

Abstract
Goodpasture's, or anti-glomerular basement membrane (GBM), disease presents with rapidly progressive glomerulonephritis, caused by autoimmunity to a component of the GBM, the non-collagenous domain of the α3 chain of type IV collagen [α3(IV)NC1]. To investigate the mechanisms of inflammation in glomerulonephritis and to test new approaches to treatment, animal models of glomerulonephritis, termed experimental autoimmune glomerulonephritis (EAG), have been developed in susceptible strains of rats and mice. This review article describes how these models of EAG have been developed over the past three decades, discusses the evidence for the involvement of both humoral and cell-mediated immunity in the induction and pathogenesis of glomerulonephritis in these models and highlights recent, emerging data that have identified potential candidate genes that may control the genetic susceptibility in these different strains of rats and mice. The identification of these susceptibility genes has lead to a better understanding of the genetic basis of this model of anti-GBM disease, which may be relevant to the immunopathogenesis of Goodpasture's disease, and more generally to the progression from autoimmunity to target-organ damage.
Keywords: anti-GBM disease, experimental autoimmune glomerulonephritis, glomerular basement membrane, non-collagenous domain of the alpha 3 chain of type IV collagen, strain differences
Introduction to anti-GBM disease
Glomerulonephritis is one of the most common causes of established renal failure worldwide. The exact cause of most types of glomerulonephritis is not known, but immunological mechanisms are clearly involved (Salama & Pusey 2002; Pusey 2003). Current forms of treatment are only effective in certain cases and have considerable side effects. Goodpasture's, or anti-glomerular basement membrane (GBM), disease is an autoimmune disorder characterized by rapidly progressive glomerulonephritis and lung haemorrhage (Goodpasture 1919; Wilson & Dixon 1973). The disease is caused by autoimmunity to a component of the GBM, the non-collagenous domain of the α3 chain of type IV collagen, α3(IV)NC1 (Turner et al. 1992; Saus et al. 1998) (Figure 1).
Figure 1.
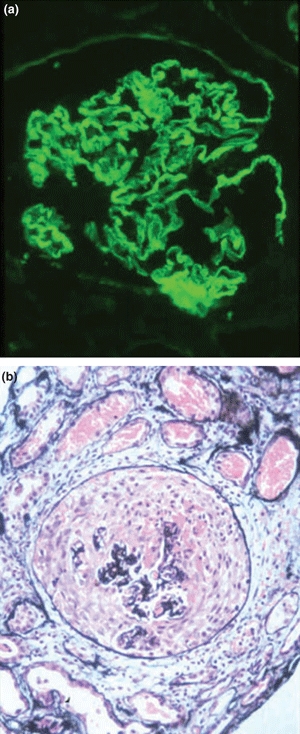
A renal biopsies from a patient with Goodpasture's disease showing (a) strong linear staining for IgG along the glomerular basement membrane (Direct immunofluorescence; ×300), and (b) severe crescentic nephritis with crescent formation (Periodic Acid-Schiff; ×300). Courtesy of Professor D.J. Evans.
Anti-GBM disease is strongly associated with the MHC class II allele, HLA-DRB1*1501, which is found 3.5 times more in patients than in controls (Fisher et al. 1997). HLA DRB1*03 and HLADRB1*04 are also found at higher frequencies in patients than in controls. The alleles HLA DRB1*07 and HLA DRB1*01 are found at lower frequencies in patients, suggesting that they confer a protective effect (Phelps & Rees 1999). However, HLA-DRB1*1501 alone is not sufficient to explain the susceptibility to this rare disease. It is thought that in addition, non-MHC genes and environmental factors must play a role in the progression from susceptibility to overt disease (Salama & Pusey 2002).
Animal models of anti-GBM disease
To investigate the mechanisms of autoimmunity and inflammation in glomerulonephritis and to test approaches to specific immune intervention, animal models of Goodpasture's disease have been developed in various species. The term ‘experimental autoimmune glomerulonephritis’ (EAG) is used to describe those models induced by immunization with antigenic material from the GBM.
This type of model was first described by Steblay, who observed that sheep immunized with human GBM developed anti-GBM antibodies and crescentic nephritis (Steblay 1962). More recently, animal models of EAG have been induced in susceptible strains of rats and mice by immunization with preparations of bovine and rat GBM (Sado et al. 1984; Pusey et al. 1991; Bolton et al. 1993; Kalluri et al. 1997; Reynolds et al. 1998) or with preparations of recombinant human or rat α3(IV)NC1 (Sado et al. 1998; Ryan et al. 2001; Hopfer et al. 2003). These studies have demonstrated that EAG shares many features in common with the human disease, in that the renal pathology is very similar and that the autoimmune response involves anti-GBM antibodies with the same specificity for the main target antigen, α3(IV)NC1 (Ryan et al. 2001; Reynolds et al. 2003).
