

Abstract
The need for novel insights into the mechanisms of progression of renal disease has become urgent during the last several years because of the increasing incidence of chronic renal disease worldwide. Independent of the underlying disease, the subsequent progression of renal fibrosis is characterized mainly by both an exaggerated synthesis and abnormal accumulation of extracellular matrix proteins produced by mesenchymal cells within the kidney. These cells are mainly myofibroblasts deriving from a variety of renal cells such as vascular smooth muscle, mesangial, resident stem, tubular epithelial, vascular endothelial cells or pericytes. The appearance of myofibroblasts is a reversible process, as suggested by studies in experimental models showing regression of renal fibrosis during therapy with antagonists and/or blockers of the renin–angiotensin system. An additional factor that can also affect the mechanisms of progression/regression of fibrosis is the plasticity of podocytes controlling glomerular filtration.
Keywords: angiotensin II, endothelial–mesenchymal transition, epithelial-mesenchymal transition, podocyte, TGFbeta, vascular smooth muscle cell
Introduction
Current estimates place the number of patients suffering from renal disease at 500 million. This number increases regularly all over the world, and a significant proportion of these patients progress towards end-stage renal disease (ESRD). During the last 25 years, the incidence of ESRD doubled in the United States and in Europe (Valderrabano & Berthoux 1996; Anonymous 1998), mainly because of population ageing, prevention of the early deaths from cardiovascular diseases and greater incidence of diabetes. The prevention and/or reversal of chronic kidney disease (CKD) are major public health objectives. Current treatments based mainly on inhibition of the renin–angiotensin system are only partially effective (Mauer et al. 2009). Thus, a better understanding of the mechanisms involved in this pathology will provide important information to identify novel targets and to design new therapies.
Progression of CKD and renal fibrosis
End-stage renal disease results from a continuous decline of renal function associated with the excessive accumulation of extracellular matrix proteins such as collagen type IV (already present in the normal kidney) and collagens type I and III (absent in the normal kidney) and with structural changes within all renal compartments (vascular, glomerular and tubulo-interstitial). Fibroblasts, key cells for collagen synthesis and stabilization, play a major role in the development of the renal fibrosis. Traditionally, it was considered that fibroblasts found in the renal parenchyma derived from a resident population. In fact, the cellular source of this abnormal production of collagen is variable, and different types of renal cells can present themselves in an activated fibroblast phenotype (myofibroblasts) in response to a variety of stimuli [such as migration of inflammatory cells into the renal parenchyma, inappropriate immune response, abnormal activity of the renin–angiotensin system (Wynn et al. 2007)]. In the last several years, a process – well established in tumour progression – called epithelial-to-mesenchymal transition (EMT) was proposed as a major mechanism contributing to CKD (Harris & Neilson 2006; Burns et al. 2007). In addition, an endothelial-to-mesenchymal transition (EndMT) was also suggested as an alternative mechanism to vascular remodelling during hypertensive disease. Table 1 summarizes the most commonly used proteins to characterize normal or disease phenotype of renal cells. In the following paragraphs, we will briefly describe studies regarding the origin and the reversibility of renal fibrosis in experimental models.
Table 1.
Representative examples of proteins characterizing the normal or activated (pro-fibrotic) phenotype of renal cells
| Cell type | Normal phenotype | Pro-fibrotic (activated) phenotype |
|---|---|---|
| Vascular smooth muscle | α-SMA | Collagen I, III, VCAM-1, vimentin |
| Endothelial | VWF, PECAM-1 | E-selectin, FSP-1, ICAM-1 |
| Mesangial | Desmin | α-SMA, collagen I |
| Podocyte | Nephrin, Podocin | Loss of nephrin, podocin and foot processes |
| Pericyte | α-SMA, NG 2 | FSP-1, collagen I, |
| Tubular epithelial | E-cadherin, ZO-1 | Vimentin, FSP-1 |
| Fibroblast | FSP-1, HSP47 | α-SMA, vimentin |
VCAM-1, vascular cell adhesion protein 1; VWF, von Willebrand factor; PECAM-1, platelet endothelial cell adhesion molecule; FSP-1, fibroblast-specific protein 1; ICAM-1, intercellular adhesion molecule 1; NG2, nerve/glial antigen 2; ZO-1, zona occludens; HSP47, heat-shock protein 47; SMA, smooth muscle actin.
