

Abstract
Pseudomonas elastase (LasB), a metalloprotease virulence factor, is known to play a pivotal role in pseudomonal infection. LasB is secreted at the site of infection, where it exerts a proteolytic action that spans from broad tissue destruction to subtle action on components of the host immune system. The former enhances invasiveness by liberating nutrients for continued growth, while the latter exerts an immunomodulatory effect, manipulating the normal immune response. In addition to the extracellular effects of secreted LasB, it also acts within the bacterial cell to trigger the intracellular pathway that initiates growth as a bacterial biofilm. The key role of LasB in pseudomonal virulence makes it a potential target for the development of an inhibitor as an antimicrobial agent. The concept of inhibition of virulence is a recently established antimicrobial strategy, and such agents have been termed “second-generation” antibiotics. This approach holds promise in that it seeks to attenuate virulence processes without bactericidal action and, hence, without selection pressure for the emergence of resistant strains. A potent inhibitor of LasB, N-mercaptoacetyl-Phe-Tyr-amide (Ki = 41 nM) has been developed, and its ability to block these virulence processes has been assessed. It has been demonstrated that thes compound can completely block the action of LasB on protein targets that are instrumental in biofilm formation and immunomodulation. The novel LasB inhibitor has also been employed in bacterial-cell-based assays, to reduce the growth of pseudomonal biofilms, and to eradicate biofilm completely when used in combination with conventional antibiotics.
INTRODUCTION
Bacterial toxins, with enzymatic activity on mammalian tissues, include some of the most toxic substances known. Tetanus toxin, botulinum toxin, and anthrax toxin are key examples, each of which is a zinc-metalloprotease virulence factor secreted by its respective bacterial strain (8). Pseudolysin is the key zinc metalloprotease virulence factor secreted by the opportunistic pathogen Pseudomonas aeruginosa and is also known as LasB or pseudomonas elastase (32).
This virulence factor is highly toxic, causing tissue damage and invasion, processing components of the immune system to cause immunomodulation (58), and acting intracellularly to initiate bacterial biofilm growth (20). These three collective virulence mechanisms of LasB are potentially of great significance in the progression to a chronic infection. First, the direct tissue destruction in the host liberates nutrients for bacterial growth, accelerating the general assault on host tissues. This also contributes to an excess of proteolytic activity at the site of infection that upsets the balance of proteolysis in the host.
Second, the action of LasB on components of the immune system and the immunomodulation that results manipulate the host immune system into a destructive inflammatory cycle (28). Third, LasB initiates the biofilm pathway through activation of nucleoside diphosphate kinase (NDK) within the bacterial cell (20). Once formed, biofilms are highly resistant to the immune response and to antibiotics. The inflammatory response raised against the biofilm matrix is ineffectual in clearing the biofilm and instead perpetuates the inflammatory cycle in the host (17, 29, 34, 43). The biofilm also releases planktonic bacterial cells, again contributing to the inflammatory response and maintaining the infection (18).
Chronic pseudomonal infections are therefore characterized by a protracted self-perpetuating “vicious cycle” of host-derived inflammation and tissue destruction that is well defined and that impedes the normal clearance of the bacteria (47). The balance is tipped toward an environment with excess immune, inflammatory, oxidative, and proteolytic activity, which in turn triggers further inflammation and destruction. The influence of LasB is a potential underlying trigger of this sustained host-derived inflammatory environment that persists during chronic intractable infections by P. aeruginosa.
Chronic infections in the cystic fibrosis (CF) lung are perhaps the most commonly cited example of this self-amplifying exacerbation process, with P. aeruginosa recognized as the key pathogen. Exacerbations of CF are chronic biofilm-based infections and are characterized by a sustained alteration in the balance of host inflammation (44). However, several other chronic pseudomonal infections are also characterized by misdirected and chronic inflammatory and immune responses that show homology to the vicious cycle in the CF lung. These include leg ulcers, burn infections, septicemia, keratitis, and pneumonia (2, 15, 16, 45, 48, 49, 62).
The collective virulence mechanisms of LasB, therefore, implicate the protease as a key target for virulence inhibition. The attenuation of LasB-mediated virulence could simultaneously inhibit the destructive action of LasB in the host and exert an antibiofilm effect in the bacterial cell. It is hoped that this antivirulence mechanism might prevent the subtle mechanisms by which LasB is proposed to evade and manipulate the host immune system in the establishment of a chronic infection.
Inhibition of bacterial virulence factors has been suggested previously and has gained momentum recently as an antimicrobial strategy that is nondestructive to the bacteria. It has recently been proposed that such virulence inhibitors could constitute a second-generation class of antibiotics (59). By attenuating virulence mechanisms without challenging bacterial-cell viability directly, these second-generation antibiotic agents would potentially place little or no pressure on the bacterial cell for the emergence of resistant strains (9, 59).
In order to conduct a comprehensive investigation of LasB inhibition, a dipeptide library was designed to include every possible variation of the 20 naturally occurring amino acids at the P′1 and P′2 positions. This yielded a library of 202 (400) compounds produced by solid-phase synthesis and subsequently tested against purified LasB using a fluorogenic substrate in a spectrofluorometric assay.
These compounds have been evaluated for the ability to block the in vitro activity of LasB on two of its in vivo targets: immunoglobulin G (IgG) as a human immune component and NDK of the bacterial biofilm pathway. LasB inhibitors have been assessed for antibiofilm activity, alone and in synergy with antimicrobials. These combinations were tested for the ability both to inhibit the formation of growing biofilm and to eradicate established biofilms.
MATERIALS AND METHODS
Chemistry.
Inhibitors were synthesized according to a dipeptide template shown in Fig. 1, with all possible variations of naturally occurring amino acids present. This methodology is based upon the procedures detailed previously by our group (33). The mercaptoacetyl metal-chelating functionality was incorporated onto the N terminus of the resin-bound dipeptide through the use of an S-trityl-protected thioglycolic acid intermediate. This was coupled using standard peptide synthesis procedures, employing O-benzotriazole-N,N,N′,N′-tetramethyl-uronium-hexafluorophosphate (HBTU) and 1-hydroxybenzotriazole (HOBt) as coupling reagents (26).
Fig. 1.

(a) N-Alpha-mercaptoacetyl dipeptide inhibitor template. Four hundred inhibitors were synthesized, with the dipeptide template allowing all possible combinations of 20 naturally occurring amino acid side chains. These variable regions are indicated by “X” in this structure and refer to the P′1 and P′2 positions on the inhibitor molecule when read from left to right (Fig. 2). (b) Structure of the first lead inhibitor identified in this study, HS-CH2-CO-Trp-Tyr-NH2 (Ki = 40.5 nM). (c) Structure of the second lead inhibitor, HS-CH2-CO-Phe-Tyr-NH2 (Ki = 41 nM).
Inhibitors were synthesized at a scale of 0.5 mmol by automated peptide synthesis on an Advanced Chemtech 396 Automated Peptide Synthesizer. The synthesis of the target inhibitor templates proceeded without difficulty, except in those sequences containing very hydrophobic amino acids. In these instances, the syntheses were repeated using a CEM Liberty microwave-assisted peptide synthesizer with coupling conditions of 18 W, 75°C, and 300 s. The identity and purity of all peptide products were confirmed using reversed-phase high-performance liquid chromatography (HPLC) and electrospray mass spectrometry.
