

Abstract
Coeliac disease is a widespread, lifelong disorder for which dietary control represents the only accepted form of therapy. There is an unmet need for non-dietary therapies to treat this condition. Most ongoing and emerging drug discovery programmes are based on the understanding that coeliac disease is caused by an inappropriate T-cell-mediated immune response to dietary gluten proteins. Recent genome-wide association studies lend further support to this pathogenic model. The central role of human leukocyte antigen genes has been validated, and a number of new risk loci have been identified, most of which are related to the biology of T cells and antigen-presenting cells. Here we review the status of potential non-dietary therapies under consideration for coeliac disease. We conclude that future development of novel therapies will be aided by the identification of new, preferably non-invasive, surrogate markers for coeliac disease activity.
Keywords: Coeliac disease, gluten, T cell, HLA, transglutaminase, drug, therapy
Introduction
Coeliac disease is a common, lifelong disorder with an incidence of about 1:100, and awareness of this condition is increasing. The disease is precipitated in genetically susceptible individuals by the ingestion of wheat gluten and related cereal proteins derived from barley and rye [1, 2]. The coeliac lesion is characterised by villous atrophy, crypt hyperplasia and infiltration of inflammatory cells, both in the epithelium and in the lamina propria of the small intestine. The disease can be considered to be due to food hypersensitivity as well as being an autoimmune condition. On ingestion of gluten, patients with coeliac disease typically develop disease-specific autoantibodies that recognise the enzyme transglutaminase 2 (TG2) [3]. It is striking that both the intestinal lesions and the autoantibodies are reversibly dependent on oral gluten exposure [4, 5]. At present, a lifelong gluten-free diet is the only accepted form of treatment for coeliac disease. Thus there is an unmet need for alternative therapies [6–9]. In this article we will review the current status of potential non-dietary therapies under consideration for coeliac disease.
Pathogenesis of Coeliac Disease
Intestinal enteropathy in coeliac disease is caused by a combination of genetic and environmental factors. Gluten is clearly the most critical environmental component, whereas both human leukocyte antigen (HLA) and non-HLA genes are predisposing hereditary factors. Recent genome-wide association studies have underscored the fact that HLA is the single most important genetic factor [10]. The vast majority of patients with coeliac disease carry a variant of HLA-DQ2, (DQ2.5; DQA1*05/DQB1*02), and most of the remaining patients carry HLA-DQ8 (DQA1*03/DQB1*0302). HLA-DQ2 and HLA-DQ8 predispose patients to coeliac disease by preferential presentation of gluten peptides to CD4 helper T cells in the lamina propria. These T cells become activated upon recognition of gluten peptides and produce many different cytokines, of which interferon-γ (IFN-γ) predominates [11]. In turn, a cascade of inflammatory reactions is triggered, leading eventually to the hallmark small intestinal lesion.
Although genome-wide analysis has also identified numerous non-HLA loci associated with coeliac disease, each individual locus contributes very modestly to the overall genetic risk [10, 12]. Moreover, a majority of these non-HLA loci harbour genes involved in the biology of T cells and antigen presenting cells [13]. Together, these genetic findings have validated the most well-accepted model for coeliac disease pathogenesis, which originated from the identification of HLA susceptibility alleles and their role in gluten antigen presentation to T cells [2]. The emerging picture from the genetics of coeliac disease also bodes well for ongoing drug discovery programmes, as most are based on the assumption that gluten-reactive T cells play a central role in controlling disease onset and severity. Furthermore, the loci discovered in genome-wide association studies also reveal additional potential targets for coeliac disease drug discovery.
A number of distinct disease-specific T cell epitopes are known to exist in dietary gluten. A common feature of these epitopes is the presence of multiple proline and glutamine residues. The high proline content renders the peptides resistant to breakdown by gastric, pancreatic and intestinal digestive proteases [14]. As a result, an elevated intestinal concentration of potentially immunoreactive peptides is maintained following a gluten-containing meal. At the same time, some glutamine residues in these gluten peptides are catalytically deamidated by TG2 [15, 16]. Deamidation enhances their immunogenicity due to high-affinity interactions between the modified residues and specific pockets in the ligand-binding sites of HLA-DQ2 and HLA-DQ8 [17, 18].
Whereas the HLA-mediated response to gluten antigens in patients with coeliac disease is well understood, three features of gluten enteropathy in coeliac disease remain unclear at present. Firstly, dietary gluten reversibly increases small intestinal permeability in patients with coeliac disease. It has been proposed that enhanced intestinal permeability is the consequence of increased expression of zonulin [19]. Alternatively, or in addition, this phenomenon may be caused by the cytokines (especially IFN-γ) produced by activated CD4 T cells [11]. Secondly, dietary gluten also appears to activate the innate immune system in patients with coeliac disease, leading to production of interleukin-15 (IL-15) both in the lamina propria and in the epithelium. In turn, IL-15 causes aberrant MIC expression on enterocytes and increased NKG2D expression on intra-epithelial lymphocytes. Upon engagement, these proteins trigger the killing of the enterocytes [20, 21]. However, the molecular basis of this innate immune response to dietary gluten has not yet been defined [22]. Thirdly, gluten consumption also induces anti-TG2 autoantibody production in those with coeliac disease [3]. The cause and pathogenic consequences of such autoantibody production remain unclear. Any or all of these lines of investigation may reveal important new targets for coeliac disease therapy.
Potential non-dietary therapies
Based on our current understanding of coeliac disease pathogenesis, several therapeutic modes of action can be considered for this unmet medical need. We will first discuss pharmacological mechanisms for which an investigational new drug (IND) has either already been identified, or there is good reason to anticipate that one may be identified within the next few years. Then we will focus on therapeutic modes of action that are being investigated by the pharmaceutical industry in the context of other disorders, and could have relevance to coeliac disease therapy.
The drug pipeline for coeliac disease
For several pharmacological mechanisms, results from in vitro, ex vivo and/or in vivo studies have already set the stage for the identification of one or more IND candidates. Broadly speaking, these approaches to the development of a drug for the treatment of coeliac disease fall into three categories. In a few cases, a generic drug could be repurposed via reformulation for potential use in coeliac disease. In other situations, IND candidates have already been advanced into human clinical trials. The third category includes modes of action for which a compelling pharmacological case can be made, and lead molecules have also been identified. However, the identification of an IND candidate is hampered by the lack of a suitable animal model for coeliac disease.
Glucocorticosteroids with low systemic bioavailability
Budesonide is an example of a topically active glucocorticosteroid with low oral bioavailability. As a result, systemic exposure to this anti-inflammatory agent is insignificant, and its pharmacological effects are localised to the gut mucosa. Budesonide is used for the treatment of asthma and inflammatory bowel disease. Pilot studies in patients with refractory [23, 24] and non-refractory [25] coeliac disease have demonstrated that budesonide may provide clinical benefit in both groups of patients. Oral budesonide may also have acceptable safety characteristics for use in patients with active coeliac disease; for example, 6 mg budesonide has been administered daily to patients with primary biliary cirrhosis for up to 3 years with no change in budesonide pharmacokinetics and only minor changes in bone mineral density [26]. However, because the predominant use of oral budesonide is for illnesses of the lower intestine, available formulations of this generic drug are unsuitable for coeliac disease. Pending development of a new budesonide formulation that targets the drug to the upper intestine, controlled clinical trials are warranted to investigate its safety and efficacy in patients with coeliac disease.
