

This work introduces a bioinformatics approach that identifies positive and negative coordination of gene expression between sets of genes, or entire gene networks, in response to environmental or developmental cues. The approach is illustrated by a case study that identifies distinct expression behavior of the energy-associated gene network in response to different biotic and abiotic stresses.
Abstract
The expression pattern of any pair of genes may be negatively correlated, positively correlated, or not correlated at all in response to different stresses and even different progression stages of the stress. This makes it difficult to identify such relationships by classical statistical tools such as the Pearson correlation coefficient. Hence, dedicated bioinformatics approaches that are able to identify groups of cues in which there is a positive or negative expression correlation between pairs or groups of genes are called for. We herein introduce and discuss a bioinformatics approach, termed Gene Coordination, that is devoted to the identification of specific or multiple cues in which there is a positive or negative coordination between pairs of genes and can further incorporate additional coordinated genes to form large coordinated gene networks. We demonstrate the utility of this approach by providing a case study in which we were able to discover distinct expression behavior of the energy-associated gene network in response to distinct biotic and abiotic stresses. This bioinformatics approach is suitable to a broad range of studies that compare treatments versus controls, such as effects of various cues, or expression changes between a mutant and the control wild-type genotype.
INTRODUCTION
Organisms respond to external cues (biological perturbations) by synchronized changes in the expression levels of multiple genes, which together integrate into specific phenotypic outputs. The development of microarray technology, together with the development of a variety of bioinformatics approaches, has enabled the analysis of the simultaneous response of gene networks to various developmental, physiological, or external cues at the systems biology level (Loraine, 2009; Orlando et al., 2009; Sreenivasulu et al., 2010). As sessile organisms, plants adjust to environmental stresses through highly compound changes in gene expression programs. The model plant Arabidopsis thaliana is highly suitable for systems biology studies in which large data sets of microarray expression results have accumulated, particularly through use of the Affymetrix GeneChip array ATH1, which contains more than 22,400 unique probe sets representing ~24,000 genes. Thousands of ATH1 arrays have been used to monitor the expression of the Arabidopsis transcriptome in various genetic backgrounds and under a variety of biological conditions, particularly stress-associated cues (for example, see Craigon et al., 2004). This enormous resource has also been used for systems biology analyses using a variety of approaches, including bioinformatics approaches (for example, see Van Norman and Benfey, 2009).
Bioinformatics analyses of microarrays generally use basic statistical tests, such as t test, Pearson correlation, and analysis of variance, as well as various grouping algorithms, such as clustering, to elucidate groups of genes with similar expression behavior over multiple experiments. However, these techniques possess limitations with respect to elucidating genes with coregulated expression because such coregulated expression generally occurs only in a subset of the cues (biological perturbations) under study. Several published bioinformatics approaches, such as Mutual Information (Wells et al., 1996) and Biclustering (Van Mechelen et al., 2004; Dharan and Nair, 2009), have been developed to address this limitation. We present here an additional simple, intuitive, and user-friendly bioinformatics method that can be used by scientists without extensive bioinformatics expertise. Our method, which is based on our previously reported Gene Coordination approach (Less and Galili, 2009; Less et al., 2010), is a statistical approach that can distinguish, for each pair of genes, between different biological perturbations in which their response (significant stimulation or suppression of expression) is either positively or negatively correlated (positive and negative coordination) or not correlated at all. We have now developed the Gene Coordination tool further to enable the assembly of the highly coordinated genes into large cliques and clusters, possessing similar expression response to biological perturbations. We have used this approach to analyze the expression behavior of the entire set of Arabidopsis genes encoding metabolic enzymes and transcription factors (TFs) to multiple external cues, including biotic and abiotic stresses as well as hormonal and nutritional cues.
DATA SOURCE AND BIOINFORMATICS METHOD
Data Source
Genes encoding enzymes were identified and collected using the AraCyc database (http://www.arabidopsis.org/biocyc/index.jsp). These included 1838 different genes, which are represented by 1726 unique probe sets in the Affymetrix ATH1 microarray. In addition, we also collected all of the genes indicated to encode TFs (a total of 1632 genes represented by 1597 probe sets) according to the database of Arabidopsis TFs (http://datf.cbi.pku.edu.cn). Expression data were obtained from the Nottingham Arabidopsis Stock Centre (http://affymetrix.arabidopsis.info/AffyWatch.html), which contains hundreds of publicly available expression profiles. To ensure reliability of the data, we focused on well-documented experiments containing at least two replicates for both treatment and control in which the treatment could be described as a relatively short-term response to some external cue. Overall, our selected data set contained 758 microarrays representing 211 different biological perturbations, which in total included 3323 unique probe sets.