Plasticity of vascular smooth muscle and glomerular cells during the progression and reversal of nephroangiosclerosis
Models of hypertension-induced renal disease are used traditionally as the experimental basis to study mechanisms of nephroangiosclerosis. In this pathology, the development of tubulo-interstitial fibrosis is secondary to the vascular and glomerular lesions. This ‘diffusion’ of fibrosis in the tubulo-interstitial space was attributed to the excess of proteins filtered through a pathological glomerular barrier. This excessive protein leakage alters tubular reabsorption and in the long term induces fibrotic lesions in the tubular epithelium (Remuzzi et al. 2006). During nephroangiosclerosis, the shift of the equilibrium between vasoconstrictor and pro-fibrotic factors (angiotensin II, endothelin) and vasodilators (nitric oxide, C natriuretic peptide) is responsible for the change in the phenotype of the mesangial and arteriolar smooth muscle cells (Ardaillou et al. 1999; Belmadani et al. 2008), following a pathological process common to all types of contractile cells (Berk et al. 2007). In the experimental model of hypertension induced by nitric oxide deficiency, the essential role of the angiotensin II in these structural changes is attested by the protective effect of the AT1 receptor antagonists against the development of nephroangiosclerosis. In this model, the excessive activity of the renin–angiotensin system is attributable to an overexpression of angiotensin-converting enzyme, which is not compensated by the concomitant decrease in AT1 receptor expression in the renal cortex (Vandermeersch et al. 2003). Angiotensin II facilitates the migration of inflammatory cells by stimulating the production of chemokines (such as MCP-1) and induces directly the synthesis of the collagens by activating several transduction pathways: endothelin, PAI-1, PDGF, TGFbeta and/or EGF (Berk 1999; Klahr & Morrissey 2000; Fogo 2001; Modlinger et al. 2004; Ruiz-Ortega et al. 2006) (Figure 1). The activation of PDGF or EGF receptors stimulates in turn the MAP/ERK kinase pathway and the synthesis of the transcription factor AP1, whereas the activation of TGFbeta leads to the stimulation of SMADs.
Figure 1.
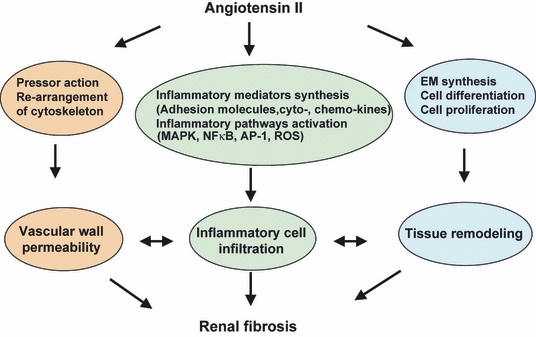
Angiotensin II can induce simultaneously a variety of effects on cells such as contractility, inflammation, differentiation, proliferation, apoptosis or extracellular matrix gene activation, which are capable of interacting with each other, and according to the specific environmental, hormonal or homoeostatic conditions within the kidney can lead (or not) to the development of fibrosis.
The involvement of these pathways in the development of renal fibrosis was clearly established by pharmacological experiments. AT1 receptor antagonists or angiotensin converting enzyme (ACE) inhibitors, ETA/B or epidermal growth factor receptor (EGFR) receptor antagonists, and inhibitors of MAP/ERK kinase phosphorylation prevented the activation of collagen type I gene in renal vessels, glomeruli and renal cortex (Chatziantoniou et al. 1998; Boffa et al. 1999; Tharaux et al. 2000; Flamant et al. 2003). Blocking of the TGFbeta/SMAD pathway has a similar anti-collagenic effect (Fakhouri et al. 2001). The anti-fibrotic action of AT1 blockers is independent of their pressure-lowering effect, because this curative effect is not reproduced by other vasodilators such as hydralazine (Boffa et al. 2003). Furthermore, administration of endothelin or EGF receptor antagonists, which are devoid of anti-hypertensive action, inhibited the development of renal fibrosis (Boffa et al. 2001; François et al. 2004).
However, stopping the excessive synthesis of extracellular matrix is not alone a sufficient condition for the reversal of renal disease (Boffa et al. 2003). Two additional conditions must be fulfilled in parallel: degradation of the excessive matrix by stimulating the activity of matrix metalloproteinases (MMPs) (Boffa et al. 2003), and recovery or return towards a normal cell phenotype. Among the various renal cells, glomerular podocytes have received particular attention because they are highly differentiated epithelial cells incapable of proliferating. Several investigators have advanced the hypothesis that damage of podocytes is the no-return point beyond which renal fibrosis becomes irreversible (Adamczak et al. 2003). Nevertheless, data from our laboratory indicate that changes in podocyte phenotype are reversible, in a novel progressive model of nephroangiosclerosis in mice. These mice express ectopically a renin transgene at a constant rate (six- to eightfold over endogenous levels) by genetic clamping. When these mice were treated with an AT1 receptor antagonist at an advanced phase of renal disease (12-month-old mice), proteinuria decreased to almost normal levels. This reversal of proteinuria was attributable to the de novo synthesis of proteins specific to the glomerular barrier (nephrin, podocin), reappearance of foot processes and restoration of slit diaphragms (Huby et al. 2009). It would be interesting to investigate whether the appearance of the pathological phenotype of podocytes is attributable to a direct action of angiotensin II in these cells or whether it is an adaptive consequence of the well-known angiotensin II-induced phenotype changes in mesangial cells. It will also be important to investigate whether the blockade of angiotensin II action in podocytes initiated the restoration of the normal phenotype or whether the reversal was attributable to the disappearance of myofibroblasts in glomeruli.
Plasticity of the tubular epithelial cells and epithelial to mesenchymal (and mesenchymal to epithelial) transition: myth or reality?