Oral proteases for gluten detoxification
It is now well accepted that the most immunotoxic gluten peptides are also highly resistant to breakdown by pepsin, pancreatic proteases and intestinal brush border membrane peptidases [14, 27]. This unusual stability is principally due to two factors: the inability of gastric and pancreatic endoproteases to cleave after proline or glutamine residues and the inability of dipeptidyl peptidase IV and dipeptidyl carboxypeptidase I in the brush border membrane to cleave long peptides. Together, these two features lead to the accumulation of long, metastable intermediates in the small intestinal lumen, which in turn elicit an HLA-DQ2- or HLA-DQ8-restricted T-cell response in patients with coeliac disease. Therefore, it is anticipated that co-administration of exogenous proline-and/or glutamine-specific proteases with food could provide therapeutic benefit to patients by accelerating gluten detoxification (Figure 1) [14, 27]. This possibility has subsequently gained support from a range of in vitro, in vivo animal, and ex vivo human studies [28–36], and has led to the introduction of at least two drug candidates, ALV003 [37] and AN-PEP, into clinical trials (Table 1). It has also led to the identification of STAN1, a combination of over-the-counter dietary enzymes with modest gluten detoxification capacity [38]; this cocktail is undergoing clinical evaluation (Table 1). A key question that must be addressed for all such experimental therapies is the gluten dose that can be effectively detoxified in vivo by a given enzyme dose.
Figure 1. The coeliac lesion in the proximal small intestine.
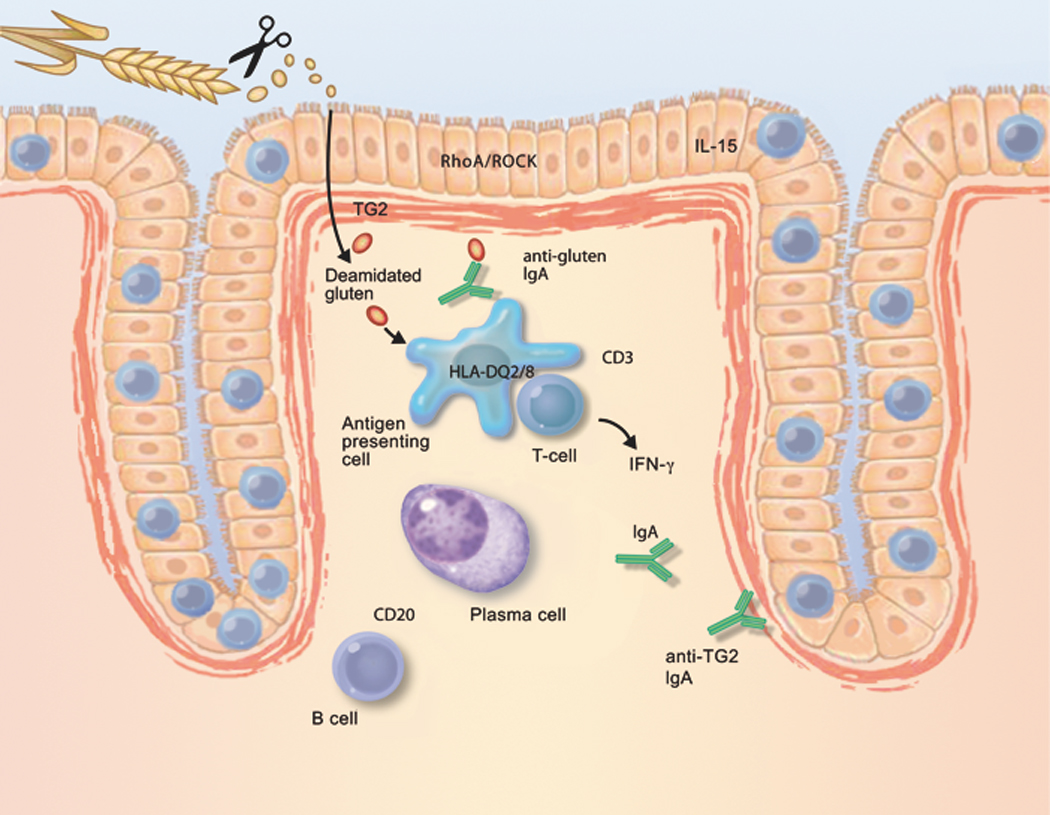
Schematic depiction of factors that contribute to the development of coeliac disease, and that could be novel therapeutic targets. Long, proline-rich fragments of gluten survive digestion by luminal and brush-border enzymes; as a result, they are able to gain access to the lamina propria. Gluten-sequestering polymers and oral proteases may reduce the exposure of the immune system to immunogenic gluten peptides. Similar effects may be derived from zonulin antagonists or RhoA/ROCK inhibitors, all of which reduce epithelial permeability. Most gluten peptides that survive gastrointestinal breakdown are excellent substrates for TG2. The resulting deamidated products are recognised by CD4-positive T cells, when bound to HLA-DQ2 or HLA-DQ8 molecules on the cell surface of antigen-presenting cells. Therefore, TG2 inhibitors and HLA blockers are candidates for future coeliac disease therapy. Alternatively, activation of gluten-reactive T cells may be suppressed by peptide vaccines or by anti-CD3 treatment. Upon activation, gluten-reactive CD4-positive T cells produce IFN-γ, which is a major contributor to the development of the coeliac lesion. IFN-γ is also produced by intra-epithelial T cells. Therefore, anti-IFN-γ therapy could be considered a drug candidate. Similarly, IL-15, produced by either mononuclear cells in the lamina propria or by enterocytes themselves, attracts T cells with the capacity to kill enterocytes. IL-15 production is stimulated by gluten in the intestine in coeliac disease. Therefore, compounds that neutralise the effect of IL-15 are interesting drug candidates. Finally, B cells receive help from T cells to differentiate into plasma cells, which then produce autoantibodies against TG2. Because the interaction with CD20-positive B cells may amplify the anti-gluten T-cell response, anti-CD20 antibodies could be useful for the treatment of coeliac disease.
Table I.