Kinetic screening.
Each peptide was screened at 50 μM for inhibitory activity against purified pseudomonas elastase. Pseudolysin was obtained from Elastin Products Company (MO) and was stored in aliquots of 100 μg/ml stock solution in assay buffer containing 0.05 M Tris-HCl, 2.5 mM CaCl2, 1% (vol/vol) dimethyl formamide (DMF), pH 7.2. When required, these were diluted for use at a final concentration of 1 ng per 100-μl well. Inhibition was monitored using a microtiter-based fluorimetric assay following the hydrolysis of the fluorogenic substrate aminobenzoyl-Ala-Gly-Leu-Ala-p-nitro-benzyl-amide at 400 μM (42). The change in fluorescence was monitored using a BMG Fluostar Optima fluorescence microplate reader at an excitation wavelength of 330 ± 10 nm and an emission wavelength of 460 ± 10 nm. Inhibitor sequences exhibiting activity against LasB were then investigated further over an extended concentration range in order to determine the Ki for each. Peptides were prepared as stock solutions in DMF at 100 times the required concentrations for use at 1 in 100 dilution. This produced the correct working concentration, with only 1% overall organic solvent present in the well. A typical dose-response relationship for HS-CH2-CO-Y-V-NH2 is shown in Fig. S1 in the supplemental material, demonstrating the competitive nature of the inhibition.
Ki values were calculated using a derivation of the Michaelis-Menten equation (61). A Km value for this substrate, required for the calculation, was experimentally determined as 0.24 μM using a Lineweaver-Burk Plot. Potent inhibitors of Ki less than approximately 1 μM did not fit the standard linear transformation required for determination of Ki via Michaelis-Menten kinetics. This applied to the top 4 inhibitors in this library, so these compounds were treated via “tight-binding” kinetics by analysis with Morrison plots.
Recombinant expression of nucleoside diphosphate kinase.
The gene for pseudomonal NDK, coding for the 16-kDa inactive form of the kinase enzyme, was obtained from the genome of the reference strain, PA01. A hexahistidine tag was included for purification and detection of the expressed protein. It had been proposed previously that NDK is activated by LasB by processing it from the inactive 16-kDa form to the 12-kDa active kinase (20). As a result, part of the overall protein structure would be lost during activation, potentially including the N-terminal His tag.
Two separate sets of primers were therefore designed and synthesized by Eurofins MWG Operon (Ebersberg, Germany). One contained an N-terminal hexahistidine tag, and another incorporated both an N-terminal hexahistidine tag and a C-terminal Flag tag in order to investigate the location of the cleaved fragment. Both variants described above were expressed, and on subsequent detection, the N-terminally His-tagged NDK variant was confirmed to have retained the His tag on the active 12-kDa fragment, so it was used in all subsequent steps.
Primers.
The DNA sequence for NDK from P. aeruginosa PA01 was retrieved from PubMed for primer design. A hexahistidine tag was also included in the sequence at the 3′ end to aid detection of the expressed protein. The gene products were amplified by PCR and incorporated into the pQE30 Xa vector, using Bam and Sal restriction enzymes. The primers were as follows: NDK forward primer incorporating a His tag at the 3′ end, TTTTTTGGATCCGCACTGCAACGCACCCTGTCCATCATC; NDK reverse primer, TTTTTTGTCGACTCAGCGAATGCGCTCGCAGACTTCG.
NDK expression.
Escherichia coli strain TOP10 transformed with the plasmid was grown on agar plates containing ampicillin at 50 μg/ml, and three colonies of the transformed E. coli were retained. A single colony was obtained from one of these samples and used to inoculate 100 ml of nutrient broth supplemented with ampicillin, which was grown for a further 24 h at 37°C.
Isopropyl-β-d-thiogalactopyranoside (IPTG) at a concentration of 0.5 mM was then used to induce NDK expression. The cells were incubated for a further 3 h, and then the culture was centrifuged at 15,000 × g for 5 min. The pellet was resuspended in medium, pelleted at 15,000 × g for 2 min, and then resuspended in distilled water. Twenty-second bursts of sonication were performed for this concentrated solution and repeated 20 times at 100 mHz while the solution was cooled on ice. The lysate was centrifuged at 15,000 × g for 2 min to separate the cell lysate and debris. The supernatant of the crude cell lysate containing recombinant NDK was filtered and retained for analysis by gel electrophoresis and Western blot analysis.
Gel electrophoresis and Western blotting.
Polyacrylamide gel electrophoresis (SDS-PAGE) was carried out on both NDK and IgG to demonstrate the action of LasB on these proteins and its subsequent prevention by LasB inhibitors. Samples of IgG and NDK underwent a 1-h incubation with LasB (500 ng per lane) at 37°C, with and without inhibitors. This was followed by electrophoresis on a 4 to 12% Tris polyacrylamide gel at 200 V, 120 mA, and 25 W for 35 min. Gels containing IgG (12 μg per lane) were stained using Coomassie brilliant blue stain, with IgG visible as a dual band consisting of the light chain at 28 kDa and the heavy chain at 54 kDa.
Gels containing NDK were transferred to nitrocellulose membranes for Western blot analysis, using a Bio-Rad Trans-Blot SD semidry transfer cell, for 80 min at a constant setting of 0.11 mA and voltage fixed at up to 200 V. Nonspecific binding sites were blocked by 1-h incubation with 5% skim milk protein in phosphate-buffered saline. Protein bands were labeled using anti-His antibody conjugated to horseradish peroxidase at 1 in 10,000 in 10 ml of 0.5% skim milk protein in phosphate-buffered saline containing 0.1% Tween. Subsequent detection of antibody-bound protein was achieved using Millipore chemiluminescent horseradish peroxidase substrate, with development on Kodak Biomax photographic film.
MBEC assay.
Sterile minimum biofilm eradication concentration (MBEC) plates were obtained from Innovotec Inc., Edmonton, AB, Canada, and the standard method was followed (7). The MBEC plate was placed in a sealed container with a source of humidity, using an inoculum of 5 × 105 CFU/ml P. aeruginosa PA01, accession number 10548. The plate was placed in an orbital incubator at 37°C at 100 rpm to generate a shear force. Assays measuring biofilm formation were conducted over 24 h from the introduction of this inoculum to the plate, along with the test agents. Established biofilms for eradication testing were first grown for 48 h before the 24-h test period of challenge with the relevant agents.
Antimicrobials were prepared in sterile distilled water, while LasB inhibitors were prepared in dimethyl formamide (DMF). Inhibitors in DMF were prepared as stock solutions at 100 times the requisite concentrations and added to test wells at 1% (vol/vol) for 100-fold dilution. This achieved the required inhibitor concentration while keeping the DMF concentration at or below 1% of the total well contents. DMF was also added to all control wells, and six replicates of each test well were included.
