

Abstract
Dendritic cells are not only the master regulators of adaptive immunity, but also participate profoundly in innate immune responses. Much has been learned about their basic immunological functions and their roles in various diseases. Comparatively little is still known about their role in renal disease, despite their obvious potential to affect immune responses in the kidney, and immune responses that are directed against renal components. Kidney dendritic cells form an abundant network in the renal tubulointerstitium and constantly survey the environment for signs of injury or infection, in order to alert the immune system to the need to initiate defensive action. Recent studies have identified a role for dendritic cells in several murine models of acute renal injury and chronic nephritis. Here we summarize the current knowledge on the role of kidney dendritic cells that has been obtained from the study of murine models of renal disease.
Keywords: cytokines, dendritic cells, glomerulonephritis
Dendritic cells (DCs) exist in all lymphatic and in nearly all non-lymphatic tissues (Steinman et al. 2003). As professional antigen presenting cells (APCs), their main task is the activation and regulation of T cells. Furthermore, they mediate the communication between the innate and the adaptive immune system (Steinman et al. 2005). DCs are specialized in taking up antigens in tissues, processing them and presenting them, after migration into lymphatic tissues, on MHC class I and II molecules to cytotoxic CD8+ T cells (CTL) and CD4+ T helper cells (Th cells) respectively. Depending on the activation status of the DCs, T cells become activated or are tolerized. That status results from recognition of pathogen-associated molecular patterns by means of a great variety of receptors, such as toll-like-receptors, that DCs use to sense whether an antigen was encountered in infectious or dangerous context. Various subsets of DCs exist that differ in their lineage, migratory properties, tissue distribution and their ability to activate Th cells and/or CTL. This complex topic has recently been reviewed elsewhere (Shortman & Naik 2007; Heath & Carbone 2009; Geissmann et al. 2010).
Relatively little is known about the role of DCs in renal diseases, despite abundant information on their roles in diseases affecting other organs. Cells with phenotypic characteristics of DCs have been described inside the kidneys of humans (Markovic-Lipkovski et al. 1990; Cuzic et al. 1992) and rodents more than 15 years ago (Austyn et al. 1994; Kaissling & Le Hir 1994; Roake et al. 1995; Kaissling et al. 1996). However, because of difficulties in identifying and in isolating these cells from the kidney, relatively little was known about their functional role until recently. Moreover, due to the expression of the F4/80-molecule, all APCs in the kidney were initially categorized as macrophages (Hume & Gordon 1983). However, F4/80 is specific for macrophages only in the spleen, whereas DCs in non-lymphoid tissues, including the kidney, express this marker too. Morphological and functional analysis showed that the tubulointerstitial stellate-shaped F4/80+ cells mostly co-express the murine DC-marker CD11c and possess the functionality of conventional tissue DCs (Kruger et al. 2004). By confocal laser-microscopy it was shown that kidney dendritic cells (kDCs) form an extensive anatomic network that spans the entire tubulointerstitium and encloses all nephrons (Soos et al. 2006). Functional investigations have revealed that kDCs in the steady-state maintain renal homeostasis (Kurts et al. 1997; Lukacs-Kornek et al. 2008). In transplantation, the tolerogenic properties of passenger leucocytes, most likely DCs, have long been known to induce a certain degree of transplantation tolerance (Ko et al. 1999). The role of renal DCs in transplantation has recently been reviewed elsewhere (Rogers et al. 2009). Studies in murine models of kidney diseases showed that kDCs accumulate in inflamed organs, secrete different cytokines and thereby either attenuate or aggravate renal injury (Figure 1). It is currently unclear why DCs are either anti- or pro-inflammatory, depending on the disease model used. In this review, we summarize the presently available knowledge on the functions of murine kDCs in the steady-state and in models of acute or chronic kidney injury.
Figure 1.
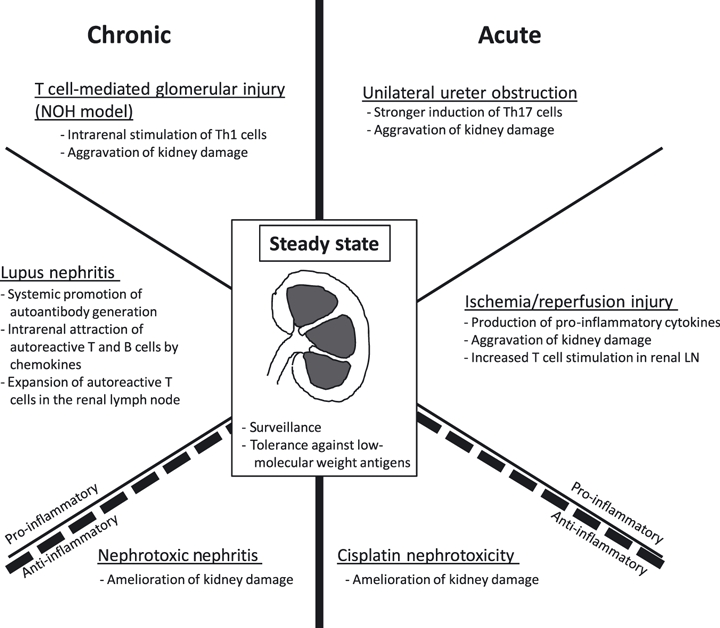
Functions of dendritic cells in various models of renal disease.
Kidney DCs in the steady-state
Murine kDCs can be reliably identified by expression of both CD11c and MHC class II (one marker alone is insufficient). Most of them express the fractalkine receptor CX3CR1 (Soos et al. 2006), F4/80 and the subtype marker CD11b at low levels (Kruger et al. 2004), which is characteristic of conventional tissue DCs (Table 1) (Shortman & Naik 2007; Merad et al. 2008; Heath & Carbone 2009; Geissmann et al. 2010). A subset of 5–15% shows the phenotype CD11c+ CD103+ ± CD11b– F4/80LO (Ginhoux et al. 2009), which characterizes tissue DCs related to the CD8α+ DCs in lymphatic tissues (Hildner et al. 2008). Their role in the kidney is unclear. Table 1 summarizes murine DC subsets that have been identified in the kidney.
Table 1.