Overview over non-dietary therapies for coeliac disease
| Mode of action | Compound | Compound class |
Company/ university |
Status |
|---|---|---|---|---|
| Topical steroid | Budesonide | Small molecule | Generic drug | Approved |
| Rho kinase inhibition | Fasudil | Small molecule | Generic drug | Approved |
| Glutenase | ALV003 | Enzyme | Alvine, USA | Phase II |
| Glutenase | AN-PEP | Enzyme | DSM, Netherlands | Phase I + II |
| Glutenase | STAN1 (enzyme supplements) | Enzyme | Heim Pal Childrens Hospital, Hungary | Phase I + II |
| Zonulin antagonist | AT-1001 | Peptide | Alba, USA | Phase II |
| CCR9 antagonist | CCX282-B | Small molecule | ChemoCentryx, USA | Phase II |
| Immune modulation | Necator americanus | Parasite | Princess Alexandra Hospital, Australia | Phase II |
| Peptide vaccination | Nexvax2 | Peptide | Nexpep, Australia | Phase I |
| Anti-IL-15 | AMG 714 | Monoclonal antibody | Amgen, USA | Phase II in RA1, psoriasis |
| Anti-IFN-γ | Fontolizumab | Monoclonal antibody | PDL and Biogen Idec, USA | Phase II in IBD1, (discont.) |
| Anti-CD3 | Visilizumab | Monoclonal antibody | Facet, USA | Phase II in UC1,, GvHD1, |
| Anti-CD3 | Teplizumab | Monoclonal antibody | MacroGenics, USA | Phase II in T1D1, |
| Anti-CD3 | Otelixizumab | Monoclonal antibody | Tolerx, USA | Phase III in T1D1, |
| Anti-CD20 | Rituximab | Monoclonal antibody | Biogen Idec, USA | Approved |
| Anti-CD20 | Tositumab | Monoclonal antibody | GlaxoSmithKline, USA | Approved |
| Anti-CD20 | Ibritumomab | Monoclonal antibody | Spectrum, USA | Approved |
| RhoA inhibition | BA-210 | Recombinant protein | Alseres, USA | Phase II in spinal cord injury |
| TG2 inhibitor | Dihydroisoxazoles | Small molecule | Stanford University, USA | Discovery |
| TG2 inhibitor | ZED-101 | Small molecule | Zedira, Germany | Discovery |
| TG2 inhibitor | Cinnamoyl triazoles | Small molecule | University of Montreal, Canada | Discovery |
| HLA-DQ2 blocker | Dimeric analogue of gluten peptide | Peptide | Stanford University, USA & University of Oslo, Norway | Discovery |
| HLA-DQ2 blocker | Azidoproline analogue of gluten peptide | Peptide | Leiden University, Netherlands | Discovery |
| Gluten tolerisation | Genetically modified Lactococus lactis | Bacteria | ActoGeniX, Belgium | Discovery |
| Gluten-sequestering polymers | P(HEMA-co-SS) | Polymer resin | University of Montreal, Canada | Discovery |
RA: Rheumatoid arthritis; IBD: inflammatory bowel disease; UC: ulcerative colitis; GvHD: graft versus host disease; T1D: type 1 diabetes,
Gluten-sequestering polymers
An alternative strategy for blocking the immunotoxic manifestations of dietary gluten in the gut lumen of patients with coeliac disease relies on the use of an oral polymeric resin, poly(hydroxyethyl methacrylate-co-styrene sulfonate) (P(HEMA-co-SS) (Figure 1). In vitro studies have demonstrated that P(HEMA-co-SS) can bind to gluten proteins under simulated gastric and intestinal conditions [39, 40]. The polymer also reduced the mucosal toxicity induced by oral gavage of gliadin in HLA-DQ8 transgenic mice that had been systemically sensitised with gliadin [41]. While awaiting safety testing, P(HEMA-co-SS) could be tested in the foreseeable future in controlled clinical trials involving patients with coeliac disease (Table 1). Again, the gluten dose that can be effectively detoxified in vivo by a given dose of polymer must be established.
Zonulin antagonists
Zonulin is overexpressed in intestinal tissue of patients with coeliac disease compared to healthy controls [19]. When exposed to gliadin peptides, intestinal biopsies from patients in remission showed a sustained zonulin release and an increase in trans-epithelial permeability, whereas a less-pronounced effect was observed in biopsies from controls [42]. Pilot studies in patients with coeliac disease [43], and the related disorder dermatitis herpetiformis [44], revealed a correlation between serum zonulin levels and intestinal permeability. Recently, zonulin was identified as prehaptoglobin-2 [45]. The zonulin antagonist AT-1001, based on what now appears to be an erroneous partial sequence of zonulin [46], is undergoing Phase II clinical trials in patients with coeliac disease [47] (Table 1).
Inhibitors of TG2
As summarised above, TG2 plays a critical role in coeliac disease pathogenesis by unmasking gluten-derived T-cell epitopes via deamidation. Therefore, TG2 may be a suitable target for non-dietary therapy (Figure 1). The observation that TG2-null mice do not have any gross physiological, developmental or reproductive characteristics [48, 49] lends further support to the hypothesis that TG2 inhibitors may be well tolerated in humans. The structure–activity relationships of a number of different classes of small molecule TG2 inhibitors have been analysed. These include both active site-directed irreversible inhibitors such as thiadiazoles [50], epoxides [51], α,β-unsaturated amides [51] and dihydroisoxazoles [52, 53], as well as reversible inhibitors such as thienopyrimidines [54], cinnamoyl compounds [55], β-aminoethyl ketones [56] and acylidine oxindoles [57]. Until recently, a key barrier to further lead optimisation has been the absence of an appropriate animal model to quantify TG2 inhibitory activity of drug candidates in the upper small intestine (i.e. the primary site of the disease). Now that it has been shown that Toll-like receptor-3-mediated inflammation of the mouse small intestine is associated with rapid TG2 activation [58], this may provide a suitable assay system to compare the in vivo activity of alternative compounds. In the next few years, it is probable that one or more TG2 inhibitors will undergo proof-of-concept clinical evaluation in coeliac disease. Parenthetically, it should be noted that aberrant TG2 activity has been implicated in the pathogenesis of a number of unrelated disorders including certain neurological and renal diseases as well as some cancers [59–61]. Therefore, inhibitors of human TG2 could also be of use in the treatment of other disorders. Whereas this treatment modality could be assessed with virtually any TG2 inhibitor that enters human clinical trials, the activity of the ideal drug candidate for coeliac disease should presumably be localised to the small intestinal mucosa due to high first-pass metabolism.
Blockers of HLA-DQ-mediated T-cell activation
HLA (i.e. HLA-DQ2 or HLA-DQ8) is a necessary factor for developing coeliac disease, but insufficient by itself. Because homozygosity for HLA alleles is not associated with apparent increased susceptibility to infections, blocking of HLA-DQ2- or HLA-DQ8-mediated presentation of gluten peptides is an attractive therapeutic target in coeliac disease (Figure 1). Although the feasibility of blocking HLA-mediated peptide presentation has previously been explored for other HLA-associated diseases, such programmes were abandoned principally due to the difficulty of delivery of HLA-blocking compounds into the affected organs (i.e. central nervous system in multiple sclerosis or joints in rheumatoid arthritis). By contrast, topical delivery of HLA blockers to the small intestine in coeliac disease should be considerably easier. Indeed, lead peptide blockers have been engineered using gliadin-derived T-cell epitopes as scaffolds [62, 63] (Table 1), but these blockers have only shown moderate efficacy in inhibiting gluten-induced T-cell activation. One reason for this could be the relatively modest binding affinity of the gluten-derived T-cell epitopes. Effort has thus been focused on identifying optimal HLA-DQ2 binders [64, 65]. These ligands bind with at least 50-fold better affinity to HLA-DQ2 than the immunodominant the gluten epitope DQ2-α-I, but this is probably still insufficient to completely suppress the activation of gluten-reactive T cells in the intestine in coeliac disease, raising some concern regarding the viability of this approach.
