

Abstract
It is well recognized that voltage-gated calcium (Ca2+) channels modulate the function of peripheral and central pain pathways by influencing fast synaptic transmission and neuronal excitability. In the past, attention focused on the modulation of different subtypes of high-voltage-activated-type Ca2+ channels; more recently, the function of low-voltage-activated or transient (T)-type Ca2+ channels (T-channels) in nociception has been well documented. Currently, available pain therapies remain insufficient for certain forms of pain associated with chronic disorders (e.g. neuropathic pain) and often have serious side effects. Hence, the identification of selective and potent inhibitors and modulators of neuronal T-channels may help greatly in the development of safer, more effective pain therapies. Here, we summarize the available information implicating peripheral and central T-channels in nociception. We also discuss possible future developments aimed at selective modulation of function of these channels, which are highly expressed in nociceptors.
Keywords: low-threshold Ca2+ channel, nociception, pain killers, dorsal root ganglion, dorsal horn, thalamus, diabetes
The function of T-channels in the pain pathway
Pain is often a result of the activation of neurons (nociceptors) that can sense damaging stimuli by acute events subsequent to the injury of peripheral tissue such as the skin and internal organs. This form of pain, called nociceptive pain, is self-protective and typically evokes actions aimed at limiting or avoiding further tissue damage. In contrast, neuropathic pain, which results from primary dysfunction of peripheral nociceptive, as well as non-nociceptive nerves or central nervous system (‘central pain’), typically outlasts the initial stage of injury and frequently leads to debilitating disorders that are not amenable to conventionally available drug therapies. Many in vitro and in vivo studies have identified the functions of diverse subtypes of voltage-gated Ca2+ channels (VGCCs) in both peripheral and central sensory pathways to nociception. These channels can contribute to the development of both nociceptive and neuropathic pain. In this review, we summarize the most recent evidence linking T-type Ca2+ channels (T-channels) to central and peripheral pain processing. We also discuss the potential development of pain therapies aimed at these channels, which are abundantly expressed in nociceptors.
Molecular pharmacology of T-channels
Based on the membrane potential at which they gate ion currents, VGCCs are classified as high-voltage activated (HVA) or sustained currents, and low-voltage activated (LVA) or transient currents (T-type) (Catterall, 2000). Based on their sensitivity to pharmacological agents, HVA channels are further classified into at least five subtypes: L-, N-, P-, Q- and R-. While HVA calcium channels function in fast synaptic transmission in the central nervous system (CNS) (Miller, 1998; Catterall, 2000), it is thought that T-currents have a unique function in neuronal excitability (Llinas, 1988; Huguenard, 1996; Perez-Reyes, 2003). This notion is based on the unique ability of T-currents to become activated after a small depolarization of the cell membrane, which allows them to function at near-resting membrane potentials. Neuronal T-channels have been shown to promote Ca2+-dependent burst firing and low-amplitude intrinsic neuronal oscillations, as well as Ca2+ entry and amplification of weak dendritic synaptic signals in a myriad of CNS neurons. Moreover, the function of T-channels can be altered by pathological conditions such as absence epilepsy, which, in turn, can decrease the threshold for the initiation of seizure activity (Kim et al., 2001; Song et al., 2004; Zhang et al., 2004). Cloning of pore-forming α1 subunits of T-channels has demonstrated the existence of at least three subtypes, α1G (Cav3.1) (Perez-Reyes et al., 1998), α1H (Cav3.2) (Cribbs et al., 1998) and α1I (Cav3.3) (Lee et al., 1999a). Post-translational modifications such as alternative splicing of all three genes encoding the α1 subunit of CaV3 can occur in tissues (Monteil et al., 2000; Chemin et al., 2001b; Murbartián et al., 2002; 2004; Latour et al., 2004; Emerick et al., 2006; Zhong et al., 2006), further contributing to the diversity of native T-currents. Indeed, such modifications may underlie the biophysical and pharmacological heterogeneity of T-currents previously described in native neuronal, cardiac and endocrine cells (Herrington and Lingle, 1992; Todorovic and Lingle, 1998).
