassier DE, MacIntosh BR, Herzog W. J Appl Physiol. 1999;86:1445–1457. doi: 10.1152/jappl.1999.86.5.1445. [DOI] [PubMed] [Google Scholar]
- 92.Vandenboom R, Grange RW, Houston ME. AJP - Cell Physiology. 1993;265:C1456–1462. doi: 10.1152/ajpcell.1993.265.6.C1456. [DOI] [PubMed] [Google Scholar]
- 93.Rassier DE, Tubman LA, MacIntosh BR. Braz J Med Biol Res. 1999;32:121–129. doi: 10.1590/s0100-879x1999000100018. [DOI] [PubMed] [Google Scholar]
- 94.Metzger JM, Greaser ML, Moss RL. J Gen Physiol. 1989;93:855–883. doi: 10.1085/jgp.93.5.855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Davis JS, Satorius CL, Epstein ND. Biophys J. 2002;83:359–370. doi: 10.1016/S0006-3495(02)75175-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Patel JR, Diffee GM, Huang XP, Moss RL. Biophys J. 1998;74:360–368. doi: 10.1016/S0006-3495(98)77793-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Brown IE, Loeb GE. Journal of Muscle Research and Cell Motility. 1999;20:443–456. doi: 10.1023/a:1005590901220. [DOI] [PubMed] [Google Scholar]
- 98.Gittings W, Huang J, Smith IC, Quadrilatero J, Vandenboom R. Journal of Muscle Research and Cell Motility. 2011 doi: 10.1007/s10974-011-9239-8. in press. [DOI] [PubMed] [Google Scholar]
- 99.Butler TM, Siegman MJ, Mooers SU, Barsotti RJ. Science. 1983;220:1167–1169. doi: 10.1126/science.6857239. [DOI] [PubMed] [Google Scholar]
- 100.Grange RW, Cory CR, Vandenboom R, Houston ME. Am J Physiol. 1995;269:C713–724. doi: 10.1152/ajpcell.1995.269.3.C713. [DOI] [PubMed] [Google Scholar]
- 101.Grange RW, Vandenboom R, Xeni J, Houston ME. J Appl Physiol. 1998;84:236–243. doi: 10.1152/jappl.1998.84.1.236. [DOI] [PubMed] [Google Scholar]
- 102.Abbate F, Sargeant AJ, Verdijk PWL, de Haan A. Journal of Applied Physiology. 2000;88:35–40. doi: 10.1152/jappl.2000.88.1.35. [DOI] [PubMed] [Google Scholar]
- 103.MacIntosh BR, Bryan SN. Pflugers Arch. 2002;443:804–812. doi: 10.1007/s00424-001-0746-0. [DOI] [PubMed] [Google Scholar]
- 104.MacIntosh BR, Taub EC, Dormer GN, Tomaras EK. Pflugers Arch. 2008;456:449–458. doi: 10.1007/s00424-007-0374-4. [DOI] [PubMed] [Google Scholar]
- 105.MacIntosh BR, Willis JC. Journal of Applied Physiology. 2000;88:2088–2096. doi: 10.1152/jappl.2000.88.6.2088. [DOI] [PubMed] [Google Scholar]
- 106.Geeves MA, Lehrer SS. Results Probl Cell Differ. 2002;36:111–132. doi: 10.1007/978-3-540-46558-4_10. [DOI] [PubMed] [Google Scholar]
- 107.Vandenboom R, Claflin DR, Julian FJ. J Physiol. 1998;511(Pt 1):171–180. doi: 10.1111/j.1469-7793.1998.171bi.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Allen DG, Lamb GD, Westerblad H. Physiological Reviews. 2008;88:287–332. doi: 10.1152/physrev.00015.2007. [DOI] [PubMed] [Google Scholar]
- 109.Gordon DA, Enoka RM, Stuart DG. The Journal of Physiology. 1990;421:569–582. doi: 10.1113/jphysiol.1990.sp017962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Vandenboom R, Houston ME. Can J Physiol Pharmacol. 1996;74:1315–1321. [PubMed] [Google Scholar]
- 111.Tubman LA, MacIntosh BR, Maki WA. Pflugers Arch. 1996;431:882–887. doi: 10.1007/s004240050081. [DOI] [PubMed] [Google Scholar]