Subsets of murine DCs
| Murine DC subsetPhenotype | Origin; presence in the kidney; functionality |
|---|---|
| DC subsets occurring in the kidney | |
| Conventional (myeloid) CD8− DC: CD11c+ CD11b+ CD8−, F4/80+ in tissues, in lymphatics DCIR+ | Likely pre-DC-derived, present in the kidney (Kruger et al. 2004); antigen transport from tissues to LNs, activation of Th cells |
| Conventional CD103+ DC: CD11c+ CD11b− CD8+ CD103+ CD205−, Langerin+ | Pre-DC-derived, present in the kidney (Ginhoux et al. 2009); antigen transport from tissues to LNs, activation or tolerization of CD8+ and CD4+ T cells, cross-presentation |
| Inflammatory DC: CD11c+ CD11b+ CD8− F4/80+ Gr-1+ | Monocyte-derived; present in the kidney (Heymann et al. 2009); proinflammatory functions and regulation of infiltrating Th cells |
| Plasmacytoid DC: CD11cint CD11b− CD8− B220+ Gr-1+ | Precursor distinct from that of conventional DCs; present in the human kidney (Woltman et al. 2007); produce IFN-α in viral infections |
| Follicular DC: CD11c− CD20− CD21+ CD35+ | Probably not of haematopoietic origin; described only in Lupus nephritis (Turner et al. 2008); foster B cell responses |
| DC subsets not described in the kidney | |
| Conventional CD8+ DC: CD11c+ CD11b− CD8+ CD205+ | Pre-DC-derived; present in lymphatic tissues only, reported in the renal LN (Scholz et al. 2008); activation of CD8+ and CD4+ T cells, cross-presentation |
| Langerhans cell: CD11c+ CD11b+ CD8− Langerin+ | Monocyte-derived; present in the skin only; transport antigen to cutaneous LNs, probably T cell activation |
CD, cluster of differentiation; DC, dendritic cell; IFN, interferon; LN, lymph node; MR, mannose receptor.
kDCs have a turnover of about 14 days in homeostasis, as determined by bone marrow-transplantation experiments (Leszczynski et al. 1985; Dong et al. 2005). Tissue DCs may generally derive from circulating pre-DCs (proposed to give rise to the CD103+ subset (Ginhoux et al. 2009)), from Gr1+ inflammatory monocytes or Gr1lo‘patrolling’ monocytes (Liu et al. 2007; Geissmann et al. 2010; ). Both monocytes subsets are recruited to the inflamed kidney (Table 1) (Li et al. 2008) and can be identified within the organ by co-expression of CD11c, MHC class II and Gr1 (Heymann et al. 2009), until they downregulate Gr1. The DC precursor in the steady-state has not been identified yet.
Although experimental evidence for a functional role of the tubulointerstitial kDCs network is still quite limited, its location suggested early on that it may serve the surveillance of the renal parenchyma. The antigens captured by DCs within the kidney include tubular (Kurts et al. 1997; Dong et al. 2005; ) and glomerular (Heymann et al. 2009) autoantigens, and small molecular weight antigens that are constitutively filtrated in glomeruli (Dong et al. 2005; Lukacs-Kornek et al. 2008). There is evidence that upon encountering maturation stimuli, kDCs migrate to the renal lymph nodes (Roake et al. 1995) where they activate specific T cells (Edgtton et al. 2008), consistent with the canonical life-cycle of DCs extrapolated from the paradigm of Langerhans cells in the skin (Steinman et al. 2003; Shortman & Naik 2007; Heath & Carbone 2009; Geissmann et al. 2010). Additionally, small filterable antigens constitutively reach DCs resident in the renal lymph node by bulk drainage, and are captured there and, in the absence of pathogen-associated molecular patterns, are used to tolerize harmful T cells (Lukacs-Kornek et al. 2008). Such antigens may include food-derived antigens or autoantigens that are produced intermittently, like certain hormones, which are not readily available in the thymus for efficient negative selection of autoreactive T cells. The ability of the kidney to tolerize the immune system against innocuous small-molecular weight antigens may be of general relevance, but this notion remains to be formally demonstrated.
Kidney DCs in acute renal injury
Kidney DCs in ischaemia reperfusion injury
Ischaemia-reperfusion injury (IRI) is relevant especially for kidney transplantation. Early after ischaemic injury, kidney-resident F4/80+ CD11c+ DCs have been identified as the earliest source of TNF-α (Dong et al. 2005). Later, also hypoxic endothelial cells produced this cytokine (Schlichting et al. 2006). TNF-α caused influx of circulating immune cells, especially pro-inflammatory DC subsets, monocytes/macrophages and Th cells (Ysebaert et al. 2000), and directly contributed to kidney parenchymal damage. TNF-α receptors are expressed by many cells types, and upon ligation may initiate renal epithelial apoptosis (Dong et al. 2007).
Within 24 h following acute ischaemia, the T cell stimulatory capacity of kDCs increased markedly (Dong et al. 2005). Th cells have been demonstrated to aggravate IRI (Yokota et al. 2002; Burne-Taney et al. 2003; Ysebaert et al. 2004). T cells are important effectors also in other renal disease and in transplant rejection (Kurts et al. 2007; Rogers et al. 2009), suggesting that conditions of renal hypoxia might contribute to disease progression by activating kDCs to stimulate T cells. Indeed, hypoxia and the transcription factor HIF-1α have recently been shown to potently activate DC function when combined with LPS stimulation (Jantsch et al. 2008).
In contrast to these pro-inflammatory functions, myeloid cells of the kidney, likely kDC, have recently been proposed to attenuate IRI by production of single Ig IL-1-related receptor (SIGIRR) (Lech et al. 2009), an immunosuppressive mediator that attenuates also murine lupus models (discussed below) (Lech et al. 2008, 2010). Mice deficient for this mediator showed aggravated IRI. This could be reversed by treatment with clodronate liposomes (Clo-Lip), which effectively depletes phagocytic cells of the mononuclear phagocyte system from all tissues (Van Rooijen & Sanders 1994) including kDCs (Dong et al. 2005). It remains to be formally demonstrated that kDCs expressing SIGIRR actively suppress IRI and if so, how this may be reconciled with the pro-inflammatory TNF-α-mediated role of kDCs in IRI.
Kidney DCs in unilateral ureteral obstruction
The model of unilateral uretral obstruction (UUO) generates progressive renal fibrosis, which is considered a common feature of progressive renal diseases (Nagler 1973). The severity of fibrosis correlates with infiltration of activated macrophages (Eddy 1995), which secrete profibrotic and pro-inflammatory cytokines such as TGF-β and TNF-α respectively (Kitamoto et al. 2009). These macrophages induce apoptosis of tubular epithelial cells via TNF-α and TGF-β and produce reactive oxygen species that aggravate renal tubular injury (Misseri et al. 2005). Also endothelial cells undergo apoptosis, and renal fibroblasts differentiate into myofibroblasts which promotes the deposition of extracellular matrix leading to fibrosis (Iwano et al. 2002; Zeisberg et al. 2008).
F4/80+ kDCs are a potent source of TNF-α during the early stage of UUO (Dong et al. 2008). DC depletion with Clo-Lip attenuated obstruction-induced tubular apoptosis and renal fibrosis, and TNF-α and TGF-β were less elevated in obstructed kidneys (Kitamoto et al. 2009). T cells are thought to play a role in UUO as well, as evidenced by their accumulation at the cortico-medullary junction and in the cortex, where they colocalized with F4/80+ kDCs (Dong et al. 2008). Although Clo-Lip treatment did not reduce the number of T cells, it attenuated their secretion of IFN-γ and IL-17 (Dong et al. 2008). However, it has recently been shown that Clo-Lip generally depletes renal F4/80+ cells (Kitamoto et al. 2009), and hence not only DCs but also macrophages. Thus, although these findings clearly demonstrated that F4/80+ phagocytes contribute to progressive renal fibrosis, it cannot be distinguished at present whether this was due to F4/80+ macrophages and/or to F4/80+ kDCs.