Induction of EAG in rats
Immunization with preparations of GBM
The MHC class II has been shown to have a direct effect on strain susceptibility to anti-GBM glomerulonephritis in rats. In humans, the gene structure of the MHC region is called the HLA. The equivalent MHC region in the rat is called the RT1 region. Strain-dependent susceptibility to the development of EAG was first demonstrated in rats by Sado et al. (1986). In this study, they showed that Wistar Kyoto (WKY) and SHR rats immunized with bovine GBM in Freund's complete adjuvant (FCA) developed severe glomerulonephritis with marked pulmonary haemorrhage, while F344 and Lewis (LEW) rats developed mild glomerulonephritis with only petechial pulmonary haemorrhage. This suggested that differences in the strain of experimental animals influenced the severity of renal damage.
Parallel studies by Pusey et al. (1991) also demonstrated a strain-dependent phenomenon. Brown Norway (BN) (RT1-n) rats immunized with rat GBM in FCA developed circulating and deposited anti-GBM antibodies, albuminuria and variable focal segmental necrotizing glomerulonephritis, while PVG (RT1-c) and DA (RT1-a) rats developed only circulating and deposited antibody, but no nephritis, and LEW (RT1-l) and WAG (RT1-u) rats showed virtually no response. In additional studies, it has also been shown that two genetically different substrains of WKY rat, differing only at the MHC, vary in their susceptibility to the induction of EAG (Reynolds et al. 1998). WKY (RT1-l) rats immunized with rat GBM in FCA developed severe crescentic nephritis, while WKY (RT1-k) rats developed milder nephritis.
Immunization with preparations of recombinant α3(IV)NC1
However, the induction of EAG in WKY rats using preparations of GBM, of which α3(IV)NC1 is only a minor component, it is not an ideal model for assessing the specificity of the autoimmune response. For this reason, several groups cloned, sequenced and expressed the NC1 domain of the α3 chain of type IV collagen (Sado et al. 1998; Ryan et al. 2001). EAG has now therefore been induced in WKY rats with preparations of recombinant human and rat α3(IV)NC1. This was first described by Sado et al. (1998) who reported that a recombinant form of human α3(IV)NC1 was capable of inducing severe crescentic nephritis in WKY rats. More recently, it was also shown that immunization of WKY rats with recombinant rat α3(IV)NC1 resulted in the development of circulating anti-α3(IV)NC1 antibodies, deposition of IgG on the GBM, severe focal necrotizing glomerulonephritis with crescent formation and a glomerular infiltration of T cells and macrophages (Ryan et al. 2001; Reynolds et al. 2005) (Figure 2).
Figure 2.
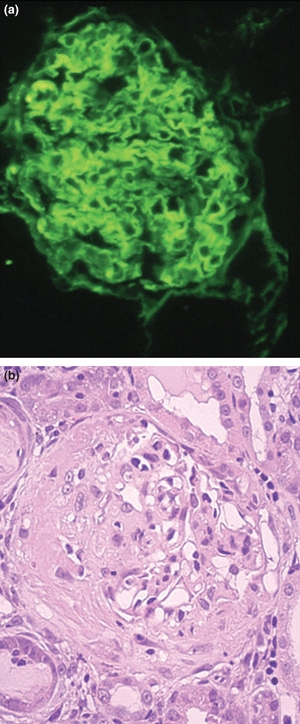
Kidney tissue at day 28 from WKY rats with experimental autoimmune glomerulonephritis showing (a) strong linear deposits of IgG on the glomerular basement membrane (Direct immunofluorescence; ×300), and (b) severe crescentic glomerulonephritis with large areas of fibrinoid necrosis (Haematoxylin and Eosin; ×300).
Immunization with immunodominant peptides
In attempts to identify immunodominant regions within α3(IV)NC1, several recent studies have demonstrated that synthetic peptides derived from α3(IV)NC1 can induce glomerulonephritis in WKY rats (Luo et al. 2002; Wu et al. 2003; Reynolds et al. 2008a). Recent studies from our group have identified a 15-mer immunodominant peptide, pCol(24–38), from the N-terminus of rat α3(IV)NC1, which contains the major B- and T-cell epitopes in EAG and which can induce crescentic nephritis (Reynolds et al. 2008a). Previous studies by Luo et al. showed that a 24-mer synthetic peptide, pCol(28–51), from the N-terminus of human α3(IV)NC1 was capable of inducing glomerulonephritis, although this was mild and inconsistent (Luo et al. 2002), while Wu et al. (2003) showed that a 13-mer peptide, pCol(28–40), containing a T-cell epitope from rat α3(IV)NC1 induced severe crescentic glomerulonephritis. A summary of the history of susceptibility to EAG in the rat is shown in Table 1.