Bioinformatics Approach
Our present research was based on a novel Gene Coordination approach, developed previously (Less and Galili, 2009), which includes two basic central aspects: (1) calculating the significance of expression differences (cue/control; statistical significance defined as P < 0.05 based on a t test) for each of the studied Arabidopsis genes in response to individual time points derived from all of the different cues and selection of genes having significant expression changes in response to one or more cues; and (2) defining a significant positive or negative coordination between individual pairs of genes based on the number of biological perturbations that were either coexpressed or oppositely expressed relative to the background distribution, respectively. We further searched for groups of coexpressed genes that possess among themselves: (1) significant positive coordination in more than 16 biological perturbations (see Supplemental Figure 1A online; background distribution threshold calculated as coexpression of pairs of genes in up to 16 biological perturbations); and (2) a nonsignificant negative coordination in less than three biological perturbations (see Supplemental Figure 1B online; background distribution threshold calculated as opposite expression in up to 16 biological perturbations).
Our bioinformatics approach consisted of three steps. The first step included the assembly of the individual genes into all possible pairs of coexpressed genes. The second step included a stepwise joining of additional genes into each of the pairs of coexpressed genes, based on the same approach, resulting altogether in 141 different cliques having overlaps in up to 20% of their genes and possessing an average size of ~55 probe sets (genes) per clique. The final step included clustering of the different cliques according to their expression coordination in a way that pairs of cliques possessing a relatively high positive coordination and a relatively low negative coordination will fall into the same cluster. The entire three-step bioinformatics approach is described in detail in Methods and also is illustrated schematically in Figure 1.
Figure 1.

Schematic Diagram of the Gene Coordination Approach.
EXTRACTION OF BIOLOGICAL INFORMATION
Principal Expression Coordination of Genes Encoding All Arabidopsis Enzymes and TFs in Response to Various Stress, Nutrition, and Hormone-Associated Cues
Our approach yielded three distinct clusters, each of which possesses high positive expression coordination and almost no negative expression coordination among genes within each cluster (Figure 2; the three clusters are presented within the three different squares with black borders). Analysis of the final clusters also revealed two interesting observations: (1) the cliques of cluster 1 showed a high negative coordination and no positive coordination to the cliques of cluster 3 in response to specific cues; and (2) the cliques of cluster 2 exhibited positive and negative expression coordination to cliques of cluster 1 and cluster 3 in response to specific cues. Notably, even though the bioinformatics process did not include specific efforts to eliminate overlaps of the same gene(s) in more than one cluster, the actual clustering results revealed that only two genes appeared in more than one cluster.
Figure 2.

The Full Coordination Matrix of Genes Encoding TFs and Metabolic Enzymes.
Genes were classified into three clusters (clusters 1 to 3) based on their expression behavior (see text). The coordination scale is provided on the right, and the Euclidian distance between the different cliques composing the entire matrix is indicated on the left. Euclidian distance is a mathematical distance function between two objects, which in this case measures the relative similarity (or difference) between different cliques with respect to the overall changes in the expression levels of their genes in response to the different stress cues. Gene expression raw data analysis was performed using the robust multichip analysis algorithm, and a t test was used to calculate the P value of the expression change of each probe set in each biological perturbation. For gene coordination calculation, each expression change possessing a P value of <0.05 was considered to be a significant change. C1, C2, and C3, clusters 1, 2, and 3, respectively.
Next, we further analyzed the principal gene expression patterns of the three different clusters, based on the original expression ratios calculated directly from the Nottingham Arabidopsis Stock Centre (http://affymetrix.arabidopsis.info/AffyWatch.html). This was performed in two steps: (1) assembling the genes of each one of the three clusters together; and (2) calculating the percentage of genes showing significant upregulation (red dotted lines) or significant downregulation (blue dotted lines) for each one of the 211 biological perturbations. The results of this analysis are illustrated in Figure 3, clusters 1 to 3. In addition, for each of the given cues (such as salt stress; cues are indicated on top of Figure 3), we ordered the results from left to right to include increasing time points of the same cue in shoots, followed by increasing time points of the same cue in roots. The different clusters showed distinct principal expression patterns, namely: (1) genes of cluster 1 (Figure 3, top) are apparently mostly upregulated as parts of coordinated groups in response to the different biological perturbations; (2) genes in cluster 3 (Figure 3, bottom) are apparently largely downregulated as parts of coordinated groups in response to the different biological perturbations; and (3) genes in cluster 2 (Figure 3, middle) are principally distinct from those of clusters 1 and 3 by being downregulated or upregulated as parts of coordinated groups in response to the different cues, particularly abiotic and biotic stresses, respectively (Figure 3, middle; abiotic and biotic stresses are marked by bars below and on top of the graph). Interestingly, even though UV-B light is actually an abiotic stress, this stress yields in general similar expression patterns to the various biotic stresses rather than to the other abiotic stresses in respect to the genes grouped in cluster 2. Notably, most of the hormone treatments had quite minor effects on the expression of the highly coordinated genes in the three different clusters (Figure 3, treatments on the right side). This is likely due to the relatively low dose and short time exposures (usually not more than 3 h) of the plants to the different hormones (http://affymetrix.arabidopsis.info/AffyWatch.html), which may be insufficient to generate significant responses.
Figure 3.

Characteristics of the Three Different Clusters.
Clusters 1 to 3 are given in the top, middle, and bottom panels, respectively. The different cues are indicated on top. Red graphs and blue graphs represent the percentages of upregulated and downregulated genes, respectively, as indicated on the y axis in the left hand side. For each of the given cues, the individual diamond-shaped dots are ordered from left to right to include increasing time points of the same cue in shoots, followed by increasing time points of the same cue in roots.