Kidney DCs in cisplatin nephrotoxicity
Renal tubular cells are particularly susceptible to toxic damage, for example in response to certain drugs, and undergo necrotic cell death. Cisplatin reliably induces such acute tubular necrosis in murine systems. The role of DCs in cisplatin nephrotoxicity has recently been addressed using CD11c-DTR mice that express the diphtheria toxin receptor in CD11c+ cells (Jung et al. 2002). Injection of diphtheria toxin into these mice permits conditional (albeit systemic) ablation of CD11c+ DCs. However, CD11c is expressed at lower levels also by activated CTL and NK cells, as well as by distinct macrophages like those in alveoli or in the splenic marginal zone (Probst et al. 2005), which are depleted to some extent as well. By contrast, plasmacytoid DCs express only low CD11c levels and are not targeted in these mice (Sapoznikov et al. 2007). Despite these restrictions, CD11c-DTR mice permit conclusions on the in vivo role of DCs with reasonable accuracy (Bar-On & Jung 2010). Using these animals, it has recently been shown that kDC depletion aggravates renal injury in cisplatin nephrotoxicity (Tadagavadi & Reeves 2010). Local inflammatory effects by dying DCs within the kidney were excluded by mixed bone-marrow chimeras. The underlying molecular mechanisms have not been clarified, but may relate to increased expression of inducible costimulatory ligand (ICOS-L) on DCs, which can suppress T cells (Akbari et al. 2002). These findings point towards an anti-inflammatory role of kDCs in cisplatin nephrotoxicity.
Kidney DCs in chronic renal disease
Systemic DCs in Lupus nephritis
Lupus nephritis is a serious complication of systemic lupus erythematosus (SLE), which occurs in the majority of lupus patients at some point (Foster 2007). It is initiated by glomerular deposition of immune complexes, followed by complement activation and production of pro-inflammatory cytokines and chemokines, which leads to renal inflammation. The immune complexes often contain auto-antibodies against self nucleoproteins, likely derived from dying cells. Numerous animal models with lupus-like symptoms are being used, including MRL/lpr, NZB/W F1, NZM2328, and B6/TC mice. These mice carry allelic variants or mutations in genes relevant in the immune system, which render them susceptible for autoimmune diseases (Foster 2007). Alternatively, injection of hydrocarbon oil induces a lupus nephritis-like disease, which is useful when knockout mice are required. Whilst much attention has been focussed at the roles of T and B lymphocytes, those of DCs have only recently been addressed. Observations in patients (Fiore et al. 2008) and in murine models indicate both systemic roles, in the induction of autoantibodies, and local roles, in intrarenal inflammation that compromises organ function.
Production of auto-antibodies against nuclear self antigens requires activation of autoreactive Th cells that stimulate B cells, a systemic event that occurs in lymphatic organs like the spleen. Since DCs phagocytose apoptotic and necrotic cells and are able to present self antigens (Albert et al. 1998; Inaba et al. 1998), they are likely to be the APC population that breaks Th cell-tolerance to self antigens in SLE. Increased numbers of DCs in thymus and spleen have been suggested to play a role in breaking central and peripheral immune tolerance respectively (Ishikawa et al. 2001). In aged BWF1 mice that develop lupus nephritis, thymic and splenic DCs produced high levels of the chemokine BLC (CXCL13) that attracts B cells (Ishikawa et al. 2001). Additionally, Georgiev et al. showed that injection of syngeneic bone marrow-derived DCs, which had been exposed to necrotic or apoptotic cells, induced high levels of IgG1 autoantibodies in wild-type C57/BL6 mice, whereas injection of macrophages did not, indicating that DCs and not macrophages can break self tolerance (Georgiev et al. 2005).
Immunogens in SLE are most probably derived from dying cells. Apoptosis and thus provision of immunogens for autoantibody production in lupus is triggered by type I interferons, which are mainly produced by plasmacytoid DCs. Type I interferon production can be stimulated by circulating chromatin-containing immune complexes from lupus patients (Baechler et al. 2004). Furthermore, type I interferon in the serum of lupus patients can convert circulating monocytes into DCs that present autoantigens (Blanco et al. 2001). These observations support the notion that DC subsets are differentially involved in lupus pathogenesis.
A pro-inflammatory role of DCs in lupus pathogenesis was further supported by studies in B6.TC mice, which produce antinuclear nephritogenic auto-antibodies and develop glomerulonephritis. In these mice, DCs accumulated in lymphoid organs due to greater production in the bone marrow and to reduced apoptosis. Additionally, DCs showed reduced expression of co-stimulatory molecules and inhibited the function of regulatory T cells (Tregs) by production of high levels of IL-6 (Wan et al. 2007), suggesting that abnormal DC maturation might contribute to disease progression.
A hint at protective roles of DCs came from studies in SIGIRR-deficient mice. This is an immunosuppressive mediator expressed by DCs and macrophages. Mice that lacked SIGIRR showed aggravated hydrocarbon oil-induced lupus (Lech et al. 2008, 2010). These findings demonstrate that DCs not only break tolerance in SLE, but also mediate certain immuno-suppressive functions in lupus pathogenesis. It is possible that the suppressive functions are lost in manifest lupus nephritis.
Kidney DCs in lupus nephritis
Lupus nephritis to date is the only condition in which DCs have been found to infiltrate glomeruli (Bagavant et al. 2006; Tucci et al. 2008). The Th1–driving cytokine, IL-12, is produced by macrophages and DCs within glomeruli in this condition, illustrating their ability to provide a pro-inflammatory microenvironment (Tucci et al. 2009). In the tubulointerstitium, kDC numbers in nephritic BWF1 mice were increased and production of BLC was noted. Co-stimulatory blockade together with cyclophosphamide reduced the number of renal CD11c+ and in turn reduced B and Th cell infiltration and nephritis (Ishikawa et al. 2001; Schiffer et al. 2003), suggesting that kDCs attract effector immunocytes into the kidney. Additionally, in MRL-Fas(lpr) mice, DCs secreted the pro-inflammatory mediator high mobility group box protein (HMGB-1) via p38 mitogen-activated protein kinase (MAPK) activation. Inhibition of p38 MAPK decreased numbers of CD11c+ cells in the kidney and in the spleen, reduced HMGB-1 protein in the kidney and improved kidney pathology (Iwata et al. 2009), identifying a potential therapeutic approach. In NZM2328 mice, progression of acute lupus nephritis towards chronic glomerulonephritis was accompanied by T cell activation and expansion in the renal lymph nodes (Bagavant et al. 2006). The authors did not rule out the possibility that T cells might also be stimulated by DCs inside the kidney. Taken together, these findings support the notion that kidney-resident DCs might promote renal infiltration by effector immunocytes, i.e. T cells, B cells or macrophages and thereby contribute to the progression to renal failure (Bagavant & Fu 2009). Further studies to directly address the role of kDCs in models of this important nephritis form are necessary.