Gluten tolerisation
Protein-based desensitisation therapy is used to treat allergic diseases [66]. For example, peptide-based vaccination by intradermal injection of cat allergen peptides has shown efficacy in the treatment of cat allergy [67]. In patients with this allergy, CD4-positive T cells with regulatory capacity are induced by peptide vaccination, implying that induction of regulatory T cells is the likely mode of action of this treatment [68]. With the identification of the major HLA-DQ2-restricted T-cell epitopes from wheat, barley and rye [69, 70], a key prerequisite for peptide-based desensitisation is now in place, and two fundamental questions can be addressed with a candidate vaccine against coeliac disease (Figure 1). Firstly, do its peptide constituents tolerise gluten-reactive T cells in the intestinal mucosa of patients with coeliac disease? And secondly, because a synthetic peptide vaccine can only include a finite number of antigens, to what extent does this immune tolerance extend to other less immunodominant epitopes? Clinical trials have been initiated with Nexvax2, a prototypical vaccine based on a set of gluten peptides that are recognised by HLA-DQ2 in an immunodominant manner (Table 1). As is the case with HLA-blocking strategies, a separate vaccine must be designed for HLA-DQ8-positive coeliac disease, because HLA-DQ2- and HLA-DQ8-restricted T-cell epitopes are largely non-overlapping [71].
An alternative delivery system for tolerogenic gluten antigens is a genetically modified Lactococus lactis bacterium. Oral administration of an immunodominant, HLA-DQ8-restricted T-cell epitope in this engineered vehicle led to induced suppression of local and systemic T-cell responses to the corresponding gliadin peptide in a transgenic mouse model, along with the induction of Foxp3+ regulatory cells [72]. Here too, it remains to be established whether T-cell responses to other gluten epitopes can be suppressed via this approach.
Other therapeutic targets
Beyond the drug pipeline for coeliac disease summarised above, a number of promising pharmacological mechanisms are being actively targeted in the context of other medical conditions. Some of these approved or experimental drugs could have relevance to coeliac disease therapy. Here it is important to differentiate between target validation and drug development per se. As long as an animal model that captures the signature features of coeliac disease is unavailable, validation of a new mode of action may be most effectively achieved via an experimental drug, even one that is unsuitable for long-term chronic use by most patients with coeliac disease. If successful, such an exercise in target validation would prepare for a next-generation drug that acts upon the same target albeit with a more acceptable safety–efficacy profile for coeliac disease therapy. Alternatively (or additionally), it could lead to a more focused effort to identify a pharmacologically superior drug target in the same pathway. Below we discuss some examples of such sentinel drug development programmes.
Rho/Rho kinase inhibition
As discussed above, patients with coeliac disease undergo a rapid increase in intestinal permeability upon exposure to dietary gluten, and at least one modulator of this phenomenon (AT-1001) is already undergoing clinical studies. IFN-γ is another important trigger of epithelial permeability [73]. Pharmacological analysis with T84 intestinal epithelial cells has shown that this increase in intestinal permeability is dependent on Rho kinase (ROCK) activity [74]. In addition to regulating tight junction structure and function [75], RhoA and ROCK are known to regulate axon growth [76]. The benefits of blocking RhoA or ROCK have been well documented for the treatment of spinal cord injury. For example, fasudil, an inhibitor of ROCK, is an approved drug in Japan for the treatment of cerebral vasospasm, and the RhoA inhibitor BA-210 is in clinical trials (Table 1). Whereas the side effects of fasudil make it inappropriate for chronic, long-term use in patients with coeliac disease, the drug could be used to establish whether ROCK inhibition can reverse gluten-dependent increase in intestinal permeability in these patients (Figure 1). If so, a number of next-generation ROCK inhibitors are under active evaluation for other indications such as asthma, pulmonary hypertension and multiple sclerosis [77], and some of these drugs may hold greater promise for coeliac disease therapy.
Anti-IFN-γ therapy
IFN-γ is the main pro-inflammatory cytokine produced by gluten reactive CD4-positive T cells of the coeliac disease lesion. Therefore, anti-IFN-γ therapy would not only be invaluable for validating the role of these T cells, but may also prove useful in some circumstances for the treatment of coeliac disease (Figure 1). Pharmacologically relevant doses of the anti-IFN-γ antibody fontolizumab (Table 1) were well tolerated by patients with inflammatory bowel disease [78]; however, further product development was discontinued, presumably because of the lack of efficacy in this indication. The exploratory use of fontolizumab in patients with coeliac disease may be justifiable, pending availability of additional data.
Anti-CD3 therapy
The CD3 protein complex is a co-receptor for the T-cell receptor. Several anti-CD3 monoclonal antibodies are undergoing clinical evaluation in type 1 diabetes and ulcerative colitis (Table 1) [79–81]. Anti-CD3 therapy seems to be mainly effective in the context of a primed and ongoing immune response, and the mode of action of anti-CD3 therapy is likely to involve both the elimination of effector T cells and the induction of regulatory T cells [82]. For reasons discussed above, anti-CD3 therapy could yield insights into the pathogenic significance of T cells in coeliac disease, and may even prove to be clinically useful in the management of this disease under certain circumstances (Figure 1).
Anti-CD20 therapy
Selective B-cell depletion with anti-CD20 therapy has shown clinical benefit in many autoimmune diseases such as rheumatoid arthritis [83], multiple sclerosis [84] and type 1 diabetes [85], suggesting that B cells play an important, and in some diseases an unexpected, role in the pathogenesis of these HLA associated disorders. A possible explanation for this is that, by presenting disease-associated antigens to T cells, B cells act as an amplifying loop for the T-cell response. B cells may also play a critical role in the pathogenesis of coeliac disease, as both gluten-specific and TG2-specific B cells should be able to present gluten antigens to T cells in this disorder [86] (Figure 1). However, it is now known that the steady-state generation of immunoglobulin A plasmablasts in the gut mucosa is not abrogated by anti-CD20 treatment despite peripheral B-cell depletion [87]. Therefore, if mucosally produced antibodies play a pathogenic role in coeliac disease, anti-CD20 therapy may not be effective. Several monoclonal antibodies against CD20 have already been approved for clinical use (Table 1).
Anti-IL-15 therapy
As discussed above, the cytokine IL-15 appears to play a central role in the innate immune response to gluten in coeliac disease, and may be particularly important in refractory disease. Moreover, antibody-mediated blockade of IL-15 reverses intestinal damage in transgenic mice that over-express IL-15 in enterocytes [88]. Therefore, anti-IL-15 therapy warrants investigation in patients with coeliac disease (Figure 1). An anti-IL-15 monoclonal antibody, AMG 714, is undergoing Phase II clinical studies in rheumatoid arthritis and psoriasis (Table 1).
CCR9 antagonists
Lymphocytes homing to the gut mucosa have specific receptors such as the chemokine receptor CCR9 and the α4β7 integrin. Interfering with the homing of gluten-specific T cells could therefore curb the immune response to gluten in coeliac disease. CCX282-B is a selective, orally bioavailable antagonist of human CCR9 that affects lymphocyte migration, and is undergoing clinical trials in Crohn’s disease [89]. A Phase II trial has also been undertaken in coeliac disease (Table 1). If the drug is effective, the possibility of an increased risk of gastrointestinal infections must be carefully monitored, because this mode of action is not antigen specific.
Hookworm
Infestation with the hookworm Necator americanus has been suggested as a possible treatment for autoimmune diseases. The rationale behind this therapy is that the increased prevalence of autoimmune disorders in developed countries is closely paralleled by a disappearance of intestinal parasites. These parasites are thought to play an important role in modulating the host’s immune system. A Phase II clinical trial has been undertaken with this hookworm in coeliac disease (Table 1).
Monitoring the treatment effect
Although biopsy of the small bowel is the gold standard to assess the effects of dietary or non-dietary treatments for coeliac disease, it is extremely challenging to implement in the context of controlled clinical trials (especially large-scale trials), because it requires invasive and expensive endoscopic procedures and is subjective. Non-invasive alternatives to small bowel biopsy are therefore urgently needed in order to propel coeliac disease drug development efforts.
