

Abstract
Interstitial fibrosis, associated with extensive accumulation of extracellular matrix constituents in the cortical interstitium, is directly correlated to progression of renal disease. The earliest histological marker of this progression is the accumulation in the interstitium of fibroblasts with the phenotypic appearance of myofibroblasts. These myofibroblasts are contractile cells that express alpha smooth muscle actin and incorporate it into intracellular stress fibres. Although fibroblasts are histologically visible in normal kidneys, there are relatively few of them and proximal tubular epithelial cells predominate. In progressive disease, however, the interstitium becomes filled with myofibroblasts. In this review, we will examine the phenotype and function of fibroblasts and myofibroblasts in the cortical interstitium and the processes that may modulate them.
Keywords: differentiation, EMT, extracellular matrix, fibroblast, fibrocyte, hyaluronan, myofibroblast
Introduction
Renal fibrosis, whether the origin is inflammatory or immunological (e.g. pyelonephritis or lupus nephritis), obstructive (e.g. kidney stones), metabolic (diabetic nephropathy) or systemic [e.g. nephrogenic systemic fibrosis (NSF)], inevitably progresses to end-stage renal disease with progressive, irreversible decline in renal function. Glomerular inflammation, mesangial expansion and sclerosis have all been considered important factors in the development of chronic kidney disease (CKD). In the majority of patients with CKD, however, the progression of renal insufficiency is most closely correlated to the degree of tubular atrophy and interstitial fibrosis.
Fibrosis can be considered aberrant wound healing, in which there is progression rather than resolution of scarring following injury and fibroblasts are central to this process. Fibroblasts have distinct phenotypes depending on the disease and site from which they are isolated. Fibroblasts in the interstitium of kidneys with chronic progressive disease take on a contractile myofibroblastic phenotype and are responsible for the formation of the fibrillar collagen-rich extracellular matrix (ECM) that fills the interstitium leading to nephron loss and declining kidney function. The presence of myofibroblasts is, therefore, recognized as a predictor of fibrotic progression in both experimental models and human renal diseases.
What are fibroblasts?
Fibroblasts are mesenchymal cells that display a spindle-shaped morphology and are ubiquitous in tissues and organs throughout the body. They are the source of many of the constituents of the ECM and so are essential for the maintenance of normal tissue architecture. They also synthesise a variety of proteolytic enzymes and inhibitors, which enables them to control the assembly and turnover of the ECM. Fibroblasts from the renal cortical interstitium typify this and for many years have been known to have an extensive endoplasmic reticulum with a high capacity for protein synthesis (Lemley & Kriz 1991).
Fibroblasts display a large degree of heterogeneity depending on their anatomical site of isolation and their degree of activation (Rodemann & Muller 1990, 1991; Muller & Rodemann 1991; Rodemann et al. 1991; Muller et al. 1995). Unlike many other tissues, the normal renal cortex contains relatively few fibroblasts, with perhaps only two or three positioned in a perivascular or peritubular location when viewed in biopsy sections. These cells stain positively for the intermediate filament protein vimentin, but, while they do not stain for the smooth muscle marker desmin, they are weakly positive for alpha smooth muscle actin (α-SMA) (Alpers et al. 1994; Clayton et al. 1997). There are relatively few specific markers; however, for these cells and once in culture, it is difficult to distinguish fibroblasts from, for example, mesangial cells or smooth muscle cells. Several research groups, including our own, have described patterns of marker expression that can be used for identification, particularly in vitro (Knecht et al. 1991; Muller & Rodemann 1991; Rodemann & Muller 1991; Rodemann et al. 1991; Clayton et al. 1997; Strutz et al. 2001); nevertheless, cortical fibroblasts have not been extensively researched either in vivo or in vitro.
That interstitial fibroblasts in the kidney have an endocrine role has been known for some time. Maxwell et al. described a population of cells in the interstitium of the cortex and outer medulla with the appearance of fibroblast-like type I interstitial cells and that these were the source of erythropoietin (EPO) (Maxwell et al. 1993). Regulation of EPO production by the kidneys is central to the control of erythropoiesis, and EPO controls erythropoiesis by regulating the survival, proliferation and differentiation of erythroid progenitor cells. Thus, the presence of normal interstitial fibroblasts is essential for homoeostasis and protection against anaemia. In a subsequent study examining EPO expression in a variety of models of renal injury, Maxwell et al. found a marked reduction in interstitial cells expressing EPO, or able to induce EPO when given a hypoxic challenge (Maxwell et al. 1997). There were, however, cells present, even in severely injured areas, that could be induced to express EPO and this suggested that myofibroblasts may also have an endocrine function, although reduced compared to fibroblasts.
Opinion is still divided on the origin of the resident fibroblast in the renal cortex. There is some evidence that fibroblasts derived from bone marrow may make up as much as 12% of the interstitial population of the normal kidney (Iwano et al. 2002). Furthermore, in a disease context (chronic allograft rejection), this number increased to 30% (Grimm et al. 2001), clearly confirming the potential of this route for populating the cortex. Classical studies, however, indicate that resident interstitial fibroblasts are derived from the uninduced mesenchyme in the embryonic kidney (Ekblom & Weller 1991). Whatever the source of the normal resident fibroblasts, however, it is clear that their numbers increase in disease, and they may be activated by a variety of cytokines, growth factors, particularly transforming growth factor (TGF) β1 or ECM constituents to differentiate into myofibroblasts.
What are myofibroblasts?
Myofibroblasts are terminally differentiated cells, rarely found in non-pathological situations that are responsible for the synthesis and accumulation of interstitial ECM components such as type I and III collagens and fibronectin during wound healing and at sites of scarring and fibrosis. Myofibroblasts were identified initially in the granulation tissue of healing wounds (Gabbiani et al. 1971; Majno et al. 1971). They are contractile cells expressing many of the morphological and structural features of smooth muscle cells, with flattened and irregular morphology and well-developed cell–ECM interactions and intercellular gap junctions (Vaughan et al. 2000). In particular, they have abundant expression of α-SMA and incorporate it into stress fibres. The classical description of the differentiation of the myofibroblast from resident fibroblasts involves their passing through a proto-myofibroblastic stage (Desmouliere et al. 2005). This process is poorly understood, but the importance of mechanical factors is becoming increasingly apparent (Hinz & Gabbiani 2003a,b;, Hinz et al. 2004; Tomasek et al. 2002, 2006) (Figure 1). The proto-myofibroblast phenotype is characterized by the increased expression of fibronectin (Hinz & Gabbiani 2003a,b; Hinz et al. 2001a,b;, Hinz et al. 2007) and specifically the expression of the alternately spliced ED-A isoform, which is not expressed by fibroblasts (Ffrench-Constant et al. 1989). Proto-myofibroblasts are distinct from myofibroblasts and do not express the classical marker of the myofibroblast phenotype, α-SMA (Hinz et al. 2001a,b, 2003; Tomasek et al. 2002). The expression of ED-A fibronectin has been shown to precede that of α-SMA, and inhibition of the ED-A domain of cellular fibronectin inhibits the TGF-β1-dependent induction of α-SMA (Serini et al. 1998). The proto-myofibroblast is therefore intermediate in the process of myofibroblastic differentiation.
Figure 1.
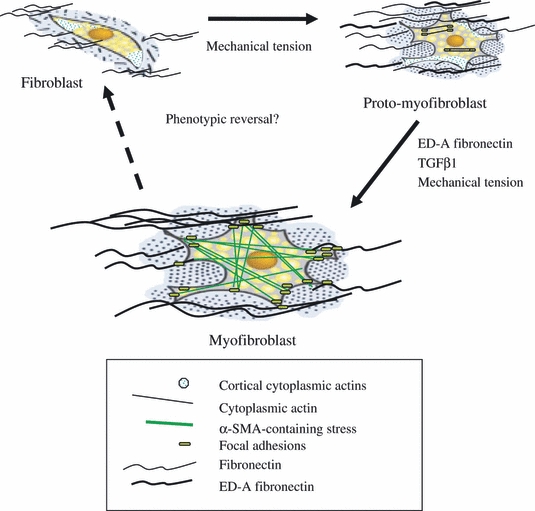
Following an increase in mechanical tension, fibroblasts become activated and acquire a migratory phenotype termed the proto-myofibroblast. Proto-myofibroblasts are characterized by the presence of stress fibres containing filamentous actins, and synthesis of ED-A fibronectin. In the presence of prolonged mechanical tension, ED-A fibronectin, and TGF-β1 further differentiation occurs to a contractile phenotype, termed a differentiated myofibroblast, characterized by the expression of alpha smooth muscle actin (adapted from Tomasek et al. 2002).