For recovery of the biofilm, the MBEC lid was removed and the pegs were suspended in a series of 3 rinse plates (physiological saline) for 2 min per wash to remove planktonic bacteria. The pegs were then immersed in a plate containing 1% universal neutralizing solution in medium supplemented with 20 g/liter saponin and 10 g/liter Tween 80, adjusted to pH 7.0 using NaOH (7).
The pegs were immersed in this medium supplemented with neutralizing solution, without detachment from the lid. For sonication, the pegs were placed in a sterile 96-well plate containing neutralizing solution in medium. The pegs were either sonicated as part of the intact lid of the MBEC plate or detached, and where appropriate, serial dilution was performed for enumeration of cell numbers using the technique of Miles et al. (36).
Samples of the fluid medium from the 96-well plate after removal of the lid were also enumerated by Miles et al. counting for determination of planktonic cell numbers. The dilution range from 1 in 10−3 to 1 in 10−8 was plated and grown for the planktonic samples. For the lower cell numbers of the biofilm samples, 10−1 to 10−5 were plated and grown. For each test sample in the original plate, 6 of the dilutions were plated out at 6 spots per dilution, totalling 36 spots per sample.
Crystal violet staining was also employed as a means to quantify the biofilm biomass. Assays were conducted on 96-well plates, following the formats described above. The crystal violet staining was carried out as a modification of the method of Stepanović et al. (54). After the growth period of 24 h, the contents were removed, and the wells were washed three times with 250 μl of sterile PBS solution. Adherent biomass was fixed to the wells by adding methanol for 10 min and then removing and air drying it for 10 min.
The biofilm biomass was then stained with 180 μl of crystal violet stain prepared at 1% in 1:4 ethanol-distilled water. The crystal violet stain was removed after 10 min, and the plate was rinsed well. The wells were destained with 33% acetic acid in distilled water, and the biomass was determined by measuring the dye content of this solution spectrophotometrically, using a Biotek EL808 spectrophotometric plate reader at 570 nm.
RESULTS AND DISCUSSION
Screening of LasB inhibitors.
The kinetic data for the inhibitor library are summarized in Table 1. Micromolar Ki values are displayed, and the data are tabulated according to the amino acid residues populating the P′1 and P′2 positions. Solid-phase peptide synthesis constructs the peptide molecule C to N terminally, and hence, the P′2 residue was the first coupled onto the resin. Inhibitors were therefore synthesized in 20 batches of 20 arranged in libraries of 20 compounds, each with a common residue at P′2 (shown in Table S1 and Fig. S2 in the supplemental material, along with their respective Ki values).
Table 1.
Top 20 LasB inhibitors ranked by Ki value
| Rank | Structure | Ki (μM) |
|---|---|---|
| 1 | HS-CH2-CO-Trp-Tyr-NH2 | 0.0405 |
| 2 | HS-CH2-CO-Phe-Tyr-NH2 | 0.041 |
| 3 | HS-CH2-CO-Phe-Gln-NH2 | 0.754 |
| 4 | HS-CH2-CO-Tyr-Val-NH2 | 0.768 |
| 5 | HS-CH2-CO-Trp-Ile-NH2 | 1.03 |
| 6 | HS-CH2-CO-Trp-Phe-NH2 | 1.10 |
| 7 | HS-CH2-CO-Ile-Leu-NH2 | 1.26 |
| 8 | HS-CH2-CO-Ile-Val-NH2 | 1.83 |
| 9 | HS-CH2-CO-Ile-Tyr-NH2 | 2.06 |
| 10 | HS-CH2-CO-Tyr-Tyr-NH2 | 2.08 |
| 11 | HS-CH2-CO-Phe-Met-NH2 | 2.82 |
| 12 | HS-CH2-CO-Tyr-Arg-NH2 | 2.95 |
| 13 | HS-CH2-CO-Ile-Thr-NH2 | 3.41 |
| 14 | HS-CH2-CO-His-Ala-NH2 | 3.62 |
| 15 | HS-CH2-CO-Met-Tyr-NH2 | 3.65 |
| 16 | HS-CH2-CO-Trp-Leu-NH2 | 3.66 |
| 17 | HS-CH2-CO-Ile-Gln-NH2 | 3.73 |
| 18 | HS-CH2-CO-Met-Lys-NH2 | 3.86 |
| 19 | HS-CH2-CO-Trp-Val-NH2 | 4.07 |
| 20 | HS-CH2-CO-Phe-Ile-NH2 | 4.47 |
Two compounds with particularly high activity against LasB were produced, as shown in Fig. 1. HS-CH2-CO-Phe-Tyr-NH2 (Ki = 41 nM) and HS-CH2-CO-Trp-Tyr-NH2 (Ki = 40.5 nM) had potencies an order of magnitude greater than that of the next-nearest compound (HS-CH2-CO-Phe-Gln-NH2 [Ki = 754 nM]). These two compounds are structurally similar, and when tested at 100 μM, they were without effect on MMP2, and MMP9 and caused only ∼6.0% inhibition of MMP8 in comparison to the control, indicating specificity for the LasB protease.
Since the two lead compounds have comparable potencies, HS-CH2-CO-Phe-Tyr-NH2 (Ki = 41 nM) was used as the lead compound in biofilm assays. It was judged to possess a more “drug-like” character according to commonly used measures, such as the Lipinski rule of five (31). However, the abilities of a number of other LasB inhibitors to block biofilm production/formation were also assessed for comparative purposes. Sequences with slightly lower inhibitory potency against LasB were included, such as mercaptoacetyl-Val-Ile-amide (Ki = 9.0 μM) and mercaptoacetyl-Trp-Phe-amide (Ki = 1.1 μM).
The presence of tyrosine at P′2 on both inhibitor structures demonstrates the preference for aromatic residues at this position and suggests contact with a hydrophilic region of the active site through the hydroxyl group of tyrosine. Bulky aromatic groups at P′1 were also favored, and some dipeptide templates containing combinations of aromatic and aliphatic residues also appeared in the top 20 inhibitors, as listed in Table 1.
The active-site binding properties of LasB are known, with the S′1 subsite generally accepted as the principal determinant of specificity, with a preference for bulky aromatic residues at this position (41, 42). In accordance with this, Phe, Trp, and Tyr show particular success at P′1. Initial inspection of the inhibition data for the full library indicates that certain residues at the P′1 position can produce consistently good inhibitors (see Fig. S2 in the supplemental material, left axis). For example, the groups corresponding to P′1 Ile, Phe, Trp, and Tyr, and to a lesser extent Met and Val, show definite prominence.
Table 1 reveals that Trp, and perhaps also Tyr, was the only pharmacophore at P′1 that consistently produced good inhibitors almost irrespective of the P′2 residue. This was true even when it was coupled to an acidic residue at P′2, which generally produced poor inhibitors in other cases. This suggests that the contacts that Trp or Tyr make within the S′1 site are a predominant factor in inhibitor binding and that the P′1 residue in general could be considered the principal determinant of specificity.
Investigation of the P′2 position again shows that the effective inhibitors occupy sets that are largely confined to the same P′2 residues. In particular, Ile, Phe, Leu, Val, Gln, and Tyr, and to a lesser extent Arg, Lys, Asp, and Thr, are predominant. This binding pocket again shows a strong preference for bulky hydrophobic residues, both aromatic and aliphatic. However, the P′2 residue has a much less consistent effect on inhibitor binding (see Fig. S3 and S4 in the supplemental material).