The transition from an epithelial to a mesenchymal phenotype (EMT) normally takes place in several phases during embryonic development. This process is also observed during tumour progression. In renal physiopathology, the role of EMT in the development of the renal fibrosis remains a highly controversial hypothesis (Iwano et al. 2002; Kalluri & Neilson 2003; Liu 2004; Faulkner et al. 2005; Nangaku 2006; Lin et al. 2008; Cook 2010; Humphreys et al. 2010). In the normal kidney, fibroblasts are scarce; their number explodes during the progression of interstitial fibrosis. Several hypotheses have been proposed to explain their origin: resident fibroblasts, fibrocytes, circulating cells derived from bone marrow, and/or tubular epithelial cells or pericytes undergoing metaplasia (Lin et al. 2008). The development of renal fibrosis is then accelerated by the loss of function of specialized cells such as electrolyte transport for the tubular epithelial cells and vascularization for endothelial cells and pericytes (Nangaku 2006).
Epithelial-to-mesenchymal transition was mainly studied in experimental unilateral ureteral obstruction (UUO). Initial studies suggested that 30% of interstitial fibroblasts originated from tubular epithelial cells (Iwano et al. 2002). According to these studies, EMT follows several steps. In the beginning, the expression of protein constituents of tight junctions (E-cadherin, ZO-1) is decreased in epithelial cells because of the action of pro-fibrotic growth factors (TGFbeta, FGF-2, EGF). As result, epithelial cells lose their intercellular contacts and polarity and undergo cytoskeleton reshaping by expressing proteins specific to mesenchyma such as fibroblast-specific protein-1 (FSP-1), alpha smooth muscle actin (SMA), fibronectin and collagens I and III. In addition, abnormal activity of MMPs leads to breaks in the tubule basement membrane, thus facilitating the escape and migration of myofibroblasts into the interstitium (Kalluri & Neilson 2003). However, other studies are very critical of the existence and/or the importance of EMT in renal fibrosis (Faulkner et al. 2005; Cook 2010; Humphreys et al. 2010). Specifically, mice were genetically modified using cre/lox techniques to label proteins exclusively in renal tubular epithelial cells (Humphreys et al. 2010). Two models of injury were induced: UUO and ischaemia–reperfusion injury (these models of injury have been used in the majority of studies that supported the EMT hypothesis). Although renal injury associated with the development of interstitial fibrosis was evident in both models, the authors did not observe any labelled tubular epithelial cells in the interstitium or any tubular cell that expressed fibroblast characteristics. In addition, it appeared that the abnormal collagen production in the interstitium was originating mainly from perivascular cells (Humphreys et al. 2010). These results imply that that EMT did not occur, at least in these two models. Of course these findings do not exclude the possibility of cell–cell communication between injured tubular epithelial cells and perivascular cells to induce fibrosis.
During normal development, cellular plasticity is also observed in the opposite direction: mesenchymal cells are transformed to epithelial cells and participate in the formation of epithelium via a mesenchymal-to-epithelial transition (MET) (Horster et al. 1999). The existence of physiological MET suggests that in pathology MET could be induced by the action of anti-fibrotic agents opposing to the action TGFbeta. Among them, the most efficient appears to be the hepatic growth factor (HGF) (Liu & Yang 2006) and certain members of the bone morphogenetic proteins family (BMPs). The anti-fibrotic efficiency of the HGF was observed in a model of renal ablation (Yang et al. 2003) and a model of renal allograft rejection (Herrero-Fresnada et al. 2006). Hepatic growth factor antagonizes almost every cellular pro-fibrotic effect of TGFbeta. Thus, HGF preserves tubular epithelial cells from apoptosis, inhibits activation of myofibroblasts and interstitial fibroblasts and reduces extracellular matrix formation. In addition, HGF and its receptor c-Met are involved in neovascularization during atherosclerosis by inducing pericyte migration and endothelial cell plasticity (Liu et al. 2007). Very little is known regarding the role of peritubular angiogenesis during regression of renal fibrosis. It will be thus interesting to study whether neovascularization can be involved in renal repair and whether a part of this process is dependent on HGF-induced pericyte migration.