Fibroblasts differentiate to proto-myofibroblasts in response to increasing tension in the surrounding ECM. Cell culture studies using hydrated collagen lattices to model interactions between cells and the ECM have shown that mechanical tension is essential for the induction and also the subsequent maintenance of the proto-myofibroblast phenotype (Tomasek et al. 2002). The actin cytoskeleton of proto-myofibroblasts contains stress fibres formed of cytoplasmic β and γ actin microfilaments, associated with contractile proteins such as non-muscle myosin (Tomasek et al. 2002; Hinz & Gabbiani 2003a,b;). As a result of the increased mechanical tension, focal adhesions, at the ends of stress fibres, evolve to form larger mature focal adhesions, containing integrins, actin binding proteins and kinases such as FAK (Geiger & Bershadsky 2001; Geiger et al. 2001). Mature focal adhesions respond to mechanical tension in the ECM and transmit contractile force through ECM proteins such as fibrillar collagens and fibronectin (Hinz 2006, 2009), resulting in a further increase in mechanical tension, which in combination with TGF-β, released from other resident cells or infiltrating inflammatory cells, triggers differentiation to the myofibroblast phenotype (Arora & McCulloch 1999; Hinz 2007).
The actin cytoskeleton undergoes reorganization and redistribution, causing stress fibres to run parallel to the cell periphery and leading to further evolution of focal adhesions. Mature focal adhesions become supermature focal adhesions, containing tensin, α-SMA, extracellular ED-A fibronectin and α5β1 integrin. Alpha-SMA is incorporated into stress fibres and has been shown to increase the contractile activity of fibroblasts both in vitro and in vivo (Dugina et al. 2001; Hinz 2009; Hinz et al. 2003).
The contractile force generated by myofibroblasts in healing wounds in the skin is essential for efficient closure and healing of the wound, although intercellular communication between myofibroblasts and other cells in the wound (e.g.: keratinocytes) is also important (Gailit et al. 1994). In most wounds, myofibroblasts do not persist; they are cleared by apoptosis (Chipev et al. 2000; Smith & Liu 2002; Gabbiani 2003; Moulin et al. 2004; Darby & Hewitson 2007). Their persistence, however, is associated with excessive ECM deposition leading to loss of tissue structure, aberrant or pathological would healing and the development of scarring. The repair of injured tissue requires repopulation of the tissue by endogenous cells. This cannot occur unless there is remodelling of the ECM to reconstitute an approximation of the original tissue architecture. Extracellular matrix remodelling requires the activity of proteinases, and renal myofibroblasts produce several members of the plasminogen/plasmin and matrix metalloproteinase families that are involved (Liu 2006). During the process of ECM accumulation, the natural inhibitors of these proteinases (plasminogen activator inhibitor-1 and tissue inhibitor of metalloproteinase-1) are often elevated in animal models of fibrosis, so contributing to the accumulation of the fibrotic ECM. This matrix alters the normal structure of the tissue and thereby detrimentally affects function. If the myofibroblasts are not cleared, this leads to progressive organ failure (Lane et al. 2002).
Myofibroblasts in the renal cortex
Once detectable, myofibroblasts are prognostic indicators of fibrotic expansion and progressive tubular atrophy, leading to end-stage organ failure. Intervening in their activity or preventing their accumulation is a major focus of many research groups.
The ‘classical’ source of myofibroblasts in kidney pathology could be said to be that outlined above: differentiation of endogenous fibroblasts under the influence of growth factors. Numerous growth factors, cytokines and hormones have been studied as potential mediators in the development of fibrosis. These include TGF-β, connective tissue growth factor, fibroblast growth factor (FGF), platelet-derived growth factor, interleukin-1 (IL-1), tumour necrosis factor (TNF)-α, angiotensin II and aldosterone. Of these, however, TGF-β is the principal mediator implicated in regulating fibrosis, and its aberrant expression has been documented in a wide variety of fibrotic disorders. Furthermore, TGF-β is known to have a direct effect on the turnover of the ECM and has been shown to be essential for myofibroblastic differentiation in a variety of systems including the kidney and in each of these it fulfils a similar role (Ina et al. 2002; Howell & McAnulty 2006; Uhal et al. 2007; Werner et al. 2007).
Transforming growth factor-β is a multifunctional cytokine, originally isolated from platelets (Sporn & Roberts 1990). In mammals, there are three isoforms, but the form most implicated in renal fibrosis is TGF-β1. TGF-β1 is synthesized as an inactive pro-peptide that, following translation, is secreted as an inactive complex, associated with latency-associated peptide (LAP), and is then sequestered in the ECM. Activation of TGF-β1 occurs through cleavage of the LAP and release of bioactive TGF-β1, which is then free to bind to cell-surface receptors (Piek et al. 1999). TGF-β1 binds to at least three membrane proteins referred to as receptor types I, II and III. The type I and II receptors are trans-membrane serine–threonine kinases that transduce signals and initiate gene transcription (Ebner et al. 1993), while the type III receptor (betaglycan) is a membrane-anchored proteoglycan that sequesters TGF-β1 and presents it to the type I and II receptors (Lopez-Casillas et al. 1994). Gene transcription is triggered by phosphorylation of Smad transcription factors and their subsequent translocation to the nucleus (Shi & Massague 2003). There are also, however, Smad-independent pathways triggered by TGF-β1, which include mitogen-activated protein (MAP) kinases, Rho-GTPases and Protein kinase B (Attisano & Wrana 2002).
Several of our own studies investigated the TGF-β-triggered differentiation of fibroblasts in some detail (Clayton et al. 2001; Evans et al. 2003; Thomas et al. 2003). These were originally intended as a study to find additional biomarkers of myofibroblasts that may have been useful as diagnostic tools in combination with α-SMA. This work, however, suggested a wholesale alteration in the receptors and mechanisms that control the cellular responses to growth factors and other mediators following myofibroblastic differentiation. They described the identification and expression of cell-surface heparan sulphate (HS) proteoglycans on renal and lung fibroblasts and demonstrated that these were essential for the control of the proliferative response of these cells to FGF-2. While both fibroblasts and myofibroblasts responded to platelet-derived growth factor, the latter, unlike the former, did not proliferate to FGF-2. A response was acquired, however, when myofibroblasts were incubated with FGF-2 in the presence of HS chains or heparin. The mechanism underlying this was shown to involve changes in the expression of glycosaminoglycans (GAG), particularly cell-surface HS proteoglycans that mediated the interaction of FGF-2 with its signalling receptors. There was no difference in FGF-2 binding affinity between the 2 cell-types, but the HS-GAG chains secreted by myofibroblasts had twice the binding capacity of those from fibroblasts. Thus, it was likely that the difference in response to FGF-2 was because of differences in FGF-2 sequestration and receptor interaction with FGF-2–HS complexes.
Clinically, this was an important finding in the light of studies by Strutz et al. (Strutz 2009b, Strutz et al. 2000), in which a role was described for FGF-2 in the differentiation process of immortalized renal fibroblasts. In addition, other findings implicate a role for FGF-2 in high glucose-altered molecular signalling in the pathogenesis of diabetic renal disease (Vasko et al. 2009). The change in HS expression may be an important mechanism involved in this pathogenesis, it is also likely, however, to be important for interactions with other growth factors and chemokines, many of which also bind to HS structures on the cell surface. These are subsequently either presented to their signalling receptors or are sequestered by soluble or ECM HS proteoglycans and prevented from reaching their receptors.
Phage-display technology (van Kuppevelt et al. 2001) has now allowed more detailed profiling of HS expression in the kidney (Lensen et al. 2005) and identified its aberrant profile in diabetic nephropathy (Wijnhoven et al. 2007). This supports the clinical significance of the expression of particular HS-GAG structures demonstrated by Morita et al. (1994) who examined the expression of HS-GAG chains in renal biopsy sections. While total HS expression was increased in both the glomerulus and the tubulo-interstitium, FGF-2 binding was only increased on cells in the interstitium, suggesting that the expression of specific cell-surface HS proteoglycans may play a major role in controlling fibrotic events in the interstitium.