Due to the extreme variability of symptoms associated with coeliac disease, symptom scores will probably be unsuitable for this purpose. Serum antibodies have high disease specificity but inadequate sensitivity, and are therefore unsuitable for monitoring the effects of low-to-moderate gluten consumption. By contrast, measures of intestinal permeability such as the lactulose:mannitol excretion ratio are sensitive but not specific. A detailed discussion of current surrogate endpoints for clinical trials of experimental therapeutic agents is beyond the scope of this review. Recent studies have led to the observation that disease-specific T cells enter the bloodstream after short-term gluten challenges [70, 90, 91], and can be adopted for early (proof-of-concept) clinical trials of new drug candidates [37]. Surrogate markers, which reflect key features of coeliac disease pathogenesis and are suitable for larger and/or longer clinical trials, are also needed.
Conclusions
The pathogenic disease model on which most ongoing drug discovery programmes for coeliac disease are based has been confirmed by recent genetic studies. This encouraging trend could gain momentum in the coming years, if one or more early clinical trials yield promising results. Notwithstanding this trend, drug development for coeliac disease is hampered by the lack of good surrogate markers of disease activity. The development of such markers should therefore be a priority.
Acknowledgements
L.M.S. is supported by grants from the Research Council of Norway, the European Commission FP7 programme, the South-Eastern Norway Regional Health Authority, the Norwegian Foundation for Health and Rehabilitation Research and the Juvenile Diabetes Research Foundation. C.K. is supported by a grant from the National Institutes of Health (R01 DK 063158) for research on coeliac disease.
Footnotes
Conflict of Interest Statement
L.M.S. is a member of the scientific advisory boards of Alvine Pharmaceuticals, ActoGeniX and Flamentera AG. C.K. is a stockholder and Director of Alvine Pharmaceuticals and Flamentera AG.
References
- 1.Green PH, Cellier C. Celiac disease. N Engl J Med. 2007;357:1731–1743. doi: 10.1056/NEJMra071600. [DOI] [PubMed] [Google Scholar]
- 2.Sollid LM. Coeliac disease: dissecting a complex inflammatory disorder. Nat Rev Immunol. 2002;2:647–655. doi: 10.1038/nri885. [DOI] [PubMed] [Google Scholar]
- 3.Dieterich W, Ehnis T, Bauer M, Donner P, Volta U, Riecken EO, Schuppan D. Identification of tissue transglutaminase as the autoantigen of celiac disease. Nat Med. 1997;3:797–801. doi: 10.1038/nm0797-797. [DOI] [PubMed] [Google Scholar]
- 4.Dieterich W, Laag E, Schopper H, et al. Autoantibodies to tissue transglutaminase as predictors of celiac disease. Gastroenterology. 1998;115:1317–1321. doi: 10.1016/s0016-5085(98)70007-1. [DOI] [PubMed] [Google Scholar]
- 5.Sulkanen S, Halttunen T, Laurila K, et al. Tissue transglutaminase autoantibody enzyme-linked immunosorbent assay in detecting celiac disease. Gastroenterology. 1998;115:1322–1328. doi: 10.1016/s0016-5085(98)70008-3. [DOI] [PubMed] [Google Scholar]
- 6.Sollid LM, Khosla C. Future therapeutic options for celiac disease. Nat Clin Pract Gastroenterol Hepatol. 2005;2:140–147. doi: 10.1038/ncpgasthep0111. [DOI] [PubMed] [Google Scholar]
- 7.Anderson RP. Coeliac disease: current approach and future prospects. Intern Med J. 2008;38:790–799. doi: 10.1111/j.1445-5994.2008.01741.x. [DOI] [PubMed] [Google Scholar]
- 8.Lerner A. New therapeutic strategies for celiac disease. Autoimmun Rev. 2010;9:144–147. doi: 10.1016/j.autrev.2009.05.002. [DOI] [PubMed] [Google Scholar]
- 9.Schuppan D, Junker Y, Barisani D. Celiac disease: from pathogenesis to novel therapies. Gastroenterology. 2009;137:1912–1933. doi: 10.1053/j.gastro.2009.09.008. [DOI] [PubMed] [Google Scholar]
- 10.van Heel DA, Franke L, Hunt KA, et al. A genome-wide association study for celiac disease identifies risk variants in the region harboring IL2 and IL21. Nat Genet. 2007;39:827–829. doi: 10.1038/ng2058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nilsen EM, Lundin KEA, Krajci P, Scott H, Sollid LM, Brandtzaeg P. Gluten specific, HLA-DQ restricted T cells from coeliac mucosa produce cytokines with Th1 or Th0 profile dominated by interferon gamma. Gut. 1995;37:766–776. doi: 10.1136/gut.37.6.766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dubois PC, Trynka G, Franke L, et al. Multiple common variants for celiac disease influencing immune gene expression. Nat Genet. 2010;42:295–302. doi: 10.1038/ng.543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Trynka G, Wijmenga C, van Heel DA. A genetic perspective on coeliac disease. Trends Mol Med. 2010;16:537–550. doi: 10.1016/j.molmed.2010.09.003. [DOI] [PubMed] [Google Scholar]
- 14.Shan L, Molberg, Parrot I, et al. Structural basis for gluten intolerance in celiac sprue. Science. 2002;297:2275–2279. doi: 10.1126/science.1074129. [DOI] [PubMed] [Google Scholar]
- 15.Molberg, McAdam SN, Körner R, et al. Tissue transglutaminase selectively modifies gliadin peptides that are recognized by gut-derived T cells. Nat Med. 1998;4:713–717. doi: 10.1038/nm0698-713. [DOI] [PubMed] [Google Scholar]
- 16.van de Wal Y, Kooy Y, van Veelen P, Pena S, Mearin L, Papadopoulos G, Koning F. Selective deamidation by tissue transglutaminase strongly enhances gliadin-specific T cell reactivity. J Immunol. 1998;161:1585–1588. [PubMed] [Google Scholar]
- 17.Kim CY, Quarsten H, Bergseng E, Khosla C, Sollid LM. Structural basis for HLA-DQ2-mediated presentation of gluten epitopes in celiac disease. Proc Natl Acad Sci USA. 2004;101:4175–4179. doi: 10.1073/pnas.0306885101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Henderson KN, Tye-Din JA, Reid HH, et al. A structural and immunological basis for the role of human leukocyte antigen DQ8 in celiac disease. Immunity. 2007;27:23–34. doi: 10.1016/j.immuni.2007.05.015. [DOI] [PubMed] [Google Scholar]
- 19.Fasano A, Not T, Wang W, Uzzau S, Berti I, Tommasini A, Goldblum SE. Zonulin, a newly discovered modulator of intestinal permeability, and its expression in coeliac disease. Lancet. 2000;355:1518–1519. doi: 10.1016/S0140-6736(00)02169-3. [DOI] [PubMed] [Google Scholar]
