

This work presents the generation of a predictive model describing the DNA recognition specificity of the LEAFY floral transcription factor. The model is used to predict in vivo regulatory interactions between LEAFY and its target genes from mere inspection of various plant genome sequences.
Abstract
Despite great advances in sequencing technologies, generating functional information for nonmodel organisms remains a challenge. One solution lies in an improved ability to predict genetic circuits based on primary DNA sequence in combination with detailed knowledge of regulatory proteins that have been characterized in model species. Here, we focus on the LEAFY (LFY) transcription factor, a conserved master regulator of floral development. Starting with biochemical and structural information, we built a biophysical model describing LFY DNA binding specificity in vitro that accurately predicts in vivo LFY binding sites in the Arabidopsis thaliana genome. Applying the model to other plant species, we could follow the evolution of the regulatory relationship between LFY and the AGAMOUS (AG) subfamily of MADS box genes and show that this link predates the divergence between monocots and eudicots. Remarkably, our model succeeds in detecting the connection between LFY and AG homologs despite extensive variation in binding sites. This demonstrates that the cis-element fluidity recently observed in animals also exists in plants, but the challenges it poses can be overcome with predictions grounded in a biophysical model. Therefore, our work opens new avenues to deduce the structure of regulatory networks from mere inspection of genomic sequences.
INTRODUCTION
New technologies rapidly deliver whole-genome sequences from a wide variety of organisms at low cost, but functional annotation of these genomes remains a major challenge. Whereas conserved protein sequences are easily identified, transcriptional cis-regulatory modules can be evolutionarily fluid (Wilson and Odom, 2009; Schmidt et al., 2010; Weirauch and Hughes, 2010). Several recent studies revealed significant divergence in binding profiles of transcription factor (TF) homologs between vertebrate species (Mikkelsen et al., 2010; Schmidt et al., 2010). This divergence is due to the nature of cis-elements, which are small and degenerate motifs that can change rapidly and are thus difficult to detect by simple DNA sequence comparison (Wasserman and Sandelin, 2004; Ward and Bussemaker, 2008; Badis et al., 2009; Wilson and Odom, 2009). Whereas it is possible to study the genome-wide binding profile of TFs to DNA experimentally using chromatin immunoprecipitation (ChIP), a more streamlined functional analysis of genomes requires methods to predict variable cis-elements accurately directly from DNA sequences.
To address this problem, we focused on the genetic circuitry downstream of the LEAFY (LFY), a TF with a central role in the evolution and development of flowers (Liu et al., 2009; Moyroud et al., 2010). In Arabidopsis thaliana, LFY directly activates the expression of several floral homeotic MADS box genes, including AGAMOUS (AG), APETALA1 (AP1), and AP3 (Parcy et al., 1998; Busch et al., 1999; Wagner et al., 1999; Lohmann et al., 2001; Lamb et al., 2002), while repressing the shoot program by downregulating genes such as TERMINAL FLOWER1 (TFL1) (Liljegren et al., 1999; Ratcliffe et al., 1999; Parcy et al., 2002). From the small number of known LFY DNA binding sites, only a poorly defined 7-bp consensus sequence, CCANTG[G/T], has been previously deduced (Busch et al., 1999; Lamb et al., 2002). The three-dimensional structure of the LFY DNA binding domain has revealed contacts over 19 bp, suggesting considerably greater specificity (Hamès et al., 2008). Our aim was to capture this specificity in a predictive tool capable of detecting LFY binding sites from plant genomic sequences and ultimately tackle evolutionary questions. Here, we show how a biophysical model, built on biochemical ground and optimized using genome-wide in vivo binding data, can predict the evolution of the relationship between LFY and AG homologs, despite extensive variation in the sequences and positions of binding sites.
RESULTS
A Model for LEAFY DNA Binding Specificity
We determined the DNA binding preferences of the LFY DNA binding domain (DBD) using high-throughput systematic evolution of ligands by exponential enrichment (Selex) (Figure 1A) (Zhao et al., 2009). Alignment of the 494 unique sequences obtained revealed a 19-bp motif (Figure 1C), in good agreement with the three-dimensional structure of LFY DBD complexed with DNA (Hamès et al., 2008). This motif displays the previously established 7-bp consensus as the core. From the alignment, we deduced an asymmetric (ASY) position-specific scoring matrix (PSSM) (Wasserman and Sandelin, 2004) (Figure 1C; see Supplemental Table 1 online). Using this matrix with any 19-bp DNA fragment, scores can be calculated that should be proportional to the logarithm of the affinity of LFY DBD for this fragment. We used quantitative multifluorescence relative affinity (QuMFRA) assays (Man and Stormo, 2001) to measure the relative affinity of LFY DBD for 48 different oligonucleotides. We found that the ASY matrix scores correlated well with experimentally measured DNA binding affinities (Pearson correlation, r2 = 0.59) (Figure 1C). Since the LFY DBD binds DNA as a symmetric homodimer (Hamès et al., 2008), we sought to improve the PSSM by imposing symmetry. With the corresponding SYM matrix (Figure 1D), r2 increased to 0.69. To improve the matrix predictive power further, we analyzed the dependence between nucleotide positions: simple PSSMs assume that different positions contribute independently to the overall binding, but this condition is not always satisfied (Benos et al., 2002). For LFY, we indeed observed nonindependent triplets at two symmetric positions and in the center of the alignment (Figure 2). We modeled this dependence using the frequency of trinucleotides (Figure 1E). The resulting SYM-T matrix further increased r2 to 0.81. Notably, whereas the SYM-T matrix was well correlated with experimental DNA binding affinities, the simple presence or absence of the 7-bp consensus motif in the oligonucleotides tested was a poor predictor of binding, confirming the usefulness of the PSSM approach (Figures 1B to 1E).
Figure 1.

Optimization of the LFY Binding Site Model.
(A) Enrichment of DNA sequences bound by LFY over different Selex cycles.
(B) Binding of LFY to different sequences, either from AG or AP1 genes, or synthetic (S), with varying numbers of mismatches to the previously recognized consensus LFY binding motif.
(C) to (E) Comparison of experimentally determined and predicted scores (see Methods) for different DNA sequences with the three PSSMs (asymmetric [ASY], symmetric [SYM], and symmetric with triplets [SYM-T]), illustrated below by their logos. Open and closed circles represent sequences with or without the CCANTG[G/T] consensus, respectively.
[See online article for color version of this figure.]
Figure 2.

Detection of Dependence between Positions of the LFY Binding Sites.
Alignment of the 494 Selex sequences was analyzed with enoLOGOS software (Workman et al., 2005). The mutual information of each pair of positions of the alignment is displayed as a gray-scale-coded matrix plot below the logo corresponding to the SYM PSSM. Dependence is detected between positions 4, 5, and 6 or 14, 15, and 16 (lateral triplets) and, to a lesser extent, between positions 9, 10, and 11 (central triplet).
[See online article for color version of this figure.]
In Vivo Validation of the LFY Model by ChIP-seq
To test how well the in vitro–determined DNA binding specificity correlated with in vivo binding, we performed a ChIP experiment with LFY-specific antibodies followed by short read sequencing (ChIP-seq). The genomic regions enriched in plants that overexpressed LFY (35S:LFY) compared with wild-type seedlings were ordered using the rank product from two ChIP-seq replicates. In parallel, we used a biophysical model to compute the predicted occupancy (POcc) of these genomic regions by LFY (Granek and Clarke, 2005; Ward and Bussemaker, 2008). Such a model uses a PSSM to estimate the scores of all binding sites present on a large DNA fragment and then integrates these scores to compute the POcc value. The regions identified in ChIP-seq were ranked according to their POcc. We found a good correlation between the prediction and the experimental ChIP-based ranking. Moreover, we observed that the correlation increased from the ASY (Spearman’s rank correlation coefficient, 0.44) and the SYM (0.45) to the SYM-T matrix (0.53).