The X-ray crystal structure of the active site is shown in Fig. 2. The S′1 and S′2 sites can be seen to be predominantly hydrophobic, with a deep pocket at S′1 and a shallower site at S′2. Areas of hydrophilic character can be seen immediately beyond the nonpolar S′2 pocket, extending in all directions. Inhibitors containing extended/elongated P′2 side chains, and containing ionizable groups, could make favorable interactions with these polar regions just adjacent to the S′2 pocket. Accordingly, the particularly frequent appearance of tyrosine at the P′2 position in the top 10 inhibitors might be a consequence of this.
Fig. 2.

LasB active site determined by X-ray crystallography, with the inhibitor-binding region outlined in green. The zinc atom can be seen as a blue ball, and the positions of the S′1 and S′2 binding pockets, which bind the P′1 and P′2 amino acid residues of the inhibitor, respectively, are shown. Red shading denotes hydrophobic regions, while blue shading can be seen in hydrophilic areas of the protein surface. The image was produced using UCSF Chimera, from the Resource for Biocomputing, Visualization, and Informatics, University of California, San Fancisco, CA.
Although the pKa of the phenolic grouping of tyrosine is approximately 10.0, making it relatively difficult to form its conjugate base at physiological pH, it is conceivable that electrostatic interactions could take place between this inhibitor residue and a strongly basic residues at this location on the peptidase. Similarly, glutamine possesses a long side chain, with a strong dipole at its end.
Proteolytic targets of LasB.
LasB is a broad-spectrum metalloprotease, and the range of host molecules degraded during infection reflects this (40). Secreted LasB is known to degrade immunoglobulins, complement factors, and cytokines involved in normal immunity, manipulating the host immune system during infection (28). The immunomodulatory effect is widespread, from activation of the Hageman factor/prekallikrien-kinin cascade to degradation of fibrinogen, leukotaxis and the resulting protease release, and inactivation of host antiproteases and the disabling of cell surface receptors, such as PARs (15, 19, 27, 30, 39). LasB is also known to process many biomolecule components of the host immune system, such as IgG (10).
Host proteinase inhibitors, such as α2-macroglobulin, α1-antitrypsin, inter-α-inhibitor, and secretory leukocyte protease inhibitor (SLPI), are also inactivated by LasB. These antiproteases normally exert an inhibitory influence on host neutrophil elastase, so the silencing of these antiproteolytic effects alters the balance toward excess proteolysis in the host. This excess proteolysis accelerates the destruction of host antiproteases and so exerts a positive-feedback effect and creates a vicious cycle of proteolysis.
Each of the many components of the host immune system that are processed by LasB potentially leads to similar proinflammatory consequences. These include reduction of bacterial clearance, leukotaxis, generation of excessive proteolytic activity, and tissue destruction. It is perhaps noteworthy that inhaled α1-antitrypsin, a natural LasB inhibitor, significantly reduces airway inflammation in CF (14). In addition to this, inhibitors of this virulence factor have previously been demonstrated to protect host corneal tissue from the destruction that characterizes pseudomonal eye infection (23).
Mice immunized with LasB peptide suffered less severe lung infection under either P. aeruginosa or Burkholderia cepacia challenge (52). Similarly, Dulon, using a commercially available pseudolysin inhibitor, was able to significantly reduce the PAR-2 deactivation occurring on exposure to P. aeruginosa (12). The key role of pseudolysin in mediating both the defense and virulence of this opportunistic pathogen (3) suggests the possibility that inhibitors of LasB might prevent the establishment and perpetuation of chronic infections caused by P. aeruginosa.
The presence of biofilms at the site of infection also contributes to the sustained and ineffectual inflammation during chronic infection (13, 17, 18, 29, 34, 35, 43), making eradication of biofilm a desirable goal in a therapeutic context. The role of LasB as an essential mediator of alginate synthesis and biofilm formation is based on activation of NDK by proteolytic processing (21). LasB converts NDK from 16 kDa to its 12-kDa active form by removal of a 4-kDa fragment, which then generates GTP for production and secretion of alginate exopolysaccharide (20).
NDK is therefore the natural LasB substrate in the biofilm pathway, so the processing of NDK by LasB was monitored, as shown in Fig. 3 and 4. IgG has also been chosen as a representative of the various components of the host immune system degraded by LasB (Fig. 5). The ability of LasB inhibitors to prevent the processing of these two proteinaceous LasB substrates was therefore evaluated in the assays shown in Fig. 3 to 5.
Fig. 3.
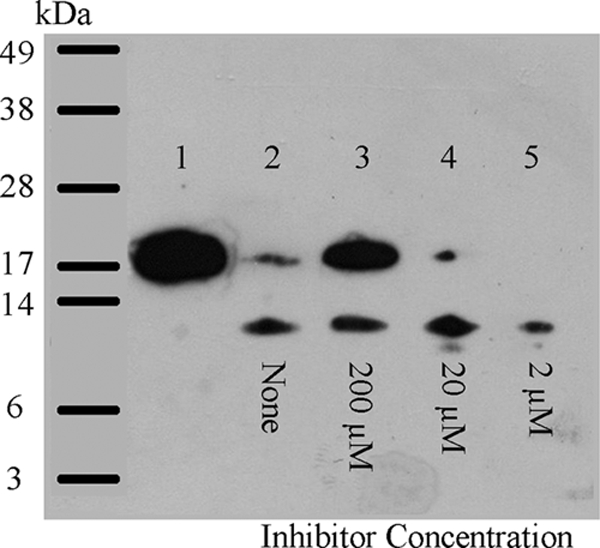
Inhibition of LasB-catalyzed activation of recombinant NDK by HS-CH2-CO-Trp-Phe-NH2 (Ki = 1.1 μM). Samples of recombinant NDK (5 μg per lane) were incubated with LasB (0.5 μg per lane) for 60 min at 37°C in the absence and presence of inhibitor and were then subjected to SDS-PAGE, followed by Western blotting. Lane 1, NDK alone; lane 2, NDK plus LasB; lanes 3 to 5, like lane 2 but with 200 μM, 20 μM, and 2 μM HS-CH2-CO-Trp-Phe-NH2, respectively.
Fig. 5.

Inhibition of LasB-catalyzed degradation of human IgG by HS-CH2-CO-Phe-Tyr-NH2 (Ki = 41 nM). IgG was used at 10 μg per lane and appears as a dual band consisting of the light chain (28 kDa) and the heavy chain (54 kDa). LasB was added at 0.5 μg per lane, with a 1-h incubation at 37°C. Lane 1, IgG (negative control); lane 2, IgG plus LasB (positive control); lanes 3 to 7, like lane 2 but including HS-CH2-CO-Phe-Tyr-NH2 across the indicated range of concentrations from 25 μM to 1.6 μM.