In electrophysiological experiments, T-currents, because of their unique biophysical properties, can be relatively easy identified; that is, these currents have characteristically slow voltage-dependent activation and almost complete inactivation, which typically generate a criss-crossing pattern when the family of currents is generated at progressively more positive voltages (Figure 1A). In addition, T-currents have several-fold slower deactivation (transition from open to closed states) than any HVA subtype does (Figure 1B). However, in spite of the progress made in recent years, pharmacological tools for the study of T-currents are still very limited (see also other reviews by McGivern, 2006 and Lory and Chemin, 2007). While there are many natural toxins or venom components that could be used to study the multiple HVA currents, only recently have substances been identified that can selectively and potently block T-currents. For example, a scorpion toxin, kurtoxin, which is a potent submicromolar blocker and gating modifier of cloned T-currents, also, with similar potency, affects voltage-gated sodium currents (Chuang et al., 1998) and native HVA Ca2+ currents (Sidach and Mintz, 2002). Traditional compounds previously used to investigate T-channels, such as ethosuximide (Coulter et al., 1989) and amiloride (Tang et al., 1988), can block T-currents, but only at fairly high concentrations [inhibitory concentrations that block 50% of the maximal current (IC50s) in excess of 100 µM]. At such concentrations, these compounds are also likely to block other ion channels, thus limiting their value for determining the function of T-channels both in vitro and in vivo.
Figure 1.
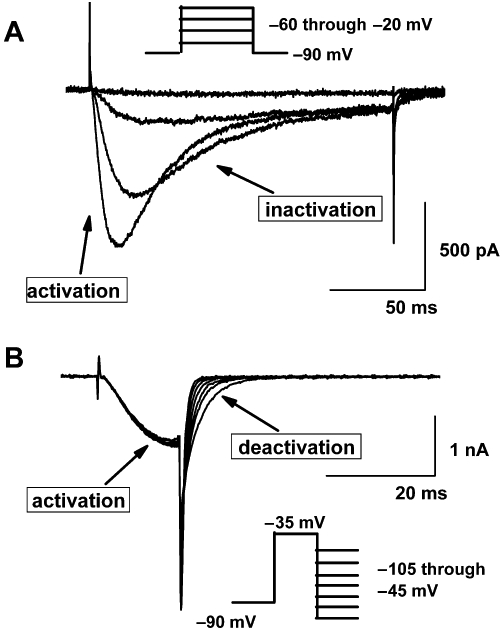
(A) A family of inward T-currents evoked in small, acutely dissociated dorsal root ganglion (DRG) cells from holding potential of −90 mV with 200 ms-long depolarizing steps of −60 mV through −40 mV. Note that current waveforms at more positive potentials exhibit faster activation and inactivation kinetics as evidence by ‘criss-crossing’ pattern. (B) Typical slow deactivation of tail currents in another small DRG cell generated with a short 15 ms-long depolarizing steps from −90 mV to −35 mV, followed by incremental hyperpolarizing steps of 10 mV starting from −105 mV through −45 mV. Insets on both panels show voltage stimulation protocol used to generate these currents.
Mibefradil, marketed by Hofmann-La Roche (Basel, Switzerland), is a peripherally acting antihypertensive drug. Even at low micro- and nanomolar concentrations, mibrefradil has been shown preferentially to block T-channels over HVA channels in vascular smooth muscle (Ertel et al., 1997) and cerebellar Purkinje cells (McDonough and Bean, 1998) and thus, for a while, was considered to be a promising selective T-channel blocker. Further studies have shown that mibefradil exhibits voltage-dependent inhibition of T-currents in acutely dissociated small dorsal root ganglia (DRG) cells in vitro by stabilizing the channel into inactivated states (Todorovic and Lingle, 1998). This is important since binding to inactivated states is an important property of drugs that modulate ion channels as it can provide selectivity to their action, and it implies that drugs preferentially affects actively-firing neurons. Consistent with this, mibefradil has moderate analgesic properties in healthy rats (Todorovic et al., 2001a) but completely reverses symptoms of neuropathic pain from chronic constrictive injury (CCI) of sciatic nerve in rats in vivo (Dogrul et al., 2003). Particularly exciting was the fact that the drug was marketed in Europe as an antihypertensive agent and was well tolerated, presumably due to poor penetration into the CNS. Unfortunately, it was soon withdrawn from the market due to drug–drug interactions. Moreover, studies have since indicated that this molecule blocks not only L-type HVA-type Ca2+ currents, but also other voltage-gated currents (e.g. INa+ and IK+) at low µM concentrations, thus casting doubt on its selectivity and usefulness in selectively studying T-channels (Jimenez et al., 2000; McNulty and Hanck, 2004; Coste et al., 2007). However, an interesting compound (NNC 55-0396) was recently generated by modifying the molecule of mibefradil that preserves its potency in blocking T-channels while greatly diminishing its potency on HVA currents (Huang et al., 2004). Towards this end, R(-) efonidipine has been shown to exhibit greater selectivity for T-type versus HVA currents in hippocampal neurons, although with more than 10-fold lower potency when compared with mibefradil (Shin et al., 2008). Thus, it is still possible that more potent and selective peripherally acting analogues of mibefradil will be developed and will prove useful not only in studies of the function of T-channels, but in the development of clinical analgesics.