Elucidation of Biological Processes Enriched in Genes That Appear in Clusters 1 to 3
Elucidation of highly coordinated regulatory and metabolic gene networks was performed using the PageMan enrichment tool (http://mapman.mpimp-golm.mpg.de/pageman/; Usadel et al., 2006; Table 1). The most enriched networks of cluster 1, whose genes are mainly upregulated in most analyzed cues, were associated with: (1) hormone metabolism (abscisic acid and ethylene) and hormonal signal transduction; and (2) transcription regulation (APETALA2/Ethylene-responsive elements and WRKY TFs). The most enriched network of cluster 2, whose genes are principally downregulated in abiotic stresses while upregulated in biotic stresses and UV-B light, was the compound energy network, including glycolysis and the pentose phosphate pathway, tricarboxylic acid (TCA) cycle, mitochondrial electron transport, and ATP biosynthesis (from now on this network is defined as energy-associated metabolism). In addition, cluster 2 was strongly enriched in the category of amino acid biosynthesis, particularly the Lys and Met branches of the Asp family pathway, whose catabolism is associated with energy production (Arruda et al., 2000; Galili, 2002; Angelovici et al., 2009, 2010a, 2010b; Araujo et al., 2010), as well as Glu, Arg, Ser, and Gly that are associated with photorespiration. The most significantly enriched networks in cluster 3, whose genes are mainly downregulated in most analyzed cues, included amino acid metabolism (Asp family and branched-chain amino acids), photosynthesis and its associated tetrapyrrole biosynthesis pathway, starch metabolism, lipid metabolism (particularly fatty acid synthesis), and C1 metabolism.
Table 1.
Overrepresented Regulatory, Metabolic, and Hormonal Categories of Each Cluster
| Levels | Count | P Value | Levels | Count | P Value |
| Cluster 1 | Cluster 3 | ||||
| 1. Hormone metabolism | 47 | 1E-04 | 1. Amino acid metabolism | 65 | 1E-05 |
| 1.2 Abscisic acid | 9 | 1E-03 | 1.2 Synthesis | 53 | 6E-07 |
| 1.2 Ethylene | 23 | 2E-06 | 1.2.3 Asp family | 18 | 2E-05 |
| 1.2.3 Signal transduction | 17 | 4E-08 | 1.2.3.4 Misc. homoserine | 5 | 8E-03 |
| 2. RNA | NE | 1.2.3 Branched-chain group | 9 | 1E-03 | |
| 2.2 Transcription regulation | NE | 1.2 Degradation | NE | ||
| 2.2.3 APETALA2/Ethylene-responsive element | 32 | 3E-05 | 1.2.3 Ser-Gly-Cys group | 6 | 2E-03 |
| 2.2.3 WRKY domain | 26 | 1E-07 | 1.2.3.4 Gly | 4 | 3E-02 |
| 3. Stress | NE | 2. Photosynthesis | 47 | 1E-22 | |
| 3.2 Abiotic | NE | 2.2 Light reaction | 27 | 3E-14 | |
| 3.2.3 Heat | 5 | 2E-03 | 2.2.3 Photosystem II | 14 | 9E-10 |
| Cluster 2 | 2.2.3.4 Polypeptide subunits | 14 | 9E-10 | ||
| 1. Glycolysis | 18 | 2E-06 | 2.2.3 Photosystem I | 13 | 4E-09 |
| 1.2 Enolase | 3 | 1E-03 | 2.2.3.4 Polypeptide subunits | 13 | 4E-09 |
| 2. Oxidative pentose phosphate | 10 | 1E-04 | 2.2 Photorespiration | 6 | 6E-03 |
| 2.2 Oxidative pentose phosphate | 6 | 4E-03 | 2.2.3 Gly cleavage | 3 | 1E-02 |
| 3. TCA | 19 | 4E-08 | 2.2 Calvin cycle | 14 | 6E-08 |
| 3.2 TCA | 17 | 4E-09 | 2.2.3 GAP | 4 | 3E-03 |
| 3.2.3 Pyruvate DH | 5 | 5E-03 | 3. Tetrapyrrole synthesis | 22 | 7E-08 |
| 3.2.3 Succinyl-CoA ligase | 3 | 1E-03 | 3.2 Mg chelatase | 4 | 3E-03 |
| 3.2.3 Succinate dehydrogenase | 3 | 1E-03 | 4. Major CHO metabolism | 23 | 2E-02 |
| 4. Mitochondrial electron transport/ATP synthesis | 27 | 1E-16 | 4.2 Synthesis | 23 | 3E-03 |
| 4.2 NADH-DH | 15 | 1E-11 | 4.2.3 Starch synthesis | 1E-03 | |
| 4.2.3 Complex I | 4 | 2E-04 | 4.2.3 Starch degradation | 9E-03 | |
| 4.2.3 Cytochrome C reductase | 6 | 5E-05 | 5. Secondary metabolism | NE | |
| 4.2.3 Cytochrome C oxidase | 6 | 1E-04 | 5.2 Isoprenoids | 26 | 6E-04 |
| 5. Amino acid metabolism | 63 | 8E-19 | 5.2.3 Nonmevalonate pathway | 11 | 3E-03 |
| 5.2 Synthesis | 55 | 3E-21 | 5.2.3 Carotenoids | 6 | 2E-03 |
| 5.2.3 Glu family | 5 | 1E-04 | 6. Lipid metabolism | 46 | 4E-03 |
| 5.2.3.4 Arg | 6 | 2E-05 | 6.2 FA synthesis/elongation | 16 | 1E-03 |