Daily administration of BMP-7 reversed glomerular injury in a model of genetic renal disease (Zeisberg et al. 2003a), in diabetes (Sugimoto et al. 2007) and in a model of nephrotoxic serum nephritis (Zeisberg et al. 2003b). In addition, BMP-7 or HGF administration induced MET and a partial regression during UUO (Klahr & Morrissey 2003; Yang & Liu 2003). Our laboratory produced data showing that MET occurs spontaneously in the UUO model of UUO after correction of the initiating event. Thus, the expression of proteins characterizing myofibroblasts decreased, and inversely, proteins typical of a normal tubular epithelial phenotype reappeared gradually following reconstruction of the ligated ureter (manuscript submitted). We have also studied the reversibility of tubulo-interstitial fibrosis secondary to nephroangiosclerosis, in the model of mice overexpressing renin (Huby et al. 2009). In these mice, the expression of megalin and E-cadherin decreased, whereas that of the FSP-1 was increased with age and preceded the appearance of renal tubulo-interstitial fibrosis. Treatment with an AT1 receptor antagonist restored the normal tubular phenotype independently of the blood pressure. Interestingly, these changes appear to be controlled by altered expression of uterine sensitization-associated gene-1 (USAG-1) and noggin, two endogenous antagonists of BMPs. Both proteins are several-fold more abundant in aged renin transgenic mice. This upregulation inhibited the anti-fibrotic action of BMPs, and thus, a relatively moderate increase in the expression of TGFbeta and its pro-fibrotic cofactors was enough to produce renal fibrosis (Figure 2). During AT1 receptor blockade, the expression of USAG-1 and noggin returned to normal levels, and thus, BMPs were again efficient at antagonizing the action of TGFbeta and reversing fibrosis (Huby et al. 2009) (Figure 2). It will be interesting to investigate whether blockade of endogenous antagonists of BMPs promotes reversal of renal fibrosis in other experimental models as well.
Figure 2.
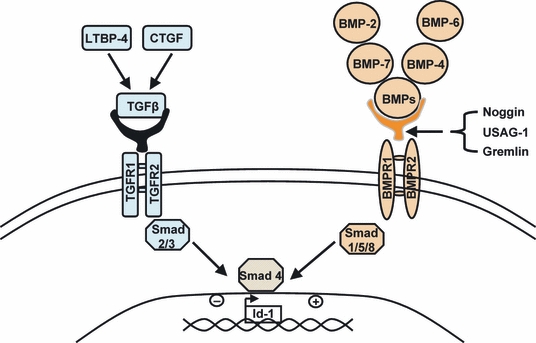
The complexity of the roles of different members of the TGFbeta/bone morphogenetic proteins (BMP) superfamily in the development of renal fibrosis. Latent TGFbeta binding protein-4 (LTBP-4) and connective tissue growth factor (CTGF) act as cofactors of TGFbeta leading to the activation of TGF receptors, the stimulation of SMAD2 and SMAD3, and the formation of the SMAD 2/3/4 complex. This complex inhibits the transcription factor Id-1 and promotes the induction of mesenchymal phenotype. In contrast, BMPs antagonize the effect of TGFbeta by stimulating SMADs 1/5/8, which capture SMAD4, inhibit the formation of the SMAD 2/3/4 complex and induce the epithelial phenotype. New players in this interaction are the endogenous inhibitors of BMPs, noggin, uterine sensitization–associated gene-1 (USAG-1) and gremlin which block the interaction of BMPs with their receptors and inactivate the SMAD 1/5/8 cascade. As result, SMAD4 is free to interact with SMAD2 and SMAD3, and thus, the final outcome is a shift towards the pro-fibrotic action of TGFbeta.
Endothelial cell plasticity: what about an endothelial-to-mesenchymal transition?
Endothelial cells play a key role in several important physiological processes, including leucocyte recruitment and regional blood flow regulation. A functional impairment of the resident endothelium is consequently capable of contributing to the progression of chronic kidney injury through multiple pathways. Over recent years, interest has been focused on the capacity of altered endothelial cells to acquire functional and structural characteristics of mesenchymal cells. This so-called endothelial–mesenchymal transition (EndMT) has been studied largely in physiological conditions, particularly during embryonic development of the heart (Markwald et al. 1977). Increasing evidence suggests that EndMT may occur postnatally and can have a significant role in a variety of pathologies, including kidney diseases.
In vitro, endothelial cells of different origins can spontaneously acquire a mesenchymal phenotype (Arciniegas et al. 1992; Frid et al. 2002). Similar to epithelial–mesenchymal transition, EndMT is highly dependent upon the equilibrium between the TGFbeta and BMPs pathways. Endothelial cells isolated from human skin and treated with TGFbeta showed a reduced expression of endothelial markers, with a parallel increase in FSP-1, alpha SMA and type I collagen (Liebner et al. 2004; Arciniegas et al. 2005; Kitao et al. 2009).
EndMT has also emerged as a novel important mechanism involved in cancer progression with up to 40% of cancer-associated fibroblasts shown to originate from EndMT (Zeisberg et al. 2007a). The same team of investigators studied EndMT during the development of cardiac fibrosis in a transgenic strain of mice that allows tracking of cells of endothelial origin (Tie2-Cre;ROSA-STOP-lacZ transgenic mice) (Zeisberg et al. 2007b). In the fibrotic heart, the presence of fibrotic markers (FSP-1, alpha SMA) was co-localized with the expression of lacZ, suggesting the endothelial origin of heart myofibroblasts. Interestingly, mice treated with human recombinant BMP-7 were significantly protected against both EndMT and the development of cardiac fibrosis.