Kidney DCs in other glomerulonephritis models
DCs have been histologically detected in periglomerular infiltrates in human glomerulonephritis and in various murine models thereof (Janssen et al. 1998; Kruger et al. 2004; Fujinaka et al. 2007; Woltman et al. 2007; Segerer et al. 2008; Tucci et al. 2009). Nephrotoxic nephritis (NTN) is a model for human crescentic glomerulonephritis, which is characterized by rapidly progressive glomerular damage and crescentic cellular infiltrates. It is induced by injection of a nephrotoxic antiserum, generated in sheep by vaccination with murine kidney cortex (Assmann et al. 1985; Lang et al. 2005; Tipping & Holdsworth 2006). Upon injection, sheep antibodies are taken up by DCs in lymphatic organs, and are used to activate specific Th1 cells and B cells. Due to their specificity, the antibodies bind to cortical antigens within the kidney. This allows local DCs and macrophages to capture and present them to infiltrating Th1 cells, which leads to an inflammatory response of the DTH type, with macrophages as the main effector cells (Tipping & Holdsworth 2006; Kurts et al. 2007).
We previously used CD11c-DTR mice to show that early depletion of DCs in NTN (days 4 and 10 after disease induction) aggravated renal damage, indicating a protective role of kDCs (Scholz et al. 2008). kDCs from nephritic mice stimulated T cell proliferation more potently than those from healthy mice and induced IL-10 and IFN-γ expression in Th cells. IL-10 is known to attenuate NTN whereas conflicting results have been reported for the role of IFN-γ (Tipping et al. 1997; Kitching et al. 1999; Ring et al. 1999; Timoshanko et al. 2002). The induction of IL-10 may have been mediated by ICOS-L expressed by kDCs in nephritic mice. Blockade of ICOS-L has been shown to aggravate NTN (Odobasic et al. 2006), reminiscent of the aggravation after early DC ablation (Scholz et al. 2008). A similar protective role for DCs involving up-regulation of ICOS-L-expression has recently been shown for cisplatin induced nephropathy, a model for acute kidney injury discussed above (Tadagavadi & Reeves 2010). Naturally occurring Tregs are known to attenuate NTN, by suppressing effector T cells in lymphatic organs (Wolf et al. 2005). Also here, a role of DCs in induction of such Tregs is likely, but has not been formally shown.
Contrasting these findings, we have recently shown a pro-inflammatory role for renal DCs in a model of T cell-mediated glomerular injury. In that study, we used transgenic NOH mice that express the model antigens ovalbumin and hen egg lysozyme in glomerular podocytes (Heymann et al. 2009). Co-injection of antigen-specific CD8+ and CD4+ T cells into these mice resulted in periglomerular mononuclear infiltrates and inflammation of parietal epithelial cells, resembling the histological picture in rapid-progressive glomerulonephritis in humans. DCs played two roles in the NOH model: First, they cross-presented glomerular antigen in the renal lymph node and activated CTL, which in turn infiltrated the kidney and released more glomerular antigen. Second, DCs in the kidney presented these antigens to Th cells, which resulted in intrarenal cytokine and chemokine production and in recruitment of macrophages, monocyte-derived DCs and more CTL. Depletion of DCs in nephritic mice by employing the CD11c-DTR transgenic system disbanded the periglomerular infiltrates, indicating that kidney DCs were required for its maintenance (Heymann et al. 2009). Hence, kidney DCs can link glomerular injury and tubulointerstitial infiltration, which is important since spreading of glomerular injury to the tubulointerstitium critically determines kidney function (Bohle et al. 1996).
Open questions for future studies
Recent studies revealed that kDCs are centrally involved in various models of kidney disease, including some that are not primarily immune-mediated (Table 2, Figure 1). This is not surprising given their key roles in diseases affecting for example the intestinal tract, the lung or the skin. However, recent data are conflicting, and at present it is completely unclear why kDCs were protective in some renal disease models and pro-inflammatory in others. One possible explanation is that the methods currently used to ablate DCs in mice are not completely specific for DCs, let alone for those in the kidney (Probst et al. 2005; Bennett & Clausen 2007; Kitamoto et al. 2009). Thus, some effects attributed to kDCs, which have been extrapolated from experiments that used such techniques, may in fact be due to functions of DCs in tissues other than the kidney, or even to non-DCs, such as macrophages that are targeted by Clo-Lip and to some extent also in CD11c-DTR mice (Probst et al. 2005; Bennett & Clausen 2007; Kitamoto et al. 2009). An alternative explanation may come from considering the DC maturation state. As described above, kDCs possess a tolerizing phenotype under steady-state-conditions, which seems reasonable since kDCs constantly encounter foreign antigen and would cause permanent inflammation if they were pro-inflammatory. Persistent inflammatory conditions, for example in chronic glomerulonephritis, might mature the kDCs, which thereby become immunogenic.
Table 2.
Functions of DCs in animal models
| Model | DC function | Citation |
|---|---|---|
| Steady state | ||
| Healthy mice | Surveillance of the tubulointerstitium | Soos et al. (2006) |
| Healthy mice | T cell tolerance against innocuous small-molecular weight antigens | Lukacs-Kornek et al. (2008) |
| Acute renal injury | ||
| Ischaemia/reperfusion injury | Production of pro-inflammatory cytokines, attenuation of damage after Clo-Lip, increased T cell stimulation in renal LN | Dong et al. (2005, 2007) |
| Unilateral ureter obstruction (UUO) | Stronger induction of Th17 cells; attenuation of damage after Clo-Lip | Dong et al. (2008), Kitamoto et al. (2009) |
| Cisplatin nephrotoxicity | Aggravation of kidney damage after depletion in CD11c-DTR mice | Tadagavadi and Reeves (2010) |
| Chronic renal disease | ||
| Lupus nephritis | Systemic promotion of autoantibody generation; Suppression of such responses by SIGIRR; intrarenal attraction of autoreactive T and B cells by chemokines; expansion of autoreactive T cells in the renal lymph node | Ishikawa et al. (2001), Bagavant et al. (2006). Lech et al. (2008, 2010) |
| Nephrotoxic Nephritis (NTN) (model of crescentic glomerulonephritis) | Aggravation of kidney damage after early depletion using CD11c-DTR mice | Scholz et al. (2008) |
| T cell-mediated glomerular injury (NOH model) | Intrarenal stimulation of Th1 cells; attenuation of kidney damage after depletion using CD11c-DTR mice | Heymann et al. (2009) |
There are numerous models of renal disease in which the role of kDCs has not been studied sufficiently or not at all, for example experimental autoimmune glomerulonephritis (EAG), a murine model for anti-GBM nephritis, or rat models of nephritis. The lack of suitable antibodies specific for rat DC subsets, and the unavailability of transgenic rats that allow conditional depletion of DCs have thus far precluded addressing their role in these animals. This is regrettable, because many relevant models for human nephritis exist only in rats, such as Adriamycin-, Heymann- and Thy1-nephritis. But at least the Clo-Lip depletion technique (Van Rooijen & Sanders 1994) should be applicable.