As further validation, we performed a receiver operating characteristic (ROC) analysis (Hanley and McNeil, 1982) comparing the 1564 regions most strongly enriched in ChIP (false discovery rate [FDR] < 0.1 in each of two independent replicates, meaning that the FDR is lower than 0.01 on the whole experiment for each gene selected; see Supplemental Data Set 1A online) with a set of random nonbound negative regions. In this analysis, we compared the percentage of regions whose POcc is higher than a given threshold in bound and unbound fragments sets. The area under the curve (ROC AUC) quantifies the tradeoff of specificity and sensitivity of the model as the POcc threshold varies. We evaluated the performance of two versions of the biophysical model: a first one that integrates all sites present on the fragment and a second one (hit-based model) that selects binding sites with a score higher than a cutoff value (Roider et al., 2007). With a ROC AUC value of 0.865 (Figure 3), the second model was best, but both of our models performed very well compared with other studies where ROC AUC values higher than 0.85 are found for <15% of the TFs studied (Granek and Clarke, 2005; Roider et al., 2007).
Figure 3.

Comparison of the Different Models for Prediction of in Vivo LFY Binding Sites.
ROC curves for LFY-bound and unbound sequences, using a biophysical model taking all sites (black line) into account or only those with a SYM-T matrix score higher than −23 (gray line).
LFY Directly Binds to Key Genes Regulating Flower Development
The most highly ranked ChIP-enriched fragment was in the 3′ region of the TFL1 gene, which is repressed by LFY and has important regulatory elements downstream of the transcribed region (Ratcliffe et al., 1999; Kaufmann et al., 2010). The strong binding observed in ChIP is explained by the presence of a cluster of LFY binding sites missing the CCANTG[G/T] consensus but detected by the SYM-T model (Figure 4B). Another very highly ranked region was present in the promoter of the well-characterized target AP1 (Parcy et al., 1998; Wagner et al., 1999), which also showed a second peak due to the presence of a binding site in its first intron (Figure 4A). These two results strongly suggest that LFY represses TFL1 both directly, as proposed before based on experiments with an activated form of LFY (Parcy et al., 2002), and indirectly, through AP1 activation (Kaufmann et al., 2010). For both AP1 and TFL1 as for most of the regions examined, the similarity between the ChIP-seq profiles and the computed binding site landscapes was striking (Figure 4), underscoring the predictive power of the SYM-T binding model.
Figure 4.

Examples of LFY-Bound Regions Identified by ChIP-seq.
Noncoding and coding sequences in exons are shown on top as open and closed boxes, respectively. ChIP-seq read coverage combined from both strands is shown in the middle. The bottom panels show the scores of binding sites (computed with the SYM-T model) and the presence of the CCANTG[G/T] consensus (indicated by arrows). AP1 (A), TFL1 (B), AG (C), and SEP4 (D).
[See online article for color version of this figure.]
The ChIP experiment also identified binding of LFY to regulatory regions of numerous floral regulator genes, such as AG (Busch et al., 1999) and SEPALLATA4 (Figures 4C and 4D) but also LFY itself (suggesting autoregulation) and GLABROUS INFLORESCENCE STEMS (Gan et al., 2006) (see Supplemental Figure 1 online; Table 1). Bound regions were also found in genes related to gibberellins and auxin signaling, two hormones known to be important for flower development (see Supplemental Figure 1 online; Table 1). Among the 2677 genes adjacent to the 1564 bound regions (see Supplemental Data Sets 1B and 1C online), 320 genes have an altered expression in lfy mutants (Schmid et al., 2003) and 54 (out of 445 genes; P value = 0.025) are deregulated in LFY-GR–overexpressing plants (William et al., 2004) (see Supplemental Data Set 1C online), including nine of the 15 genes previously considered as LFY direct targets by William et al. (2004). We expect many of the genes that are both bound and regulated to represent bona fide LFY direct target genes. In most cases, our model identified the LFY binding sites potentially responsible for the signal observed in ChIP (Figure 4; see Supplemental Figure 1 online).
Table 1.
Examples of Genes Bound by LFY
| Gene | Primary Gene Symbol | Rank | POcc | Best Site |
| Flowering | ||||
| At5G03840 | underline>TFL1/underline> | 1 | 0.01803 | −13.60 |
| At2G45660 | underline>SOC1/underline> | 770 | 0.0098 | −12.49 |
| At2G39250 | SCHNARCHZAPFEN | 1029 | 0.0070 | −13.95 |
| At3G58070 | GLABROUS INFLORESCENCE STEMS | 259 | 0.0034 | −19.26 |
| 288 | 0.0225 | −10.25 | ||
| 1482 | 0.0024 | −19.25 | ||
| At1G01183 | miR156 | 8 | 0.0085 | −13.61 |
| At4G35900 | FD | 796 | 0.0037 | −15.69 |
| At4G01500 | NGATHA4 | 725 | 0.0081 | −13.49 |
| At1G25560 | TEM1 | 6 | 0.0048 | −17.12 |
| 498 | 0.0026 | −19.40 | ||
| At4G25520 | SLK1 | 68 | 0.0042 | −16.63 |
| At2G45190 | FILAMENTOUS FLOWER | 503 | 0.0030 | −16.45 |
| Floral meristem specification | ||||
| At5G61850 | LFY | 1269 | 0.0156 | −10.82 |
| At3G57130 | BOP1 | 165 | 0.0476 | −7.82 |
| 1400 | 0.0026 | −18.83 | ||
| At2G41370 | BOP2 | 166 | 0.0051 | −16.05 |
| 556 | 0.0034 | −20.12 | ||
| 671 | 0.0112 | −11.83 | ||
| At5G18560 | PUCHI | 574 | 0.0046 | −18.27 |
| Floral organ specification and development | ||||
| At1G69120 | APETALA1 | 19 | 0.0609 | −7.33 |
| 1216 | 0.055 | −14.22 | ||
| At4G18960 | AG | 888 | 0.0104 | −14.28 |
| At3G54320 | WRINKLED1 | 815 | 0.0041 | −14.95 |
| At1G24260 | SEPALLATA3 | 25 | 0.0096 | −16.15 |
| 829 | 0.0023 | −20.59 | ||
| At2G03710 | SEPALLATA4 | 983 | 0.0049 | −14.70 |
| At1G31140 | GORDITA | 1421 | 0.0403 | −8.20 |
| At5G02030 | PENNYWISE | 1221 | 0.0043 | −14.77 |
| At3G63530 | BIG BROTHER | 940 | 0.0110 | −12.23 |
| 989 | 0.0016 | −19.54 | ||
| At5G67060 | HECATE1 | 527 | 0.0068 | −13.97 |
| At4G36260 | STYLISH 2 | 520 | 0.0071 | −13.81 |
| At5G07280 | EMS1 | 772 | 0.0044 | −14.70 |
| At3G02000 | ROXY1 | 367 | 0.0084 | −14.60 |
| At2G28056 | miR172 | 1213 | 0.0054 | −16.79 |
| At2G28610 | PRESSED FLOWER | 424 | 0.0166 | −12.23 |
| At4G37750 | AINTEGUMENTA | 462 | 0.0024 | −20.34 |
| 1460 | 0.0034 | −18.39 | ||
| At5G10510 | AINTEGUMENTA-like 6 | 1653 | 0.0056 | −14.89 |
| At1G01510 | ANGUSTIFOLIA 3 | 723 | 0.0019 | −21.45 |
| Gibberellins | ||||
| At5G15230 | GASA4 | 231 | 0.0095 | −12.69 |
| 431 | 0.0062 | −13.83 | ||
| At4G25420 | GA2OX1 | 1427 | 0.0086 | −13.52 |
| At1G30040 | GA2OX2 | 1045 | 0.0074 | −13.94 |
| At3G63010 | GID1B | 350 | 0.0068 | −14.69 |
| 425 | 0.0052 | −14.82 | ||
| 1536 | 0.0025 | −17.42 | ||
| At1G15550 | GA3OX1 (GA4) | 1573 | 0.0056 | −17.72 |
| At1G80340 | GA3OX2 | 879 | 0.0052 | −15.90 |
| Auxin | ||||
| At1G19840 | SAUR-like | 263 | 0.0416 | −8.21 |
| At1G19850 | MONOPTEROS | 289 | 0.0217 | −10.15 |
| At2G01420 | PIN4 | 235 | 0.0029 | −17.34 |
| 999 | 0.0053 | −15.03 | ||
| At3G62980 | TIR1 | 100 | 0.0107 | −12.78 |
| 110 | 0.0033 | −17.03 | ||
| At5G11320 | YUCCA4 | 510 | 0.0061 | −15.82 |
| 1261 | 0.0055 | −14.73 | ||
| At1G04240 | SHY2 | 1350 | 0.0043 | −15.32 |
| At2G34650 | PINOID | 225 | 0.0040 | −15.83 |
| At1G29430 | Auxin-responsive (SAUR-like) | 212 | 0.0228 | −9.85 |
| 438 | 0.0151 | −11.53 | ||
| Cytokinins | ||||
| AT1G59940 | ARR3 | 1088 | 0.0204 | −10.13 |
For a selection of genes expressed in floral tissues or dependent on LFY, the table indicates the rank from the ChIP-seq experiments (Rank), the POcc value, and the score of the best LFY binding site. Binding profiles are shown in Figure 4 or Supplemental Figure 1 online for the genes with underlined names.