Table 1.
Summary of the history of susceptibility to experimental autoimmune glomerulonephritis (EAG) in the rat
| Strain | Immunogen | Severity of EAG |
|---|---|---|
| WKY | Bovine GBM | +++ |
| SHR | Bovine GBM | +++ |
| F344 | Bovine GBM | +/− |
| LEW | Bovine GBM | +/− |
| BN | Rat GBM | ++ |
| PVG | Rat GBM | + |
| DA | Rat GBM | + |
| LEW | Rat GBM | − |
| WAG | Rat GBM | − |
| WKY (RT1-l) | Rat GBM | +++ |
| WKY (RT1-k) | Rat GBM | ++ |
| WKY | Human α3(IV)NC1 | +++ |
| WKY | Rat α3(IV)NC1 | +++ |
| LEW | Rat α3(IV)NC1 | − |
| WKY | Human pCol(28–51) | ++ |
| WKY | Rat pCol(28–40) | +++ |
| WKY | Rat pCol(24–38) | +++ |
α3(IV)NC1, the non-collagenous domain of the alpha 3 chain of type IV collagen; GBM, glomerular basement membrane; pCol(24–38), a 15-mer immunodominant peptide from the N-terminus of rat α3(IV)NC1.
Induction of EAG in mice
Immunization with preparations of GBM
The MHC class II has also been shown to have a direct effect on strain susceptibility to anti-GBM glomerulonephritis in mice. The MHC region in the mouse is called the H-2 region. Strain-dependent susceptibility to the development of EAG was first demonstrated in mice by Kalluri et al. (1997). In this study, they reported that SJL (H-2s) mice immunized with purified bovine α3(IV)NC1 dimers were particularly susceptible to the development of glomerulonephritis, while C57/BL/6 (H-2b) and DBA/2 (H-2d) developed mild nephritis and CBA (H-2k), A/J (H-2a) and AKR (H-2k) mice were resistant. However, our group and others have found it difficult to reproduce these studies, possibly due to the differences in the antigenic preparations.
Immunization with preparations of recombinant α3(IV)NC1
In subsequent studies by this group, it was shown that immunization of DBA/1 mice with recombinant human α3(IV)NC1 resulted in the development of severe crescentic nephritis by 11 weeks, while C57/BL/6 and AKR mice developed a chronic disease course resulting in comparable kidney injury to DBA/1 mice within 6 months, and NOD mice revealed only minor glomerular abnormalities (Hopfer et al. 2003). Owing to the inconsistency and the long time frame to develop glomerulonephritis in these mouse models of EAG, functional susceptibility studies in mice have been rather limited. However, it has been shown recently that C57/BL/6 mice deficient in IL-23 were protected from the development of EAG, when compared with wild-type controls (Ooi et al. 2009). Interestingly, recombinant mouse α3(IV)NC1 was used in this study to induce EAG in the C57/BL/6 wild types, rather than recombinant human α3(IV)NC1.
In recent studies, our group has tried to induce crescentic nephritis in the CD1 mouse, which has previously been shown to be particularly susceptible to nephrotoxic nephritis (Lloyd et al. 1997). Our preliminary results have demonstrated that immunization of CD1 mice with recombinant rat α3(IV)NC1 resulted in the development of circulating and deposited anti-GBM antibodies, albuminuria and focal proliferative glomerulonephritis by week 6 after immunization, which progresses to severe crescentic glomerulonephritis by week 12 (Reynolds et al. 2008b) (Figure 3). In this study, we have also demonstrated that anti-IL6 monoclonal antibody was effective in reducing the severity of glomerulonephritis in this model of EAG in the CD1 mouse (Reynolds et al. 2008b), demonstrating that it can be used for therapeutic studies. A summary of the history of susceptibility to EAG in the mouse is shown in Table 2.
Figure 3.
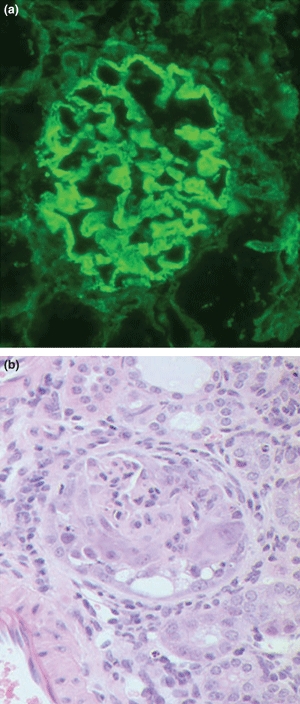
Kidney tissue at week 12 from CD1 mice with experimental autoimmune glomerulonephritis (EAG) showing (a) strong linear deposits of IgG along the glomerular basement membrane (Direct immunofluorescence; ×300), and (b) marked segmental necrosis of the glomerular tuft with crescent formation (Haematoxylin and Eosin; ×300).