In the kidney, an EndMT-like process has been observed after UUO, in a model of eNOS inhibition, in streptozotocin-induced nephropathy and in a model of Alport syndrome (Col4a3-deficient mice) (O'riordan et al. 2007; Zeisberg et al. 2008; Li et al. 2009). We have evaluated the modifications of endothelial phenotype during the progression of angiotensin II-induced nephropathy in Sprague–Dawley rats and observed a limited co-expression of FSP-1 with the endothelial marker RECA1, suggesting the existence of EndMT in peritubular renal capillaries. Importantly, features consistent with EndMT were observed at early stages of the renal disease, before the onset of significant proteinuria or fibrosis.
These studies imply that EndMT can be associated with CKD at least in experimental models. Furthermore, they suggest that endothelial cells that acquire mesenchymal characteristics may contribute to early pathogenetic mechanisms of fibrogenesis. Together with the broader consequences of the impairment of resident endothelial function, EndMT can be an additional mechanism involved in the progression of renal fibrosis. Additional studies are needed to better understand the molecular mechanisms and local consequences of EndMT. These investigations could lead to the development of therapeutic strategies specifically targeting this novel aspect of endothelial dysfunction.
Conclusion
During the last decade, important advancements have been made regarding the mechanisms of progression and reversal of CKD. The capacity of renal resistance vessels, mesangial cells and podocytes to undergo remodelling, to acquire fibroblastic phenotype and to return to normal during therapy is also well documented in several experimental models. These studies suggest that regression of renal fibrosis is not attributable to a single cell type's function, but to a simultaneous improvement in the various cell types involved in development of CKD (Figure 3). The renin–angiotensin system appears to be a key player in this pathological process. The shift in the regulation and relative expression between pro and anti-fibrotic members of TGFbeta/BMPs superfamily is also a very important factor. We consider that the controversial hypothesis of an EMT in tubular epithelium is losing ground, whereas the endothelial-to-mesenchymal transition hypothesis is still open for discussion. In addition, several interconnected questions remain to be thoroughly investigated: How to define the no-return point of CKD? Why is blockade of the renin–angiotensin system not as efficient in humans as in experimental models? Is it a question of time or of dose? Do we have to associate additional targets to angiotensin blockers? Can cell therapy be an option? Is it possible to use and/or activate stem cells locally as suggested by recent studies (Hopkins et al. 2009)?
Figure 3.
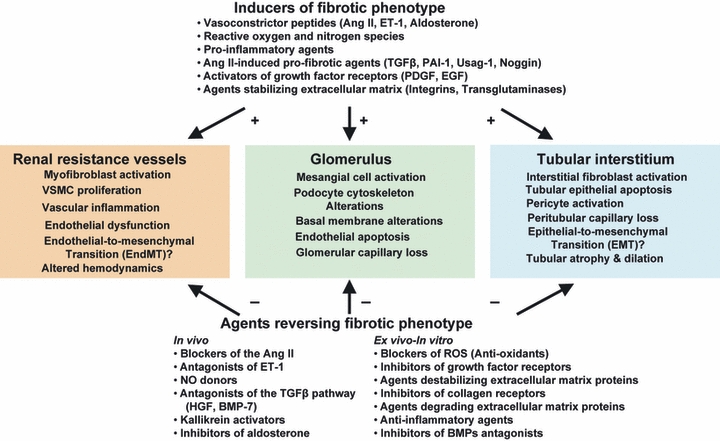
Cellular mechanisms involved in the phenotype changes during progression or regression of renal fibrosis. Pro-fibrotic agents (such as angiotensin II, endothelin or TGFbeta) modify the renal morphology and function by shifting the gene expression of proteins towards a fibrotic state in all renal compartments. Data from a variety of experimental models show that renal fibrosis is reversible and that is possible to retrieve a normal phenotype following therapy with the agents listed in the lower part of the figure.
The hope of improving the care of millions of patients affected by chronic renal failure fully justifies continuing our quest for answers to the above questions.
Acknowledgments
This work was funded by grants of the Institut National de la Sante et de la Recherche Medicale (INSERM) and the University Pierre et Marie Curie (Paris VI).