Analysis of the LEAFY-AG Link over Large Evolutionary Distances
A major motivation for developing predictive DNA binding models is the functional annotation of genomes from nonmodel organisms. For a proof of concept, we examined the large intron of AG homologs, since this region is known to be important for AG regulation in various species and contains several conserved motifs (Sieburth and Meyerowitz, 1997; Busch et al., 1999; Davies et al., 1999; Hong et al., 2003; Causier et al., 2008). AG belongs to a small subfamily of MADS box genes (Ferrario et al., 2004; Zahn et al., 2006). A first duplication led to the formation of the AG and AGL11 lineages at the base of the angiosperms, and a second duplication in ancestral core eudicots yielded the euAGAMOUS (euAG) and PLENA (PLE) lineages (Kramer et al., 2004) (Figure 5A). All these proteins have similar DNA binding and protein–protein interaction profiles, and it is thought that they evolved specific functions primarily through diversification of their expression patterns (Ferrario et al., 2004; Zahn et al., 2006). Sequence similarity and genomic position are therefore not sufficient to predict functional equivalence with AG in other species.
Figure 5.

Prediction of LFY Occupancy of the Large Intron of AG Homologs Using the SYM-T Model.
(A) Schematic phylogeny of AG homologs after Kramer et al. (2004).
(B) and (C) POcc of AG homologs in monocots (B) and eudicots (C). A star indicates gene expression during early floral stages, and a circle indicates later expression. Expression data come from the references listed in Supplemental Table 2 online.
As the structural models indicated that the LFY-DNA interface is highly conserved in angiosperms (Moyroud et al., 2009), we applied our threshold-based biophysical model to the large intron of AG subfamily members of multiple angiosperm species. In both A. thaliana and its relative Arabidopsis lyrata, the predicted occupancy by LFY is much higher for the AG second intron than for that of SHATTERPROOF (SHP1 and SHP2, belonging to the PLE lineage) and SEEDSTICK (STK; belonging to the AGL11 lineage) genes (Figure 5C). This prediction is validated by functional analyses in A. thaliana demonstrating that LFY is responsible for the early induction of the AG gene (Parcy et al., 1998; Busch et al., 1999; Lohmann et al., 2001) but is not involved in regulating SHP or STK genes, which play later roles in fruit and ovule development (Liljegren et al., 2000; Colombo et al., 2010). Consistent with this, only AG, but not SHP or STK, was found to be a LFY target in our ChIP-seq experiments. Conversely, in several eudicots, such as Antirrhinum majus or Solanum lycopersicum, genes from the PLE clade were found to have the highest POcc compared with euAG or STK genes (Figure 5C). Our analysis thus predicts that they should be regulated by LFY. This prediction has indeed been validated in A. majus, where the SHP ortholog PLE was shown to be activated by the LFY ortholog FLORICAULA and to have an AG-like function (Davies et al., 1999; Causier et al., 2005). In other eudicot species, where less functional data is available, we observed a good agreement between a high POcc by LFY and the expression of the corresponding genes during early stages of flower development, when LFY is active (Figure 5C; see Supplemental Table 2 online).
We also examined AG and AGL11 orthologs from grasses, which are monocots. In all species examined, our model predicts much higher DNA occupancy by LFY for both AG orthologs compared with those of AGL11 (Figure 5B). This prediction is validated by expression data and functional analyses demonstrating that, in grasses, AG genes are both expressed before AGL11 orthologs and share the C-function (see Supplemental Table 2 online) (Thompson and Hake, 2009). Also, genetic analyses have suggested that ZFL1/2, the LFY maize (Zea mays) orthologs, regulate AG genes expression (Bomblies et al., 2003).
Detection of cis-Element Fluidity in AG Introns
Whereas our model correctly predicts global LFY occupancy in the large introns of AG homologs, we observed that the binding site landscapes are highly variable between these genes (Figure 6; see Supplemental Figures 2 to 4 online). In some cases, such as Bd-AG and Vv-AG2, there is a single binding site of very high affinity (corresponding to the AG2 LFY binding site in A. thaliana; Busch et al., 1999), whereas in others, such as At-AG, Al-AG, Os-MADS58, or PMADS3, this site is present but has a lower affinity that is compensated for through the action of multiple other sites (Figure 6). We experimentally verified the predicted high affinity for LFY for some of these additional binding sites (AG1, AG4, and AG5 from A. thaliana AG) (Figure 1B). We also detected their presence in multiple Brassicaceae species (see Supplemental Figure 4 online), strongly suggesting that they are functionally relevant.
Figure 6.

Distribution of LFY Binding Sites in AG-Like Genes.
LFY binding sites with a score higher than −20 are shown in eudicots (PLENA and euAG lineages) and monocots (AG lineage). The score scale is shown in each panel; the best binding sites correspond to the less negative score values. Stars mark the LFY binding site AG2, which can be located with confidence in most introns thanks to a nearby conserved sequence (see Supplemental Figure 2 online). Gene and species names are indicated on the right.
[See online article for color version of this figure.]
Next, we aligned the introns of AG homologs using the DIALIGN program (Morgenstern, 2004), which allows identification of local sequence similarities in divergent sequences. The highest-affinity binding site (corresponding to AG2 in A. thaliana) can be detected in alignments, but the sequence conservation is fairly low with many more regions of higher conservation spread throughout the intron (see Supplemental Figures 2 to 4 online). The other LFY binding sites cannot be identified based on sequence conservation alone, even in plants belonging to the same family such as the Brassicaceae (see Supplemental Figures 2 to 4 online). These results illustrate the fluidity of binding sites and the difficulty of detecting them by sequence alignment, in agreement with recent comparative genome-wide analyses of TF binding sites in vertebrates (Mikkelsen et al., 2010; Schmidt et al., 2010). The strength of a biophysical model is to overcome cis-element plasticity and detect regulatory links despite extensive sequence variation.
DISCUSSION
In this work, we built a model for DNA recognition by the LFY TF. The core tools we used (PSSMs and biophysical models) were developed and validated for bacterial and animal TFs (Wasserman and Sandelin, 2004) and have rarely been used in plant studies. The originality of our work resides in the fact that we have incorporated structural information (to impose the PSSM symmetry) and the dependence between nucleotides, thereby generating an improved model with high predictive power both for in vitro and in vivo binding (Figures 1 to 3). The fact that the PSSM built in vitro using LFY DBD explains very well the ChIP-seq results obtained with the full-length LFY protein strongly suggests that LFY DBD contains most of the DNA binding specificity.