Electrophoretic analysis of both IgG and NDK demonstrated that inhibitors of LasB were indeed able to prevent degradation/processing of these proteins, respectively. The recombinant NDK prepared in the present study migrates as a band of apparent molecular mass just slightly greater than 16 kDa (Fig. 3), which is slightly larger than that reported for the native NDK protein, owing to the presence of the hexahistidine tag. It can be seen from Fig. 4 that it was possible to completely block the action of LasB on NDK. Inhibitors with a range of potencies against LasB produced a gradient of inhibition of the LasB-mediated processing of NDK. The degree of processing of the proteinaceous substrate was in relatively good agreement with the Ki values, as determined using the synthetic fluorogenic substrate.
Fig. 4.

Comparison of the rank potencies of inhibitors to block the LasB-catalyzed activation of recombinant NDK. Samples of recombinant NDK (5 μg per lane) were incubated with LasB (0.5 μg per lane) for 60 min at 37°C in the presence of each inhibitor (used at a fixed concentration of 200 μM) and were then subjected to SDS-PAGE, followed by Western blotting. Lane 1, NDK (negative control); lane 8, NDK plus LasB (positive control). Lanes 2 to 7 were like lane 8 but with each inhibitor as indicated: lane 2, HS-CH2-CO-Phe-Ile-NH2 (Ki = 4.5 μM); lane 3, HS-CH2-CO-Ile-Val-NH2 (Ki = 1.8 μM); lane 4, HS-CH2-CO-Ile-Ile-NH2 (Ki = 6.5 μM); lane 5, HS-CH2-CO-Trp-Phe-NH2 (Ki = 1.1 μM); lane 6, HS-CH2-CO-Arg-Ser-NH2 (Ki = 462 μM); lane 7, HS-CH2-CO-His-Cys-NH2 (Ki = 1840 μM).
The lead LasB inhibitor (HS-CH2-CO-Phe-Tyr-NH2 [Ki = 41 nM]) successfully protects IgG from LasB-catalyzed degradation (Fig. 5). It can be seen that the native IgG protein remains intact in the presence of the inhibitor, and this has been demonstrated across a range of concentrations. The absence of degradation products can be seen at concentrations above 1.6 μM. The protective effect of these inhibitors on IgG provides evidence in principle that they could be used to block the action of the virulence factor on components of the host immune system.
Antibiofilm activity.
The effects of LasB inhibitors on the development of growing P. aeruginosa PA01 biofilms are summarized in Fig. 6. In order to assess any antibiofilm effect, inhibitors were incorporated into the growth media from the beginning of the assay. LasB inhibitor was therefore present throughout all stages of biofilm development. The subsequent effects on biofilm growth have been monitored either by enumeration of viable cells or by staining the biofilm biomass and reading the absorbance at 570 nm.
Fig. 6.

P. aeruginosa PA01 cell-based assays growing biofilms in the presence of the lead LasB inhibitor, HS-CH2-CO-Phe-Tyr-(NH2) (Ki = 0.041 μM). (A) Bactericidal effects on planktonic P. aeruginosa after 48 h. (B) Antibiofilm effect expressed as reduction in viable biofilm cells after 48 h. (C) Antibiofilm effect expressed as reduction in biofilm biomass after 24 h of growth. Standard deviations are shown by the error bars, and the data were analyzed by one-way analysis of variance (ANOVA) with Dunnett's multiple-comparison test for comparison of reductions in biofilm relative to the control. *, P < 0.05; ***. P < 0.001.
The effect of the lead inhibitor on planktonic populations was assessed to rule out any underlying bactericidal effect that might give a false positive in antibiofilm assays by retarding planktonic growth. After challenge with 100 μM HS-CH2-CO-Phe-Tyr-NH2 (Ki = 0.041 μM), the structure of which is given in Fig. 1, there was no significant reduction in planktonic cell numbers.
The data from Fig. 6B demonstrate that biofilm formation was significantly reduced in the presence of the lead LasB inhibitor. It was established that the peptide exhibited a dose-dependent reduction in biofilm growth at 1 μM and 100 μM concentrations, with the higher inhibitor concentration causing an approximately 2-log cycle, or 99%, reduction in biofilm cell numbers. The reduction was statistically significant at 1 and 100 μM concentrations (P < 0.001).
A reduction in biofilm formation (cell numbers) of approximately 4 log cycles relative to the control was seen, equating to approximately 99.99% reduction in viable biofilm cells (P < 0.001), and was statistically significant at all concentrations tested.
The maximum biofilm reduction was achieved at an inhibitor concentration of 40 μM and did not increase at higher concentrations. The remaining level of biofilm growth at 40 μM and above most likely represents the rate at which the biofilms are reseeded by a saturated planktonic culture or the background level of bacterial cells that are adherent but are prevented from forming biofilms by the inhibitor.
This residual growth was perhaps an unavoidable limitation in testing the antibiofilm effect of a nonbactericidal compound. However, crystal violet staining of the biofilm provided an alternative to enumeration of viable cells (Fig. 6C). The effect of the lead inhibitor was assessed at concentrations up to 100 μM conducted over a shorter test period of only 24 h. This assessed effects on the developing biofilm, potentially before saturation and before reseeding from the planktonic culture.
Twenty-four hours of growth and crystal violet staining demonstrates that the lead inhibitor had an increased antibiofilm effect at concentrations above 40 μM in less mature biofilms. At an inhibitor concentration of 100 μM, a maximum of 71.7% reduction in mean biofilm biomass was achieved relative to the control. The assays in Fig. 6, therefore, demonstrate that the antibiofilm effect is dependent upon both the potency and the concentration of the LasB inhibitors, validating the key role of LasB in biofilm formation.
Combination with antimicrobials.
The reduction in biofilm growth achieved by LasB inhibitors is significant and presents an opportunity for combination of such agents with antimicrobials. The known sensitivity of Pseudomonas to ciprofloxacin (CIP), for example, coupled with the poor penetration of the antimicrobial into biofilms due to binding to extracellular polysaccharide, makes biofilm inhibition a rational adjunctive antimicrobial strategy with the antibiotic (5, 50, 55).
A limitation of the study of antibiotic combinations in an antibiofilm capacity was the difficulty of distinguishing the effects of the various agents. The antibiofilm effect of the LasB inhibitors would be difficult to distinguish from the bactericidal effect of the antibiotics, since the latter would reduce biofilm by reducing planktonic cell numbers. Additionally, since planktonic P. aeruginosa is much more susceptible than biofilm cells to antimicrobials, the combination was tested for its ability to eradicate biofilms that are established, rather than those that are developing.
A concentration of the antibiotic combination that would challenge established biofilms significantly without causing eradication was selected. This was determined by a checkerboard assay, and 50 μg/ml of each agent was employed as the working concentration. Biofilms were then challenged using the antibiotic combination, with and without the LasB inhibitor, to determine the ability of the various combinations to eradicate established biofilms.
Typical antibiotics to which P. aeruginosa is sensitive include CIP and gentamicin (GEN). Combination treatment with these two agents is known to deliver significant bactericidal activity, but their action is impeded by the presence of biofilm. It was proposed that the antibiofilm effect of the LasB inhibitor would reduce the architectural integrity and barrier properties of the biofilm and enhance the efficacy of the antibiotic combination against established biofilms.