Divalent cations such as nickel (Ni2+), at concentrations up to 30 µM (Lee et al., 1999b), as well as low micromolar concentrations of zinc (Zn2+) and copper (Cu2+), preferentially inhibit the CaV3.2 isoform of T-channels in preference to other T-channel isoforms and HVA channels (Todorovic and Lingle, 1998; Nelson et al., 2007a,b; Traboulsie et al., 2007). This is particularly important, given that metal ions such as Zn2+ and Cu2+ may be endogenously present in neural tissue and may act as natural modulators of T-channel function. Some agents used in human medicine are particularly interesting in that, at clinically relevant concentrations, they may alter the function of T-channels. Nitrous oxide (N2O; laughing gas) is a potent analgesic but a weak anaesthetic; at clinically relevant concentrations up to 80%, it selectively inhibits T-type over HVA-type Ca2+ currents in acutely dissociated DRG neurons (Todorovic et al., 2001b). Like Ni2+, N2O selectively blocks the α1H (CaV3.2), but not the α1G (CaV3.1) or α1I (CaV3.3) isoforms of T-channels (Todorovic et al., 2001b; Joksovic et al., 2005). In contrast, recent study showed that N2O may also inhibit in smaller extent recombinant CaV3.1 currents (Bartels et al., 2009). The apparent reason for inconsistency with the above mentioned study is not obvious. However, it is possible that different methods of application of N2O in these studies contributed to this discrepancy. However, it is well established at these concentrations N2O also blocks the neuronal N-methyl-D-aspartate (NMDA) subtype of glutamate receptors (Jevtović-Todorovićet al., 1998) and activates two-pore domain background potassium conductance (Gruss et al., 2004).
Neuroleptics are another major group of clinically used drugs that potently block T-channels at the same concentrations as those used for other cellular targets such as D2 dopamine receptors. Santi et al. (2002) showed that two neuroleptics, diphenylbutylpiperidine pimozide and penfluoridol, are particularly potent blockers of recombinant T-channels, with estimated IC50 values of about 30–50 nM and 70–100 nM, respectively.
Some neuroactive steroids with a 5α configuration at the steroid A, B ring fusion, could be promising pharmacological agents for the study of T-channels. For example, (+)-ECN [(3β, 5α, 17β)-17-hydroxyestrane-3-carbonitrile] is a potent, albeit partial voltage-dependent blocker of T-channels in acutely dissociated rat sensory neurons (IC50 of 300 nM). It has no significant effect on other voltage-gated channels (e.g. Na+, K+ or HVA-type Ca2+ channels) or ligand-gated ion channels [e.g. glutamate and gamma amino-butyric acid (GABA)-gated channels; Todorovic et al., 1998]. The selective pharmacological actions of ECN and related structural steroid analogues provide a unique opportunity to test their role in peripheral somatic pain transmission in vivo. When peripheral thermal nociception was studied by locally injecting a steroid into the peripheral receptive fields of a rat hind paw, it was found that the peripheral analgesic effects of 5α-reduced neuroactive steroids depended on their T-channel-blocking potential (Pathirathna et al., 2005a). For example, structural modifications, which eliminated T-channel blocking capacity, resulted in almost the complete loss of steroid-induced analgesic activity in vivo. On the other hand, ECN was the most potent and enantioselective peripheral analgesic in the thermal pain paradigm.