References
- Adamczak M, Gross ML, Krtil J, et al. Reversal of glomerulosclerosis after high-dose enalapril treatment in subtotally nephrectomized rats. J. Am. Soc. Nephrol. 2003;14:2833–2842. doi: 10.1097/01.asn.0000095248.91994.d3. [DOI] [PubMed] [Google Scholar]
- Anonymous. Incidence and prevalence of ESRD – United States Renal Data System. Am. J. Kidney Dis. 1998;32(Suppl. 1):S38–S49. doi: 10.1053/ajkd.1998.v32.pm9713406. [DOI] [PubMed] [Google Scholar]
- Arciniegas E, Sutton AB, Allen TD, Schor AM. Transforming growth factor beta 1 promotes the differentiation of endothelial cells into smooth muscle-like cells in vitro. J. Cell Sci. 1992;103:521–529. doi: 10.1242/jcs.103.2.521. [DOI] [PubMed] [Google Scholar]
- Arciniegas E, Neves CY, Carrillo LM, Zambrano EA, Ramírez R. Endothelial-mesenchymal transition occurs during embryonic pulmonary artery development. Endothelium. 2005;12:193–200. doi: 10.1080/10623320500227283. [DOI] [PubMed] [Google Scholar]
- Ardaillou R, Chansel D, Chatziantoniou C, Dussaule JC. Mesangial AT1 receptors: expression, signaling, and regulation. J. Am. Soc. Nephrol. 1999;10(Suppl. 11):S40–S46. [PubMed] [Google Scholar]
- Belmadani S, Zerfaoui M, Boulares HA, Palen DI, Matrougui K. Microvessel vascular smooth muscle cells contribute to collagen type I deposition through ERK1/2 MAP kinase, alphavbeta3-integrin, and TGF-beta1 in response to ANG II and high glucose. Am. J. Physiol. Heart Circ. Physiol. 2008;295:H69–H76. doi: 10.1152/ajpheart.00341.2008. Epub 2008 May 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berk BC. Angiotensin II. J. Am. Soc. Nephrol. 1999;10(Suppl. 11):S62–S68. [PubMed] [Google Scholar]
- Berk BC, Fujiwara K, Lehoux S. ECM remodeling in hypertensive heart disease. J. Clin. Invest. 2007;117:568–575. doi: 10.1172/JCI31044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boffa JJ, Tharaux PL, Placier S, Ardaillou R, Dussaule JC, Chatziantoniou C. Angiotensin II activates collagen type I gene in the renal vasculature of transgenic mice during inhibition of nitric oxide synthesis: evidence for an endothelin-mediated mechanism. Circulation. 1999;100:1901–1908. doi: 10.1161/01.cir.100.18.1901. [DOI] [PubMed] [Google Scholar]
- Boffa JJ, Tharaux PL, Dussaule JC, Chatziantoniou C. Regression of renal vascular fibrosis by endothelin receptor antagonism. Hypertension. 2001;37:490–496. doi: 10.1161/01.hyp.37.2.490. [DOI] [PubMed] [Google Scholar]
- Boffa JJ, Ying L, Placier S, Stefanski A, Dussaule JC, Chatziantoniou C. Regression of renal vascular and glomerular fibrosis: role of angiotensin II receptor antagonism and metalloproteinases. J. Am. Soc. Nephrol. 2003;14:1132–1144. doi: 10.1097/01.asn.0000060574.38107.3b. [DOI] [PubMed] [Google Scholar]
- Burns WC, Kantharidis P, Thomas MC. The role of tubular epithelial-mesenchymal transition in progressive kidney disease. Cells Tissues Organs. 2007;18:222–231. doi: 10.1159/000101323. [DOI] [PubMed] [Google Scholar]
- Chatziantoniou C, Boffa JJ, Ardaillou R, Dussaule JC. Nitric oxide inhibition induces early activation of type I collagen gene in renal resistance vessels and glomeruli in transgenic mice: role of endothelin. J. Clin. Invest. 1998;101:2780–2789. doi: 10.1172/JCI2132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook HT. The origin of renal fibroblasts and progression of kidney disease. Am. J. Pathol. 2010;173:22–24. doi: 10.2353/ajpath.2010.090898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fakhouri F, Placier S, Ardaillou R, Dussaule JC, Chatziantoniou C. Angiotensin II activates collagen type I gene in the renal cortex and aorta of transgenic mice through interaction with endothelin and TGF-beta. J. Am. Soc. Nephrol. 2001;12:2701–2710. doi: 10.1681/ASN.V12122701. [DOI] [PubMed] [Google Scholar]
- Faulkner JL, Szcykalski LM, Springer F, Barnes JL. Origin of interstitial fibroblasts in an accelerated model of angiotensin II-induced renal fibrosis. Am. J. Pathol. 2005;167:1193–1205. doi: 10.1016/S0002-9440(10)61208-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flamant M, Tharaux PL, Placier S, et al. Epidermal growth factor transactivation mediates the tonic and fibrogenic effects of endothelin in the aortic wall of transgenic mice. FASEB J. 2003;17:327–329. doi: 10.1096/fj.02-0115fje. Epub 02-0115fje, Dec 2002. [DOI] [PubMed] [Google Scholar]
- Fogo AB. Renal fibrosis and the renin-angiotensin system. Adv. Nephrol. Necker Hosp. 2001;31:69–87. [PubMed] [Google Scholar]