Biofilms established by 48-h growth were therefore challenged with a range of concentrations of the lead LasB inhibitor, in combination with two antibiotics used at a fixed concentration. The lead inhibitor at 100 μM concentration, in combination with the antimicrobial combination at 50 μg/ml, produced complete eradication of the biofilm (Fig. 7).
Fig. 7.

MBEC assays showing eradication of established (48-h) biofilms, which were challenged for 24 h with a range of concentrations of HS-CH2-CO-Phe-Tyr-(NH2) (Ki = 0.041 μM) in combination with 50 μg/ml of CIP and GEN. (A) Antibiofilm effect expressed as the minimum dilution at which no growth occurs, with biofilm eradication indicated by sterility of the undiluted sample. (B) Antibiofilm effect expressed as the viable biofilm cell count. Standard deviations are shown by the error bars, and the data were analyzed by one-way ANOVA with Dunnett's multiple-comparison test for comparison of reductions in biofilm relative to the control. **, P < 0.01; ***, P < 0.001.
The antibiofilm effect of the lead LasB inhibitor confirms the crucial roles of LasB and NDK in pseudomonal biofilm formation. It is known that NDK mutants that cannot produce GTP do not produce alginate, while NDK overexpression completely restores biofilm formation (56). The essential role of LasB in biofilm growth has been demonstrated in a similar manner, as LasB overexpression in mucoid (biofilm-forming) and nonmucoid strains results in increased alginate production in various contexts (20).
The introduction of plasmids encoding LasB into nonmucoid strains, for example, results in alginate formation comparable to that observed in mucoid strains. Similarly, introduction of the LasB gene to normal mucoid strains on a multicopy plasmid enhances alginate secretion beyond normal levels (20, 21, 56). The antibiofilm effect demonstrated here confirms the essential role of the LasB protease as a signaling molecule in the alginate production pathway and reinforces the suitability of the protease as a potential target for a small-molecule inhibitor of biofilm formation (20).
The dipeptide mercaptoacetyl inhibitor scaffold was chosen for its reasonable drug likeness and known suitability for inhibition of metalloproteases. Successful examples of similar drug scaffolds in a therapeutic context include the antihypertensive captopril, which is a Pro-Ala analog incorporating a mercapto group. Perhaps more relevant to the antibiotic drug class, if less structurally analogous to the inhibitor template used in this study, penicillin also employs a dipeptide scaffold and is an analog of d-Ala–d-Ala (51).
The use of LasB inhibitors in a therapeutic context was suggested previously to prevent the tissue destruction associated with pseudomonal infection. Specific inhibitors directing P′1 Phe dipeptide templates against LasB have been employed successfully in rabbit models of corneal keratitis (1, 4, 22, 25, 53). Phosphoramidon has also been investigated for its protective effect in corneal ulcer caused by P. aeruginosa (22).
Additionally, mercaptoacetyl dipeptide-based LasB inhibitors have previously been patented for use in eye drop formulations to prevent the corneal melting that occurs in pseudomonal keratitis (24). The lead compound identified in this study therefore serves as a useful starting point for the further development of a therapeutic agent capable of blocking the virulence mechanisms of tissue destruction, immunomodulation, and biofilm formation that are caused by LasB in pseudomonal infection.
It is proposed that inhibition of LasB would serve to block the immunomodulation and tissue destruction that are the other major virulence processes caused by LasB. Accordingly, it was found that the inhibitors were able to offer IgG complete protection against the effects of LasB in vitro. It could also be anticipated that LasB inhibition in vivo would prevent the tissue destruction that facilitates bacterial growth by release of essential nutrients, such as iron, from host tissues (11, 37, 63).
As virulence inhibitors, the compounds employed in this study act on the principle of blocking virulence processes without bactericidal activity, as potential second-generation antibiotic agents. It is this factor that makes inhibition of virulence an attractive antimicrobial strategy, as this approach would not pose a direct challenge to cell viability and hence would not exert selection pressure for the emergence of strains carrying resistance to the antibiotic agent (9, 46).
Conclusions.
It can be seen from the inhibitor studies that LasB demonstrates a strong preference for large aromatic residues at both the P′1 and P′2 positions, which is in accordance with previously published work from our group (6). The P′1 residue has been confirmed as the most important structural parameter for inhibitors of LasB, as illustrated by the structure-function relationship. The X-ray crystal structure of the LasB active site illustrates that the S′2 site has a much more open conformation than that at S′1. This perhaps helps to explain the lower specificity for the amino acid residue in the P′2 position of the inhibitor molecule.
Microbiological screening confirmed that LasB inhibitors did achieve a significant antibiofilm effect against live pseudomonal biofilms. Significant reductions in both viable biofilm cells and total biofilm biomass were achieved when growing biofilms were challenged (P < 0.001). A maximum of approximately 99.99% reduction in viable biofilm cells was achieved, or 71.7% reduction in mean biofilm biomass. The correlation between the degree of antibiofilm effect and both the inhibitor potency and concentration provides validation of a direct antibiofilm effect due to inhibition of LasB during biofilm formation.
Total eradication of the biofilm was not achieved using LasB inhibitors alone. However, this effect was not expected, due to the lack of direct toxicity to the bacterial cell, permitting constant reseeding of the biofilm by the planktonic population. The complete eradication of established biofilms by the lead LasB inhibitor, in combination with antibiotics, supports the potential of LasB inhibition as a therapeutic strategy in chronic biofilm-based infections involving P. aeruginosa (59).
Bacterial proteases play a pivotal role during infection (49, 57), and the multifaceted role of LasB in pseudomonal virulence is no exception (28, 38, 49, 58, 60). LasB is a key factor in the biofilm pathway, in invasiveness, and in immunomodulation, each of which is instrumental in the establishment of a chronic pseudomonal infection. As a virulence factor at the apex of many complex processes, the inhibition of LasB would therefore serve to attenuate the virulence mechanisms of the pathogen. Inhibitory activity of these compounds has also been demonstrated against related zinc metalloproteases, such as ZapA from Proteus mirabilis (unpublished data). LasB inhibition can therefore be advocated in a variety of pathologies of pseudomonal infection, with potential for combination with conventional antibiotic treatment.
Supplementary Material
ACKNOWLEDGMENT
We thank the Department for Education and Learning (DEL), Northern Ireland (http://www.delni.gov.uk/), for funding this work.
Footnotes
Supplemental material for this article may be found at http://aac.asm.org/.
Dedicated to the memory of John Lynas, deceased January 2001.
Published ahead of print on 28 March 2011.