Another addition to the potentially useful T-channel-selective blockers is a novel neuroactive steroid with a 5β configuration at the steroid A, B ring fusion, [(3β, 5β, 17β)-3-hydroxyandrostane-17-carbonitrile] (3βOH), which is an effective voltage-dependent blocker of T-currents in acutely isolated DRG cells (with an IC50 of 2.8 µM at a holding potential of −90 mV and IC50 0.8 µM at holding potentials of −70 mV). However, this steroid has little effect on HVA-type Ca2+ voltage-gated Na+ and K+ channels or GABAA-gated channels at concentrations from 1 to 10 µM (Todorovic et al., 2004a). To evaluate the ability of 5β-reduced neuroactive steroids to induce analgesia in vivo, we injected various doses of each of these steroids into the peripheral receptive fields of sensory neurons. We found that 5β-reduced neuroactive steroids induce potent dose- and time-dependent anti-nociceptive responses in rats. These responses correlated closely with the ability of the steroids to block T-currents in acutely dissociated rat sensory neurons.
In further search for T-channel selective compounds, Merck and Co (Whitehouse Station, NJ, USA) has synthesized and examined the in vitro effects of 3,5-dichloro-N-[1-(2,2-dimethyl-tetrahydro-pyran-4-ylmethyl)-4-fluoro-piperidin-4-ylmethyl]-benzamide (TTA-P2) and related derivatives of 4-aminomethyl-4-fluoropiperdine on T-currents in recombinant cells (Shipe et al., 2008). It was found that these compounds are highly selective, state-independent and potent blockers of all recombinant T-channel isoforms (IC50s ranging from 20 to 100 nM), which makes them some of the best new tools available for the study of T-channels. However, the potency and selectivity of these drugs in native neurons, including those in pain pathways, have not been well studied. Of note, a related compound and state-dependent blocker of T-channels TTA-A2 [2-(4-Cyclopropylphenyl)-N-((1R)-1-{5-[(2,2,2-trifluoroethyl)oxo]-pyridin-2-yl}ethyl)acetamide], when given systemically, suppressed in vivo active waking and promoted slow-wave sleep in wild-type mice, but not mice with double knockout of CaV3.1 and CaV3.3 (Kraus et al., 2010). Thus, CNS side effects of pan-T-channel blockers that promote sedation may impede the use of these drugs as clinical analgesics.
Modulation of T-type channels
Recent evidence indicates that T-channels may be potently modulated by various classes of endogenous and experimental agents.
Redox modulation
T-channels in peripheral sensory or DRG neurons can be selectively and potently modulated by various reducing and oxidizing agents, including the endogenous sulphur-containing amino acid L-cysteine. Recent studies have shown that T-currents in acutely dissociated sensory neurons are significantly enhanced when T-channels are in a reduced state. However, these currents are largely inhibited when T-channels are in an oxidized state, strongly suggesting the importance of putative redox-sensitive sites on peripheral T-type channels (Todorovic et al., 2001a). This, in turn, can profoundly affect cellular excitability by enhancing Ca2+ influx during action potential of at least three different subtypes of nociceptors (Nelson et al., 2005; 2007b; Jagodic et al., 2007); it can also affect the excitability of cells of the reticular thalamic nucleus (Joksovic et al., 2006). A computational modelling study has demonstrated that an increase in the cellular excitability of putative nociceptive ‘T-rich’ cells in DRGs can be achieved by hyperpolarizing gating shifts in T-channels by L-cysteine (Nelson et al., 2005). Thus, both an increase in the amplitude of T-current and alterations in the gating parameters of T-channels in nociceptors may increase the probability of spike firing. Further studies using Markov-state models have shown that L-cysteine induces gating alterations of T-currents in nociceptors by increasing the transition rate from resting to open, and decreasing the transition rate from open to inactivated states (Nelson et al., 2010).