- 20.Hüüe S, Mention JJ, Monteiro RC, et al. A direct role for NKG2D/MICA interaction in villous atrophy during celiac disease. Immunity. 2004;21:367–377. doi: 10.1016/j.immuni.2004.06.018. [DOI] [PubMed] [Google Scholar]
- 21.Meresse B, Chen Z, Ciszewski C, et al. Coordinated induction by IL15 of a TCR-independent NKG2D signaling pathway converts CTL into lymphokine-activated killer cells in celiac disease. Immunity. 2004;21:357–366. doi: 10.1016/j.immuni.2004.06.020. [DOI] [PubMed] [Google Scholar]
- 22.Jabri B, Sollid LM. Tissue-mediated control of immunopathology in coeliac disease. Nat Rev Immunol. 2009;9:858–870. doi: 10.1038/nri2670. [DOI] [PubMed] [Google Scholar]
- 23.Daum S, Ipczynski R, Heine B, Schulzke JD, Zeitz M, Ullrich R. Therapy with budesonide in patients with refractory sprue. Digestion. 2006;73:60–68. doi: 10.1159/000092639. [DOI] [PubMed] [Google Scholar]
- 24.Brar P, Lee S, Lewis S, Egbuna I, Bhagat G, Green PH. Budesonide in the treatment of refractory celiac disease. Am J Gastroenterol. 2007;102:2265–2269. doi: 10.1111/j.1572-0241.2007.01380.x. [DOI] [PubMed] [Google Scholar]
- 25.Ciacci C, Maiuri L, Russo I, et al. Efficacy of budesonide therapy in the early phase of treatment of adult coeliac disease patients with malabsorption: an in vivo/in vitro pilot study. Clin Exp Pharmacol Physiol. 2009;36:1170–1176. doi: 10.1111/j.1440-1681.2009.05211.x. [DOI] [PubMed] [Google Scholar]
- 26.Rautiainen H, Farkkila M, Neuvonen M, et al. Pharmacokinetics and bone effects of budesonide in primary biliary cirrhosis. Aliment Pharmacol Ther. 2006;24:1545–1552. doi: 10.1111/j.1365-2036.2006.03155.x. [DOI] [PubMed] [Google Scholar]
- 27.Hausch F, Shan L, Santiago NA, Gray GM, Khosla C. Intestinal digestive resistance of immunodominant gliadin peptides. Am J Physiol Gastrointest Liver Physiol. 2002;283:G996–G1003. doi: 10.1152/ajpgi.00136.2002. [DOI] [PubMed] [Google Scholar]
- 28.Piper JL, Gray GM, Khosla C. Effect of prolyl endopeptidase on digestive-resistant gliadin peptides in vivo. J Pharmacol Exp Ther. 2004;311:213–219. doi: 10.1124/jpet.104.068429. [DOI] [PubMed] [Google Scholar]
- 29.Marti T, Molberg O, Li Q, Gray GM, Khosla C, Sollid LM. Prolyl endopeptidase-mediated destruction of T cell epitopes in whole gluten: chemical and immunological characterization. J Pharmacol Exp Ther. 2005;312:19–26. doi: 10.1124/jpet.104.073312. [DOI] [PubMed] [Google Scholar]
- 30.Pyle GG, Paaso B, Anderson BE, et al. Effect of pretreatment of food gluten with prolyl endopeptidase on gluten-induced malabsorption in celiac sprue. Clin Gastroenterol Hepatol. 2005;3:687–694. doi: 10.1016/s1542-3565(05)00366-6. [DOI] [PubMed] [Google Scholar]
- 31.Bethune MT, Strop P, Tang Y, Sollid LM, Khosla C. Heterologous expression, purification, refolding, and structural-functional characterization of EP-B2, a self-activating barley cysteine endoprotease. Chem Biol. 2006;13:637–647. doi: 10.1016/j.chembiol.2006.04.008. [DOI] [PubMed] [Google Scholar]
- 32.Siegel M, Bethune MT, Gass J, et al. Rational design of combination enzyme therapy for celiac sprue. Chem Biol. 2006;13:649–658. doi: 10.1016/j.chembiol.2006.04.009. [DOI] [PubMed] [Google Scholar]
- 33.Gass J, Vora H, Bethune MT, Gray GM, Khosla C. Effect of barley endoprotease EP-B2 on gluten digestion in the intact rat. J Pharmacol Exp Ther. 2006;318:1178–1186. doi: 10.1124/jpet.106.104315. [DOI] [PubMed] [Google Scholar]
- 34.Stepniak D, Spaenij-Dekking L, Mitea C, et al. Highly efficient gluten degradation with a newly identified prolyl endoprotease: implications for celiac disease. Am J Physiol Gastrointest Liver Physiol. 2006;291:G621–G629. doi: 10.1152/ajpgi.00034.2006. [DOI] [PubMed] [Google Scholar]
- 35.Gass J, Vora H, Hofmann AF, Gray GM, Khosla C. Enhancement of dietary protein digestion by conjugated bile acids. Gastroenterology. 2007;133:16–23. doi: 10.1053/j.gastro.2007.04.008. [DOI] [PubMed] [Google Scholar]
- 36.Mitea C, Havenaar R, Drijfhout JW, Edens L, Dekking L, Koning F. Efficient degradation of gluten by a prolyl endoprotease in a gastrointestinal model: implications for coeliac disease. Gut. 2008;57:25–32. doi: 10.1136/gut.2006.111609. [DOI] [PubMed] [Google Scholar]
- 37.Tye-Din JA, Anderson RP, Ffrench RA, et al. The effects of ALV003 pre-digestion of gluten on immune response and symptoms in celiac disease in vivo. Clin Immunol. 2010;134:289–295. doi: 10.1016/j.clim.2009.11.001. [DOI] [PubMed] [Google Scholar]
- 38.Ehren J, Moron B, Martin E, Bethune MT, Gray GM, Khosla C. A food-grade enzyme preparation with modest gluten detoxification properties. PLoS One. 2009;4:e6313. doi: 10.1371/journal.pone.0006313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liang L, Pinier M, Leroux JC, Subirade M. Interaction of alpha-gliadin with poly(HEMA-co-SS): structural characterization and biological implication. Biopolymers. 2009;91:169–178. doi: 10.1002/bip.21109. [DOI] [PubMed] [Google Scholar]
- 40.Liang L, Pinier M, Leroux JC, Subirade M. Interaction of alpha-gliadin with polyanions: design considerations for sequestrants used in supportive treatment of celiac disease. Biopolymers. 2010;93:418–428. doi: 10.1002/bip.21352. [DOI] [PubMed] [Google Scholar]
- 41.Pinier M, Verdu EF, Nasser-Eddine M, David CS, Vezina A, Rivard N, Leroux JC. Polymeric binders suppress gliadin-induced toxicity in the intestinal epithelium. Gastroenterology. 2009;136:288–298. doi: 10.1053/j.gastro.2008.09.016. [DOI] [PubMed] [Google Scholar]
- 42.Drago S, El AR, Di PM, et al. Gliadin, zonulin and gut permeability: Effects on celiac and non-celiac intestinal mucosa and intestinal cell lines. Scand J Gastroenterol. 2006;41:408–419. doi: 10.1080/00365520500235334. [DOI] [PubMed] [Google Scholar]
- 43.Duerksen DR, Wilhelm-Boyles C, Veitch R, Kryszak D, Parry DM. A comparison of antibody testing, permeability testing, and zonulin levels with small-bowel biopsy in celiac disease patients on a gluten-free diet. Dig Dis Sci. 2010;55:1026–1031. doi: 10.1007/s10620-009-0813-5. [DOI] [PubMed] [Google Scholar]