Among the various methods available to build PSSMs, reiterative in vitro selection of binding sites followed by PCR (Selex) is particularly well suited: for TFs with large binding sites such as LFY, it is superior to the use of defined microarrays (Badis et al., 2009), which are limited in their complexity and cannot be reasonably used for binding sites larger than 11 nucleotides. Also, Selex allows the capture of important specificities that are not detected using ChIP experiments, such as the dependence between nucleotides. As illustrated in this and other studies (Figures 1 and 4; see Supplemental Figure 1 online), PSSMs (derived from Selex or ChIP experiments) are far superior to consensus sequences, which show poor predictive power and provide only binary information that cannot be incorporated into biophysical models.
To validate the in vitro–generated model, we performed a ChIP-seq experiment on seedlings constitutively expressing LFY. This experiment confirmed the quality of our model but is not sufficient to establish that all identified bound regions indeed correspond to genuine target genes. Still, many expected candidates, such as AP1, AG, or TFL1, have been identified with high confidence and the expression of several genes with bound regions changes in lfy mutants or plants overexpressing the LFY-GR inducible LFY protein (Wagner et al., 1999; Schmid et al., 2003). Combining the ChIP experiment with the biophysical model predictions allowed us to identify numerous previously unknown LFY binding sites that cannot be detected with the 7-bp consensus sequence (Figure 4; see Supplemental Figure 1 online). The good agreement observed in many cases between the location of these sites and the ChIP-seq peaks illustrates the capacity of our model to position the LFY binding sites correctly in genomic DNA sequence. Some cases remain where the model does not easily explain the in vivo LFY binding, suggesting that LFY might possess other modes of DNA binding (through contacts with another TF, for example).
We also used the LFY binding model to search the whole A. thaliana genome for high scoring binding sites or for regions with a high POcc (see Supplemental Table 3 online). Among the 100 highest-scoring sites in the genome, ~25% were found to be bound in ChIP-seq, and it is likely that this percentage would increase if the ChIP-seq experiments were performed with inflorescence tissues. This result further corroborates the unique performance of this model when applied to the whole genome. Lowering the score or the POcc threshold identifies numerous regions that were not bound in the ChIP-seq experiments (see Supplemental Table 3 online). A major cause for this discrepancy is probably the accessibility of DNA. As shown in other systems, the incorporation of DNA accessibility estimated from chromatin marks or nucleosome positioning is likely to improve the prediction of bound sites further (Whitington et al., 2009; Won et al., 2010).
The results we obtained in vitro and in A. thaliana plants demonstrate that our model is highly predictive and can be used to address evolutionary questions. We analyzed the relationship between LFY and one of its target genes (AG) in various species. We showed that the computation of the predicted occupancy (POcc), which integrates the influence of numerous binding sites over a large DNA region, enables us to predict the relationship between LFY and members of the AG subfamily solely based on genomic sequence analysis. The case of the grasses is particularly striking: in all species examined, the two AG paralogs show much higher POcc values than the AGL11 genes do (Figure 5). Based on the presence of one LFY consensus site in a single rice (Oryza sativa) AG paralog (Causier et al., 2008), it had been previously proposed that the regulation of AG by LFY could predate the divergence between monocots and dicots. We now confirm this hypothesis based on the analysis of eight AG genes from monocots. The power of the POcc computation is also illustrated in angiosperms: for all AG-like genes, we found a good agreement between expression during early flower meristem development (when LFY is active) and high POcc of the AG large intron by LFY. Our analysis could even differentiate between the functional homologs of A. thaliana AG in species such as A. majus or S. lycopersicum where a functional shift has occurred so that the SHP orthologs (PLE and TAGL1, respectively) participate in AG-like function.
In addition to the global analysis based on POcc computation, the examination of the distribution of individual LFY binding sites in AG introns also yielded interesting insights. In the Brassicaceae, the family to which A. thaliana belongs, a previous study analyzed the AG large second intron by phylogenetic shadowing, identifying several conserved regions (Hong et al., 2003). One of these regions included a conserved site (AG3; see Supplemental Figure 4 online) that exhibited the 7-bp consensus sequence CCANTG[G/T] and was therefore proposed to be a LFY binding site. We have now shown that it is not a bona fide LFY binding site (Figure 1). Conversely, our LFY PSSM identified a previously unrecognized site (AG5), for which we confirmed a high affinity of LFY in vitro (Figure 1). Neither this site nor the previously identified AG4 site (Hong et al., 2003) was bound in our ChIP experiment in seedlings, presumably because of their closed chromatin conformation: analysis of the H3K27 trimethylation repressive marks indeed has shown that in A. thaliana seedlings, only a short region encompassing the AG1 and AG2 sites is in open configuration (Zhang et al., 2007). Still, the presence in most Brassicaceae examined of the AG5 high-affinity site (with little sequence conservation of the site itself) (see Supplemental Figure 4 online), together with AG4 analysis in A. thaliana (Hong et al., 2003), strongly support their functional importance.
Comparing more distant species (Figure 6; see Supplemental Figures 2 and 3 online) revealed that the LFY/AG transcriptional link was conserved despite extensive variation in number, position, sequence, and affinity of individual binding sites. Several recent studies in animals have observed considerable variation in TF binding profiles between species. However, these differences do not seem to be systematically associated with changes in target gene expression (Odom et al., 2007; Wilson and Odom, 2009; Dowell, 2010; Kasowski et al., 2010; Weirauch and Hughes, 2010). A recent study examining TF binding in vertebrate genomes showed that conserved regulatory interactions do not increase sequence constraints (Schmidt et al., 2010). Therefore, cis-elements must be fluid; they can vary without necessarily compromising transcriptional regulation. This property represents an obstacle for approaches based on sequence conservation, such as genomic shadowing or phylogenetic footprinting (Wasserman and Sandelin, 2004). Our study shows that this fluidity also exists in plants but can be overcome using an integrative biophysical model, which detects regulatory interactions despite extensive cis-element plasticity.
As more plant genome sequences become available, it is essential to be able to derive functional information from direct examination of primary sequences. Our work illustrates the potential of biophysical models to predict regulatory interactions. Thanks to its relatively large binding site with high information content, LFY presents key advantages to pioneer such an approach. Nevertheless, it should be possible to generalize this type of analysis to other TFs provided that the PSSM have been established: biophysical models can easily incorporate cooperativity and competition between TFs and can be efficiently applied to combinations of TFs with smaller individual binding sites (Granek and Clarke, 2005). The case of heterodimeric TFs, such as MADS box factors, is obviously more complex: PSSMs could be derived from Selex procedures adapted to heterodimeric complexes or from ChIP experiments, but in the latter case, they would represent a mixture of the different complexes present in the tissue. Once successfully generalized to various types of TF, our strategy represents a powerful approach for both the functional annotation of genomes of nonmodel species and the prediction of regulatory network evolution directly from primary DNA sequences. It can be efficiently coupled to genome-wide expression data or comparison between species (Ward and Bussemaker, 2008; Yeo et al., 2009). In particular, it will be interesting to analyze genomic sequences from basal angiosperms, once available, to understand the origin of the regulation of A, B, and C genes by LFY, a central part of the network leading to the emergence and development of flowers (Theissen and Melzer, 2007; Moyroud et al., 2010).
METHODS
Plant Materials
Wild-type plants were of the Columbia-0 accession. 35S:LFY has been described before (Nilsson et al., 1998). Seedlings were grown under long-day photoperiods at 23°C on Murashige and Skoog plates.