One particular HS proteoglycan that has a central role in the accumulation of myofibroblasts in areas of fibrosis is betaglycan or TGFβ receptor III. As described earlier, it is central to the presentation of TGFβ to its signalling receptors. It also, however, when released from the cell membrane, acts as an inhibitor of TGFβ function by sequestering TGFβ away from the cell surface. Thus, using a recombinant form of soluble betaglycan (SBG), Juarez et al. (2007) demonstrated that SBG was a renoprotective agent that neutralized TGFβ action in a mouse model of diabetic nephropathy. Because SBG has a high affinity for all TGFβ isoforms, the authors suggested that SBG could be a successful therapy for the long-term treatment of renal disease and other pathologies in which TGFβ plays a pathophysiological role. Proteoglycans have a protein core that is covalently substituted with GAG side chains. Although TGFβ binds to the betaglycan core protein rather than the HS chains, Eickelberg et al. (2002) reported that the ratio of TGFβRI to TGFβRII and signalling from the receptor complex was regulated by the HS moiety on betaglycan, with smaller chains or their absence favouring the optimal formation of the signalling complex, whereas larger chains inhibited TGF-β-induced cellular responses. Development of differentially HS-substituted SBG might allow for selectivity in the effect of anti-TGFβ treatment that could be used therapeutically.
The GAG most often associated with wound healing is the non-sulphated linear polysaccharide hyaluronan (Gailit et al. 1994). Hyaluronan or hyaluronic acid (HA) is ubiquitous and in vivo is present as a high molecular mass component of most extracellular matrices. In addition to the biological functions associated with its viscoelastic properties, HA regulates cellular function through its interactions with cell-surface receptors (principally CD44) as well as through the generation of cell surface and pericellular HA matrices in association with HA binding proteins (hyaladherins) (Day 1999; Day & Prestwich 2002; Lesley et al. 2002; Melrose et al. 2002; Milner et al. 2006). Hyaluronic acid therefore plays important roles maintaining homoeostasis, as a ECM scaffold, and in tissue repair and regeneration. Although HA is not a major constituent of the normal renal cortex, it accumulates in the corticointerstitium following acute inflammatory injury and in chronic fibrotic diseases (Wells et al. 1993, 1999, Sibalic et al. 1997; Feusi et al. 1999; Lewington et al. 2000; Sano et al. 2001; Lewis et al. 2008).
Our studies examining the TGFβ-dependent activation of cells have shown that HA potently modulates the response of proximal tubular epithelial cells to TGFβ (Ito et al. 2004a,b,c;). In addition, HA accumulated in pericellular matrices around myofibroblasts following differentiation induced by TGFβ. Because Zoltan-Jones et al. (2003) had demonstrated that the endogenous expression of HA was essential for mesenchymal transformation of certain epithelial cell-lines, we reasoned that the changes observed in fibroblast differentiation may also be causally related.
Subsequent studies (Meran et al. 2007, 2008; Simpson et al. 2009a,b; Webber et al. 2009a,b;) have revealed a complex inter-relationship between the HA pericellular matrix and interactions between the epidermal growth factor receptor (EGFR) and the TGF receptor. This is summarized in Figure 2.
Figure 2.
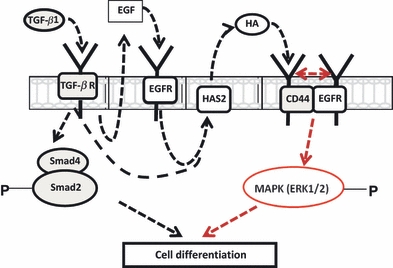
TGF-β1-dependent phenotypic activation triggers two distinct but cooperative pathways that involve TGF-R/Smad2 activation and EGF-mediated EGF-R/ERK mitogen-activated protein kinase activation. HAS2-dependent HA synthesis is also initiated by EGF/EGFR binding. The subsequent binding of HA to CD44 facilitates CD44 association with the EGFR. This association triggers phosphorylation of ERK1 and ERK2, which promotes cellular differentiation when combined with Smad activation (adapted from Simpson et al. 2009b). EGFR, epidermal growth factor receptor; HA, hyaluronic acid.
Alternative origins of myofibroblasts
Fibrocytes
There are other potential sources for myofibroblasts in the diseased kidney than tissue resident fibroblasts. These sources involve circulating cells of bone marrow origin (fibrocytes) that migrate into the interstitium and contribute to the pathogenesis of renal disease (Bucala et al. 1994; Quan et al. 2004; Wada et al. 2007). This model has been suggested as important in several systems and has been widely examined in NSF (Bucala 2008). NSF occurs predominantly but not exclusively in patients with renal impairment (Grobner & Prischl 2007). It is characterized by pigmented fibrotic lesions on the skin of the trunk and limbs and fibrosis in internal organs, including heart and lungs, liver and muscle (Kribben et al. 2009). It is a rapidly progressive disabling disease, and currently, there are few therapeutic options as with most fibrotic diseases. In patients on renal replacement therapy, NSF is linked to exposure to gadolinium-containing contrast media used in magnetic resonance imaging (High et al. 2007; Marckmann & Skov 2009).
Circulating fibrocytes may be major mediators of NSF, particularly when internal organs are involved (Ortonne et al. 2004). Fibrocytes are fibroblast precursors expressing CD34 and procollagen. They migrate into areas of tissue damage and have been identified in a number of fibrotic conditions (Quan et al. 2004, 2006; Quan & Bucala 2007) and within NSF skin lesions (Ortonne et al. 2004). Circulating fibrocytes express CD34; however, this expression is reduced on their surface, as they differentiate and become more specialized (Bucala 2008). This decrease is accelerated following stimulation with TGF-β1. TGF-β1 is produced by fibrocytes and is likely to participate in the differentiation events occurring in fibrosis (Chesney & Bucala 1997; Wada et al. 2007). An average of 32% of all α-SMA-positive myofibroblasts involved in ECM synthesis in the postischaemic interstitium are derived from the bone marrow (Broekema et al. 2007). It therefore follows that the rest of the myofibroblasts present were derived from another source. There may, however, be interactions between infiltrating fibrocytes and resident cells that influence progression and fibrocyte-derived TGF may be central to this.
The involvement of fibrocytes in renal disease is not universally acknowledged. Other groups have also examined whether interstitial myofibroblasts are derived from fibrocytes in a variety of models of renal injury. For example, using obstructive models of nephritis, both Lin et al. (2008) and Roufosse et al. (2006) failed to find a contribution of circulating cells to the synthesis of collagen in the injured kidney. Thus, although circulating fibrocytes seem to be effectors in other organs such as the heart, lung and skin (Abe et al. 2001; Schmidt et al. 2003; Haudek et al. 2006), their role as mediators in the kidney is still debatable, and there is one other hypothesis that should be explored.
Epithelial to mesenchymal transition
Another potential hypothesis, developed over the last 20 years, to describe an alternative mechanism for myofibroblasts accumulating in the renal cortex, has been the transition of tubular epithelial cells to a mesenchymal phenotype (epithelial to mesenchymal transition – EMT), reviewed in (Strutz & Neilson 2003; Strutz 2009a,b;). In response to injury, it is proposed that epithelial cells undergo major morphological changes, losing epithelial characteristics such as polarity and the expression of junctional markers, while inducing fibroblast markers (fibroblast-specific protein or FSP-1, vimentin and α-SMA) (Strutz et al. 1995; Ng et al. 1998; Zeisberg et al. 2001; Iwano et al. 2002). The cells begin to express stress fibres and migrate along and then through the basement membrane to become (myo)fibroblasts in the interstitium.