| 5.2.3 Asp family | 15 | 3E-07 | 6.2.3 Pyruvate DH | 4 | 1E-02 |
| 5.2.3.4 Met | 7 | 1E-04 | 6.2 FA desaturation | 5 | 6E-04 |
| 5.2.3.4 Lys | 4 | 4E-03 | 7. Hormone metabolism | NE | |
| 5.2.3 Ser-Gly-Cys group | 9 | 9E-05 | 7.2 Brassinosteroid | 11 | 2E-02 |
| 5.2.3.4 Ser | 5 | 1E-04 | 7.2.3 Synthesis/degradation | 9 | 2E-02 |
| 5.2.3 Aromatic amino acid | 13 | 4E-05 | 7.2.3.4 Sterols | 8 | 6E-04 |
| 5.2.3.4 Chorismate | 6 | 6E-04 | 8. Nucleotide metabolism | NE | |
| 5.2.3.4 Trp | 5 | 5E-03 | 8.2 Salvage | 7 | 1E-02 |
| 6. Nucleotide metabolism | 32 | 1E-10 | 8.2 Deoxynucleotide | 5 | 6E-04 |
| 6.2 Synthesis | 17 | 4E-10 | 9. C1 metabolism | 12 | 6E-04 |
| 6.2.3 Pyrimidine | 6 | 3E-04 | 10. RNA | NE | |
| 6.2.3 Purine | 9 | 3E-06 | 10.2 Transcription regulation | NE | |
| 7. Protein | 24 | 1E-06 | 10.2.3 TCP | 7 | 2E-02 |
| 7.2 Amino acid activation (tRNA ligase) | 16 | 5E-09 | |||
| 7.2 Targeting | 4 | 2E-04 | |||
| 7.2.3 Mitochondria | 3 | 1E-03 | |||
| 8. Cell wall | NE | ||||
| 8.2 Precursor synthesis | 13 | 4E-05 | |||
| 8.2.3 UGD | 3 | 5E-03 | |||
| 9. C1 metabolism | 7 | 7E-03 |
Functional categories that are overrepresented in the list of genes were clustered. Overrepresentation analysis was performed by the PageMan enrichment tool (http://mapman.mpimp-golm.mpg.de/pageman/). Only functional categories with more than three genes are shown. The background genes are available in Supplemental Table 1, and the genes from each cluster are available in Supplemental Table 2. The elaborated analysis is available in Supplemental Table 3. NE, not enriched.
SELECTED EXAMPLES OF THE TRANSLATION OF DATA EXTRACTED FROM THIS BIOINFORMATICS APPROACH INTO METABOLIC REGULATION
A Bioinformatics-Based Viewpoint on Metabolic Regulation
The adjustment and reorganization of metabolism play a central regulatory role in the adaptation of plants to external cues, particularly to biotic and abiotic stresses. Yet, our current understanding of principal regulatory aspects associated with the reorganization of biological gene networks at the systems biology level is limited due to insufficient availability of suitable bioinformatics approaches. For example, the Pearson correlation approach can elucidate a significant negative or positive expression correlation between two genes only if there is such a correlation in the entire set of biological perturbations under study. However, in nature, different genes may be positively correlated, negatively correlated, or not correlated at all in response to different biological perturbations. In addition, the commonly used clustering approach forces individual genes to belong only to a single cluster. Yet, in biological reality, a given gene can possess a variety of functions and, as a consequence, also possess numerous expression correlation patterns with different sets of genes in response to different biological perturbations; hence, other approaches are needed, such as, for example, the Fuzzy clustering approach (http://reference.wolfram.com/applications/fuzzylogic/Manual/12.html). The three-step bioinformatics approach that we describe in this article identifies three major patterns of gene expression behaviors with minimal gene overlaps between them, which possess distinct functions in metabolic regulation. The separation of the genes into these three major patterns of gene expression is based on their real expression patterns in which: (1) genes of cluster 1 are mostly upregulated as coordinated groups in response to the various cues, although some of the genes of cluster 1 may be downregulated in a relatively noncoordinated manner in response to some cues; (2) genes of cluster 3 are for the most part downregulated as coordinated groups in response to the various cues, although some of the genes of cluster 3 may be upregulated in a relatively noncoordinated manner in response to some cues; and (3) genes of cluster 2 are principally both upregulated and downregulated as coordinated groups in response to different cues.