Systematic Evolution of Ligands by Exponential Enrichment
Selection Cycles
In vitro selection of aptamers was performed with fluorescent 81-mers and a recombinant version of the DNA binding domain of Arabidopsis thaliana LFY protein (LFY DBD) produced and purified as previously described (Hamès et al., 2008)
Initially, a random sequence library was synthesized by PCR amplification (98°C for 1 min and 30 s followed by 20 cycles of 98°C for 10 s, 55°C for 25 s, and 72°C for 15 s) with Phusion DNA polymerase (Ozyme) using 81-mers [5′-TGGAGAAGAGGAGAGATCTAGC(N)30CTCTAGATCTTGTTCTTCTTCGATTCCGG-3′] as template with a fluorescent forward primer (SElex-F, TAMRA 5′-TGGAGAAGAGGAGAGATCTAG-3′) and a nonlabeled reverse primer (SElex-R, 5′-CCGGAATCGAAGAAGAACAA-3′) (Sigma-Aldrich). The size of the PCR products was verified on 3% agarose gels stained with SYBR Safe (Invitrogen), and double-stranded DNA (dsDNA) concentration was measured using SYBR green (Invitrogen) and a microplate reader (Safire2; TECAN) according to the manufacturer’s instructions.
For each selection cycle, 200 nM LFY-C was mixed to 10 nM fluorescent dsDNA (81-mers) in 225 μL Selex buffer (20 mM Tris, pH 8, 250 mM NaCl, 2 mM MgCl2, 5 mM TCEP, 10 μg/mL dIdC, and 1% glycerol). After a 2-min incubation on ice, 25 μL Ni Sepharose 6 fast flow (GE Healthcare), previously equilibrated in Selex buffer without TCEP, was added to the reaction mix to immobilize the DNA/protein complexes via the His tag of the protein. After 30 min incubation at 4°C on a rotating wheel, the reaction mix was loaded on an Ultrafree-MC centrifugal filter unit (Millipore) and centrifuged for 1 min at 500g at 4°C to eliminate the unbound DNA. Four washes were subsequently made by adding 300 μL of Selex buffer without dIdC on top of the filter unit followed by 1 min centrifugation at 500g at 4°C. Finally, the Ni Sepharose was resuspended in 100 μL water and transferred into a clean tube. Selected 81-mers were amplified by PCR as described above, using 2 μL of the Ni-Sepharose solution as template. PCR products were quantified as described before, and the selection cycle was repeated seven times, using each time the newly synthesized fluorescent DNA as a library.
The whole selection process has been performed twice independently.
Enrichment Evaluation
An electrophoretic mobility shift assay (Hamès et al., 2008) was used to estimate the enrichment for 81-mers with a high affinity for LFY DBD through the successive selection cycles: 10 nM 81-mers library of each cycle was incubated with 200 nM LFY DBD in 20 μL binding buffer. Electrophoresis and gel analysis was performed as described for QuMFRA assays, and libraries that gave a visible shift were selected for sequencing (cycles 3 to 7) using the 454 technology (Cogenics). More than 2500 sequences were obtained.
These sequences yielded 494 unique sequences, which were aligned with the MEME software version 4.3.0 (Bailey and Elkan, 1994) (http://meme.sdsc.edu/meme4_3_0/cgi-bin/meme.cgi) using the default parameters with either no constraints or with the symmetry imposed. This alignment was subsequently analyzed with the enoLOGOS software to identify dependence between nucleotides (Workman et al., 2005). For PSSM generation, frequencies of individual nucleotides and/or triplets were derived from the alignments and used to calculate, at each position i of the motif, the weight (W) associated to each nucleotide (or triplet) n according to: Wn,i = ln(fn,i/fmax,i), where fn,i is the frequency of nucleotide n at position i, and fmax,i is the maximal frequency observed at position i. When fn,i = 0, a pseudocount value (Wasserman and Sandelin, 2004) of 0.001 was applied.
QuMFRA Assay
QuMFRA assays were performed as described by Liu and Stormo (2005). Complementary single-stranded oligonucleotides were annealed in annealing buffer (10 mM Tris, pH 7.5, 150 mM NaCl, and 1 mM EDTA). The resulting dsDNA with a protruding G was fluorescently labeled by end-filling: 4 pmol of dsDNA was incubated with 1 unit of Klenow fragment polymerase (Ozyme) and 8 pmol Cy5-dCTP (GE Healthcare) (dsDNA samples) or Cy3-dCTP (dsDNA reference) in 1× Klenow buffer during 2 h at 37°C, followed by 10 min enzyme inactivation at 65°C. Sequences used as references or as samples are listed in Supplemental Table 4 online.
Binding reactions were performed in 20 μL binding buffer (20 mM Tris-HCl, pH 7.5, 150 mM NaCl, 1% glycerol, 0.25 mM EDTA, 2 mM MgCl2, 28 ng/mL fish sperm DNA [Roche], and 1 mM DTT) using 10 nM Cy3-dsDNA, 10 nM to 30 nM Cy5-dsDNA, and 500 nM or 1 μM LFY DBD. After 10 min incubation on ice, the binding reactions were loaded onto native 6% polyacrylamide gels and 0.5× TBE (45 mM Tris, 45 mM boric acid, and 1 mM EDTA, pH 8) and electrophoresed at 90 V for 90 min at 4°C.
Gels were scanned on a Typhoon 9400 scanner (Molecular Dynamics), and signals were quantified using ImageQuant software (Molecular Dynamics). Relative dissociation constants were calculated according to Man and Stormo (2001): for each gel lane, the fluorescent intensities of the bound and unbound fractions at both emission wavelengths were quantified and the background signal was subtracted. The resultant fluorescence intensities (FIcor) were used to calculate the relative dissociation constant (KDRel) given by Equation (1):
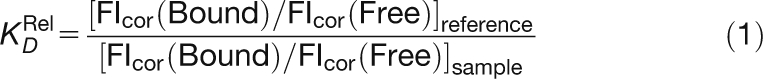 |
The relative dissociation constant of each dsDNA was measured at least three times independently, and the average value was used as KDRel for comparison to the scores.
Experimental scores from Figures 1C to 1E are defined as ln(KDRel/KDRelmax), with KDRelmax corresponding to KDRel of the dsDNA with the highest affinity for LFY DBD.
Cross-Linking, Chromatin Isolation, and ChIP-seq
The entire experiment from seed sowing through deep sequencing was performed twice to produce independent biological replicates. ChIP-seq (Yant et al., 2010) was performed with an antibody raised in rabbit (#4028) against the LFY C-terminal amino acids 223 to 424 (BioGenes). Briefly, 15-d-old 35S:LFY and Columbia-0 (control) seedlings were harvested and fixed as described previously (Gomez-Mena et al., 2005). Frozen tissue was ground, filtered three times through Miracloth (Calibrochem), and washed as described previously with buffers M1, M2, and M3 (Gomez-Mena et al., 2005). Nuclear pellets were resuspended in sonic buffer as described (1 mM PEFA BLOC SC [Roche Diagnostics] was substituted for PMSF), split into technical duplicate samples, and sonicated with a Branson sonifier at continuous pulse (output level 3) for eight rounds of 2 × 6 s and allowed to cool on ice between rounds. Immunoprecipitation reactions were performed by incubating chromatin with 2.5 μL anti-LFY serum overnight at 4°C as described (Gomez-Mena et al., 2005). The immunoprotein-chromatin complexes were captured by incubating with protein A-agarose beads (Santa Cruz Biotechnology), followed by consecutive washes in immunoprecipitation buffer and then elution as described (Gomez-Mena et al., 2005). Immunoprotein-DNA was then incubated consecutively in RNase A/T1 mix (Fermentas) and Proteinase K (Roche Diagnostics) as described after which DNA was purified using Minelute columns (Qiagen) (Gomez-Mena et al., 2005). ChIP samples were tested for enrichment by quantitative PCR, and deep sequencing libraries were produced by standard Illumina protocols.