The expression of the macrophage marker F4/80 by most of the renal CD11c+ MHC II+ DCs implies that there must be considerable overlap between kDCs and previously described renal macrophage subsets (Lin et al. 2009), but the exact extent of this overlap is unknown. At present there is an ongoing debate about when a mononuclear phagocyte may be classified as a macrophage or a DC (Steinman & Idoyaga 2010). Extreme points of view have been taken (Hume 2008) and contradicted (Heath & Carbone 2009). This controversy appears somewhat semantic, given that both the DC and the macrophage field have been developing for a long time, albeit more or less separate from each other. A mononuclear phagocyte may well fulfill the current criteria of both DC and macrophage. These terms may merely represent differentiation extremes in a function continuum of mononuclear phagocytes and drawing a clear demarcation in mutual consent between scientists of the DC and macrophage fields will be difficult. And even if possible, it is unclear whether this would help understanding kidney function or disease. Assessing functional abilities, such as the capacity to activate T cells, to phagocytose or to produce antibacterial effector molecules, appears a more purposeful approach to APC classification (Kruger et al. 2004). This should be kept in mind before embarking on extensive studies aimed at subdividing kDCs or macrophages into numerous subsets solely defined by their expression of surrogate markers like CD11c, F4/80, Gr-1, Mac-1 or DC-SIGN, whose functional roles are unclear. In the DC field, several overlapping nomenclatures for subsets currently exist and one commonly used underlies Table 1 (Shortman & Naik 2007). However, general consensus on a DC subset classification has yet to be reached (Steinman & Idoyaga 2010).
Of supreme importance is the clarification of the role of kDCs in human nephritis. Determining DC subset markers, and perhaps even more so, of activation markers, may open new opportunities for immunohistological analysis of renal disease. First steps towards this aim have been made by histological studies examining kidney sections of nephritic patients (Woltman et al. 2007; Fiore et al. 2008; Segerer et al. 2008; Tucci et al. 2009). Antibodies specific for the various subsets of human DCs have recently become available, eg. BDCA-1, -2 or -3, or DC-SIGN (Autissier et al. 2010). However, these markers are not expressed by murine DCs, so that knowledge gained in animal experiments may not easily be translated to human DCs. Nevertheless, a systematic appraisal of DC numbers, their tissue distribution pattern, subtype and, most importantly, their functional state may shed new light on the involvement of DCs in kidney disease and open new diagnostic opportunities. Finally, the development of drugs that suppress pro-inflammatory DC responses may permit novel therapeutic avenues in kidney disease.
Acknowledgments
We apologize to all colleagues whose work could not be cited due to space restrictions. The authors are supported by the German Research foundation (DFG grants Ku1063/5 and Ku1063/6, Sonderforschungsbereiche 704 and 645, Transregio 57, Klinische Forschergruppe 228), the German Academic Exchange service (DAAD) and the German National Academic Foundation (Studienstiftung des deutschen Volkes).
References
- Akbari O, Freeman GJ, Meyer EH, et al. Antigen-specific regulatory T cells develop via the ICOS-ICOS-ligand pathway and inhibit allergen-induced airway hyperreactivity. Nat. Med. 2002;8:1024–1032. doi: 10.1038/nm745. [DOI] [PubMed] [Google Scholar]
- Albert ML, Sauter B, Bhardwaj N. Dendritic cells acquire antigen from apoptotic cells and induce class I- restricted CTLs. Nature. 1998;392:86–89. doi: 10.1038/32183. [DOI] [PubMed] [Google Scholar]
- Assmann KJ, Tangelder MM, Lange WP, Schrijver G, Koene RA. Anti-GBM nephritis in the mouse: severe proteinuria in the heterologous phase. Virchows Arch. A. Pathol. Anat. Histopathol. 1985;406:285–299. doi: 10.1007/BF00704298. [DOI] [PubMed] [Google Scholar]
- Austyn JM, Hankins DF, Larsen CP, Morris PJ, Rao AS, Roake JA. Isolation and characterization of dendritic cells from mouse heart and kidney. J. Immunol. 1994;152:2401–2410. [PubMed] [Google Scholar]
- Autissier P, Soulas C, Burdo TH, Williams KC. Evaluation of a 12-color flow cytometry panel to study lymphocyte, monocyte, and dendritic cell subsets in humans. Cytometry A. 2010;77:410–419. doi: 10.1002/cyto.a.20859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baechler EC, Gregersen PK, Behrens TW. The emerging role of interferon in human systemic lupus erythematosus. Curr. Opin. Immunol. 2004;16:801–807. doi: 10.1016/j.coi.2004.09.014. [DOI] [PubMed] [Google Scholar]
- Bagavant H, Fu SM. Pathogenesis of kidney disease in systemic lupus erythematosus. Curr. Opin. Rheumatol. 2009;21:489–494. doi: 10.1097/BOR.0b013e32832efff1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagavant H, Deshmukh US, Wang H, Ly T, Fu SM. Role for nephritogenic T cells in lupus glomerulonephritis: progression to renal failure is accompanied by T cell activation and expansion in regional lymph nodes. J. Immunol. 2006;177:8258–8265. doi: 10.4049/jimmunol.177.11.8258. [DOI] [PubMed] [Google Scholar]
- Bar-On L, Jung S. Defining dendritic cells by conditional and constitutive cell ablation. Immunol. Rev. 2010;234:76–89. doi: 10.1111/j.0105-2896.2009.00875.x. [DOI] [PubMed] [Google Scholar]
- Bennett CL, Clausen BE. DC ablation in mice: promises, pitfalls, and challenges. Trends Immunol. 2007;28:525–531. doi: 10.1016/j.it.2007.08.011. [DOI] [PubMed] [Google Scholar]
- Blanco P, Palucka AK, Gill M, Pascual V, Banchereau J. Induction of dendritic cell differentiation by IFN-alpha in systemic lupus erythematosus. Science. 2001;294:1540–1543. doi: 10.1126/science.1064890. [DOI] [PubMed] [Google Scholar]
- Bohle A, Muller GA, Wehrmann M, Mackensen-Haen S, Xiao JC. Pathogenesis of chronic renal failure in the primary glomerulopathies, renal vasculopathies, and chronic interstitial nephritides. Kidney Int. Suppl. 1996;54:S2–S9. [PubMed] [Google Scholar]