L-cysteine induces potent hyperalgesia in rats when injected into the peripheral receptive fields of sensory neurons in vivo (Todorovic et al., 2001a). This was recently confirmed by the demonstration that hyperlagesia induced by local peripheral injections of L-cysteine is present only in wild-type mice, not CaV3.2-knock-out mice (Nelson et al., 2007b). Furthermore, molecular mutagenesis studies have shown that histidine 191 in repeat I of CaV3.2 is critical for the action of reducing agents that chelate off endogenously bound divalent metal ions (e.g. Zn2+), which appears to exert tonic inhibition of T-channels both in vitro and in vivo (Nelson et al., 2007b; Kang et al., 2010). A related phenomenon was recently described for gaseous transmitter hydrogen sulfide (H2S), an endogenous compound formed from L-cysteine. It was found that H2S facilitates CaV3.2 currents in vitro, probably via chelation of zinc, consequently leading to both peripheral and central sensitization of acute somatic (Kawabata et al., 2007; Maeda et al., 2009) and visceral (Matsunami et al., 2009; Nishimura et al., 2009) nociception, as well as maintenance of chronic neuropathic pain (Takahashi et al., 2010). Thus, reducing agents and other endogenous metal chelators present in the body (e.g. albumin, histidine, H2S) may serve as natural agonists for the CaV3.2 T-channels.
Surprisingly, further experiments with H191 demonstrated that this site is crucial for the inhibitory modulation of peripheral and central T-channels by another endogenous reducing agent, L-ascorbic acid (ascorbate, vitamin C). Ascorbate, a ubiquitous redox agent, may act as both an antioxidant and a prooxidant. At nanomolar concentrations (IC50 10 nM for human and 25 nM for rat channels), ascorbate selectively inhibited recombinant CaV3.2-based T-currents but, even at 1000-fold higher concentrations, had no significant effect on CaV3.1 or CaV3.3 T-current (Nelson et al., 2007b). At physiologically relevant concentrations, ascorbate selectively inhibited native DRG and thalamic T-currents and diminished T-channel-dependent burst firing in reticular thalamic cells. This highly subtype-specific effect was achieved via metal-catalysed oxidation of critical metal-binding and CaV3.2-unique H191 residue. Furthermore, using recombinant CaV3.2 channels and CaV3.2 knockout-mice, it was recently demonstrated that N2O-induced analgesia in mice formalin test is likely mediated by metal-catalysed oxidation of H191 of CaV3.2 (Orestes et al., 2010). This strongly suggests that the development of novel inhibitors of T-channels may focus on the H191 of repeat I of T-channels.
Phosphorylation and G-protein modulation
Recent evidence from experiments involving recombinant T-channels indicates that G-protein β−γ subunits selectively inhibit the function of CaV3.2 channels by interacting with the intracellular loop connecting domain II and II of CaV3.2 (Wolfe et al., 2003) and by decreasing single-channel open probability without affecting voltage-dependent kinetics of the channel (DePuy et al., 2006). In contrast, corticotrophin-releasing factor interacts with G-protein β−γ subunits to selectively inhibit CaV3.2 channels by causing hyperpolarizing shift in half-inactivation potential (Tao et al., 2008). Furthermore, activation of Gαq/11-coupled muscarinic acetylcholine receptors leads to their interaction with domains II and IV of human CaV3.3, selectively inhibiting CaV3.3 (Hildebrand et al., 2007).