ChIP-seq Analysis
Standard Illumina base calling software was used to base call the 40- to 42-nucleotide sequence reads. We used SHORE (Ossowski et al., 2008) as a platform for further analysis. The obtained reads were quality filtered, and low-quality bases at the 3′ end were pruned as described (Ossowski et al., 2008). GenomeMapper (Schneeberger et al., 2009) was used for mapping to the TAIR9 genome, allowing for up to four mismatching nucleotides and no gaps.
To proceed, the mapped data were subjected to a heuristic for removal of duplicate sequence reads, which were assumed to be uninformative for the detection of enriched loci. A threshold was applied limiting the number of 5′ ends mapping to the same position on the same strand. To retain the power to discriminate between multiple strongly enriched regions, the threshold for any particular position was varied depending on the coverage in close vicinity, such that the variance of the number of reads per position would roughly equal its mean in a 30-bp sliding window.
We further applied a two-step procedure to identify regions significantly enriched in the positive sample when compared with the control. First, potentially enriched regions were identified based on the positive samples only. These sites were then directly compared with the corresponding control sample regions to assess statistical significance.
For estimation of the depth of coverage for each position in the genome, all positive sample reads mapping to unique positions were extended in 3′ direction to 130 bp, corresponding to half the experimentally observed approximate DNA fragment size, while discarding all other reads. To detect possible peak sites, a 2-kb wide sliding window was applied to the coverage graph in single base steps. In each step a P value was assigned to the coverage value at the central base using a one-sided Poisson test, with the distribution parameter set to the average coverage within the sliding window. Only positions with coverage >0 were included in the calculation of the average, assuming all other positions to be inaccessible to the experiment. Finally, any consecutive stretch of positions with P value <0.05 and length >130 bp was retained as a potentially enriched site. To reduce further the number of regions to be considered, each was checked for unwarranted high average coverage in the control sample. A potential peak in the positive sample was discarded if the coverage mean in the control sample in the corresponding region was larger than the median average control coverage plus a tolerance of three standard deviations in all peak regions.
For assignment of final P values to each candidate region, in each replicate a one-sided binomial test was applied to the number of reads mapping to the region in the positive sample, with the distribution parameter N set to the joint read count for the site for the positive and the corresponding control samples. To estimate the probability parameter for the test, from now on called r, we computed a scaling factor s for the control sample and the chromosome containing the considered region. The complete chromosome sequence was subdivided into 400-bp bins, and for each bin, the positive sample and the control sample read counts were recorded. Then, s was chosen such that the median ChIP sample read count for all bins equaled the median control sample read count multiplied by s. From this the binomial test parameter, r was calculated as r = s/(s + 1).
Finally, FDRs were obtained through the Benjamini-Hochberg correction method. To establish a ranking of peak regions across replicates, the rank product over the per-replicate FDR ranks was used.
Biophysical Model for LFY-DNA Binding
We used POcc (Roider et al., 2007), defined as the expected number of bound TF molecules for a given TF matrix of length W and a DNA sequence of length L, as given by Equation (2), where KA,s is the relative equilibrium association constant for sites.
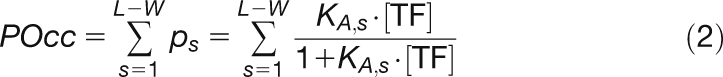 |
KA,s is the inverse of the relative equilibrium dissociation constant (1/KD,s) and was calculated thanks to the correlation curve in Figure 1, as given by Equation 3:
We found that a = 1.6349 and b = −3.9647 for the ASY PSSM, a = 1.8031 and b = 0.4133 for the SYM PSSM, and a = 2.5663 and b = 0.3598 for the SYM-T PSSM, and we used [TF] equal to the KD for the optimal site (score = 0), resulting in ps-opt = 0.5 (Granek and Clarke, 2005; Roider et al., 2007).
In the analyses presented in Figures 3 and 5, we used a variant of POcc in which only binding sites with a score higher than a threshold t = −23 are considered (Roider et al., 2007).
POcc was calculated for all peaks in ChIP experiment (~20,000). The correlation between ChIP and POcc ranking while using different PSSM was measured with the Spearman’s rank correlation coefficient. This is a nonparametric measure of statistical dependence between the two variables ChIP and POcc. First, the n raw values (ChIPi and Pocci) were converted to ranks (xi and yi). Second, the differences, di = xi − yi, between the ranks of each observation on the two variables were calculated. The Spearman’s rho (i.e., the correlation coefficient) was then given by Equation 4:
 |
Selection of Bound Peaks Set and Unbound Genomic Set
To perform ROC analysis, the bound DNA set was composed of all peaks with FDR < 0.1 in both ChIP experiments, resulting in 1564 peaks. The peaks were ranked using the rank product from both ChIP-seq replicates. The unbound set was generated by randomly selecting 1564 sequences from the A. thaliana genome that did not overlap with bound fragments and with the same size distribution as the bound set.
Data Processing
Various scripts in Python (www.python.org; v2.6.4) were written for automatic data processing, including PSSM score calculation, POcc determination, and ROC-AUC estimation.
Microarray Data Source
Microarray data was retrieved from Gene Expression Omnibus data sets (www.ncbi.nlm.nih.gov/geo): record GDS515 (William et al., 2004) and record GDS453 (Schmid et al., 2003). From GDS453, we used wild-type plants versus lfy12 floral transition microarrays at 0, 3, 5, and 7 d. From GDS515, we used dexamethasone versus mock treatment and dexamethasone+cycloheximide versus cycloheximide treatment in 35S:LFY-GR plants to select for potential direct targets of LFY. We selected all genes with a fold change higher than 2 in one of the conditions without attempting to calculate a statistical significance of this fold change.
The significance of the overlap between deregulated genes in the GDS515 microarray and the bound genes from the LEAFY ChIP-seq experiment was computed using a hypergeometric distribution, given by Equation 5:
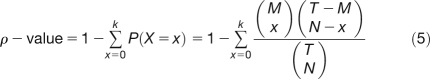 |
where M is the number of bound genes, N the number of deregulated genes in the microarray, T the total number of genes in the microarray, and k the number of genes that are both bound and deregulated. All computations were done using R software, and scripts are available upon request.
Genomic Sequence Retrieval and Analysis
For all species (except A. thaliana, Antirrhinum majus, Brachypodium distachyon, and Sorghum bicolor), the coding regions of previously identified members of the AG subfamily (see Supplemental Table 2 online for accession numbers) were retrieved from GenBank (http://www.ncbi.nlm.nih.gov) and used as BLAST queries against their respective species genome assembly to identify the corresponding genomic sequences. Coding sequences of members of the AG subfamily in Oryza sativa or Zea mays were blasted against the genomes of S. bicolor or B. distachyon to find the orthologs in these species. Plant genomes assemblies of A. thaliana, Arabidopsis lyrata, Populus trichocarpa, Carica papaya, Vitis vinifera, Prunus persica, Cucumis sativus, B. distachyon, O. sativa, S. bicolor, and Z. mays were browsed and queried at Phytozome v5.0 (http://www.phytozome.net). The S. lycopersicum genome assembly (v1.50) was browsed and queried at the Sol genomic network (http://solgenomics.net). The POcc values (t = −23) were then calculated on the longest intron of each gene, which corresponds to the first or the second intron depending on the gene. The accession numbers for the large intron of AG orthologs in Brassicaceae (Hong et al., 2003) can be found on Supplemental Table 5 online.
Intron sequences were aligned with DIALIGN software (Morgenstern, 2004), and a sliding-window analysis with a window size of 20 bp was used to estimate the mean divergence between sequences using the Jukes-Cantor model. The inverse of the mean divergence (mean conservation) is represented on Supplemental Figures 2 to 4 online.
Accession Numbers
All ChIP-seq data are freely available from the Gene Expression Omnibus database (http://www.ncbi.nlm.nih.gov/; accession number GSE24568). Sequence data from this article can be found in the Arabidopsis Genome Initiative or GenBank/EMBL databases under the following accession numbers: AY935269 (PLE), AY935268 (FARINELLI), AT4G18960 (AG), AT3G58780 (SHP1), AT2G42830 (SHP2), and AT4G09960 (STK). All other accession numbers are listed in Table 1 and Supplemental Tables 2 and 5 online.