- Burne-Taney MJ, Kofler J, Yokota N, Weisfeldt M, Traystman RJ, Rabb H. Acute renal failure after whole body ischemia is characterized by inflammation and T cell-mediated injury. Am. J. Physiol. Renal. Physiol. 2003;285:F87–94. doi: 10.1152/ajprenal.00026.2003. [DOI] [PubMed] [Google Scholar]
- Cuzic S, Ritz E, Waldherr R. Dendritic cells in glomerulonephritis. Virchows Arch B Cell Pathol. Incl. Mol. Pathol. 1992;62:357–363. doi: 10.1007/BF02899704. [DOI] [PubMed] [Google Scholar]
- Dong X, Swaminathan S, Bachman LA, Croatt AJ, Nath KA, Griffin MD. Antigen presentation by dendritic cells in renal lymph nodes is linked to systemic and local injury to the kidney. Kidney Int. 2005;68:1096–1108. doi: 10.1111/j.1523-1755.2005.00502.x. [DOI] [PubMed] [Google Scholar]
- Dong X, Swaminathan S, Bachman LA, Croatt AJ, Nath KA, Griffin MD. Resident dendritic cells are the predominant TNF-secreting cell in early renal ischemia-reperfusion injury. Kidney Int. 2007;71:619–628. doi: 10.1038/sj.ki.5002132. [DOI] [PubMed] [Google Scholar]
- Dong X, Bachman LA, Miller MN, Nath KA, Griffin MD. Dendritic cells facilitate accumulation of IL-17 T cells in the kidney following acute renal obstruction. Kidney Int. 2008;74:1294–1309. doi: 10.1038/ki.2008.394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eddy AA. Interstitial macrophages as mediators of renal fibrosis. Exp. Nephrol. 1995;3:76–79. [PubMed] [Google Scholar]
- Edgtton KL, Kausman JY, Li M, et al. Intrarenal antigens activate CD4+ cells via co-stimulatory signals from dendritic cells. J. Am. Soc. Nephrol. 2008;19:515–526. doi: 10.1681/ASN.2007030386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiore N, Castellano G, Blasi A, et al. Immature myeloid and plasmacytoid dendritic cells infiltrate renal tubulointerstitium in patients with lupus nephritis. Mol. Immunol. 2008;45:259–265. doi: 10.1016/j.molimm.2007.04.029. [DOI] [PubMed] [Google Scholar]
- Foster MH. T cells and B cells in lupus nephritis. Semin. Nephrol. 2007;27:47–58. doi: 10.1016/j.semnephrol.2006.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujinaka H, Nameta M, Kovalenko P, et al. Periglomerular accumulation of dendritic cells in rat crescentic glomerulonephritis. J. Nephrol. 2007;20:357–363. [PubMed] [Google Scholar]
- Geissmann F, Manz MG, Jung S, Sieweke MH, Merad M, Ley K. Development of monocytes, macrophages, and dendritic cells. Science. 2010;327:656–661. doi: 10.1126/science.1178331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Georgiev M, Agle LM, Chu JL, Elkon KB, Ashany D. Mature dendritic cells readily break tolerance in normal mice but do not lead to disease expression. Arthritis Rheum. 2005;52:225–238. doi: 10.1002/art.20759. [DOI] [PubMed] [Google Scholar]
- Ginhoux F, Liu K, Helft J, et al. The origin and development of nonlymphoid tissue CD103+ DCs. J. Exp. Med. 2009;206:3115–3130. doi: 10.1084/jem.20091756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heath WR, Carbone FR. Dendritic cell subsets in primary and secondary T cell responses at body surfaces. Nat. Immunol. 2009;10:1237–1244. doi: 10.1038/ni.1822. [DOI] [PubMed] [Google Scholar]
- Heymann F, Meyer-Schwesinger C, Hamilton-Williams EE, et al. Kidney dendritic cell activation is required for progression of renal disease in a mouse model of glomerular injury. J. Clin. Invest. 2009;119:1286–1297. doi: 10.1172/JCI38399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hildner K, Edelson BT, Purtha WE, et al. Batf3 deficiency reveals a critical role for CD8alpha+ dendritic cells in cytotoxic T cell immunity. Science. 2008;322:1097–1100. doi: 10.1126/science.1164206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hume DA. Macrophages as APC and the dendritic cell myth. J. Immunol. 2008;181:5829–5835. doi: 10.4049/jimmunol.181.9.5829. [DOI] [PubMed] [Google Scholar]
- Hume DA, Gordon S. Mononuclear phagocyte system of the mouse defined by immunohistochemical localization of antigen F4/80. Identification of resident macrophages in renal medullary and cortical interstitium and the juxtaglomerular complex. J. Exp. Med. 1983;157:1704–1709. doi: 10.1084/jem.157.5.1704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inaba K, Turley S, Yamaide F, et al. Efficient presentation of phagocytosed cellular fragments on the major histocompatibility complex class II products of dendritic cells. J. Exp. Med. 1998;188:2163–2173. doi: 10.1084/jem.188.11.2163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishikawa S, Sato T, Abe M, et al. Aberrant high expression of B lymphocyte chemokine (BLC/CXCL13) by C11b+CD11c+ dendritic cells in murine lupus and preferential chemotaxis of B1 cells towards BLC. J. Exp. Med. 2001;193:1393–1402. doi: 10.1084/jem.193.12.1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwano M, Plieth D, Danoff TM, Xue C, Okada H, Neilson EG. Evidence that fibroblasts derive from epithelium during tissue fibrosis. J. Clin. Invest. 2002;110:341–350. doi: 10.1172/JCI15518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwata Y, Furuichi K, Sakai N, et al. Dendritic cells contribute to autoimmune kidney injury in MRL-Faslpr mice. J. Rheumatol. 2009;36:306–314. doi: 10.3899/jrheum.080318. [DOI] [PubMed] [Google Scholar]
- Janssen U, Ostendorf T, Gaertner S, et al. Improved survival and amelioration of nephrotoxic nephritis in intercellular adhesion molecule-1 knockout mice. J. Am. Soc. Nephrol. 1998;9:1805–1814. doi: 10.1681/ASN.V9101805. [DOI] [PubMed] [Google Scholar]
- Jantsch J, Chakravortty D, Turza N, et al. Hypoxia and hypoxia-inducible factor-1 alpha modulate lipopolysaccharide-induced dendritic cell activation and function. J. Immunol. 2008;180:4697–4705. doi: 10.4049/jimmunol.180.7.4697. [DOI] [PubMed] [Google Scholar]
- Jung S, Unutmaz D, Wong P, et al. In vivo depletion of CD11c(+) dendritic cells abrogates priming of CD8(+) T cells by exogenous cell-associated antigens. Immunity. 2002;17:211–220. doi: 10.1016/s1074-7613(02)00365-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaissling B, Le Hir M. Characterization and distribution of interstitial cell types in the renal cortex of rats. Kidney Int. 1994;45:709–720. doi: 10.1038/ki.1994.95. [DOI] [PubMed] [Google Scholar]