It recently has been proposed that a form of modulation involving phosphorylation reactions may modify native and recombinant T-currents, thus having an important effect on the function of T-channels. For example, recombinant CaV3.2 T-channels are stimulated by Ca2+/calmodulin-dependent kinase II (Wolfe et al., 2002) via phosphorylation of a critical Serine1198 residue in the II-III intracellular loop of CaV3.2 (Welsby et al., 2003) and modulated by protein kinase A (Chemin et al., 2007b), by phosphorylation of the Serine1107 residue on the II-III intracellular loop of CaV3.2 (Hu et al., 2009). Furthermore, all three isoforms of recombinant T-channels can be stimulated by protein kinase C (PKC) (Park et al., 2006; Chemin et al., 2007b; Joksovic et al., 2010), presumably by phosphorylation reactions, although the specific amino acid residues targeted in the channel have not yet been identified. Interestingly, stimulation of PKC with phorbol esters inhibits T-currents in small DRG cells in culture (Schroeder et al., 1990), and thalamo-cortical (TC) cells in intact brain slices (Cheong et al., 2008). Iftinca et al. (2007) have found that different recombinant and native T-currents may be modulated by phosphorylation reactions involving Rho-associated kinase (ROCK). Notably, this was associated with the up-regulation of T-currents in small DRG cells and a depolarizing shift in voltage-dependent activation and inactivation. In this study, site-directed mutagenesis demonstrated that the ROCK-mediated effects involve two distinct phosphorylation consensus sites consisting of serines and threonines in the domain II-III intracellular linker. However, the physiological relevance of G-proteins and phosphorylation reactions that may modify native T-currents and their contribution to the function of the cells expressing these channels has not been conclusively demonstrated. It is tempting, nonetheless, to speculate that posttranslational modifications of T-channels in the pain pathway by phosphorylation reactions or G-protein-mediated modulation modulates pain signalling, as this has been demonstrated for other nociceptive ion channels, including HVA Ca2+ channels (reviewed in Bhave and Gereau, 2004).
Endocannabinoids and arachidonic acid
Local release and accumulation of arachidonic acid metabolites in the peripheral tissues may be involved in direct sensitization of peripheral nociceptors during inflammatory reactions. Interestingly, arachidonic acid and related compounds have been shown to modulate recombinant T-channel isoforms. For example, arachidonic acid inhibits, with similar potency, both recombinant CaV3.1 (IC50 3 µM; Talavera et al., 2004) and CaV3.2 T-channels (10 µM blocks about 80% of current; Zhang et al., 2000). Further, an arachidonic acid-related compound and anandamide, an endogenous endocannabinoid agonist, were recently shown to cause direct inhibition of all three isoforms of recombinant T-type channels, with CaV3.2 (IC50 330 nM) and CaV3.3 (IC50 1.1 µM) more potently inhibited than CaV3.3 was (IC50 4.1 µM; Chemin et al., 2001a; 2007a;). Importantly, Barbara et al. (2009) have provided functional evidence directly implicating endogenous lipoamino-acid inhibition of T-channels in DRG cells in nociception. They found that the inhibitory effects on T-current persisted in cell-free patches, strongly suggesting direct interactions of lipoamino acids with T-channels. Thus, lipoamino acid and related compounds may be important targets for the development of future pain therapies.
T-channels in primary sensory neurons of dorsal root ganglia (DRG)
The nociceptive system is based on highly regulated relay mechanisms that connect the periphery (e.g. skin, mucosa, viscera, muscles and joints) to the higher pain-processing regions in the CNS. Primary sensory or DRG neurons are bipolar cells. Their long processes originate in the periphery, whereas their somas are located in trigeminal or spinal (dorsal root) ganglia; their central process terminates in the dorsal horn of the spinal cord. There, these cells synapse with second-order nociceptive neurons.
Many different types of sensory receptors originate in the periphery; they can be classified based on the anatomy of the nerve fibres and the type of activating stimuli. Low-threshold mechanoreceptors have large myelinated fibres (Aα and Aβ), conduct fairly fast (10–100 m·s−1), and convey mechanosensations of light touch, proprioception and vibration. High-threshold mechanoreceptors have thinly myelinated (Aδ) fibres with conduction velocities ranging from 2 through 8 m·s−1; unmyelinated (C-type) sensory fibres conduct most slowly (less than 1.4 m·s−1) (Burgess and Perl, 1967; Bessou and Perl, 1969). While both Aδ and C-type sensory fibres respond to intense mechanical and noxious thermal stimuli, C fibres are also sensitive to a variety of chemical irritants, and thus are often called polymodal nociceptors. DRGs contain the somas of thin Aδ- and C-type sensory fibres, which typically are less than 31 µm in average diameter, as well as the bodies of Aα- and Aβ-type fibres, which typically are larger than 45 µm in average diameter; medium-size neurons (between 32 and 45 µm) could belong with either group (Lee et al., 1986; Scroggs and Fox, 1992).