Interaction of Energy-Associated Metabolism with Amino Acid Metabolism
The most intriguing observation is the clustering of the majority of the energy-associated network (including the oxidative pentose phosphate pathway, the TCA cycle, mitochondrial energy transport, and ATP biosynthesis) in cluster 2, implying that this network possesses distinct gene expression patterns relative to other metabolic networks, being mainly downregulated in response to abiotic stresses and upregulated in response to biotic stresses and UV-B light (Table 1, Figure 3). Even though UV-B light is considered an abiotic stress, it is metabolically distinct from other abiotic stresses with respect to the energy status, which is significantly depressed in most abiotic stresses (Baena-Gonzalez et al., 2007; Baena-Gonzalez and Sheen, 2008), but apparently not as much in UV-B light. Interestingly, genes encoding biosynthetic enzymes of a number of amino acid metabolic networks were also enriched in cluster 2. This pattern of metabolic regulation suggests that amino acid metabolism is tightly linked to respiration and energy regulation.
Response of Transcriptional, Hormonal, and Signal Transduction Networks to Biotic and Abiotic Stresses
Genes associated with transcriptional, hormonal, and signal transduction networks were profoundly enriched in cluster 1, which includes genes whose expression is induced in a coordinated manner (Table 1). The major fraction of genes in this group is associated with the metabolism of the hormone ethylene, while a smaller fraction of genes is associated with the hormone abscisic acid. These two hormones are well documented to be involved in response of plants to various stresses (Cutler et al., 2010). In addition, a number of genes associated with signal transduction as well as a large number of genes controlling stress-associated TFs of the APETALA2/Ethylene and WRKY families were also enriched in cluster 1. These results imply that exposure to stresses stimulate the expression of a large set of coordinated networks of regulatory genes controlling hormone metabolism, signal transduction cascades, and TFs that regulate the response of the plants to these stresses.
Exposure to Stress Causes a Highly Coordinated Downregulation of Gene Networks Particularly Associated with Photosynthesis, Tetrapyrrole Biosynthesis, as Well as Sugar, Lipid, and Amino Acid Metabolism
Interestingly, genes associated with amino acid metabolism (particularly the Asp family and branched-chain amino acid metabolic pathways) and sugar metabolism (particularly starch metabolism) were associated with cluster 3 (Table 1), implying that their expression is downregulated in a coordinated manner upon exposure to stress, particularly abiotic stresses. Since abiotic stresses generally cause energy deprivation, plants usually adjust to the stress-associated energy deprivation mainly by metabolizing sugars into energy and suppressing genes encoding biosynthetic enzymes of amino acids to conserve energy, and inducing genes encoding catabolic enzymes of amino acids to generate additional energy from protein degradation (Baena-Gonzalez and Sheen, 2008; Bunik and Fernie, 2009; Sulpice et al., 2009; Hey et al., 2010). In this context, genes encoding catabolic enzymes of the Asp family pathway appear in cluster 1, an observation that is supported by several reports showing that both expression and activity of the LKR/SDH gene of Lys catabolism are stimulated by abiotic stresses (Moulin et al., 2000, 2006; Stepansky and Galili, 2003; Stepansky et al., 2006).
Another metabolic pathway that was enriched in cluster 3 is the tetrapyrrole biosynthesis pathway, which leads to the synthesis of chlorophyll. Since exposure to stress generally suppresses photosynthesis to minimize photosynthesis-associated damages, the coordinated downregulation of genes associated with the tetrapyrrole biosynthesis pathway is expected (Tanaka and Tanaka, 2007).
ADVANTAGES AND UTILITY OF THE GENE COORDINATION APPROACH
In compound biological systems, expression correlation between pairs of genes may occur only under certain stages of development or upon exposure to certain external cues. In most experimental studies, the specific conditions or biological perturbations in which expression of pairs of genes is scientifically correlated is unknown at the initiation of the study and thus cannot be isolated from the other biological perturbations in which there is no expression correlation between these pairs of genes. Thus, analysis of expression correlation between pairs or even groups of genes under a broad scope of experimental conditions, in which positive or negative expression correlation between the two genes naturally occurs only under a small fraction of these conditions, is expected to yield relatively low or insignificant correlation. Such results make it difficult to decide whether to invest additional research to study the potential biological linkage between different genes. A major advantage of the present approach is that it very simply and intuitively identifies specific sets of biological perturbations in which there is significant expression correlation between pairs of genes or even between multiple genes. We termed such expression correlation under a specific set of biological perturbations as Gene Coordination because the expression of such pairs of genes may be noncorrelated or even oppositely regulated under other sets of biological perturbations.