It has been shown that a partial absence or disassembly of cell–cell junctions is a major trigger of TGF-β1-induced EMT. Masszi et al. (2004) demonstrated that β-catenin plays an important role in the TGF-β1- and cell contact-dependent regulation of the α-SMA promoter and protein expression. Their findings suggested a two-hit model in which both an initial tissue injury and TGF-β1 were required for EMT. More recently, the same group has demonstrated that disruption of cell contact is itself an initiator of signals that contribute to the EMT process through the induction of myocardin-related transcription factor (MRTF) (Masszi et al.). The model the authors present suggests both Smad3-dependent (early or mesenchymal) and Smad3-independent (late or myogenic) phases of EMT. Smad3 contributes to the loss of epithelial markers and is critical for the expression of mesenchymal markers and certain ECM proteins. It may also prepare the cells for the second phase by promoting nuclear MRTF accumulation and the synthesis of proteins such as ED-A fibronectin that enhances α-SMA expression. This phase is followed by gradual degradation of Smad3, which enables the mobilization of the myogenic phase. This switch is a prerequisite for the motile and contractile phenotype, and Smad3 is therefore a major regulator of EMT. These results also highlight that de-differentiation of cells in the mesenchymal phase can occur without full transition to motile myofibroblastic cells in the myogenic phase.
Until recently, specific lineage-tracing studies had not been performed to trace the cellular source of myofibroblastic cells, and several possible theories around EMT were more or less popular. There may, for example, be a sub-population of progenitor tubular cells that are uniquely able to differentiate into fibroblasts (Yamashita et al. 2005). Certainly, α-SMA and the fibroblast marker, FSP-1, do not always colocalize to the same cells within a population of fibroblasts (Okada et al. 2000) suggesting that they may have had different progenitor sources. It is also possible that there is a disease-specificity to the origin of the interstitial myofibroblasts. For example, while EMT has been identified in models of obstruction and polycystic kidney disease (Okada et al. 2000; Iwano et al. 2002; Yang et al. 2002), it was not observed in a protein overload model (Ikeda et al. 2004). Furthermore, Faulkner et al. (2005), in an accelerated model of angiotensin II-induced fibrosis, did not observe tubular basement membrane disruption or epithelial cell invasion of the interstitium.
That EMT can occur is certainly true of proximal tubular epithelial cells in culture when exposed to TGFβ. The cells lose the expression of epithelial characteristics and begin to express markers of mesenchymal cells such as α-SMA and fibrillar collagens (Tian & Phillips 2002). Tubular epithelial cells can also undergo these changes when damaged in vivo. Whether this is full EMT, leading to complete loss of the epithelial phenotype and adoption of a migratory fibroblastic phenotype, or whether this is simply a reversible response to acute injury is still open to debate. Lineage-tracing experiments, however, to assess the contribution of renal epithelial cells to fibrosis have been published (Humphreys et al. 2010). Using the unilateral ureteric obstruction and unilateral ischaemia-reperfusion models, the authors utilized a cre/lox labelling system to selectively label either collecting duct epithelial cells alone or all cells except the collecting duct epithelium. They found no evidence that epithelial cells had migrated through the tubular basement membrane or had differentiated into myofibroblasts in either model of injury. This was not explained by a loss of EMT-potential in the genetically altered epithelial cells; cre/lox-labelled tubular epithelial cells isolated from the mice and cultured in vitro, induced the expression of both α-SMA and FSP-1 and lost expression of the epithelial marker, E-cadherin, when exposed to TGF-β1.
Lin et al. (2008) have previously identified perivascular fibroblasts and pericytes as the major contributors to the myofibroblast population in experimental obstructive nephropathy. While they also observed recruited fibrocytes, their numbers were very small (<0.1% of all fibroblasts). Importantly, they found no evidence of tubular epithelial contribution to the myofibroblast pool. The follow-up studies by Humphreys et al. (2010), using lineage-analysis also implicated perivascular fibroblasts/pericytes, derived from FoxD1-positive metanephric mesenchymal cells as the major source of the myofibroblasts in the interstitium. The authors comment that these findings should lead to a realignment of focus towards identifying the factors that mediate the interactions of pericytes with vascular endothelial cells and their migration away from their perivascular location. They point out that there are many important interactions between pericytes and adjacent endothelial cells (see for review) that are mutually supporting and that reassessing the effect of endothelial perturbations as potential contributors to fibrosis may be an important next step towards understanding fibrotic initiation. This is covered in more detail by Dr Tharaux in this issue.
Conclusions
Research over many years has highlighted the importance of myofibroblasts in the renal cortex for progression of kidney disease. Much of this research has been directed towards examining the mechanisms that may initiate fibrosis and has seemed to highlight three separate, competing hypotheses. Several studies, however, are beginning to provide some degree of unification if not necessarily complete unanimity. While EMT, fibrocyte infiltration and fibroblast to myofibroblast differentiation may all be involved in the response to injury and in repair, it is the prevention of the accumulation of myofibroblasts that is most important. The studies of Lin et al. (2008) and Humphreys et al. (2010) indicate that new strategies to directly target the differentiation of perivascular fibroblasts/pericytes may successfully halt myofibroblast initiation, but it is myofibroblast persistence in tissues that is the major factor in the scarring process. Persistence of these cells is likely to depend on a variety of mediators ranging from growth factors (such as TGF-β1 Webber et al. 2009a,b;) released from resident or infiltrating cells, to those which influence whether the cells are cleared, for example, by apoptosis. In addition, studies have now identified endogenous antifibrotic factors, particularly hepatocyte growth factor (Liu 2004; Yang et al. 2002) and bone morphogenetic protein-7 (BMP-7) (Zeisberg et al. 2003), which antagonize the action of TGF-β1. Therefore, re-establishing a balance between pro- and antifibrotic signalling may provide a novel opportunity for designing antifibrotic therapeutic strategies. Understanding these mechanisms and the interactions that control the maintenance and function of myofibroblasts in areas of fibrosis presents the next challenge.
Acknowledgments
We are grateful to Kidney Research UK, Research into Ageing and Cardiff and Vale NHS Trust for financial support.
References