- 112.Rijkelijkhuizen JM, de Ruiter CJ, Huijing PA, de Haan A. Journal of Experimental Biology. 2005;208:55–63. doi: 10.1242/jeb.01329. [DOI] [PubMed] [Google Scholar]
- 113.Palmer BM, Moore RL. Am J Physiol. 1989;257:C1012–1019. doi: 10.1152/ajpcell.1989.257.5.C1012. [DOI] [PubMed] [Google Scholar]
- 114.Vandenboom R. Can J Appl Physiol. 2004;29:330–356. doi: 10.1139/h04-022. [DOI] [PubMed] [Google Scholar]
- 115.Franks-Skiba K, Lardelli R, Goh G, Cooke R. AJP - Regulatory, Integrative and Comparative Physiology. 2007;292:R1603–1612. doi: 10.1152/ajpregu.00499.2006. [DOI] [PubMed] [Google Scholar]
- 116.Stewart M, Franks-Skiba K, Cooke R. Journal of Muscle Research and Cell Motility. 2009;30:17–27. doi: 10.1007/s10974-008-9162-9. [DOI] [PubMed] [Google Scholar]
- 117.Karatzaferi C, Franks-Skiba K, Cooke R. AJP - Regulatory, Integrative and Comparative Physiology. 2008;294:R948–955. doi: 10.1152/ajpregu.00541.2007. [DOI] [PubMed] [Google Scholar]
- 118.Abbate F, Van Der Velden J, Stienen GJ, De Haan A. J Muscle Res Cell Motil. 2001;22:703–710. doi: 10.1023/a:1016383025358. [DOI] [PubMed] [Google Scholar]
- 119.Grange RW, Houston ME. J Appl Physiol. 1991;70:726–731. doi: 10.1152/jappl.1991.70.2.726. [DOI] [PubMed] [Google Scholar]
- 120.Houston ME, Lingley MD, Stuart DS, Grange RW. FEBS Letters. 1987;219:469–471. doi: 10.1016/0014-5793(87)80274-0. [DOI] [PubMed] [Google Scholar]
- 121.Houston ME, Grange RW. Can J Physiol Pharmacol. 1990;68:908–913. doi: 10.1139/y90-139. [DOI] [PubMed] [Google Scholar]
- 122.Houston ME, Grange RW. Can J Physiol Pharmacol. 1991;69:269–273. doi: 10.1139/y91-041. [DOI] [PubMed] [Google Scholar]
- 123.Houston ME, Green HJ, Stull JT. Pflugers Arch. 1985;403:348–352. doi: 10.1007/BF00589245. [DOI] [PubMed] [Google Scholar]
- 124.Stuart DS, Lingley MD, Grange RW, Houston ME. Can J Physiol Pharmacol. 1988;66:49–54. doi: 10.1139/y88-009. [DOI] [PubMed] [Google Scholar]
- 125.Sale DG. Exerc Sport Sci Rev. 2002;30:138–143. doi: 10.1097/00003677-200207000-00008. [DOI] [PubMed] [Google Scholar]
- 126.Sale D. British Journal of Sports Medicine. 2004;38:386–387. doi: 10.1136/bjsm.2002.003392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Adam A, De Luca CJ. Journal of Applied Physiology. 2005;99:268–280. doi: 10.1152/japplphysiol.01344.2004. [DOI] [PubMed] [Google Scholar]
- 128.Klein CS, Ivanova TD, Rice CL, Garland SJ. Neuroscience Letters. 2001;316:153–156. doi: 10.1016/s0304-3940(01)02389-8. [DOI] [PubMed] [Google Scholar]
- 129.Inglis G, Howard J, Gabriel DG, Vandenboom R. Acta Physiologica. doi: 10.1111/j.1748-1716.2010.02233.x. In press. [DOI] [PubMed] [Google Scholar]
- 130.Hamada T, Sale DG, MacDougall JD, Tarnopolsky MA. Journal of Applied Physiology. 2000;88:2131–2137. doi: 10.1152/jappl.2000.88.6.2131. [DOI] [PubMed] [Google Scholar]
- 131.Vandervoort AA, McComas AJ. Eur J Appl Physiol Occup Physiol. 1983;51:435–440. doi: 10.1007/BF00429079. [DOI] [PubMed] [Google Scholar]
- 132.Vandervoort AA, Quinlan J, McComas AJ. Experimental Neurology. 1983;81:141–152. doi: 10.1016/0014-4886(83)90163-2. [DOI] [PubMed] [Google Scholar]
- 133.Gossen ER, Allingham K, Sale DG. Can J Physiol Pharmacol. 2001;79:49–58. [PubMed] [Google Scholar]