REFERENCES
- 1. Adekoya O. A., Willassen N., Sylte I. 2006. Molecular insight into pseudolysin inhibition using the MM-PBSA and LIE methods. J. Struct. Biol. 153:129–144 [DOI] [PubMed] [Google Scholar]
- 2. Agren M. S., et al. 2000. Causes and effects of the chronic inflammation in venous leg ulcers. Acta Dermato-Venereologica 210:3–17 [PubMed] [Google Scholar]
- 3. Blackwood L. L., Stone R. M., Iglewski B. H., Pennington J. E. 1983. Evaluation of Pseudomonas aeruginosa exotoxin-A and elastase as virulence factors in acute lung infection. Infect. Immun. 39:198–201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Burns F., Paterson C., Gray R., Wells J. 1990. Inhibition of Pseudomonas aeruginosa elastase and Pseudomonas keratitis using a thiol-based peptide. Antimicrob. Agents Chemother. 34:2065–2069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Campanac C., Pineau L., Payard A., Baziard-Mouysset G., Roques C. 2002. Interactions between biocide cationic agents and bacterial biofilms. Antimicrob. Agents Chemother. 46:1469–1474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Cathcart G. R., Gilmore B. F., Greer B., Harriott P., Walker B. 2009. Inhibitor profiling of the Pseudomonas aeruginosa virulence factor LasB using N-alpha mercaptoamide template-based inhibitors. Bioorg. Med. Chem. Lett. 19:6230–6232 [DOI] [PubMed] [Google Scholar]
- 7. Ceri H., et al. 1999. The Calgary biofilm device: new technology for rapid determination of antibiotic susceptibilities of bacterial biofilms. J. Clin. Microbiol. 37:1771–1776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Cope W. G., Leidy R. B., Hodgson E. Classes of toxicants: use classes, p. 49–73 In Hodgson E. (ed.), A textbook of modern toxicology. Wiley-Blackwell, Hoboken, NJ [Google Scholar]
- 9. Crunkhorn S. 2008. Antibacterial drugs: new paths to beating bacteria. Nat. Rev. Drug Discov. 7:891 [Google Scholar]
- 10. Döring G., et al. 1985. Role of Pseudomonas aeruginosa exoenzymes in lung infections of patients with cystic fibrosis. Infect. Immun. 49:557–562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Döring G., Pfestorf M., Botzenhart K., Abdallah M. A. 1988. Impact of proteases on iron uptake of Pseudomonas aeruginosa pyoverdin from transferrin and lactoferrin. Infect. Immun. 56:291–293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Dulon S. 2005. Pseudomonas aeruginosa elastase disables proteinase-activated receptor 2 in respiratory epithelial cells. Am. J. Respir. Cell Mol. Biol. 32:411–419 [DOI] [PubMed] [Google Scholar]
- 13. Elizur A., Cannon C. L., Ferkol T. W. 2008. Airway inflammation in cystic fibrosis. Chest 133:489–495 [DOI] [PubMed] [Google Scholar]
- 14. Griese M., et al. 2007. Alpha-1-antitrypsin inhalation reduces airway inflammation in cystic fibrosis patients. Eur. Respir. J. 29:240–250 [DOI] [PubMed] [Google Scholar]
- 15. Grinnell F., Zhu M. F. 1996. Fibronectin degradation in chronic wounds depends on the relative levels of elastase, alpha 1-proteinase inhibitor, and alpha 2-macroglobulin. J. Investig. Dermatol. 106:335–341 [DOI] [PubMed] [Google Scholar]
- 16. Herrick S., et al. 1997. Up-regulation of elastase in acute wounds of healthy aged humans and chronic venous leg ulcers are associated with matrix degradation. Lab. Invest. 77:281–288 [PubMed] [Google Scholar]
- 17. Høiby N. 2002. Understanding bacterial biofilms in patients with cystic fibrosis: current and innovative approaches to potential therapies. J. Cyst. Fibros. 1:249–254 [DOI] [PubMed] [Google Scholar]
- 18. Jesaitis A. J., et al. 2003. Compromised host defense on Pseudomonas aeruginosa biofilms: characterization of neutrophil and biofilm interactions. J. Immunol. 171:4329–4339 [DOI] [PubMed] [Google Scholar]
- 19. Johnson D. A., Carter-Hamm B., Dralle W. M. 1982. Inactivation of human bronchial mucosal proteinase inhibitor by Pseudomonas aeruginosa elastase. Am. Rev. Respir. Dis. 126:1070–1073 [DOI] [PubMed] [Google Scholar]
- 20. Kamath S., Kapatral V., Chakrabarty A. M. 1998. Cellular function of elastase in Pseudomonas aeruginosa: role in the cleavage of nucleoside diphosphate kinase and in alginate synthesis. Mol. Microbiol. 30:933–941 [DOI] [PubMed] [Google Scholar]
- 21. Kamath S., Chen M., Chakrabarty A. M. 2000. Secretion of nucleoside diphosphate kinase by mucoid Pseudomonas aeruginosa 8821: involvement of a carboxy-terminal motif in secretion. J. Bacteriol. 182:3826–3831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kawaharajo K., Homma J., Aoyagi T., Umezawa H. 1982. Effect of phosphoramidon on protection against corneal ulcer caused by elastase and protease from Pseudomonas aeruginosa. Jpn. J. Exp. Med. 52:271–272 [PubMed] [Google Scholar]
- 23. Kessler E., Israel M., Landshman N. 1982. In vitro inhibition of Pseudomonas aeruginosa elastase by metal-chelating peptide derivatives. Infect. Immun. 38:716–723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kessler E., Spierer A., Blumberg S. September 1986. Opthalmic preparations. U.S. patent 4,613,587
- 25. Kessler E., Spierer A. 1984. Inhibition by phosphoramidon of Pseudomonas aeruginosa elastase injected intracorneally in rabbit eyes. Curr. Eye Res. 3:1075–1078 [DOI] [PubMed] [Google Scholar]
- 26. Klausner Y., Bodansky M. 1972. Coupling reagents in peptide synthesis. Synthesis 9:453–463 [Google Scholar]
- 27. Komori K., Nonogaki T., Nikai T. 2001. Hemorrhagic activity and muscle damaging effect of Pseudomonas aeruginosa metalloproteinase (elastase). Toxicon 39:1327–1332 [DOI] [PubMed] [Google Scholar]
- 28. Kon Y., et al. 1999. The role of Pseudomonas aeruginosa elastase as a potent inflammatory factor in a rat air pouch inflammation model. FEMS Immunol. Med. Microbiol. 25:313–321 [DOI] [PubMed] [Google Scholar]
- 29. Leid J. G., et al. 2005. The exopolysaccharide alginate protects Pseudomonas aeruginosa biofilm bacteria from IFN-gamma-mediated macrophage killing. J. Immunol. 175:7512–7518 [DOI] [PubMed] [Google Scholar]
- 30. Leidal K. G., Munson K. L., Johnson M. C., Denning G. M. 2003. Metalloproteases from Pseudomonas aeruginosa degrade human RANTES, MCP-1, and ENA-78. J. Interferon Cytokine Res. 23:307–318 [DOI] [PubMed] [Google Scholar]
- 31. Lipinski C. A., Lombardo F., Dominy B. W., Feeney P. J. 2001. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 46:3–26 [DOI] [PubMed] [Google Scholar]
- 32. Liu P. V. 1974. Extracellular toxins of Pseudomonas aeruginosa. J. Infect. Dis. 130:S94–S99 [DOI] [PubMed] [Google Scholar]