- Abe R, Donnelly SC, Peng T, Bucala R, Metz CN. Peripheral blood fibrocytes: differentiation pathway and migration to wound sites. J. Immunol. 2001;166:7556–7562. doi: 10.4049/jimmunol.166.12.7556. [DOI] [PubMed] [Google Scholar]
- Alpers CE, Hudkins KL, Floege J, Johnson RJ. Human renal cortical interstitial cells with some features of smooth muscle cells participate in tubulointerstitial and crescentic glomerular injury. J. Am. Soc. Nephrol. 1994;5:210–219. doi: 10.1681/ASN.V52201. [DOI] [PubMed] [Google Scholar]
- Arora PD, McCulloch CA. The deletion of transforming growth factor-beta-induced myofibroblasts depends on growth conditions and actin organization. Am. J. Pathol. 1999;155:2087–2099. doi: 10.1016/s0002-9440(10)65527-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Attisano L, Wrana JL. Signal transduction by the TGF-beta superfamily. Science. 2002;296:1646–1647. doi: 10.1126/science.1071809. [DOI] [PubMed] [Google Scholar]
- Broekema M, Harmsen MC, van Luyn MJ, et al. Bone marrow-derived myofibroblasts contribute to the renal interstitial myofibroblast population and produce procollagen I after ischemia/reperfusion in rats. J. Am. Soc. Nephrol. 2007;18:165–175. doi: 10.1681/ASN.2005070730. [DOI] [PubMed] [Google Scholar]
- Bucala R. Circulating fibrocytes: cellular basis for NSF. J. Am. Coll. Radiol. 2008;5:36–39. doi: 10.1016/j.jacr.2007.08.016. [DOI] [PubMed] [Google Scholar]
- Bucala R, Spiegel LA, Chesney J, Hogan M, Cerami A. Circulating fibrocytes define a new leukocyte subpopulation that mediates tissue repair. Mol. Med. 1994;1:71–81. [PMC free article] [PubMed] [Google Scholar]
- Chesney J, Bucala R. Peripheral blood fibrocytes: novel fibroblast-like cells that present antigen and mediate tissue repair. Biochem. Soc. Trans. 1997;25:520–524. doi: 10.1042/bst0250520. [DOI] [PubMed] [Google Scholar]
- Chipev CC, Simman R, Hatch G, Katz AE, Siegel DM, Simon M. Myofibroblast phenotype and apoptosis in keloid and palmar fibroblasts in vitro. Cell Death Differ. 2000;7:166–176. doi: 10.1038/sj.cdd.4400605. [DOI] [PubMed] [Google Scholar]
- Clayton A, Steadman R, Williams JD. Cells isolated from the human cortical interstitium resemble myofibroblasts and bind neutrophils in an ICAM-1 – dependent manner. J. Am. Soc. Nephrol. 1997;8:604–615. doi: 10.1681/ASN.V84604. [DOI] [PubMed] [Google Scholar]
- Clayton A, Thomas J, Thomas GJ, Davies M, Steadman R. Cell surface heparan sulfate proteoglycans control the response of renal interstitial fibroblasts to fibroblast growth factor-2. Kidney Int. 2001;59:2084–2094. doi: 10.1046/j.1523-1755.2001.00723.x. [DOI] [PubMed] [Google Scholar]
- Darby IA, Hewitson TD. Fibroblast differentiation in wound healing and fibrosis. Int. Rev. Cytol. 2007;257:143–179. doi: 10.1016/S0074-7696(07)57004-X. [DOI] [PubMed] [Google Scholar]
- Day AJ. The structure and regulation of hyaluronan-binding proteins. Biochem. Soc. Trans. 1999;27:115–121. doi: 10.1042/bst0270115. [DOI] [PubMed] [Google Scholar]
- Day AJ, Prestwich GD. Hyaluronan-binding proteins: tying up the giant. J. Biol. Chem. 2002;277:4585–4588. doi: 10.1074/jbc.R100036200. [DOI] [PubMed] [Google Scholar]
- Desmouliere A, Chaponnier C, Gabbiani G. Tissue repair, contraction, and the myofibroblast. Wound Repair Regen. 2005;13:7–12. doi: 10.1111/j.1067-1927.2005.130102.x. [DOI] [PubMed] [Google Scholar]
- Dugina V, Fontao L, Chaponnier C, Vasiliev J, Gabbiani G. Focal adhesion features during myofibroblastic differentiation are controlled by intracellular and extracellular factors. J. Cell Sci. 2001;114:3285–3296. doi: 10.1242/jcs.114.18.3285. [DOI] [PubMed] [Google Scholar]
- Ebner R, Chen RH, Lawler S, Zioncheck T, Derynck R. Determination of type I receptor specificity by the type II receptors for TGF-beta or activin. Science. 1993;262:900–902. doi: 10.1126/science.8235612. [DOI] [PubMed] [Google Scholar]
- Eickelberg O, Centrella M, Reiss M, Kashgarian M, Wells RG. Betaglycan inhibits TGF-beta signaling by preventing type I-type II receptor complex formation. Glycosaminoglycan modifications alter betaglycan function. J. Biol. Chem. 2002;277:823–829. doi: 10.1074/jbc.M105110200. [DOI] [PubMed] [Google Scholar]
- Ekblom P, Weller A. Ontogeny of tubulointerstitial cells. Kidney Int. 1991;39:394–400. doi: 10.1038/ki.1991.51. [DOI] [PubMed] [Google Scholar]
- Evans RA, Tian YC, Steadman R, Phillips AO. TGF-beta1-mediated fibroblast–myofibroblast terminal differentiation-the role of Smad proteins. Exp. Cell Res. 2003;282:90–100. doi: 10.1016/s0014-4827(02)00015-0. [DOI] [PubMed] [Google Scholar]
- Faulkner JL, Szcykalski LM, Springer F, Barnes JL. Origin of interstitial fibroblasts in an accelerated model of angiotensin II-induced renal fibrosis. Am. J. Pathol. 2005;167:1193–1205. doi: 10.1016/S0002-9440(10)61208-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feusi E, Sun L, Sibalic A, Beck-Schimmer B, Oertli B, Wuthrich RP. Enhanced hyaluronan synthesis in the MRL-Fas(lpr) kidney: role of cytokines. Nephron. 1999;83:66–73. doi: 10.1159/000045475. [DOI] [PubMed] [Google Scholar]
- Ffrench-Constant C, Van de Water L, Dvorak HF, Hynes RO. Reappearance of an embryonic pattern of fibronectin splicing during wound healing in the adult rat. J. Cell Biol. 1989;109:903–914. doi: 10.1083/jcb.109.2.903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabbiani G. The myofibroblast in wound healing and fibrocontractive diseases. J Pathol. 2003;200:500–503. doi: 10.1002/path.1427. [DOI] [PubMed] [Google Scholar]
- Gabbiani G, Ryan GB, Majne G. Presence of modified fibroblasts in granulation tissue and their possible role in wound contraction. Experientia. 1971;27:549–550. doi: 10.1007/BF02147594. [DOI] [PubMed] [Google Scholar]
- Gailit J, Welch MP, Clark RA. TGF-beta 1 stimulates expression of keratinocyte integrins during re-epithelialization of cutaneous wounds. J. Invest. Dermatol. 1994;103:221–227. doi: 10.1111/1523-1747.ep12393176. [DOI] [PubMed] [Google Scholar]
- Geiger B, Bershadsky A. Assembly and mechanosensory function of focal contacts. Curr. Opin. Cell Biol. 2001;13:584–592. doi: 10.1016/s0955-0674(00)00255-6. [DOI] [PubMed] [Google Scholar]
- Geiger B, Bershadsky A, Pankov R, Yamada KM. Transmembrane crosstalk between the extracellular matrix – cytoskeleton crosstalk. Nat. Rev. Mol. Cell Biol. 2001;2:793–805. doi: 10.1038/35099066. [DOI] [PubMed] [Google Scholar]
- Grimm PC, Nickerson P, Jeffery J, et al. Neointimal and tubulointerstitial infiltration by recipient mesenchymal cells in chronic renal-allograft rejection. N. Engl. J. Med. 2001;345:93–97. doi: 10.1056/NEJM200107123450203. [DOI] [PubMed] [Google Scholar]
- Grobner T, Prischl FC. Gadolinium and nephrogenic systemic fibrosis. Kidney Int. 2007;72:260–264. doi: 10.1038/sj.ki.5002338. [DOI] [PubMed] [Google Scholar]
- Haudek SB, Xia Y, Huebener P, et al. Bone marrow-derived fibroblast precursors mediate ischemic cardiomyopathy in mice. Proc. Natl. Acad. Sci. U S A. 2006;103:18284–18289. doi: 10.1073/pnas.0608799103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- High WA, Ayers RA, Chandler J, Zito G, Cowper SE. Gadolinium is detectable within the tissue of patients with nephrogenic systemic fibrosis. J. Am. Acad. Dermatol. 2007;56:21–26. doi: 10.1016/j.jaad.2006.10.047. [DOI] [PubMed] [Google Scholar]