Table 2.
Summary of the history of susceptibility to experimental autoimmune glomerulonephritis (EAG) in the mouse
| Strain | Immunogen | Severity of EAG |
|---|---|---|
| SJL | Purified bovine α3(IV)NC1 | +++ |
| C57/BL/6 | Purified bovine α3(IV)NC1 | + |
| DBA/2 | Purified bovine α3(IV)NC1 | + |
| CBA | Purified bovine α3(IV)NC1 | − |
| A/J | Purified bovine α3(IV)NC1 | − |
| AKR | Purified bovine α3(IV)NC1 | − |
| DBA/1 | Recombinant human α3(IV)NC1 | +++ |
| C57/BL/6 | Recombinant human α3(IV)NC1 | ++ |
| AKR | Recombinant human α3(IV)NC1 | ++ |
| NOD | Recombinant human α3(IV)NC1 | + |
| C57/BL/6 | Recombinant mouse α3(IV)NC1 | +++ |
| CD1 | Recombinant rat α3(IV)NC1 | +++ |
α3(IV)NC1, the non-collagenous domain of the alpha 3 chain of type IV collagen.
Humoral versus cell-mediated immunity in the induction of EAG
There is still controversy as to whether the development of EAG in both rats and mice is associated mainly with humoral or cell-mediated immunity. There is now mounting evidence for the involvement of both limbs of the immune response in the pathogenesis of glomerulonephritis, which is also likely to be true for the human condition.
Cell-mediated immunity
The pathogenic role of autoreactive T cells has been demonstrated in a variety of studies. T cells have been shown to be present in the glomeruli of animals with EAG (Ryan et al. 2001; Reynolds et al. 2003), and splenic T cells from these animals proliferate in response to α3(IV)NC1 (Wu et al. 2001; Hopfer et al. 2003) and can be used to transfer disease to naive recipients (Kalluri et al. 1997; Wu et al. 2002). Glomerular T cells from rats with EAG show restricted T-cell receptor CDR3 spectratypes, demonstrating that they are an oligoclonal antigen-driven population (Walters et al. 2004). More recently, it has been reported that immunodominant B- and T-cell epitopes from the N-terminus of rat α3(IV)NC1 are capable of inducing crescentic nephritis (Wu et al. 2003; Bolton et al. 2005; Reynolds et al. 2008a).
Several studies have shown that anti-T-cell immunotherapy can prevent or ameliorate disease. Anti-CD4 (Reynolds & Pusey 1994) and anti-CD8 (Reynolds et al. 2002a) monoclonal antibody therapy has been shown to be effective in reducing the severity of glomerulonephritis. Inhibition of T-cell activation by blockade of the CD28-B7 costimulatory pathway (Reynolds et al. 2000) or the CD154-CD40 costimulatory pathway (Reynolds et al. 2004) has also been shown to ameliorate EAG. In addition, oral administration of GBM antigen (Kalluri et al. 1997; Reynolds & Pusey 2001), or nasal administration of recombinant α3(IV)NC1 (Reynolds et al. 2005), or an immunodominant peptide (Reynolds et al. 2009a) induces mucosal tolerance and reduces the severity of crescentic nephritis in EAG. Recently, it has also been shown that B-cell-deficient mice immunized with α3(IV)NC1 develop proliferative glomerulonephritis with a glomerular infiltrate of T cells and macrophages, demonstrating that cell-mediated immunity is sufficient to induce EAG (Dean et al. 2005).
Humoral immunity
However, the pathogenic role of anti-GBM antibodies has also been demonstrated in a variety of passive transfer studies. Transfer of disease has been demonstrated using antibodies pooled from the serum of nephritic mice (Kalluri et al. 1997), antibodies purified from the urine of nephritic rats (Sado et al. 1989), monoclonal antibodies derived from rats with EAG (Kohda et al. 2004) and antibodies eluted from the kidney of nephritic rats (Reynolds et al. 2006) Transfer of sera from C57/BL6 mice with EAG to RAG-1 knockout mice (that lack adaptive immunity) has been shown to induce linear deposits of IgG on the GBM and proteinuria, demonstrating that humoral immunity alone was sufficient to induce EAG (Dean et al. 2005).