- Kaissling B, Hegyi I, Loffing J, Le Hir M. Morphology of interstitial cells in the healthy kidney. Anat. Embryol. (Berl). 1996;193:303–318. doi: 10.1007/BF00186688. [DOI] [PubMed] [Google Scholar]
- Kitamoto K, Machida Y, Uchida J, et al. Effects of liposome clodronate on renal leukocyte populations and renal fibrosis in murine obstructive nephropathy. J. Pharmacol. Sci. 2009;111:285–292. doi: 10.1254/jphs.09227fp. [DOI] [PubMed] [Google Scholar]
- Kitching AR, Holdsworth SR, Tipping PG. IFN-gamma mediates crescent formation and cell-mediated immune injury in murine glomerulonephritis. J. Am. Soc. Nephrol. 1999;10:752–759. doi: 10.1681/ASN.V104752. [DOI] [PubMed] [Google Scholar]
- Ko S, Deiwick A, Jager MD, et al. The functional relevance of passenger leukocytes and microchimerism for heart allograft acceptance in the rat. Nat. Med. 1999;5:1292–1297. doi: 10.1038/15248. [DOI] [PubMed] [Google Scholar]
- Kruger T, Benke D, Eitner F, et al. Identification and functional characterization of dendritic cells in the healthy murine kidney and in experimental glomerulonephritis. J. Am. Soc. Nephrol. 2004;15:613–621. doi: 10.1097/01.asn.0000114553.36258.91. [DOI] [PubMed] [Google Scholar]
- Kurts C, Kosaka H, Carbone FR, Miller JF, Heath WR. Class I-restricted cross-presentation of exogenous self-antigens leads to deletion of autoreactive CD8(+) T cells. J. Exp. Med. 1997;186:239–245. doi: 10.1084/jem.186.2.239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurts C, Heymann F, Lukacs-Kornek V, Boor P, Floege J. Role of T cells and dendritic cells in glomerular immunopathology. Semin Immunopathol. 2007;29:317–335. doi: 10.1007/s00281-007-0096-x. [DOI] [PubMed] [Google Scholar]
- Lang A, Benke D, Eitner F, et al. Heat shock protein 60 is released in immune-mediated glomerulonephritis and aggravates disease: in vivo evidence for an immunologic danger signal. J. Am. Soc. Nephrol. 2005;16:383–391. doi: 10.1681/ASN.2004040276. [DOI] [PubMed] [Google Scholar]
- Lech M, Kulkarni OP, Pfeiffer S, et al. Tir8/Sigirr prevents murine lupus by suppressing the immunostimulatory effects of lupus autoantigens. J. Exp. Med. 2008;205:1879–1888. doi: 10.1084/jem.20072646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lech M, Avila-Ferrufino A, Allam R, et al. Resident dendritic cells prevent postischemic acute renal failure by help of single Ig IL-1 receptor-related protein. J. Immunol. 2009;183:4109–4118. doi: 10.4049/jimmunol.0900118. [DOI] [PubMed] [Google Scholar]
- Lech M, Skuginna V, Kulkarni OP, et al. Lack of SIGIRR/TIR8 aggravates hydrocarbon oil-induced lupus nephritis. J. Pathol. 2010;220:596–607. doi: 10.1002/path.2678. [DOI] [PubMed] [Google Scholar]
- Leszczynski D, Renkonen R, Hayry P. Localization and turnover rate of rat renal ‘dendritic’ cells. Scand. J. Immunol. 1985;21:355–360. doi: 10.1111/j.1365-3083.1985.tb01441.x. [DOI] [PubMed] [Google Scholar]
- Li L, Huang L, Sung SS, et al. The chemokine receptors CCR2 and CX3CR1 mediate monocyte/macrophage trafficking in kidney ischemia–reperfusion injury. Kidney Int. 2008;74:1526–1537. doi: 10.1038/ki.2008.500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin SL, Castano AP, Nowlin BT, Lupher ML, Jr, Duffield JS. Bone marrow Ly6Chigh monocytes are selectively recruited to injured kidney and differentiate into functionally distinct populations. J. Immunol. 2009;183:6733–6743. doi: 10.4049/jimmunol.0901473. [DOI] [PubMed] [Google Scholar]
- Liu K, Waskow C, Liu X, Yao K, Hoh J, Nussenzweig M. Origin of dendritic cells in peripheral lymphoid organs of mice. Nat. Immunol. 2007;8:578–583. doi: 10.1038/ni1462. [DOI] [PubMed] [Google Scholar]
- Lukacs-Kornek V, Burgdorf S, Diehl L, Specht S, Kornek M, Kurts C. The kidney-renal lymph node-system contributes to cross-tolerance against innocuous circulating antigen. J. Immunol. 2008;180:706–715. doi: 10.4049/jimmunol.180.2.706. [DOI] [PubMed] [Google Scholar]
- Markovic-Lipkovski J, Muller CA, Risler T, Bohle A, Muller GA. Association of glomerular and interstitial mononuclear leukocytes with different forms of glomerulonephritis. Nephrol. Dial. Transplant. 1990;5:10–17. doi: 10.1093/ndt/5.1.10. [DOI] [PubMed] [Google Scholar]
- Merad M, Ginhoux F, Collin M. Origin, homeostasis and function of Langerhans cells and other langerin-expressing dendritic cells. Nat. Rev. Immunol. 2008;8:935–947. doi: 10.1038/nri2455. [DOI] [PubMed] [Google Scholar]
- Misseri R, Meldrum DR, Dinarello CA, et al. TNF-alpha mediates obstruction-induced renal tubular cell apoptosis and proapoptotic signaling. Am. J. Physiol. Renal. Physiol. 2005;288:F406–411. doi: 10.1152/ajprenal.00099.2004. [DOI] [PubMed] [Google Scholar]
- Nagler W. Vertebral artery obstruction by hyperextension of the neck: report of three cases. Arch. Phys. Med. Rehabil. 1973;54:237–240. [PubMed] [Google Scholar]
- Odobasic D, Kitching AR, Semple TJ, Holdsworth SR. Inducible co-stimulatory molecule ligand is protective during the induction and effector phases of crescentic glomerulonephritis. J. Am. Soc. Nephrol. 2006;17:1044–1053. doi: 10.1681/ASN.2005101022. [DOI] [PubMed] [Google Scholar]
- Probst HC, Tschannen K, Odermatt B, Schwendener R, Zinkernagel RM, Van Den BM. Histological analysis of CD11c-DTR/GFP mice after in vivo depletion of dendritic cells. Clin. Exp. Immunol. 2005;141:398–404. doi: 10.1111/j.1365-2249.2005.02868.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ring GH, Dai Z, Saleem S, Baddoura FK, Lakkis FG. Increased susceptibility to immunologically mediated glomerulonephritis in IFN-gamma-deficient mice. J. Immunol. 1999;163:2243–2248. [PubMed] [Google Scholar]
- Roake JA, Rao AS, Morris PJ, Larsen CP, Hankins DF, Austyn JM. Dendritic cell loss from nonlymphoid tissues after systemic administration of lipopolysaccharide, tumor necrosis factor, and interleukin 1. J. Exp. Med. 1995;181:2237–2247. doi: 10.1084/jem.181.6.2237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers NM, Matthews TJ, Kausman JY, Kitching RA, Coates PT. Review article: Kidney dendritic cells: their role in homeostasis, inflammation and transplantation. Nephrology (Carlton) 2009;14:625–635. doi: 10.1111/j.1440-1797.2009.01200.x. [DOI] [PubMed] [Google Scholar]