- Hinz B. Masters and servants of the force: the role of matrix adhesions in myofibroblast force perception and transmission. Eur. J. Cell Biol. 2006;85:175–181. doi: 10.1016/j.ejcb.2005.09.004. [DOI] [PubMed] [Google Scholar]
- Hinz B. Formation and function of the myofibroblast during tissue repair. J. Invest. Dermatol. 2007;127:526–537. doi: 10.1038/sj.jid.5700613. [DOI] [PubMed] [Google Scholar]
- Hinz B. Tissue stiffness, latent TGF-beta1 activation, and mechanical signal transduction: implications for the pathogenesis and treatment of fibrosis. Curr. Rheumatol. Rep. 2009;11:120–126. doi: 10.1007/s11926-009-0017-1. [DOI] [PubMed] [Google Scholar]
- Hinz B, Gabbiani G. Cell–matrix and cell–cell contacts of myofibroblasts: role in connective tissue remodeling. Thromb. Haemost. 2003a;90:993–1002. doi: 10.1160/TH03-05-0328. [DOI] [PubMed] [Google Scholar]
- Hinz B, Gabbiani G. Mechanisms of force generation and transmission by myofibroblasts. Curr. Opin. Biotechnol. 2003b;14:538–546. doi: 10.1016/j.copbio.2003.08.006. [DOI] [PubMed] [Google Scholar]
- Hinz B, Celetta G, Tomasek JJ, Gabbiani G, Chaponnier C. Alpha-smooth muscle actin expression upregulates fibroblast contractile activity. Mol. Biol. Cell. 2001a;12:2730–2741. doi: 10.1091/mbc.12.9.2730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinz B, Mastrangelo D, Iselin CE, Chaponnier C, Gabbiani G. Mechanical tension controls granulation tissue contractile activity and myofibroblast differentiation. Am. J. Pathol. 2001b;159:1009–1020. doi: 10.1016/S0002-9440(10)61776-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinz B, Dugina V, Ballestrem C, Wehrle-Haller B, Chaponnier C. Alpha-smooth muscle actin is crucial for focal adhesion maturation in myofibroblasts. Mol. Biol. Cell. 2003;14:2508–2519. doi: 10.1091/mbc.E02-11-0729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinz B, Pittet P, Smith-Clerc J, Chaponnier C, Meister JJ. Myofibroblast development is characterized by specific cell–cell adherens junctions. Mol. Biol. Cell. 2004;15:4310–4320. doi: 10.1091/mbc.E04-05-0386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinz B, Phan SH, Thannickal VJ, Galli A, Bochaton-Piallat ML, Gabbiani G. The myofibroblast: one function, multiple origins. Am. J. Pathol. 2007;170:1807–1816. doi: 10.2353/ajpath.2007.070112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howell JE, McAnulty RJ. TGF-beta: its role in asthma and therapeutic potential. Curr. Drug Targets. 2006;7:547–565. doi: 10.2174/138945006776818692. [DOI] [PubMed] [Google Scholar]
- Humphreys BD, Lin SL, Kobayashi A, et al. Fate tracing reveals the pericyte and not epithelial origin of myofibroblasts in kidney fibrosis. Am. J. Pathol. 2010;176:85–97. doi: 10.2353/ajpath.2010.090517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda Y, Jung YO, Kim H, et al. Exogenous bone morphogenetic protein-7 fails to attenuate renal fibrosis in rats with overload proteinuria. Nephron Exp. Nephrol. 2004;97:e123–e135. doi: 10.1159/000079177. [DOI] [PubMed] [Google Scholar]
- Ina K, Kitamura H, Tatsukawa S, Takayama T, Fujikura Y, Shimada T. Transformation of interstitial fibroblasts and tubulointerstitial fibrosis in diabetic nephropathy. Med. Electron. Microsc. 2002;35:87–95. doi: 10.1007/s007950200011. [DOI] [PubMed] [Google Scholar]
- Ito T, Williams JD, Al-Assaf S, Phillips GO, Phillips AO. Hyaluronan and proximal tubular cell migration. Kidney Int. 2004a;65:823–833. doi: 10.1111/j.1523-1755.2004.00457.x. [DOI] [PubMed] [Google Scholar]
- Ito T, Williams JD, Fraser D, Phillips AO. Hyaluronan attenuates transforming growth factor-beta1-mediated signaling in renal proximal tubular epithelial cells. Am. J. Pathol. 2004b;164:1979–1988. doi: 10.1016/s0002-9440(10)63758-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito T, Williams JD, Fraser DJ, Phillips AO. Hyaluronan regulates transforming growth factor-beta1 receptor compartmentalization. J. Biol. Chem. 2004c;279:25326–25332. doi: 10.1074/jbc.M403135200. [DOI] [PubMed] [Google Scholar]
- Iwano M, Plieth D, Danoff TM, Xue C, Okada H, Neilson EG. Evidence that fibroblasts derive from epithelium during tissue fibrosis. J. Clin. Invest. 2002;110:341–350. doi: 10.1172/JCI15518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juarez P, Vilchis-Landeros MM, Ponce-Coria J, et al. Soluble betaglycan reduces renal damage progression in db/db mice. Am. J. Physiol. Renal Physiol. 2007;292:F321–F329. doi: 10.1152/ajprenal.00264.2006. [DOI] [PubMed] [Google Scholar]
- Knecht A, Fine LG, Kleinman KS, et al. Fibroblasts of rabbit kidney in culture II: paracrine stimulation of papillary fibroblasts by PDGF. Am. J. Physiol. 1991;261:F292–F299. doi: 10.1152/ajprenal.1991.261.2.F292. [DOI] [PubMed] [Google Scholar]
- Kribben A, Witzke O, Hillen U, Barkhausen J, Daul AE, Erbel R. Nephrogenic systemic fibrosis: pathogenesis, diagnosis, and therapy. J. Am. Coll. Cardiol. 2009;53:1621–1628. doi: 10.1016/j.jacc.2008.12.061. [DOI] [PubMed] [Google Scholar]
- van Kuppevelt TH, Jenniskens GJ, Veerkamp JH, ten Dam GB, Dennissen MA. Phage display technology to obtain antiheparan sulfate antibodies. Methods Mol. Biol. 2001;171:519–534. doi: 10.1385/1-59259-209-0:519. [DOI] [PubMed] [Google Scholar]
- Lane A, Johnson DW, Pat B, et al. Interacting roles of myofibroblasts, apoptosis and fibrogenic growth factors in the pathogenesis of renal tubulo-interstitial fibrosis. Growth Factors. 2002;20:109–119. doi: 10.1080/0897719021000006181. [DOI] [PubMed] [Google Scholar]
- Lemley KV, Kriz W. Anatomy of the renal interstitium. Kidney Int. 1991;39:370–381. doi: 10.1038/ki.1991.49. [DOI] [PubMed] [Google Scholar]
- Lensen JF, Rops AL, Wijnhoven TJ, et al. Localization and functional characterization of glycosaminoglycan domains in the normal human kidney as revealed by phage display-derived single chain antibodies. J. Am. Soc. Nephrol. 2005;16:1279–1288. doi: 10.1681/ASN.2004050413. [DOI] [PubMed] [Google Scholar]
- Lesley J, English NM, Gal I, Mikecz K, Day AJ, Hyman R. Hyaluronan binding properties of a CD44 chimera containing the link module of TSG-6. J. Biol. Chem. 2002;277:26600–26608. doi: 10.1074/jbc.M201068200. [DOI] [PubMed] [Google Scholar]
- Lewington AJ, Padanilam BJ, Martin DR, Hammerman MR. Expression of CD44 in kidney after acute ischemic injury in rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2000;278:R247–R254. doi: 10.1152/ajpregu.2000.278.1.R247. [DOI] [PubMed] [Google Scholar]
- Lewis A, Steadman R, Manley P, et al. Diabetic nephropathy, inflammation, hyaluronan and interstitial fibrosis. Histol. Histopathol. 2008;23:731–739. doi: 10.14670/HH-23.731. [DOI] [PubMed] [Google Scholar]
- Lin SL, Kisseleva T, Brenner DA, Duffield JS. Pericytes and perivascular fibroblasts are the primary source of collagen-producing cells in obstructive fibrosis of the kidney. Am. J. Pathol. 2008;173:1617–1627. doi: 10.2353/ajpath.2008.080433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y. Hepatocyte growth factor in kidney fibrosis: therapeutic potential and mechanisms of action. Am. J. Physiol. Renal Physiol. 2004;287:F7–F16. doi: 10.1152/ajprenal.00451.2003. [DOI] [PubMed] [Google Scholar]
- Liu Y. Renal fibrosis: new insights into the pathogenesis and therapeutics. Kidney Int. 2006;69:213–217. doi: 10.1038/sj.ki.5000054. [DOI] [PubMed] [Google Scholar]