It has also been shown that the differences in the characteristics of the anti-GBM antibodies between WKY and LEW rats accounted for the difference in susceptibility to EAG (Reynolds et al. 2006). This study demonstrated that anti-GBM antibodies in WKY rats were present in a higher concentration and show greater specificity for recombinant α3(IV)NC1, when compared with those in LEW rats. In addition, it was shown that passive transfer of eluted anti-GBM antibodies from kidneys of WKY rats with EAG led to a similar deposition of IgG on the GBM of both WKY and LEW rats, but resulted in the development of crescentic glomerulonephritis only in WKY rats. These findings illustrated the importance of both the autoimmune response and also the inflammatory response to deposited antibody, in the susceptibility of glomerulonephritis.
Genetic susceptibility to EAG
Genetic linkage studies in backcross animals
This observation that the WKY rat was susceptible to the development of EAG, while the LEW rat was resistant, has resulted in extensive genetic linkage studies in attempts to identify susceptibility genes that are involved in the induction and pathogenesis of disease. F1 animals, generated by crossing the WKY (responder) with the LEW (non-responder), were completely resistant to the induction of EAG, while backcross (BC1) animals, generated by crossing the WKY (responder) with the F1 (non-responder), showed a range of responses from severe crescentic glomerulonephritis to no histological evidence of disease (Reynolds et al. 2002b).
A genome-wide linkage analysis of a large cohort of BC1 animals using polymorphic microsatellite markers revealed a major quantitative trait locus (QTL) on chromosome 13 (LOD = 3.9) linked to the severity of glomerulonephritis (Cook et al. 2004). Several biological candidates were found in the region of linkage, including genes encoding the activatory Fc receptor, Fcgr3 (FcγRIII), the inhibitory Fc receptor, Fcgr2 (FcγRII), and the common γ-subunit Fcer1g (FcRγ). In parallel studies, it was shown that copy number polymorphism in Fcgr3 predisposes to the development of nephrotoxic nephritis, a related rat model of glomerulonephritis (Aitman et al. 2006). In this study, it was also shown that in humans low copy number of FCGR3B, an orthologue of rat Fcgr3, was associated with glomerulonephritis in the autoimmune disease systemic lupus erythematosus.
Induction of EAG in congenic rats
To investigate this QTL in more detail, congenic rats were created by transferring the chromosome 13 QTL region from WKY rats to LEW (LEW/WKY13 congenic) and the same region from LEW rats to WKY (WKY/LEW13 congenic). The WKY/LEW13 congenic rats showed a marked reduction in the severity of EAG, when compared with WKY controls (Reynolds et al. 2008c). By contrast, LEW/W13 congenic rats were resistant to the development of EAG, as were LEW controls. This study confirmed the importance of the chromosome 13 QTL in the development of EAG, which should lead to insights into genetic susceptibility of glomerulonephritis.
Macrophage activation and function studies in congenic rats
In further studies, Fc receptor–mediated macrophage activation and function were assessed in these WKY/LEW13 congenic rats. Bone marrow–derived macrophages (BMDM) from these animals showed a significant reduction in Fc receptor–mediated oxidation, in phagocytosis of opsonized polystyrene beads and in the level of MCP-1 production, when compared with BMDM from WKY controls (Reynolds et al. 2009b). This study suggested that genetically determined Fc receptor–mediated macrophage activation/function was also important in genetic susceptibility to glomerulonephritis. Although Fc receptors have been shown to play an important role in susceptibility to glomerulonephritis, it is likely that other genes will also be involved. In fact, several other candidate genes have already been identified, which should explain why certain strains are particularly susceptible to glomerulonephritis (Aitman et al. 2006; Behmoaras et al. 2008).
Conclusion
Strain susceptibility to the development of EAG in rats and mice has certainly been very challenging over the past 3 decades. However, with the new exciting developments and discoveries in this area in the last few years, it is now very opportune and timely to review this recent literature. Recent developments in the field of recombinant protein technology and in the synthesis of synthetic peptides have made it possible to produce more specific autoantigens in large quantities. This has led to better more consistent animal models of anti-GBM glomerulonephritis, which share many features in common with the human disease, in that the autoimmune response and renal pathology are very similar. This new development has resulted in a better understanding in the induction and pathogenesis of glomerulonephritis. In the recent genetic linkage studies described earlier, the identification of susceptibility genes involved in the induction of EAG has led to a better understanding in the genetic basis of this animal model of anti-GBM disease. This new discovery may be relevant to the immunopathogenesis of autoimmune glomerulonephritis and more generally to the progression from autoimmunity to target-organ damage.
Acknowledgments
I would like to acknowledge the contribution of many past colleagues at the Hammersmith Campus of Imperial College London to the work described here, in particular Professor Charles Pusey, who was instrumental in first introducing me to the field of experimental glomerulonephritis.