- François H, Placier S, Flamant M, et al. Prevention of renal vascular and glomerular fibrosis by epidermal growth factor receptor inhibition. FASEB J. 2004;18:926–928. doi: 10.1096/fj.03-0702fje. Epub 2004 Mar 19. [DOI] [PubMed] [Google Scholar]
- Frid MG, Kale VA, Stenmark KR. Mature vascular endothelium can give rise to smooth muscle cells via endothelial-mesenchymal transdifferentiation: in vitro analysis. Circ. Res. 2002;90:1189–1196. doi: 10.1161/01.res.0000021432.70309.28. [DOI] [PubMed] [Google Scholar]
- Harris RC, Neilson EG. Towards a unified theory of renal progression. Annu. Rev. Med. 2006;57:365–380. doi: 10.1146/annurev.med.57.121304.131342. [DOI] [PubMed] [Google Scholar]
- Herrero-Fresnada I, Torras J, Franquesa M, et al. HGF gene therapy attenuates renal allograft scarring by preventing the profibrotic inflammatory-induced mechanisms. Kidney Int. 2006;70:265–274. doi: 10.1038/sj.ki.5001510. [DOI] [PubMed] [Google Scholar]
- Hopkins C, Li J, Rae F, Little MH. Stem cell options for kidney disease. J. Pathol. 2009;217:265–281. doi: 10.1002/path.2477. [DOI] [PubMed] [Google Scholar]
- Horster MF, Braun GS, Huber SM. Embryonic renal epithelia: induction, nephrogenesis, and cell differentiation. Physiol. Rev. 1999;79:1157–1191. doi: 10.1152/physrev.1999.79.4.1157. [DOI] [PubMed] [Google Scholar]
- Huby AC, Rastaldi MP, Caron K, Smithies O, Dussaule JC, Chatziantoniou C. Restoration of podocyte structure and improvement of chronic renal disease in transgenic mice overexpressing renin. PLoS ONE. 2009;4:e6721. doi: 10.1371/journal.pone.0006721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphreys BD, Lin SJ, Kobayashi A, et al. Fate tracing reveals the pericyte and not epithelial origin of myofibroblasts in kidney fibrosis. Am. J. Pathol. 2010;176:85–97. doi: 10.2353/ajpath.2010.090517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwano M, Plieth D, Danoff TM, Xue C, Okada H, Neilson EG. Evidence that fibroblasts derive from epithelium during tissue fibrosis. J. Clin. Invest. 2002;110:341–350. doi: 10.1172/JCI15518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalluri R, Neilson EG. Epithelial-mesenchymal transition and its implications for fibrosis. J. Clin. Invest. 2003;112:1776–1784. doi: 10.1172/JCI20530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitao A, Sato Y, Sawada-Kitamura S, et al. Endothelial to mesenchymal transition via transforming growth factor-beta1/Smad activation is associated with portal venous stenosis in idiopathic portal hypertension. Am. J. Pathol. 2009;175:616–626. doi: 10.2353/ajpath.2009.081061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klahr S, Morrissey JJ. The role of vasoactive compounds, growth factors and cytokines in the progression of renal disease. Kidney Int. Suppl. 2000;75:S7–S14. [PubMed] [Google Scholar]
- Klahr S, Morrissey J. Obstructive nephropathy and renal fibrosis: the role of bone morphogenic protein-7 and hepatocyte growth factor. Kidney Int. Suppl. 2003;87:S105–S112. doi: 10.1046/j.1523-1755.64.s87.16.x. [DOI] [PubMed] [Google Scholar]
- Li J, Qu X, Bertram JF. Endothelial-myofibroblast transition contributes to the early development of diabetic renal interstitial fibrosis in streptozotocin-induced diabetic mice. Am. J. Pathol. 2009;175:1380–1388. doi: 10.2353/ajpath.2009.090096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liebner S, Cattelino A, Gallini R, et al. Beta-catenin is required for endothelial-mesenchymal transformation during heart cushion development in the mouse. J. Cell Biol. 2004;166:359–367. doi: 10.1083/jcb.200403050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin SL, Kisseleva T, Brenner DA, Duffield JS. Pericytes and perivascular fibroblasts are the primary source of collagen-producing cells in obstructive fibrosis of the kidney. Am. J. Pathol. 2008;173:1617–1627. doi: 10.2353/ajpath.2008.080433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y. Epithelial to mesenchymal transition in renal fibrogenesis: pathologic significance, molecular mechanism, and therapeutic intervention. J. Am. Soc. Nephrol. 2004;15:1–12. doi: 10.1097/01.asn.0000106015.29070.e7. [DOI] [PubMed] [Google Scholar]
- Liu Y, Yang J. Hepatocyte growth factor: new arsenal in the fights against renal fibrosis? Kidney Int. 2006;70:238–240. doi: 10.1038/sj.ki.5001661. [DOI] [PubMed] [Google Scholar]
- Liu Y, Wilkinson FL, Kirton JP, et al. Hepatocyte growth factor and c-Met expression in pericytes: implications for atherosclerotic plaque development. J Pathol. 2007;212:12–19. doi: 10.1002/path.2155. [DOI] [PubMed] [Google Scholar]