COMPARISON TO OTHER BIOINFORMATICS APPROACHES
Most bioinformatics approaches used to group genes based on gene expression data sets can be divided along two main dimensions: the type of grouping algorithm that is used to group genes with similar gene expression patterns, and the distance function, which is used to measure the similarity between two gene expression patterns. Our approach differs from most other commonly used bioinformatics methods along these two dimensions. While the most commonly used distance functions, such as Pearson correlation and Mutual Information (Wells et al., 1996), usually establish expression pattern similarity based on the expression levels alone (absolute values or log ratios), our approach integrates both the expression ratio and the significance of the expression change. In our approach, we assume that in order to understand expression relationships of gene networks, it is more important to accurately establish whether a particular gene is significantly upregulated or downregulated rather than the magnitude of its upregulation or downregulation. Moreover, in our distance function, we take into account only biological perturbations in which both genes are significantly upregulated or downregulated in order to establish positive and negative gene coordination. In the second dimension, most grouping algorithms can be divided into clustering and Biclustering approaches. The main limitation of clustering approaches is that they cluster genes along the entire data sets, while in nature, genes can be coregulated only in a limited set of biological perturbations. In those cases, distance functions calculating similarity between different expression patterns across all biological perturbations in a given data set are suboptimal, especially in large data sets containing unrelated biological perturbations. Biclustering approaches (Van Mechelen et al., 2004; Dharan and Nair, 2009) were specifically developed to resolve this limitation. In Biclustering, the algorithm is tuned to identify groups of genes having similar gene expression patterns in a subset of biological perturbations. By its nature, Biclustering can assign the same gene to multiple groups of genes, a fact that makes its output difficult to decipher by nonexpert users. Our approach lies in between these two approaches, such that our distance function takes into account only that portion of the biological perturbations that are relevant to each pair of genes. In this respect, our approach is more similar to the Biclustering approach, but our grouping algorithm clusters groups of highly coordinated genes, resulting in clusters in which the vast majority of the genes appear only in one cluster and in this respect it is more similar to traditional clustering. This improves the efficiency of extraction of new biological insights. Another recently published bioinformatics approach adapted to the analysis of complex interrelationships of biological networks is MetNetAPI (Sucaet and Wurtele, 2010). API is an adaptable application programming interface that simplifies the operation and utilization of MetNetDB (http://metnet.vrac.iastate.edu/MetNet_db.htm), a database enclosing regulatory networks of metabolism and interactions between them, occurring at the transcription, translation, and posttranslation levels in Arabidopsis plants.
SUMMARY AND FUTURE PROSPECTS
We have developed a dedicated method to elucidate the coordinated response of gene networks to external cues and have used it to analyze the response of the genes encoding the entire set of TFs and metabolic enzymes to abiotic and biotic stresses, as well as to some nutritional and short-term hormonal treatments. We show that energy-associated metabolism is regulated as a highly coordinated gene network that is largely downregulated in response to abiotic stresses and induced in response to some biotic stresses and UV-B light (cluster 2). This super gene network behaves differently from other gene networks, such as: a network containing genes encoding stress-associated TFs as well as enzymes of stress hormones metabolism (ethylene and abscisic acid) and amino acid catabolism, which is induced by biotic and abiotic stresses (cluster 1); and a network encoding mostly enzymes of amino acids and chlorophyll biosynthesis whose expression is principally suppressed by biotic and abiotic stresses (cluster 3). Since this method identifies groups of genes having coordinated stimulation or suppression of expression, these groups can be used further, employing different bioinformatics approaches that operate on groups of genes, to discover additional biological insights. For example, such groups can be used to estimate the similarity between different treatments (biological perturbations) as determined based on their effects on the expression levels of the genes within each group (Less and Galili, 2009).
METHODS
Gene Expression Analysis and Gene Coordination Calculation
Gene expression analysis and calculation of Gene Coordination was done as described previously (Less and Galili, 2009). In brief, gene expression raw data analysis was done using the robust multichip analysis algorithm, and a t test was used to calculate the P value of the expression change of each probe set in each biological perturbation. For gene coordination calculation, each expression change possessing a P value of <0.05 was considered to be a significant change. For gene coordination calculation, we transformed the gene expression matrix into a three-value matrix. In this matrix, each expression change (treatment versus control) possessing a P value of <0.05 was given the value of 1 if it includes upregulation of expression or −1 if it includes a downregulation of expression. Expression changes possessing a P value of >0.05 were assigned a 0. Since we have only applied mathematical transformation to the expression matrix, there is no need to apply a false discovery rate at this stage. Positive coordination of expression between each pair of genes was defined as the number of perturbations in which both genes possess either the value of 1 or −1 in the transformed matrix, meaning that both genes were upregulated or downregulated. By contrast, negative coordination was defined as the number of perturbations in which one gene possesses the value of 1 while the other gene possesses the value of −1 in the transformed matrix, meaning that one gene is upregulated while the other is downregulated.
Calculation of the Gene Coordination Background Model
To determine the threshold values for positive and negative coordination, we calculated the expected distribution of positive and negative coordination, assuming that there is no coordination between genes, using the following background model. First, a Coordination matrix was calculated as described previously (Less and Galili, 2009) from the transformed expression matrix of −1, 0, and +1. Second, to calculate the background distribution, we shuffled the transformed matrix (−1, 0, +1) 100 times, using Monte Carlo simulation, and recalculated positive and negative coordination values for all gene pairs. In the Monte Carlo shuffling simulation, we randomly rearranged the order of the biological perturbations for each gene independently. This procedure resulted in ~5.5 million random positive and negative coordination values per iteration. The values of 16 for positive coordination and 3 for negative coordination were chosen because values of more than 15 and less than 4 never appeared in our Monte Carlo simulation.