References
- Aitman TJ, Dong R, Vyse TJ, et al. Copy number polymorphism in fcgr3 predisposes to glomerulonephritis in rats and humans. Nature. 2006;439:851–855. doi: 10.1038/nature04489. [DOI] [PubMed] [Google Scholar]
- Behmoaras J, Bhangal G, Smith J, et al. Jund is a determinant of macrophage activation and is associated with glomerulonephritis susceptibility. Nat. Genet. 2008;40:553–559. doi: 10.1038/ng.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolton WK, May WJ, Sturgill BC. Proliferative autoimmune glomerulonephritis in rats: a model for autoimmune glomerulonephritis in humans. Kidney Int. 1993;44:294–306. doi: 10.1038/ki.1993.244. [DOI] [PubMed] [Google Scholar]
- Bolton WK, Chen L, Hellmark T, Wieslander J, Fox J. Epitope spreading and autoimmune glomerulonephritis in rats induced by a T cell epitope of Goodpasture's antigen. J. Am. Soc. Nephrol. 2005;16:2657–2666. doi: 10.1681/ASN.2004100823. [DOI] [PubMed] [Google Scholar]
- Cook PR, Evans DJ, Reynolds J, Aitman TJ, Pusey CD. Genetic analysis of experimental autoimmune glomerulonephritis. J. Am. Soc. Nephrol. 2004;15:664A. (Abstract) [Google Scholar]
- Dean EG, Wilson GRA, Li M, et al. Experimental autoimmune Goodpasture's disease: a pathogenic role for both effector cells and antibody in injury. Kidney Int. 2005;67:566–575. doi: 10.1111/j.1523-1755.2005.67113.x. [DOI] [PubMed] [Google Scholar]
- Fisher M, Pusey CD, Vaughan RW, Rees AJ. Susceptibility to Goodpasture's disease is strongly associated with HLA-DRB1 genes. Kidney Int. 1997;51:222–229. doi: 10.1038/ki.1997.27. [DOI] [PubMed] [Google Scholar]
- Goodpasture EW. The significance of certain pulmonary lesions to the etiology of influenza. Am. J. Med. Sci. 1919;158:863–870. [Google Scholar]
- Hopfer H, Maron R, Butzmann U, Helmchen U, Weiner HL, Kalluri R. The importance of cell-mediated immunity in the course and severity of autoimmune anti-glomerular basement membrane disease in mice. FASEB J. 2003;17:860–868. doi: 10.1096/fj.02-0746com. [DOI] [PubMed] [Google Scholar]
- Kalluri R, Danoff TM, Okada H, Neilson EG. Susceptibility to anti-glomerular basement membrane disease and Goodpasture's syndrome is linked to MHC class II genes and the emergence of T cell-mediated immunity in mice. J. Clin. Invest. 1997;100:2263–2275. doi: 10.1172/JCI119764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohda T, Okada S, Hayashi A, et al. High nephritogenicity of monoclonal antibodies belonging to IgG2a and IgG2b subclasses in rat anti-GBM nephritis. Kidney Int. 2004;66:177–186. doi: 10.1111/j.1523-1755.2004.00719.x. [DOI] [PubMed] [Google Scholar]
- Lloyd CM, Minto AW, Dorf ME, et al. RANTES and monocyte chemoattractant protein-1 (MCP-1) play an important role in the inflammatory phase of crescentic nephritis, but only MCP-1 is involved in crescent formation and interstitial fibrosis. J. Exp. Med. 1997;185:1371–1380. doi: 10.1084/jem.185.7.1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo AM, Fox JW, Chen L, Bolton WK. Synthetic peptides of Goodpasture's antigen in anti-glomerular basement membrane nephritis in rats. J. Lab. Clin. Med. 2002;139:303–310. doi: 10.1067/mlc.2002.123623. [DOI] [PubMed] [Google Scholar]
- Ooi JD, Phoon RK, Holdsworth SR, Kitching AR. IL-23, not IL-12, directs autoimmunity to the Goodpasture antigen. J. Am. Soc. Nephrol. 2009;20:980–989. doi: 10.1681/ASN.2008080891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phelps RG, Rees AJ. The HLA complex in Goodpasture's disease: a model for analyzing susceptibility to autoimmunity. Kidney Int. 1999;56:1638–1653. doi: 10.1046/j.1523-1755.1999.00720.x. [DOI] [PubMed] [Google Scholar]
- Pusey CD. Anti-glomerular basement membrane disease. Kidney Int. 2003;64:1535–1550. doi: 10.1046/j.1523-1755.2003.00241.x. [DOI] [PubMed] [Google Scholar]