- 33. Lynas J. F., et al. 2000. Solid-phase synthesis and biological screening of N-alpha-mercaptoamide template-based matrix metalloprotease inhibitors. Comb. Chem. High Throughput Screen. 3:37–41 [DOI] [PubMed] [Google Scholar]
- 34. Mathee K., Ciofu O., Sternberg C., Lindum P. 1999. Mucoid conversion of Pseudomonas aeruginosa by hydrogen peroxide: a mechanism for virulence activation in the cystic fibrosis lung. Microbiology 145:1349–1357 [DOI] [PubMed] [Google Scholar]
- 35. May T. B., et al. 1991. Alginate synthesis by Pseudomonas aeruginosa—a key pathogenic factor in chronic pulmonary infections of cystic fibrosis patients. Clin. Microbiol. Rev. 4:191–206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Miles A., Misra S. S., Irwin J. O. 1938. The estimation of the bactericidal power of the blood. J. Hyg. Camb. 38:732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Miller R. A., Rasmussen G. T., Cox C. D., Britigan B. E. 1996. Protease cleavage of iron-transferrin augments pyocyanin-mediated endothelial cell injury via promotion of hydroxyl radical formation. Infect. Immun. 64:182–188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Miyoshi S., Shinoda S. 1997. Bacterial metalloprotease as the toxic factor in infection. J. Toxicol. Toxin Rev. 16:177–194 [Google Scholar]
- 39. Molla A., Yamamoto T., Akaike T., Miyoshi S., Maeda H. 1989. Activation of hageman factor and prekallikrein and generation of kinin by various microbial proteinases. J. Biol. Chem. 264:10589–10594 [PubMed] [Google Scholar]
- 40. Morihara K., Tsuzuki H., Oka T., Inoue H., Ebata M. 1965. Pseudomonas aeruginosa elastase isolation, crystallization, and preliminary characterization. J. Biol. Chem. 240:3295–3304 [PubMed] [Google Scholar]
- 41. Morihara K., Tsuzuki H. 1966. Substrate specificity of elastolytic and nonelastolytic proteinases from Pseudomonas aeruginosa. Arch. Biochem. Biophys. 114:158–165 [DOI] [PubMed] [Google Scholar]
- 42. Morihara K., Tsuzuki H. 1975. Pseudomonas aeruginosa elastase: affinity chromatography and some properties as a metallo-neutral proteinase. Agric. Biol. Chem. 39:1123–1128 [Google Scholar]
- 43. Pedersen S. S., Kharazmi A., Espersen F., Hoiby N. 1990. Pseudomonas aeruginosa alginate in cystic fibrosis sputum and the inflammatory response. Infect. Immun. 58:3363–3368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Poschet J. F., Boucher J. C., Firoved A. M., Deretic V. 2001. Conversion to mucoidy in Pseudomonas aeruginosa infecting cystic fibrosis patients. Methods Enzymol. 336:65–76 [DOI] [PubMed] [Google Scholar]
- 45. Rao C. N., et al. 1995. Alpha-1-antitrypsin is degraded and nonfunctional in chronic wounds but intact and functional in acute wounds—the inhibitor protects fibronectin from degradation by chronic wound fluid enzymes. J. Investig. Dermatol. 105:572–578 [DOI] [PubMed] [Google Scholar]
- 46. Rasko D. A., et al. 2008. Targeting QseC signaling and virulence for antibiotic development. Science 321:1078–1080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Sadikot R. T., Blackwell T. S., Christman J. W., Prince A. S. 2005. Pathogen-host interactions in Pseudomonas aeruginosa pneumonia. Am. J. Respir. Crit. Care Med. 171:1209–1223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Schmidtchen A. 2000. Degradation of antiproteinases, complement and fibronectin in chronic leg ulcers. Acta Dermato-Venereologica 80:179–184 [DOI] [PubMed] [Google Scholar]
- 49. Schmidtchen A., Holst E., Tapper H., Bjorck L. 2003. Elastase-producing Pseudomonas aeruginosa degrade plasma proteins and extracellular products of human skin and fibroblasts, and inhibit fibroblast growth. Microb. Pathog. 34:47–55 [DOI] [PubMed] [Google Scholar]
- 50. Shigeta M., Tanaka G., Komatsuzawa H., Sugai M. 1997. Permeation of antimicrobial agents through Pseudomonas aeruginosa biofilms: a simple method. Chemotherapy 43:340–345 [DOI] [PubMed] [Google Scholar]
- 51. Silver L. L. 2007. Multi-targeting by monotherapeutic antibacterials. Nat. Rev. Drug Discov. 6:41–55 [DOI] [PubMed] [Google Scholar]
- 52. Sokol P. A., Kooi C., Hodges R. S., Cachia P., Woods D. E. 2000. Immunization with a Pseudomonas aeruginosa elastase peptide reduces severity of experimental lung infections due to Pseudomonas aeruginosa or Burkholderia cepacia. J. Infect. Dis. 181:1682–1692 [DOI] [PubMed] [Google Scholar]
- 53. Spierer A., Kessler E. 1984. The effect of 2-mercaptoacetyl-L-phenylalanyl-L-leucine, a specific inhibitor of Pseudomonas aeruginosa elastase, on experimental Pseudomonas keratitis in rabbit eyes. Curr. Eye Res. 3:645–650 [DOI] [PubMed] [Google Scholar]
- 54. Stepanović S., Vuković D., Dakić I., Savić B. 2000. A modified microtiter-plate test for quantification of staphylococcal biofilm formation. J. Microbiol. Methods 40:175–179 [DOI] [PubMed] [Google Scholar]
- 55. Suci P., Mittelman M., Yu F., Geesey G. 1994. Investigation of ciprofloxacin penetration into Pseudomonas aeruginosa biofilms. Antimicrob. Agents Chemother. 38:2125–2133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Sundin G., Shankar S., Chakrabarty A. 1996. Mutational analysis of nucleoside diphosphate kinase from Pseudomonas aeruginosa: characterization of critical amino acid residues involved in exopolysaccharide synthesis. J. Bacteriol. 178:7120–7128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Supuran C. T., Scozzafava A., Clare B. W. 2002. Bacterial protease inhibitors. Med. Res. Rev. 22:329–372 [DOI] [PubMed] [Google Scholar]
- 58. Tamura Y., Suzuki S., Kijima M., Takahashi T., Nakamura M. 1992. Effect of proteolytic enzyme on experimental infection of mice with Pseudomonas aeruginosa. J. Vet. Med. Sci. 54:597–599 [DOI] [PubMed] [Google Scholar]
- 59. Travis J., Potempa J. 2000. Bacterial proteinases as targets for the development of second-generation antibiotics. Biochim. Biophys. Acta 1477:35–50 [DOI] [PubMed] [Google Scholar]
- 60. Voynow J., Fischer B., Zheng S. 2008. Proteases and cystic fibrosis. Int. J. Biochem. Cell Biol. 40:1238–1245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Walker B. 1982. Ph.D thesis Queen's University of Belfast, Belfast, United Kingdom [Google Scholar]
- 62. Weckroth M., Vaheri A., Lauharanta J., Sorsa T., Konttinen Y. T. 1996. Matrix metalloproteinases, gelatinase and collagenase, in chronic leg ulcers. J. Investig. Dermatol. 106:1119–1124 [DOI] [PubMed] [Google Scholar]
- 63. Wolz C., et al. 1994. Iron release from transferrin by pyoverdin and elastase from Pseudomonas aeruginosa. Infect. Immun. 62:4021–4027 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.