Author Contributions
F.P., E.M., M.S., and L.Y. designed the experiments, and E.M., L.Y., D.P., S.B., and E.T. performed the experiments. E.G.M. and F.O. performed the data analysis with contributions from E.M., O.B., and M.M. The article was written by F.P., M.S, and D.W. with contributions from E.M. and L.Y.
Supplemental Data
The following materials are available in the online version of this article.
Supplemental Figure 1. Example of LFY-Bound Regions Identified by ChIP-seq and Analyzed for LFY Binding Sites.
Supplemental Figure 2. LFY Binding Sites in the Large Intron of AG Homologs in Eudicots.
Supplemental Figure 3. LFY Binding Sites in the Large Intron of AG Homologs in Monocots.
Supplemental Figure 4. LFY Binding Sites in the Second Intron of AG Homologs in Brassicaceae.
Supplemental Table 1. Position-Specific Scoring Matrix.
Supplemental Table 2. References for Data on the Expression of Genes of the AG Subfamily in Various Plant Species.
Supplemental Table 3. Predictions of LFY Binding at the Genomic Level.
Supplemental Table 4. Sequences of Oligonucleotides Used in LFY-DNA Interaction Studies.
Supplemental Table 5. Accession Numbers Corresponding to the Sequences of the Large Intron of AG Homologs in Brassicaceae Species.
Supplemental Data Set 1A. List of the 1564 Regions Bound by LFY in ChIP-seq Experiments.
Supplemental Data Set 1B. List of Genes (Upstream and Downstream) Adjacent to the 1564 Bound Regions in ChIP-seq Experiments.
Supplemental Data Set 1C. Overlap between Genes Bound by LFY in ChIP-seq and Genes Regulated by LFY.
Acknowledgments
We thank C. Scutt, P. Lemaire, R. Vincentelli, K. Nitta, and members of the Parcy and Schmid laboratories for discussion and A.K. Martin for help with bioinformatic analyses. This work was supported by funding from the Centre National de la Recherche Scientifique (ATIP+; F.P.), the Agence Nationale de la Recherche (ANR, Plant-TFcode; F.P.), the ANR and the Biotechnology and Biological Sciences Research Council (Flower Model; F.P.), and PhD fellowships from the University J. Fourier, Grenoble (E.M. and M.M.), FP7 Collaborative Project AENEAS (Contract KBBE-2009-226477; D.W.), ERA-NET Plant Genomics Project BLOOM-NET (SCHM 1560/7-1; M.S.), and the Max Planck Society (M.S. and D.W.).
References
- Badis G., et al. (2009). Diversity and complexity in DNA recognition by transcription factors. Science 324: 1720–1723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey T.L., Elkan C. (1994). Fitting a mixture model by expectation maximization to discover motifs in biopolymers. Proc. Int. Conf. Intell. Syst. Mol. Biol. 2: 28–36 [PubMed] [Google Scholar]
- Benos P.V., Bulyk M.L., Stormo G.D. (2002). Additivity in protein-DNA interactions: How good an approximation is it? Nucleic Acids Res. 30: 4442–4451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bomblies K., Wang R.L., Ambrose B.A., Schmidt R.J., Meeley R.B., Doebley J. (2003). Duplicate FLORICAULA/LEAFY homologs zfl1 and zfl2 control inflorescence architecture and flower patterning in maize. Development 130: 2385–2395 [DOI] [PubMed] [Google Scholar]
- Busch M.A., Bomblies K., Weigel D. (1999). Activation of a floral homeotic gene in Arabidopsis. Science 285: 585–587 [DOI] [PubMed] [Google Scholar]
- Causier B., Bradley D., Cook H., Davies B. (2008). Conserved intragenic elements were critical for the evolution of the floral C-function. Plant J. 1: 41–52 [DOI] [PubMed] [Google Scholar]
- Causier B., Castillo R., Zhou J., Ingram R., Xue Y., Schwarz-Sommer Z., Davies B. (2005). Evolution in action: Following function in duplicated floral homeotic genes. Curr. Biol. 15: 1508–1512 [DOI] [PubMed] [Google Scholar]
- Colombo M., Brambilla V., Marcheselli R., Caporali E., Kater M.M., Colombo L. (2010). A new role for the SHATTERPROOF genes during Arabidopsis gynoecium development. Dev. Biol. 337: 294–302 [DOI] [PubMed] [Google Scholar]
- Davies B., Motte P., Keck E., Saedler H., Sommer H., Schwarz-Sommer Z. (1999). PLENA and FARINELLI: Redundancy and regulatory interactions between two Antirrhinum MADS-box factors controlling flower development. EMBO J. 18: 4023–4034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dowell R.D. (2010). Transcription factor binding variation in the evolution of gene regulation. Trends Genet. 26: 468–475 [DOI] [PubMed] [Google Scholar]
- Ferrario S., Immink R.G., Angenent G.C. (2004). Conservation and diversity in flower land. Curr. Opin. Plant Biol. 7: 84–91 [DOI] [PubMed] [Google Scholar]
- Gan Y., Kumimoto R., Liu C., Ratcliffe O., Yu H., Broun P. (2006). GLABROUS INFLORESCENCE STEMS modulates the regulation by gibberellins of epidermal differentiation and shoot maturation in Arabidopsis. Plant Cell 18: 1383–1395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez-Mena C., de Folter S., Costa M.M., Angenent G.C., Sablowski R. (2005). Transcriptional program controlled by the floral homeotic gene AGAMOUS during early organogenesis. Development 132: 429–438 [DOI] [PubMed] [Google Scholar]
- Granek J.A., Clarke N.D. (2005). Explicit equilibrium modeling of transcription-factor binding and gene regulation. Genome Biol. 6: R87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamès C., Ptchelkine D., Grimm C., Thevenon E., Moyroud E., Gérard F., Martiel J.L., Benlloch R., Parcy F., Müller C.W. (2008). Structural basis for LEAFY floral switch function and similarity with helix-turn-helix proteins. EMBO J. 27: 2628–2637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanley J.A., McNeil B.J. (1982). The meaning and use of the area under a receiver operating characteristic (ROC) curve. Radiology 143: 29–36 [DOI] [PubMed] [Google Scholar]
- Hong R.L., Hamaguchi L., Busch M.A., Weigel D. (2003). Regulatory elements of the floral homeotic gene AGAMOUS identified by phylogenetic footprinting and shadowing. Plant Cell 15: 1296–1309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasowski M., et al. (2010). Variation in transcription factor binding among humans. Science 328: 232–235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufmann K., Wellmer F., Muiño J.M., Ferrier T., Wuest S.E., Kumar V., Serrano-Mislata A., Madueño F., Krajewski P., Meyerowitz E.M., Angenent G.C., Riechmann J.L. (2010). Orchestration of floral initiation by APETALA1. Science 328: 85–89 [DOI] [PubMed] [Google Scholar]
- Kramer E.M., Jaramillo M.A., Di Stilio V.S. (2004). Patterns of gene duplication and functional evolution during the diversification of the AGAMOUS subfamily of MADS box genes in angiosperms. Genetics 166: 1011–1023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamb R.S., Hill T.A., Tan Q.K., Irish V.F. (2002). Regulation of APETALA3 floral homeotic gene expression by meristem identity genes. Development 129: 2079–2086 [DOI] [PubMed] [Google Scholar]
- Liljegren S.J., Ditta G.S., Eshed Y., Savidge B., Bowman J.L., Yanofsky M.F. (2000). SHATTERPROOF MADS-box genes control seed dispersal in Arabidopsis. Nature 404: 766–770 [DOI] [PubMed] [Google Scholar]
- Liljegren S.J., Gustafson-Brown C., Pinyopich A., Ditta G.S., Yanofsky M.F. (1999). Interactions among APETALA1, LEAFY, and TERMINAL FLOWER1 specify meristem fate. Plant Cell 11: 1007–1018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C., Thong Z., Yu H. (2009). Coming into bloom: The specification of floral meristems. Development 136: 3379–3391 [DOI] [PubMed] [Google Scholar]