Assembly of Probe Sets into Cliques
Cliques of probe sets were defined as probe sets that have high positive coordination and low negative coordination between themselves. Two lists of gene pairs were used to build the cliques. The first was a positive list, containing probe set pairs having a positive coordination of more than 16 and a negative coordination of less than 3. The second list was a negative, containing probe set pairs having a negative coordination of more than 3. The first step of building the cliques used a greedy approach, which tries to add as many probe sets as possible to each probe set pair in the positive list. Each probe set that was added to a given clique needed to fulfill two criteria: (1) exhibiting positive coordination with at list 30% of the probe sets already presenting this clique; and (2) exhibiting no negative coordination with any of the probe sets already present in the clique (according to the negative list). After the elimination of identical cliques, this step resulted in 1382 different cliques, from which 1267 contained 10 probe sets or more. The attribute of our greedy approach resulted in a large number of cliques with some redundancy (each probe set was on average in ~20 cliques); therefore, the second step of generating the final cliques included the merging of closely related cliques with small distance, which means they had a relatively high degree of overlapping probe sets. For this merging, we calculated the distance between each pair of cliques based on the amount of overlapping probe sets as follows:
with D being the distance between two cliques, i and j the indexes of the different cliques, oP the number of overlapping probe sets, and S the number of probe sets in each clique. Finally, we used the above distance matrix (further distance algorithm; available within the MATLAB software) to cluster all 1267 cliques, using a distance of 0.2 as the threshold for merging of overlapping cliques. This process resulted in 141 final cliques having an average size of ~55 probe sets per clique.
Assembly of the Probe Sets into Clusters of Cliques
To elucidate further the expression coordination relationship between the different cliques, we defined a positive or a negative coordination between each pair of cliques as the mean of positive or negative coordination between all possible probe sets of each of the clique pairs:
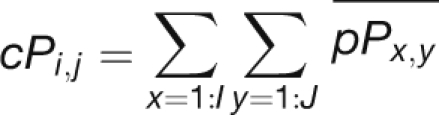 |
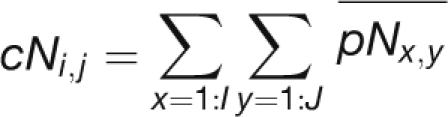 |
where cP and cN are the positive and negative coordination between cliques and pP and pN are the positive and negative coordination between probe sets. i and j are the indexes of the different cliques, and I and J are the number of probe sets in each clique.
For the clustering analysis of the final cliques, we used the following distance function:
where i and j are the indexes of the different cliques and cP and cN are the positive and negative coordination between the cliques (a constant of 20 was used to avoid negative distances).
Supplemental Data
The following materials are available in the online version of this article.
Supplemental Figure 1. Establishment of the Background Distribution for Positive and Negative Coordination between Pairs of Genes.
Supplemental Table 1. Full List of Enzymes and TFs.
Supplemental Table 2. List of Genes Separated into Cliques and Clusters.
Supplemental Table 3. The Elaborated Analysis of Overrepresented Categories of Each Cluster Analyzed by PageMan.
Acknowledgments
We thank Ron Milo for his helpful comments. This research was supported by The Israel Science Foundation (Grant 764/07) and the United States–Israel Binational Agricultural Research and Development Fund (Grant IS-3331-02). G.G. is an incumbent of the Bronfman Chair of Plant Science at the Weizmann Institute of Science.
References
- Angelovici R., Fait A., Fernie A.R., Galili G. (2010a). A seed high-lysine trait is negatively associated with the TCA cycle and slows down Arabidopsis seed germination. New Phytol. 189: 148–159 [DOI] [PubMed] [Google Scholar]
- Angelovici R., Fait A., Zhu X.H., Szymanski J., Feldmesser E., Fernie A.R., Galili G. (2009). Deciphering transcriptional and metabolic networks associated with lysine metabolism during Arabidopsis seed development. Plant Physiol. 151: 2058–2072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angelovici R., Galili G., Fernie A.R., Fait A. (2010b). Seed desiccation: A bridge between maturation and germination. Trends Plant Sci. 15: 211–218 [DOI] [PubMed] [Google Scholar]
- Araujo W.L., Ishizaki K., Nunes-Nesi A., Larson T.R., Tohge T., Krahnert I., Witt S., Obata T., Schauer N., Graham I.A., Leaver C.J., Fernie A.R. (2010). Identification of the 2-hydroxyglutarate and isovaleryl-CoA dehydrogenases as alternative electron donors linking lysine catabolism to the electron transport chain of Arabidopsis mitochondria. Plant Cell 22: 1549–1563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arruda P., Kemper E.L., Papes F., Leite A. (2000). Regulation of lysine catabolism in higher plants. Trends Plant Sci. 5: 324–330 [DOI] [PubMed] [Google Scholar]