- Markwald RR, Fitzharris TP, Manasek FJ. Structural development of endocardial cushions. Am J Anat. 1977;148:85–119. doi: 10.1002/aja.1001480108. [DOI] [PubMed] [Google Scholar]
- Mauer M, Zinman B, Gardiner R, et al. Renal and retinal effects of enalapril and losartan in type 1 diabetes. N. Engl. J. Med. 2009;361:40–51. doi: 10.1056/NEJMoa0808400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Modlinger PS, Wilcox CS, Aslam S. Nitric oxide, oxidative stress, and progression of chronic renal failure. Semin. Nephrol. 2004;24:354–365. doi: 10.1016/j.semnephrol.2004.04.007. [DOI] [PubMed] [Google Scholar]
- Nangaku M. Chronic hypoxia and tubulointerstitial injury: a final common pathway to end-stage renal failure. J. Am. Soc. Nephrol. 2006;17:17–25. doi: 10.1681/ASN.2005070757. Epub 2005 Nov 16. [DOI] [PubMed] [Google Scholar]
- O'riordan E, Mendelev N, Patschan S, et al. Chronic NOS inhibition actuates endothelial-mesenchymal transformation. Am. J. Physiol. Heart. Circ. Physiol. 2007;292:H285–H294. doi: 10.1152/ajpheart.00560.2006. [DOI] [PubMed] [Google Scholar]
- Remuzzi G, Benigni A, Remuzzi A. Mechanisms of progression and regression of renal lesions of chronic nephropathies and diabetes. J. Clin. Invest. 2006;116:288–296. doi: 10.1172/JCI27699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiz-Ortega M, Esteban V, Ruperez M, et al. Renal and vascular hypertension-induced inflammation: role of angiotensin II. Curr. Opin. Nephrol. Hypertens. 2006;15:159–166. doi: 10.1097/01.mnh.0000203190.34643.d4. [DOI] [PubMed] [Google Scholar]
- Sugimoto H, Grahovac G, Zeisberg M, Kalluri R. Renal fibrosis and glomerulosclerosis in a new mouse model of diabetic nephropathy and its regression by bone morphogenic protein-7 and advanced glycation end product inhibitors. Diabetes. 2007;56:1825–1833. doi: 10.2337/db06-1226. Epub 2007 Apr 24. [DOI] [PubMed] [Google Scholar]
- Tharaux PL, Chatziantoniou C, Fakhouri F, Dussaule JC. Angiotensin II activates collagen i gene through a mechanism involving the MAP/ER kinase pathway. Hypertension. 2000;36:330–336. doi: 10.1161/01.hyp.36.3.330. [DOI] [PubMed] [Google Scholar]
- Valderrabano F, Berthoux FC. Report on management of renal failure in Europe, XXV, 1994 end stage renal disease and dialysis report. The EDTA-ERA Registry. Nephrol. Dial. Transplant. 1996;11(Suppl. 1):2–21. doi: 10.1093/ndt/11.supp1.2. [DOI] [PubMed] [Google Scholar]
- Vandermeersch S, Stefanovic V, Hus-Citharel A, Ardaillou R, Dussaule JC, Chansel D. AT1 receptor expression in glomeruli from NO-deficient rats. Nephron Exp. Nephrol. 2003;95:e119–e128. doi: 10.1159/000074328. [DOI] [PubMed] [Google Scholar]
- Wynn TA. Common and unique mechanisms regulate fibrosis in various fibroproliferative diseases. J. Clin. Invest. 2007;117:524–529. doi: 10.1172/JCI31487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Liu Y. Delayed administration of hepatocyte growth factor reduces renal fibrosis in obstructive nephropathy. Am. J. Physiol. Renal Physiol. 2003;284:F349–F357. doi: 10.1152/ajprenal.00154.2002. [DOI] [PubMed] [Google Scholar]
- Yang J, Dai C, Liu Y. Hepatocyte growth factor suppresses renal interstitial myofibroblast activation and intercepts Smad signal transduction. Am. J. Pathol. 2003;163:621–632. doi: 10.1016/S0002-9440(10)63689-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeisberg M, Bottiglio C, Kumar N, et al. Bone morphogenic protein-7 inhibits progression of chronic renal fibrosis associated with two genetic mouse models. Am. J. Physiol. Renal Physiol. 2003a;285:F1060–F1067. doi: 10.1152/ajprenal.00191.2002. [DOI] [PubMed] [Google Scholar]
- Zeisberg M, Hanai J, Sugimoto H, et al. BMP-7 counteracts TGF-beta1-induced epithelial-to-mesenchymal transition and reverses chronic renal injury. Nat. Med. 2003b;9:964–968. doi: 10.1038/nm888. [DOI] [PubMed] [Google Scholar]
- Zeisberg EM, Potenta S, Xie L, Zeisberg M, Kalluri R. Discovery of endothelial to mesenchymal transition as a source for carcinoma-associated fibroblasts. Cancer Res. 2007a;67:10123–10128. doi: 10.1158/0008-5472.CAN-07-3127. [DOI] [PubMed] [Google Scholar]
- Zeisberg EM, Tarnavski O, Zeisberg M, et al. Endothelial-to-mesenchymal transition contributes to cardiac fibrosis. Nat. Med. 2007b;13:952–961. doi: 10.1038/nm1613. Epub 2007 Jul 29. [DOI] [PubMed] [Google Scholar]
- Zeisberg EM, Potenta SE, Sugimoto H, Zeisberg M, Kalluri R. Fibroblasts in kidney fibrosis emerge via endothelial-to-mesenchymal transition. J. Am. Soc. Nephrol. 2008;19:2282–2287. doi: 10.1681/ASN.2008050513. [DOI] [PMC free article] [PubMed] [Google Scholar]