- Sapoznikov A, Fischer JA, Zaft T, Krauthgamer R, Dzionek A, Jung S. Organ-dependent in vivo priming of naive CD4+, but not CD8+, T cells by plasmacytoid dendritic cells. J. Exp. Med. 2007;204:1923–1933. doi: 10.1084/jem.20062373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiffer L, Sinha J, Wang X, et al. Short term administration of costimulatory blockade and cyclophosphamide induces remission of systemic lupus erythematosus nephritis in NZB/W F1 mice by a mechanism downstream of renal immune complex deposition. J. Immunol. 2003;171:489–497. doi: 10.4049/jimmunol.171.1.489. [DOI] [PubMed] [Google Scholar]
- Schlichting CL, Schareck WD, Weis M. Renal ischemia-reperfusion injury: new implications of dendritic cell-endothelial cell interactions. Transplant. Proc. 2006;38:670–673. doi: 10.1016/j.transproceed.2006.01.059. [DOI] [PubMed] [Google Scholar]
- Scholz J, Lukacs-Kornek V, Engel DR, et al. Renal dendritic cells stimulate IL-10 production and attenuate nephrotoxic nephritis. J. Am. Soc. Nephrol. 2008;19:527–537. doi: 10.1681/ASN.2007060684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segerer S, Heller F, Lindenmeyer MT, et al. Compartment specific expression of dendritic cell markers in human glomerulonephritis. Kidney Int. 2008;74:37–46. doi: 10.1038/ki.2008.99. [DOI] [PubMed] [Google Scholar]
- Shortman K, Naik SH. Steady-state and inflammatory dendritic-cell development. Nat. Rev. Immunol. 2007;7:19–30. doi: 10.1038/nri1996. [DOI] [PubMed] [Google Scholar]
- Soos TJ, Sims TN, Barisoni L, et al. CX3CR1+ interstitial dendritic cells form a contiguous network throughout the entire kidney. Kidney Int. 2006;70:591–596. doi: 10.1038/sj.ki.5001567. [DOI] [PubMed] [Google Scholar]
- Steinman RM, Idoyaga J. Features of the dendritic cell lineage. Immunol. Rev. 2010;234:5–17. doi: 10.1111/j.0105-2896.2009.00888.x. [DOI] [PubMed] [Google Scholar]
- Steinman RM, Hawiger D, Liu K, et al. Dendritic cell function in vivo during the steady state: a role in peripheral tolerance. Ann. N Y Acad. Sci. 2003;987:15–25. doi: 10.1111/j.1749-6632.2003.tb06029.x. [DOI] [PubMed] [Google Scholar]
- Steinman RM, Bonifaz L, Fujii S, et al. The innate functions of dendritic cells in peripheral lymphoid tissues. Adv. Exp. Med. Biol. 2005;560:83–97. doi: 10.1007/0-387-24180-9_12. [DOI] [PubMed] [Google Scholar]
- Tadagavadi RK, Reeves WB. Renal dendritic cells ameliorate nephrotoxic acute kidney injury. J. Am. Soc. Nephrol. 2010;21:53–63. doi: 10.1681/ASN.2009040407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timoshanko JR, Holdsworth SR, Kitching AR, Tipping PG. IFN-gamma production by intrinsic renal cells and bone marrow-derived cells is required for full expression of crescentic glomerulonephritis in mice. J. Immunol. 2002;168:4135–4141. doi: 10.4049/jimmunol.168.8.4135. [DOI] [PubMed] [Google Scholar]
- Tipping PG, Holdsworth SR. T cells in crescentic glomerulonephritis. J. Am. Soc. Nephrol. 2006;17:1253–1263. doi: 10.1681/ASN.2005091013. [DOI] [PubMed] [Google Scholar]
- Tipping PG, Kitching AR, Huang XR, Mutch DA, Holdsworth SR. Immune modulation with interleukin-4 and interleukin-10 prevents crescent formation and glomerular injury in experimental glomerulonephritis. Eur. J. Immunol. 1997;27:530–537. doi: 10.1002/eji.1830270226. [DOI] [PubMed] [Google Scholar]
- Tucci M, Quatraro C, Lombardi L, Pellegrino C, Dammacco F, Silvestris F. Glomerular accumulation of plasmacytoid dendritic cells in active lupus nephritis: role of interleukin-18. Arthritis Rheum. 2008;58:251–262. doi: 10.1002/art.23186. [DOI] [PubMed] [Google Scholar]
- Tucci M, Ciavarella S, Strippoli S, Dammacco F, Silvestris F. Oversecretion of cytokines and chemokines in lupus nephritis is regulated by intraparenchymal dendritic cells: a review. Ann. N Y Acad. Sci. 2009;1173:449–457. doi: 10.1111/j.1749-6632.2009.04805.x. [DOI] [PubMed] [Google Scholar]
- Turner JE, Paust HJ, Steinmetz OM, et al. CCR5 deficiency aggravates crescentic glomerulonephritis in mice. J. Immunol. 2008;181:6546–6556. doi: 10.4049/jimmunol.181.9.6546. [DOI] [PubMed] [Google Scholar]
- Van Rooijen N, Sanders A. Liposome mediated depletion of macrophages: mechanism of action, preparation of liposomes and applications. J. Immunol. Methods. 1994;174:83–93. doi: 10.1016/0022-1759(94)90012-4. [DOI] [PubMed] [Google Scholar]
- Wan S, Xia C, Morel L. IL-6 produced by dendritic cells from lupus-prone mice inhibits CD4+ CD25+ T cell regulatory functions. J. Immunol. 2007;178:271–279. doi: 10.4049/jimmunol.178.1.271. [DOI] [PubMed] [Google Scholar]
- Wolf D, Hochegger K, Wolf AM, et al. CD4+ CD25+ regulatory T cells inhibit experimental anti-glomerular basement membrane glomerulonephritis in mice. J. Am. Soc. Nephrol. 2005;16:1360–1370. doi: 10.1681/ASN.2004100837. [DOI] [PubMed] [Google Scholar]
- Woltman AM, de Fijter JW, Zuidwijk K, et al. Quantification of dendritic cell subsets in human renal tissue under normal and pathological conditions. Kidney Int. 2007;71:1001–1008. doi: 10.1038/sj.ki.5002187. [DOI] [PubMed] [Google Scholar]
- Yokota N, Daniels F, Crosson J, Rabb H. Protective effect of T cell depletion in murine renal ischemia-reperfusion injury. Transplantation. 2002;74:759–763. doi: 10.1097/00007890-200209270-00005. [DOI] [PubMed] [Google Scholar]
- Ysebaert DK, De Greef KE, Vercauteren SR, et al. Identification and kinetics of leukocytes after severe ischaemia/reperfusion renal injury. Nephrol. Dial. Transplant. 2000;15:1562–1574. doi: 10.1093/ndt/15.10.1562. [DOI] [PubMed] [Google Scholar]
- Ysebaert DK, De Greef KE, De Beuf A, et al. T cells as mediators in renal ischemia/reperfusion injury. Kidney Int. 2004;66:491–496. doi: 10.1111/j.1523-1755.2004.761_4.x. [DOI] [PubMed] [Google Scholar]
- Zeisberg EM, Potenta SE, Sugimoto H, Zeisberg M, Kalluri R. Fibroblasts in kidney fibrosis emerge via endothelial-to-mesenchymal transition. J. Am. Soc. Nephrol. 2008;19:2282–2287. doi: 10.1681/ASN.2008050513. [DOI] [PMC free article] [PubMed] [Google Scholar]