- Baena-Gonzalez E., Rolland F., Thevelein J.M., Sheen J. (2007). A central integrator of transcription networks in plant stress and energy signalling. Nature 448: 938–942 [DOI] [PubMed] [Google Scholar]
- Baena-Gonzalez E., Sheen J. (2008). Convergent energy and stress signaling. Trends Plant Sci. 13: 474–482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunik V.I., Fernie A.R. (2009). Metabolic control exerted by the 2-oxoglutarate dehydrogenase reaction: A cross-kingdom comparison of the crossroad between energy production and nitrogen assimilation. Biochem. J. 422: 405–421 [DOI] [PubMed] [Google Scholar]
- Craigon D.J., James N., Okyere J., Higgins J., Jotham J., May S. (2004). NASCArrays: A repository for microarray data generated by NASC’s transcriptomics service. Nucleic Acids Res. 32: D575–D577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cutler S.R., Rodriguez P.L., Finkelstein R.R., Abrams S.R. (2010). Abscisic acid: Emergence of a core signaling network. Annu. Rev. Plant Biol. 61: 651–679 [DOI] [PubMed] [Google Scholar]
- Dharan S., Nair A.S. (2009). Biclustering of gene expression data using reactive greedy randomized adaptive search procedure. BMC Bioinformatics 10 (suppl. 1): S27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galili G. (2002). New insights into the regulation and functional significance of lysine metabolism in plants. Annu. Rev. Plant Physiol. Plant Mol. Biol. 7: 153–156 [DOI] [PubMed] [Google Scholar]
- Hey S.J., Byrne E., Halford N.G. (2010). The interface between metabolic and stress signalling. Ann. Bot. (Lond.) 105: 197–203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Less H., Angelovici R., Tzin V., Galili G. (2010). Principal transcriptional regulation and genome-wide system interactions of the Asp-family and aromatic amino acid networks of amino acid metabolism in plants. Amino Acids 39: 1023–1028 [DOI] [PubMed] [Google Scholar]
- Less H., Galili G. (2009). Coordinations between gene modules control the operation of plant amino acid metabolic networks. BMC Syst. Biol. 3: 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loraine A. (2009). Co-expression analysis of metabolic pathways in plants. Methods Mol. Biol. 553: 247–264 [DOI] [PubMed] [Google Scholar]
- Moulin M., Deleu C., Larher F. (2000). L-Lysine catabolism is osmo-regulated at the level of lysine-ketoglutarate reductase and saccharopine dehydrogenase in rapeseed leaf discs. Plant Physiol. Biochem. 38: 577–585 [DOI] [PubMed] [Google Scholar]
- Moulin M., Deleu C., Larher F., Bouchereau A. (2006). The lysine-ketoglutarate reductase-saccharopine dehydrogenase is involved in the osmo-induced synthesis of pipecolic acid in rapeseed leaf tissues. Plant Physiol. Biochem. 44: 474–482 [DOI] [PubMed] [Google Scholar]
- Orlando D.A., Brady S.M., Koch J.D., Dinneny J.R., Benfey P.N. (2009). Manipulating large-scale Arabidopsis microarray expression data: Identifying dominant expression patterns and biological process enrichment. Methods Mol. Biol. 553: 57–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sreenivasulu N., Sunkar R., Wobus U., Strickert M. (2010). Array platforms and bioinformatics tools for the analysis of plant transcriptome in response to abiotic stress. Methods Mol. Biol. 639: 71–93 [DOI] [PubMed] [Google Scholar]
- Stepansky A., Galili G. (2003). Synthesis of the Arabidopsis bifunctional lysine-ketoglutarate reductase/saccharopine dehydrogenase enzyme of lysine catabolism is concertedly regulated by metabolic and stress-associated signals. Plant Physiol. 133: 1407–1415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stepansky A., Less H., Angelovici R., Aharon R., Zhu X., Galili G. (2006). Lysine catabolism: An effective versatile regulator of lysine level in plants. Amino Acids 30: 121–125 [DOI] [PubMed] [Google Scholar]
- Sucaet Y., Wurtele E.S. (2010). MetNetAPI: A flexible method to access and manipulate biological network data from MetNet. BMC Res. Notes 3: 312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sulpice R., et al. (2009). Starch as a major integrator in the regulation of plant growth. Proc. Natl. Acad. Sci. USA 106: 10348–10353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka R., Tanaka A. (2007). Tetrapyrrole biosynthesis in higher plants. Annu. Rev. Plant Biol. 58: 321–346 [DOI] [PubMed] [Google Scholar]
- Usadel B., Nagel A., Steinhauser D., Gibon Y., Blasing O.E., Redestig H., Sreenivasulu N., Krall L., Hannah M.A., Poree F., Fernie A.R., Stitt M. (2006). PageMan: An interactive ontology tool to generate, display, and annotate overview graphs for profiling experiments. BMC Bioinformatics 7: 535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Mechelen I., Bock H.H., De Boeck P. (2004). Two-mode clustering methods: A structured overview. Stat. Methods Med. Res. 13: 363–394 [DOI] [PubMed] [Google Scholar]
- Van Norman J.M., Benfey P.N. (2009). Arabidopsis thaliana as a model organism in systems biology. Wiley Interdiscip. Rev. Syst. Biol. Med. 1: 372–379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wells W.M., III, Viola P., Atsumi H., Nakajima S., Kikinis R. (1996). Multi-modal volume registration by maximization of mutual information. Med. Image Anal. 1: 35–51 [DOI] [PubMed] [Google Scholar]