- Lopez-Casillas F, Payne HM, Andres JL, Massague J. Betaglycan can act as a dual modulator of TGF-beta access to signaling receptors: mapping of ligand binding and GAG attachment sites. J. Cell Biol. 1994;124:557–568. doi: 10.1083/jcb.124.4.557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majno G, Gabbiani G, Hirschel BJ, Ryan GB, Statkov PR. Contraction of granulation tissue in vitro: similarity to smooth muscle. Science. 1971;173:548–550. doi: 10.1126/science.173.3996.548. [DOI] [PubMed] [Google Scholar]
- Marckmann P, Skov L. Nephrogenic systemic fibrosis: clinical picture and treatment. Radiol. Clin. North Am. 2009;47:833–840. doi: 10.1016/j.rcl.2009.05.004. vi. [DOI] [PubMed] [Google Scholar]
- Masszi A, Speight P, Charbonney E, et al. Fate-determining mechanisms in epithelial–myofibroblast transition: major inhibitory role for Smad3. J. Cell Biol. 2010;188:383–399. doi: 10.1083/jcb.200906155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masszi A, Fan L, Rosivall L, et al. Integrity of cell–cell contacts is a critical regulator of TGF-beta 1-induced epithelial-to-myofibroblast transition: role for beta-catenin. Am. J. Pathol. 2004;165:1955–1967. doi: 10.1016/s0002-9440(10)63247-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maxwell PH, Osmond MK, Pugh CW, et al. Identification of the renal erythropoietin-producing cells using transgenic mice. Kidney Int. 1993;44:1149–1162. doi: 10.1038/ki.1993.362. [DOI] [PubMed] [Google Scholar]
- Maxwell PH, Ferguson DJ, Nicholls LG, Johnson MH, Ratcliffe PJ. The interstitial response to renal injury: fibroblast-like cells show phenotypic changes and have reduced potential for erythropoietin gene expression. Kidney Int. 1997;52:715–724. doi: 10.1038/ki.1997.387. [DOI] [PubMed] [Google Scholar]
- Melrose J, Tammi M, Smith S. Visualisation of hyaluronan and hyaluronan-binding proteins within ovine vertebral cartilages using biotinylated aggrecan G1-link complex and biotinylated hyaluronan oligosaccharides. Histochem. Cell Biol. 2002;117:327–333. doi: 10.1007/s00418-002-0392-4. [DOI] [PubMed] [Google Scholar]
- Meran S, Thomas D, Stephens P, et al. Involvement of hyaluronan in regulation of fibroblast phenotype. J. Biol. Chem. 2007;282:25687–25697. doi: 10.1074/jbc.M700773200. [DOI] [PubMed] [Google Scholar]
- Meran S, Thomas DW, Stephens P, et al. Hyaluronan facilitates transforming growth factor-beta1-mediated fibroblast proliferation. J. Biol. Chem. 2008;283:6530–6545. doi: 10.1074/jbc.M704819200. [DOI] [PubMed] [Google Scholar]
- Milner CM, Higman VA, Day AJ. TSG-6: a pluripotent inflammatory mediator? Biochem. Soc. Trans. 2006;34:446–450. doi: 10.1042/BST0340446. [DOI] [PubMed] [Google Scholar]
- Morita H, Shinzato T, David G, et al. Basic fibroblast growth factor-binding domain of heparan sulfate in the human glomerulosclerosis and renal tubulointerstitial fibrosis. Lab. Invest. 1994;71:528–535. [PubMed] [Google Scholar]
- Moulin V, Larochelle S, Langlois C, Thibault I, Lopez-Valle CA, Roy M. Normal skin wound and hypertrophic scar myofibroblasts have differential responses to apoptotic inductors. J. Cell. Physiol. 2004;198:350–358. doi: 10.1002/jcp.10415. [DOI] [PubMed] [Google Scholar]
- Muller GA, Rodemann HP. Characterization of humand renal fibroblasts in health and disease: 1 Immunophenotyping of cultured tubular epithelial cells and fibroblasts derived from kidneys with histologically proved interstitial fibrosis. Am. J. Kidney Dis. 1991;17:680–683. doi: 10.1016/s0272-6386(12)80351-9. [DOI] [PubMed] [Google Scholar]
- Muller GA, Frank J, Rodemann HP, Engler-Blum G. Human renal fibroblast cell lines (tFKIF and tNKF) are new tools to investigate pathophysiologic mechanisms of renal interstitial fibrosis. Exp. Nephrol. 1995;3:127–133. [PubMed] [Google Scholar]
- Ng YY, Huang TP, Yang WC, et al. Tubular epithelial-myofibroblast transdifferentiation in progressive tubulointerstitial fibrosis in 5/6 nephrectomized rats. Kidney Int. 1998;54:864–876. doi: 10.1046/j.1523-1755.1998.00076.x. [DOI] [PubMed] [Google Scholar]
- Okada H, Inoue T, Suzuki H, Strutz F, Neilson EG. Epithelial–mesenchymal transformation of renal tubular epithelial cells in vitro and in vivo. Nephrol. Dial. Transplant. 2000;15(Suppl 6):44–46. doi: 10.1093/ndt/15.suppl_6.44. [DOI] [PubMed] [Google Scholar]
- Ortonne N, Lipsker D, Chantrel F, Boehm N, Grosshans E, Cribier B. Presence of CD45RO+ CD34 + cells with collagen synthesis activity in nephrogenic fibrosing dermopathy: a new pathogenic hypothesis. Br. J. Dermatol. 2004;150:1050–1052. doi: 10.1111/j.1365-2133.2004.05900.x. [DOI] [PubMed] [Google Scholar]
- Piek E, Heldin CH, Ten Dijke P. Specificity, diversity, and regulation in TGF-beta superfamily signaling. FASEB J. 1999;13:2105–2124. [PubMed] [Google Scholar]
- Quan TE, Bucala R. Culture and analysis of circulating fibrocytes. Methods Mol. Med. 2007;135:423–434. doi: 10.1007/978-1-59745-401-8_28. [DOI] [PubMed] [Google Scholar]
- Quan TE, Cowper S, Wu SP, Bockenstedt LK, Bucala R. Circulating fibrocytes: collagen-secreting cells of the peripheral blood. Int. J. Biochem. Cell Biol. 2004;36:598–606. doi: 10.1016/j.biocel.2003.10.005. [DOI] [PubMed] [Google Scholar]
- Quan TE, Cowper SE, Bucala R. The role of circulating fibrocytes in fibrosis. Curr. Rheumatol. Rep. 2006;8:145–150. doi: 10.1007/s11926-006-0055-x. [DOI] [PubMed] [Google Scholar]
- Rodemann HP, Muller GA. Abnormal growth and clonal proliferation of fibroblasts derived from kidneys with interstitial fibrosis. J. Exp. Biol. Med. 1990;195:57–63. doi: 10.3181/00379727-195-43118. [DOI] [PubMed] [Google Scholar]
- Rodemann HP, Muller GA. Characterisation of human renal fibroblasts in health and disease: II. In vitro growth, differentiation, and collagen synthesis of fibroblasts from kidneys with interstitial fibrosis. Am. J. Kidney Dis. 1991;17:684–686. doi: 10.1016/s0272-6386(12)80352-0. [DOI] [PubMed] [Google Scholar]
- Rodemann HP, Muller GA, Knecht A, Norman JT, Fine LG. Fibroblasts of rabbit kidney in culture1: characterization and identification of cell-specific markers. Am. J. Physiol. 1991;261:F283–F291. doi: 10.1152/ajprenal.1991.261.2.F283. [DOI] [PubMed] [Google Scholar]
- Roufosse C, Bou-Gharios G, Prodromidi E, et al. Bone marrow-derived cells do not contribute significantly to collagen I synthesis in a murine model of renal fibrosis. J. Am. Soc. Nephrol. 2006;17:775–782. doi: 10.1681/ASN.2005080795. [DOI] [PubMed] [Google Scholar]
- Sano N, Kitazawa K, Sugisaki T. Localization and roles of CD44, hyaluronic acid and osteopontin in IgA nephropathy. Nephron. 2001;89:416–421. doi: 10.1159/000046113. [DOI] [PubMed] [Google Scholar]
- Schmidt M, Sun G, Stacey MA, Mori L, Mattoli S. Identification of circulating fibrocytes as precursors of bronchial myofibroblasts in asthma. J. Immunol. 2003;171:380–389. doi: 10.4049/jimmunol.171.1.380. [DOI] [PubMed] [Google Scholar]
- Serini G, Bochaton-Piallat ML, Ropraz P, et al. The fibronectin domain ED-A is crucial for myofibroblastic phenotype induction by transforming growth factor-beta1. J. Cell Biol. 1998;142:873–881. doi: 10.1083/jcb.142.3.873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Massague J. Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell. 2003;113:685–700. doi: 10.1016/s0092-8674(03)00432-x. [DOI] [PubMed] [Google Scholar]