- 44.Smecuol E, Sugai E, Niveloni S, et al. Permeability, zonulin production, and enteropathy in dermatitis herpetiformis. ClinGastroenterolHepatol. 2005;3:335–341. doi: 10.1016/s1542-3565(04)00778-5. [DOI] [PubMed] [Google Scholar]
- 45.Tripathi A, Lammers KM, Goldblum S, et al. Identification of human zonulin, a physiological modulator of tight junctions, as prehaptoglobin-2. Proc Natl Acad Sci USA. 2009;106:16799–16804. doi: 10.1073/pnas.0906773106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang W, Uzzau S, Goldblum SE, Fasano A. Human zonulin, a potential modulator of intestinal tight junctions. J Cell Sci. 2000;113(Pt 24):4435–4440. doi: 10.1242/jcs.113.24.4435. [DOI] [PubMed] [Google Scholar]
- 47.Paterson BM, Lammers KM, Arrieta MC, Fasano A, Meddings JB. The safety, tolerance, pharmacokinetic and pharmacodynamic effects of single doses of AT-1001 in coeliac disease subjects: a proof of concept study. Aliment Pharmacol Ther. 2007;26:757–766. doi: 10.1111/j.1365-2036.2007.03413.x. [DOI] [PubMed] [Google Scholar]
- 48.Nanda N, Iismaa SE, Owens WA, Husain A, Mackay F, Graham RM. Targeted inactivation of Gh/tissue transglutaminase II. J Biol Chem. 2001;276:20673–20678. doi: 10.1074/jbc.M010846200. [DOI] [PubMed] [Google Scholar]
- 49.De Laurenzi V, Melino G. Gene disruption of tissue transglutaminase. Mol Cell Biol. 2001;21:148–155. doi: 10.1128/MCB.21.1.148-155.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Marrano C, de Macedo P, Gagnon P, Lapierre D, Gravel C, Keillor JW. Synthesis and evaluation of novel dipeptide-bound 1,2,4-thiadiazoles as irreversible inhibitors of guinea pig liver transglutaminase. Bioorg Med Chem. 2001;9:3231–3241. doi: 10.1016/s0968-0896(01)00228-0. [DOI] [PubMed] [Google Scholar]
- 51.de Macedo P, Marrano C, Keillor JW. Synthesis of dipeptide-bound epoxides and alpha,beta-unsaturated amides as potential irreversible transglutaminase inhibitors. Bioorg Med Chem. 2002;10:355–360. doi: 10.1016/s0968-0896(01)00292-9. [DOI] [PubMed] [Google Scholar]
- 52.Choi K, Siegel M, Piper JL, et al. Chemistry and biology of dihydroisoxazole derivatives: selective inhibitors of human transglutaminase 2. Chem Biol. 2005;12:469–475. doi: 10.1016/j.chembiol.2005.02.007. [DOI] [PubMed] [Google Scholar]
- 53.Watts RE, Siegel M, Khosla C. Structure-activity relationship analysis of the selective inhibition of transglutaminase 2 by dihydroisoxazoles. J Med Chem. 2006;49:7493–7501. doi: 10.1021/jm060839a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Duval E, Case A, Stein RL, Cuny GD. Structure-activity relationship study of novel tissue transglutaminase inhibitors. Bioorg Med Chem Lett. 2005;15:1885–1889. doi: 10.1016/j.bmcl.2005.02.005. [DOI] [PubMed] [Google Scholar]
- 55.Pardin C, Pelletier JN, Lubell WD, Keillor JW. Cinnamoyl inhibitors of tissue transglutaminase. J Org Chem. 2008;73:5766–5775. doi: 10.1021/jo8004843. [DOI] [PubMed] [Google Scholar]
- 56.Ozaki S, Ebisui E, Hamada K, Goto J, Suzuki AZ, Terauchi A, Mikoshiba K. Potent transglutaminase inhibitors, aryl beta-aminoethyl ketones. Bioorg Med Chem Lett. 2010;20:1141–1144. doi: 10.1016/j.bmcl.2009.12.011. [DOI] [PubMed] [Google Scholar]
- 57.Klöck CJ, Jin X, Choi K, Khosla C, Madrid PB, Spncer A, Raimundo BC, Boardman P, Lanza G, Griffin JH. Acylidene oxoindoles: A new class of reversible inhibitors of human transglutaminase 2. Bioorg Med Chem Lett. 2011 doi: 10.1016/j.bmcl.2010.12.037. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Siegel M, Strnad P, Watts RE, Choi K, Jabri B, Omary MB, Khosla C. Extracellular transglutaminase 2 is catalytically inactive, but is transiently activated upon tissue injury. PLoS ONE. 2008;3:e1861. doi: 10.1371/journal.pone.0001861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ruan Q, Johnson GV. Transglutaminase 2 in neurodegenerative disorders. Front Biosci. 2007;12:891–904. doi: 10.2741/2111. [DOI] [PubMed] [Google Scholar]
- 60.Shweke N, Boulos N, Jouanneau C, et al. Tissue transglutaminase contributes to interstitial renal fibrosis by favoring accumulation of fibrillar collagen through TGF-beta activation and cell infiltration. Am J Pathol. 2008;173:631–642. doi: 10.2353/ajpath.2008.080025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Shao M, Cao L, Shen C, et al. Epithelial-to-mesenchymal transition and ovarian tumor progression induced by tissue transglutaminase. Cancer Res. 2009;69:9192–9201. doi: 10.1158/0008-5472.CAN-09-1257. [DOI] [PubMed] [Google Scholar]
- 62.Xia J, Bergseng E, Fleckenstein B, Siegel M, Kim CY, Khosla C, Sollid LM. Cyclic and dimeric gluten peptide analogues inhibiting DQ2-mediated antigen presentation in celiac disease. Bioorg Med Chem. 2007;15:6565–6573. doi: 10.1016/j.bmc.2007.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kapoerchan VV, Wiesner M, Overhand M, van der Marel GA, Koning F, Overkleeft HS. Design of azidoproline containing gluten peptides to suppress CD4+ T-cell responses associated with celiac disease. Bioorg Med Chem. 2008;16:2053–2062. doi: 10.1016/j.bmc.2007.10.091. [DOI] [PubMed] [Google Scholar]
- 64.Kapoerchan VV, Wiesner M, Hillaert U, et al. Design, synthesis and evaluation of high-affinity binders for the celiac disease associated HLA-DQ2 molecule. Mol Immunol. 2010;47:1091–1097. doi: 10.1016/j.molimm.2009.10.036. [DOI] [PubMed] [Google Scholar]
- 65.Jüse U, van de Wal Y, Koning F, Sollid LM, Fleckenstein B. Design of new high-affinity peptide ligands for human leukocyte antigen-DQ2 using a positional scanning peptide library. Hum Immunol. 2010;71:475–481. doi: 10.1016/j.humimm.2010.01.021. [DOI] [PubMed] [Google Scholar]
- 66.Larche M, Wraith DC. Peptide-based therapeutic vaccines for allergic and autoimmune diseases. Nat Med. 2005;11:S69–S76. doi: 10.1038/nm1226. [DOI] [PubMed] [Google Scholar]
- 67.Oldfield WL, Larche M, Kay AB. Effect of T-cell peptides derived from Fel d 1 on allergic reactions and cytokine production in patients sensitive to cats: a randomised controlled trial. Lancet. 2002;360:47–53. doi: 10.1016/s0140-6736(02)09332-7. [DOI] [PubMed] [Google Scholar]