- Liu J., Stormo G.D. (2005). Combining SELEX with quantitative assays to rapidly obtain accurate models of protein-DNA interactions. Nucleic Acids Res. 33: e141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohmann J.U., Hong R.L., Hobe M., Busch M.A., Parcy F., Simon R., Weigel D. (2001). A molecular link between stem cell regulation and floral patterning in Arabidopsis. Cell 105: 793–803 [DOI] [PubMed] [Google Scholar]
- Man T.K., Stormo G.D. (2001). Non-independence of Mnt repressor-operator interaction determined by a new quantitative multiple fluorescence relative affinity (QuMFRA) assay. Nucleic Acids Res. 29: 2471–2478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikkelsen T.S., Xu Z., Zhang X., Wang L., Gimble J.M., Lander E.S., Rosen E.D. (2010). Comparative epigenomic analysis of murine and human adipogenesis. Cell 143: 156–169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgenstern B. (2004). DIALIGN: Multiple DNA and protein sequence alignment at BiBiServ. Nucleic Acids Res. 32(Web Server issue): W33–W36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moyroud E., Kusters E., Monniaux M., Koes R., Parcy F. (2010). LEAFY blossoms. Trends Plant Sci. 15: 346–352 [DOI] [PubMed] [Google Scholar]
- Moyroud E., Tichtinsky G., Parcy F. (2009). The LEAFY floral regulators in Angiosperms: Conserved proteins with diverse roles. J. Plant Biol. 52: 177–185 [Google Scholar]
- Nilsson O., Lee I., Blázquez M.A., Weigel D. (1998). Flowering-time genes modulate the response to LEAFY activity. Genetics 150: 403–410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Odom D.T., Dowell R.D., Jacobsen E.S., Gordon W., Danford T.W., MacIsaac K.D., Rolfe P.A., Conboy C.M., Gifford D.K., Fraenkel E. (2007). Tissue-specific transcriptional regulation has diverged significantly between human and mouse. Nat. Genet. 39: 730–732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ossowski S., Schneeberger K., Clark R.M., Lanz C., Warthmann N., Weigel D. (2008). Sequencing of natural strains of Arabidopsis thaliana with short reads. Genome Res. 18: 2024–2033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parcy F., Bomblies K., Weigel D. (2002). Interaction of LEAFY, AGAMOUS and TERMINAL FLOWER1 in maintaining floral meristem identity in Arabidopsis. Development 129: 2519–2527 [DOI] [PubMed] [Google Scholar]
- Parcy F., Nilsson O., Busch M.A., Lee I., Weigel D. (1998). A genetic framework for floral patterning. Nature 395: 561–566 [DOI] [PubMed] [Google Scholar]
- Ratcliffe O.J., Bradley D.J., Coen E.S. (1999). Separation of shoot and floral identity in Arabidopsis. Development 126: 1109–1120 [DOI] [PubMed] [Google Scholar]
- Roider H.G., Kanhere A., Manke T., Vingron M. (2007). Predicting transcription factor affinities to DNA from a biophysical model. Bioinformatics 23: 134–141 [DOI] [PubMed] [Google Scholar]
- Schmid M., Uhlenhaut N.H., Godard F., Demar M., Bressan R., Weigel D., Lohmann J.U. (2003). Dissection of floral induction pathways using global expression analysis. Development 130: 6001–6012 [DOI] [PubMed] [Google Scholar]
- Schmidt D., et al. (2010). Five-vertebrate ChIP-seq reveals the evolutionary dynamics of transcription factor binding. Science 328: 1036–1040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneeberger K., Hagmann J., Ossowski S., Warthmann N., Gesing S., Kohlbacher O., Weigel D. (2009). Simultaneous alignment of short reads against multiple genomes. Genome Biol. 10: R98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sieburth L.E., Meyerowitz E.M. (1997). Molecular dissection of the AGAMOUS control region shows that cis elements for spatial regulation are located intragenically. Plant Cell 9: 355–365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theissen G., Melzer R. (2007). Molecular mechanisms underlying origin and diversification of the angiosperm flower. Ann. Bot. (Lond.) 100: 603–619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson B.E., Hake S. (2009). Translational biology: From Arabidopsis flowers to grass inflorescence architecture. Plant Physiol. 149: 38–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner D., Sablowski R.W., Meyerowitz E.M. (1999). Transcriptional activation of APETALA1 by LEAFY. Science 285: 582–584 [DOI] [PubMed] [Google Scholar]
- Ward L.D., Bussemaker H.J. (2008). Predicting functional transcription factor binding through alignment-free and affinity-based analysis of orthologous promoter sequences. Bioinformatics 24: i165–i171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wasserman W.W., Sandelin A. (2004). Applied bioinformatics for the identification of regulatory elements. Nat. Rev. Genet. 5: 276–287 [DOI] [PubMed] [Google Scholar]
- Weirauch M.T., Hughes T.R. (2010). Conserved expression without conserved regulatory sequence: The more things change, the more they stay the same. Trends Genet. 26: 66–74 [DOI] [PubMed] [Google Scholar]
- Whitington T., Perkins A.C., Bailey T.L. (2009). High-throughput chromatin information enables accurate tissue-specific prediction of transcription factor binding sites. Nucleic Acids Res. 37: 14–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- William D.A., Su Y., Smith M.R., Lu M., Baldwin D.A., Wagner D. (2004). Genomic identification of direct target genes of LEAFY. Proc. Natl. Acad. Sci. USA 101: 1775–1780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson M.D., Odom D.T. (2009). Evolution of transcriptional control in mammals. Curr. Opin. Genet. Dev. 19: 579–585 [DOI] [PubMed] [Google Scholar]
- Won K.J., Ren B., Wang W. (2010). Genome-wide prediction of transcription factor binding sites using an integrated model. Genome Biol. 11: R7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Workman C.T., Yin Y., Corcoran D.L., Ideker T., Stormo G.D., Benos P.V. (2005). enoLOGOS: A versatile web tool for energy normalized sequence logos. Nucleic Acids Res. 33(Web Server issue): W389–W392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yant L., Mathieu J., Dinh T.T., Ott F., Lanz C., Wollmann H., Chen X., Schmid M. (2010). Orchestration of the floral transition and floral development in Arabidopsis by the bifunctional transcription factor APETALA2. Plant Cell 22: 2156–2170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeo Z.X., Yeo H.C., Yeo J.K., Yeo A.L., Li Y., Clarke N.D. (2009). Inferring transcription factor targets from gene expression changes and predicted promoter occupancy. J. Comput. Biol. 16: 357–368 [DOI] [PubMed] [Google Scholar]
- Zahn L.M., Leebens-Mack J.H., Arrington J.M., Hu Y., Landherr L.L., dePamphilis C.W., Becker A., Theissen G., Ma H. (2006). Conservation and divergence in the AGAMOUS subfamily of MADS-box genes: Evidence of independent sub- and neofunctionalization events. Evol. Dev. 8: 30–45 [DOI] [PubMed] [Google Scholar]
- Zhang X., Clarenz O., Cokus S., Bernatavichute Y.V., Pellegrini M., Goodrich J., Jacobsen S.E. (2007). Whole-genome analysis of histone H3 lysine 27 trimethylation in Arabidopsis. PLoS Biol. 5: e129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y., Granas D., Stormo G.D. (2009). Inferring binding energies from selected binding sites. PLoS Comput. Biol. 5: e1000590. [DOI] [PMC free article] [PubMed] [Google Scholar]