- Pusey CD, Holland MJ, Cashman SJ, et al. Experimental autoimmune glomerulonephritis induced by homologous and isologous glomerular basement membrane in Brown-Norway rats. Nephrol. Dial. Transplant. 1991;6:457–465. doi: 10.1093/ndt/6.7.457. [DOI] [PubMed] [Google Scholar]
- Reynolds J, Pusey CD. In vivo treatment with a monoclonal antibody to T helper cells in experimental autoimmune glomerulonephritis. Clin. Exp. Immunol. 1994;95:122–127. doi: 10.1111/j.1365-2249.1994.tb06025.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds J, Pusey CD. Oral administration of glomerular basement membrane prevents the development of experimental autoimmune glomerulonephritis. J. Am. Soc. Nephrol. 2001;12:61–70. doi: 10.1681/ASN.V12161. [DOI] [PubMed] [Google Scholar]
- Reynolds J, Mavromatidis K, Cashman SJ, Evans DJ, Pusey CD. Experimental autoimmune glomerulonephritis (EAG) induced by homologous and heterologous glomerular basement membrane in two sub-strains of Wistar Kyoto rat. Nephrol. Dial. Transplant. 1998;13:44–52. doi: 10.1093/ndt/13.1.44. [DOI] [PubMed] [Google Scholar]
- Reynolds J, Tam FWK, Chandraker A, et al. CD28-B7 blockade prevents the development of experimental autoimmune glomerulonephritis. J. Clin. Invest. 2000;105:643–651. doi: 10.1172/JCI6710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds J, Norgan VA, Bhambra U, Smith J, Cook HT, Pusey CD. Anti-CD8 monoclonal antibody therapy is effective in the prevention and treatment of experimental autoimmune glomerulonephritis. J. Am. Soc. Nephrol. 2002a;13:359–369. doi: 10.1681/ASN.V132359. [DOI] [PubMed] [Google Scholar]
- Reynolds J, Cook PR, Ryan JJ, et al. Segregation of experimental autoimmune glomerulonephritis as a complex genetic trait and exclusion of Col4a3 as a candidate gene. Exp. Nephrol. 2002b;10:402–407. doi: 10.1159/000065297. [DOI] [PubMed] [Google Scholar]
- Reynolds J, Moss J, Duda MA, et al. The evolution of crescentic nephritis and alveolar haemorrhage following induction of autoimmunity to glomerular basement membrane in an experimental model of Goodpasture's disease. J. Pathol. 2003;200:118–129. doi: 10.1002/path.1336. [DOI] [PubMed] [Google Scholar]
- Reynolds J, Khan SB, Allen AR, Benjamin CD, Pusey CD. Blockade of the CD154-CD40 T cell costimulatory pathway prevents the development of experimental autoimmune glomerulonephritis. Kidney Int. 2004;66:1444–1452. doi: 10.1111/j.1523-1755.2004.00907.x. [DOI] [PubMed] [Google Scholar]
- Reynolds J, Prodromidi EI, Juggapah JK, et al. Nasal administration of recombinant rat α3(IV)NC1 prevents the development of experimental autoimmune glomerulonephritis. J. Am. Soc. Nephrol. 2005;16:1350–1359. doi: 10.1681/ASN.2004121026. [DOI] [PubMed] [Google Scholar]
- Reynolds J, Albouainain A, Duda MA, Evans DJ, Pusey CD. Strain susceptibility to active induction and passive transfer of experimental autoimmune glomerulonephritis in the rat. Nephrol. Dial. Transplant. 2006;21:3398–3408. doi: 10.1093/ndt/gfl523. [DOI] [PubMed] [Google Scholar]
- Reynolds J, Haxby J, Juggapah JK, Evans DJ, Pusey CD. Identification of a nephritogenic immunodominant B and T cell epitope in experimental autoimmune glomerulonephritis. Clin. Exp. Immunol. 2008a;155:311–319. doi: 10.1111/j.1365-2249.2008.03833.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds J, Marshall D, Shaw S, Gelinas RE, Cook HT, Pusey CD. Effect of anti-IL6 monoclonal antibody therapy in experimental autoimmune glomerulonephritis in the CD1 mouse. J. Am. Soc. Nephrol. 2008b;19:414A. (Abstract) [Google Scholar]
- Reynolds J, Tadros S, Smith J, et al. Genetic susceptibility to experimental autoimmune glomerulonephritis in congenic rats. J. Am. Soc. Nephrol. 2008c;19:414–415A. (Abstract) [Google Scholar]