- 68.Verhoef A, Alexander C, Kay AB, Larche M. T cell epitope immunotherapy induces a CD4+ T cell population with regulatory activity. PLoS Med. 2005;2:e78. doi: 10.1371/journal.pmed.0020078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Arentz-Hansen H, Körner R, Molberg, et al. The intestinal T cell response to α-gliadin in adult celiac disease is focused on a single deamidated glutamine targeted by tissue transglutaminase. J Exp Med. 2000;191:603–612. doi: 10.1084/jem.191.4.603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Tye-Din JA, Stewart JA, Dromey JA, et al. Comprehensive, quantitative mapping of T cell epitopes in gluten in celiac disease. Sci Transl Med. 2010;2 doi: 10.1126/scitranslmed.3001012. 41ra51. [DOI] [PubMed] [Google Scholar]
- 71.Tollefsen S, Arentz-Hansen H, Fleckenstein B, et al. HLA-DQ2 and -DQ8 signatures of gluten T cell epitopes in celiac disease. J Clin Invest. 2006;116:2226–2236. doi: 10.1172/JCI27620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Huibregtse IL, Marietta EV, Rashtak S, et al. Induction of antigen-specific tolerance by oral administration of Lactococcus lactis delivered immunodominant DQ8-restricted gliadin peptide in sensitized nonobese diabetic Abo DQ8 transgenic mice. J Immunol. 2009;183:2390–2396. doi: 10.4049/jimmunol.0802891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Beaurepaire C, Smyth D, McKay DM. Interferon-gamma regulation of intestinal epithelial permeability. J Interferon Cytokine Res. 2009;29:133–144. doi: 10.1089/jir.2008.0057. [DOI] [PubMed] [Google Scholar]
- 74.Utech M, Ivanov AI, Samarin SN, et al. Mechanism of IFN-gamma-induced endocytosis of tight junction proteins: myosin II-dependent vacuolarization of the apical plasma membrane. Mol Biol Cell. 2005;16:5040–5052. doi: 10.1091/mbc.E05-03-0193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Gonzalez-Mariscal L, Lechuga S, Garay E. Role of tight junctions in cell proliferation and cancer. Prog Histochem Cytochem. 2007;42:1–57. doi: 10.1016/j.proghi.2007.01.001. [DOI] [PubMed] [Google Scholar]
- 76.McKerracher L, Higuchi H. Targeting Rho to stimulate repair after spinal cord injury. J Neurotrauma. 2006;23:309–317. doi: 10.1089/neu.2006.23.309. [DOI] [PubMed] [Google Scholar]
- 77.LoGrasso PV, Feng Y. Rho kinase (ROCK) inhibitors and their application to inflammatory disorders. Curr Top Med Chem. 2009;9:704–723. doi: 10.2174/156802609789044452. [DOI] [PubMed] [Google Scholar]
- 78.Reinisch W, de Villiers W, Bene L, et al. Fontolizumab in moderate to severe Crohn's disease: a phase 2, randomized, double-blind, placebo-controlled, multiple-dose study. Inflamm Bowel Dis. 2010;16:233–242. doi: 10.1002/ibd.21038. [DOI] [PubMed] [Google Scholar]
- 79.Herold KC, Gitelman S, Greenbaum C, et al. Treatment of patients with new onset Type 1 diabetes with a single course of anti-CD3 mAb Teplizumab preserves insulin production for up to 5 years. Clin Immunol. 2009;132:166–173. doi: 10.1016/j.clim.2009.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Wiczling P, Rosenzweig M, Vaickus L, Jusko WJ. Pharmacokinetics and pharmacodynamics of a chimeric/humanized anti-CD3 monoclonal antibody, otelixizumab (TRX4), in subjects with psoriasis and with type 1 diabetes mellitus. J Clin Pharmacol. 2010;50:494–506. doi: 10.1177/0091270009349376. [DOI] [PubMed] [Google Scholar]
- 81.Sandborn WJ, Colombel JF, Frankel M, et al. Anti-CD3 antibody visilizumab is not effective in patients with intravenous corticosteroid-refractory ulcerative colitis. Gut. 2010;59:1485–1492. doi: 10.1136/gut.2009.205443. [DOI] [PubMed] [Google Scholar]
- 82.Chatenoud L. Immune therapy for type 1 diabetes mellitus-what is unique about anti-CD3 antibodies? Nat Rev Endocrinol. 2010;6:149–157. doi: 10.1038/nrendo.2009.275. [DOI] [PubMed] [Google Scholar]
- 83.Edwards JC, Szczepanski L, Szechinski J, et al. Efficacy of B-cell-targeted therapy with rituximab in patients with rheumatoid arthritis. N Engl J Med. 2004;350:2572–2581. doi: 10.1056/NEJMoa032534. [DOI] [PubMed] [Google Scholar]
- 84.Hauser SL, Waubant E, Arnold DL, et al. B-cell depletion with rituximab in relapsing-remitting multiple sclerosis. N Engl J Med. 2008;358:676–688. doi: 10.1056/NEJMoa0706383. [DOI] [PubMed] [Google Scholar]
- 85.Pescovitz MD, Bloom R, Pirsch J, Johnson J, Gelone S, Villano SA. A randomized, double-blind, pharmacokinetic study of oral maribavir with tacrolimus in stable renal transplant recipients. Am J Transplant. 2009;9:2324–2330. doi: 10.1111/j.1600-6143.2009.02768.x. [DOI] [PubMed] [Google Scholar]
- 86.Sollid LM, Molberg, McAdam S, Lundin KE. Autoantibodies in coeliac disease: tissue transglutaminase--guilt by association? Gut. 1997;41:851–852. doi: 10.1136/gut.41.6.851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Mei HE, Frolich D, Giesecke C, et al. Steady state generation of mucosal IgA+ plasmablasts is not abrogated by B cell depletion therapy with rituximab. Blood. 2010 doi: 10.1182/blood-2010-01-266536. [DOI] [PubMed] [Google Scholar]
- 88.Yokoyama S, Watanabe N, Sato N, et al. Antibody-mediated blockade of IL-15 reverses the autoimmune intestinal damage in transgenic mice that overexpress IL-15 in enterocytes. Proc Natl Acad Sci USA. 2009;106:15849–15854. doi: 10.1073/pnas.0908834106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Walters MJ, Wang Y, Lai N, et al. Characterization of CCX282-B, an orally bioavailable antagonist of the CCR9 chemokine receptor, for treatment of inflammatory bowel disease. J Pharmacol Exp Ther. 2010;335:61–69. doi: 10.1124/jpet.110.169714. [DOI] [PubMed] [Google Scholar]
- 90.Anderson RP, van Heel DA, Tye-Din JA, Barnardo M, Salio M, Jewell DP, Hill AV. T cells in peripheral blood after gluten challenge in coeliac disease. Gut. 2005;54:1217–1223. doi: 10.1136/gut.2004.059998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Ráki M, Fallang LE, Brottveit M, Bergseng E, Quarsten H, Lundin KE, Sollid LM. Tetramer visualization of gut-homing gluten-specific T cells in the peripheral blood of celiac disease patients. Proc Natl Acad Sci USA. 2007;104:2831–2836. doi: 10.1073/pnas.0608610104. [DOI] [PMC free article] [PubMed] [Google Scholar]