- Sibalic V, Fan X, Loffing J, Wuthrich RP. Upregulated renal tubular CD44, hyaluronan, and osteopontin in kdkd mice with interstitial nephritis. Nephrol. Dial. Transplant. 1997;12:1344–1353. doi: 10.1093/ndt/12.7.1344. [DOI] [PubMed] [Google Scholar]
- Simpson RM, Meran S, Thomas D, et al. Age-related changes in pericellular hyaluronan organization leads to impaired dermal fibroblast to myofibroblast differentiation. Am. J. Pathol. 2009a;175:1915–1928. doi: 10.2353/ajpath.2009.090045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson RM, Wells A, Thomas D, Stephens P, Steadman R, Phillips A. Aging fibroblasts resist phenotypic maturation because of impaired hyaluronan-dependent CD44/epidermal growth factor receptor signaling. Am. J. Pathol. 2009b;176:1215–1228. doi: 10.2353/ajpath.2010.090802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith PG, Liu M. Impaired cutaneous wound healing after sensory denervation in developing rats: effects on cell proliferation and apoptosis. Cell Tissue Res. 2002;307:281–291. doi: 10.1007/s00441-001-0477-8. [DOI] [PubMed] [Google Scholar]
- Sporn MB, Roberts AB. The transforming growth factor-betas: past, present, and future. Ann. N Y Acad. Sci. 1990;593:1–6. doi: 10.1111/j.1749-6632.1990.tb16095.x. [DOI] [PubMed] [Google Scholar]
- Strutz F. Pathogenesis of tubulointerstitial fibrosis in chronic allograft dysfunction. Clin. Transplant. 2009a;23(Suppl 21):26–32. doi: 10.1111/j.1399-0012.2009.01106.x. [DOI] [PubMed] [Google Scholar]
- Strutz F. The role of FGF-2 in renal fibrogenesis. Front. Biosci. (Schol. Ed.) 2009b;1:125–131. doi: 10.2741/S12. [DOI] [PubMed] [Google Scholar]
- Strutz F, Neilson EG. New insights into mechanisms of fibrosis in immune renal injury. Springer Semin. Immunopathol. 2003;24:459–476. doi: 10.1007/s00281-003-0123-5. [DOI] [PubMed] [Google Scholar]
- Strutz F, Okada H, Lo CW, et al. Identification and characterization of a fibroblast marker: FSP1. J. Cell Biol. 1995;130:393–405. doi: 10.1083/jcb.130.2.393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strutz F, Zeisberg M, Hemmerlein B, et al. Basic fibroblast growth factor expression is increased in human renal fibrogenesis and may mediate autocrine fibroblast proliferation. Kidney Int. 2000;57:1521–1538. doi: 10.1046/j.1523-1755.2000.00997.x. [DOI] [PubMed] [Google Scholar]
- Strutz F, Renziehausen A, Dietrich M, et al. Cortical fibroblast culture from human biopsies. J. Nephrol. 2001;14:190–197. [PubMed] [Google Scholar]
- Thomas G, Clayton A, Thomas J, Davies M, Steadman R. Structural and functional changes in heparan sulfate proteoglycan expression associated with the myofibroblastic phenotype. Am. J. Pathol. 2003;162:977–989. doi: 10.1016/S0002-9440(10)63892-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian YC, Phillips AO. Interaction between the transforming growth factor-beta type II receptor/Smad pathway and beta-catenin during transforming growth factor-beta1-mediated adherens junction disassembly. Am. J. Pathol. 2002;160:1619–1628. doi: 10.1016/s0002-9440(10)61109-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomasek JJ, Gabbiani G, Hinz B, Chaponnier C, Brown RA. Myofibroblasts and mechano-regulation of connective tissue remodelling. Nat. Rev. Mol. Cell Biol. 2002;3:349–363. doi: 10.1038/nrm809. [DOI] [PubMed] [Google Scholar]
- Tomasek JJ, Vaughan MB, Kropp BP, et al. Contraction of myofibroblasts in granulation tissue is dependent on Rho/Rho kinase/myosin light chain phosphatase activity. Wound Repair Regen. 2006;14:313–320. doi: 10.1111/j.1743-6109.2006.00126.x. [DOI] [PubMed] [Google Scholar]
- Uhal BD, Kim JK, Li X, Molina-Molina M. Angiotensin-TGF-beta 1 crosstalk in human idiopathic pulmonary fibrosis: autocrine mechanisms in myofibroblasts and macrophages. Curr. Pharm. Des. 2007;13:1247–1256. doi: 10.2174/138161207780618885. [DOI] [PubMed] [Google Scholar]
- Vasko R, Koziolek M, Ikehata M, et al. Role of basic fibroblast growth factor (FGF-2) in diabetic nephropathy and mechanisms of its induction by hyperglycemia in human renal fibroblasts. Am. J. Physiol. Renal Physiol. 2009;296:F1452–F1463. doi: 10.1152/ajprenal.90352.2008. [DOI] [PubMed] [Google Scholar]
- Vaughan MB, Howard EW, Tomasek JJ. Transforming growth factor-beta1 promotes the morphological and functional differentiation of the myofibroblast. Exp. Cell Res. 2000;257:180–189. doi: 10.1006/excr.2000.4869. [DOI] [PubMed] [Google Scholar]
- Wada T, Sakai N, Matsushima K, Kaneko S. Fibrocytes: a new insight into kidney fibrosis. Kidney Int. 2007;72:269–273. doi: 10.1038/sj.ki.5002325. [DOI] [PubMed] [Google Scholar]
- Webber J, Jenkins RH, Meran S, Phillips A, Steadman R. Modulation of TGFbeta1-dependent myofibroblast differentiation by hyaluronan. Am. J. Pathol. 2009a;175:148–160. doi: 10.2353/ajpath.2009.080837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webber J, Meran S, Steadman R, Phillips A. Hyaluronan orchestrates transforming growth factor-beta1-dependent maintenance of myofibroblast phenotype. J. Biol. Chem. 2009b;284:9083–9092. doi: 10.1074/jbc.M806989200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wells A, Larsson E, Hanas E, Laurent T, Hallgren R, Tufveson G. Increased hyaluronan in acutely rejecting human kidney grafts. Transplantation. 1993;55:1346–1349. doi: 10.1097/00007890-199306000-00025. [DOI] [PubMed] [Google Scholar]
- Wells V, Davies D, Mallucci L. Cell cycle arrest and induction of apoptosis by beta galactoside binding protein (beta GBP) in human mammary cancer cells. A potential new approach to cancer control. Eur. J. Cancer. 1999;35:978–983. doi: 10.1016/s0959-8049(99)00020-9. [DOI] [PubMed] [Google Scholar]
- Werner S, Krieg T, Smola H. Keratinocyte–fibroblast interactions in wound healing. J. Invest. Dermatol. 2007;127:998–1008. doi: 10.1038/sj.jid.5700786. [DOI] [PubMed] [Google Scholar]
- Wijnhoven TJ, Lensen JF, Wismans RG, et al. In vivo degradation of heparan sulfates in the glomerular basement membrane does not result in proteinuria. J. Am. Soc. Nephrol. 2007;18:823–832. doi: 10.1681/ASN.2006070692. [DOI] [PubMed] [Google Scholar]
- Yamashita S, Maeshima A, Nojima Y. Involvement of renal progenitor tubular cells in epithelial-to-mesenchymal transition in fibrotic rat kidneys. J. Am. Soc. Nephrol. 2005;16:2044–2051. doi: 10.1681/ASN.2004080681. [DOI] [PubMed] [Google Scholar]
- Yang Y, Gubler MC, Beaufils H. Dysregulation of podocyte phenotype in idiopathic collapsing glomerulopathy and HIV-associated nephropathy. Nephron. 2002;91:416–423. doi: 10.1159/000064281. [DOI] [PubMed] [Google Scholar]
- Zeisberg M, Strutz F, Muller GA. Renal fibrosis: an update. Curr. Opin. Nephrol. Hypertens. 2001;10:315–320. doi: 10.1097/00041552-200105000-00004. [DOI] [PubMed] [Google Scholar]
- Zeisberg M, Hanai J, Sugimoto H, et al. BMP-7 counteracts TGF-beta1-induced epithelial-to-mesenchymal transition and reverses chronic renal injury. Nat. Med. 2003;9:964–968. doi: 10.1038/nm888. [DOI] [PubMed] [Google Scholar]
- Zoltan-Jones A, Huang L, Ghatak S, Toole BP. Elevated hyaluronan production induces mesenchymal and transformed properties in epithelial cells. J. Biol. Chem. 2003;278:45801–45810. doi: 10.1074/jbc.M308168200. [DOI] [PubMed] [Google Scholar]