- Reynolds J, Abbott DS, Karegli J, Evans DJ, Pusey CD. Mucosal tolerance induced by an immunodominant peptide from rat α3(IV)NC1 in established experimental autoimmune glomerulonephritis. Am. J. Pathol. 2009a;174:2202–2210. doi: 10.2353/ajpath.2009.081041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds J, Tee J, Smith J, et al. Macrophage activation and function in experimental autoimmune glomerulonephritis. J. Am. Soc. Nephrol. 2009b;20:62A. (Abstract) [Google Scholar]
- Ryan JJ, Reynolds J, Norgan VA, Pusey CD. Expression and characterisation of recombinant rat α3(IV)NC1 and its use in the induction of experimental autoimmune glomerulonephritis. Nephrol. Dial. Transplant. 2001;16:253–261. doi: 10.1093/ndt/16.2.253. [DOI] [PubMed] [Google Scholar]
- Sado Y, Okigaki T, Takamiya H, Seno S. Experimental autoimmune glomerulonephritis with pulmonary haemorrhage in rats. The dose-effect relationship of the nephritogenic antigen from bovine glomerular basement membrane. J. Clin. Lab. Immunol. 1984;15:199–204. [PubMed] [Google Scholar]
- Sado Y, Naito I, Akita M, Okigaki T. Strain specific responses of inbred rats on the severity of experimental autoimmune glomerulonephritis. J. Clin. Lab. Immunol. 1986;19:193–199. [PubMed] [Google Scholar]
- Sado Y, Naito I, Okigaki T. Transfer of anti-glomerular basement membrane antibody-induced glomerulonephritis in inbred rats with isologous antibodies from urine of nephritic rats. J. Pathol. 1989;158:325–332. doi: 10.1002/path.1711580410. [DOI] [PubMed] [Google Scholar]
- Sado Y, Boutaud AA, Kagawa M, Naito I, Ninomiya Y, Hudson BG. Induction of anti-GBM nephritis in rats by recombinant α3(IV) NC1 and α4(IV) NC1 of type IV collagen. Kidney Int. 1998;53:664–671. doi: 10.1046/j.1523-1755.1998.00795.x. [DOI] [PubMed] [Google Scholar]
- Salama AD, Pusey CD. Immunology of anti-glomerular basement membrane disease. Curr. Opin. Nephrol. Hypertens. 2002;1:279–286. doi: 10.1097/00041552-200205000-00003. [DOI] [PubMed] [Google Scholar]
- Saus J, Wieslander J, Langeveld JPM, Quinones S, Hudson BG. Identification of the Goodpasture antigen as the α3 chain of collagen IV. J. Biol. Chem. 1998;263:13374–13380. [PubMed] [Google Scholar]
- Steblay RW. Glomerulonephritis induced in sheep by injections of heterologous glomerular basement membrane and Freund's complete adjuvant. J. Exp. Med. 1962;116:253–272. doi: 10.1084/jem.116.2.253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner N, Mason PJ, Brown R, et al. Molecular cloning of the human Goodpasture antigen demonstrates it to be the α3 chain of type IV collagen. J. Clin. Invest. 1992;89:592–601. doi: 10.1172/JCI115625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walters G, Habib A-M, Reynolds J, Wu H, Knight JF, Pusey CD. Glomerular T cells are of restricted clonality and express multiple CDR3 motifs across different Vβ T cell receptor families in experimental autoimmune glomerulonephritis. Nephron. Exp. Nephrol. 2004;98:71–81. doi: 10.1159/000080682. [DOI] [PubMed] [Google Scholar]
- Wilson CB, Dixon FJ. Anti-glomerular basement membrane antibody-induced glomerulonephritis. Kidney Int. 1973;3:74–89. doi: 10.1038/ki.1973.14. [DOI] [PubMed] [Google Scholar]
- Wu J, Hicks J, Ou C, Singleton D, Borillo J, Lou Y-H. Glomerulonephritis induced by recombinant collagen IV alpha 3 chain noncollagen domain 1 is not associated with glomerular basement membrane antibody: a potential T cell-mediated mechanism. J. Immunol. 2001;167:2388–2395. doi: 10.4049/jimmunol.167.4.2388. [DOI] [PubMed] [Google Scholar]
- Wu J, Hicks J, Borillo J, Glass WF, Lou Y-H. CD4+ T cells specific to a glomerular basement membrane antigen mediate glomerulonephritis. J. Clin. Invest. 2002;109:517–525. doi: 10.1172/JCI13876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Borillo J, Glass WF, Hicks J, Ou C-N, Lou Y-H. T-cell epitope of α3 chain of type IV collagen induces severe glomerulonephritis. Kidney Int. 2003;64:1292–1301. doi: 10.1046/j.1523-1755.2003.00227.x. [DOI] [PubMed] [Google Scholar]