In the last several years, pharmacological, electrophysiological and genetic studies have begun to demonstrate how ligand- and voltage-gated channels specify functions for subsets of sensory neurons in the pain pathway. These ion channels, if they serve as pain transducers, can activate nociceptors; they also can control transmitter release from either peripheral or central endings, or modulate the excitability of nociceptors (Caterina and Julius, 2001).
The possible functional relationship between T-channels and other ligand-gated channels thought to mediate the excitability of sensory neurons has not been determined. Most acutely dissociated small DRG cells (<31 µm in diameter) expressing T-currents are sensitive to capsaicin and have the electrical properties of classically described nociceptors with wide action potentials (Cardenas et al., 1995; Coste et al., 2007; Nelson et al., 2007b). T-channels also have been reported to co-exist with ATP- and heat-gated currents in small, acutely dissociated DRG cells (Todorovic et al., 2001a) and to co-express with GABAA channels in medium-size sensory neurons (White, 1990). T-currents are present in small- and medium-size cold- and menthol-sensitive sensory neurons (Viana et al., 2002), but their function in the excitability of cold-sensitive neurons has not been studied. We recently described novel sub-population of DRG cells (soma size 26–31 µm) that expresses robust T-currents and essentially no HVA currents (‘T-rich cells’, Nelson et al., 2005). Furthermore, most small and ‘T-rich’ DRG cells expressing T-currents are sensitive to capsaicin, express tetrodotoxin-resistant Na+ currents, are IB4 positive and generate high-threshold mechanosensory currents, all of which strongly suggest their nociceptive function (Nelson et al., 2005; 2007b; Coste et al., 2007). In addition, T-channels also are present in a subpopulation of medium-size acutely dissociated sensory neurons (soma diameter 32–45 µm) that are either putative nociceptors as they bear IB4 antigen (Jagodic et al., 2007) or that subserve touch sensation mediated by d-hair mechanoreceptors (Shin et al., 2003; Dubreuil et al., 2004). Prominent CaV3.2 T-currents in ‘T-rich’ and medium-size DRG cells generate visible after-depolarizing potential and high-frequency burst firing in current-clamp recordings. Furthermore, amplification of sub-threshold membrane depolarizations via T-channels may decrease the threshold for action potential firing in DRG cells that express T-channels (Nelson et al., 2005; 2007b; Jagodic et al., 2007). This is important since these patterns of membrane firing may contribute to symptoms of neuropathic pain such as hyperalgesia (exaggerated pain sensation) and allodynia (non-painful stimulus is perceived as painful). Furthermore, conditions associated with axonal injury may lead to a change in peripheral fibre function and a previously mechanosensitive fibre may turn into a pain conducting fibre, which is one of the physiological bases of mechanical allodynia. Table 1 summarizes the properties of T-currents and expression of other ionic currents in acutely dissociated putative nociceptive and non-nociceptive DRG neurons.
Table 1.
Summary of ionic current signatures in different subpopulations of acutely dissociated sensory neurons
| Isolated DRG soma diameter (µm) | Sensory fibre type | ITCa2+-current density | IHVACa2+- current density | INa+TTX-R-current density | ITRPV1-current density | Threshold for mechano-sensory currents | Expression of IB4 imuno | Presumed function In vivo |
|---|---|---|---|---|---|---|---|---|
| Small 15–25 | C-type Classic | ++ | ++++ | ++++ | ++++ | High | ++ | Nociceptive |
| Small 26–31 | C-type T-rich | ++++ | None | ++++ | ++ | High | +++ | Nociceptive |
| Medium 32–45 | A-δ | ++ | ++++ | ++++ | ++++ | n.a. | ++++ | Nociceptive |
| Medium 32–45 | D-hair | ++++ | ++ | + | None | Low | n.a. | Mechanosensory |
| Large >45 | A-α/β | ++ | ++++ | + | None | Low | None | Mechanosensory |
Data are based on the following references: Nelson et al. (2005), Jagodic et al. (2007), Coste et al. (2007) and Wu et al. (2004). Note that T-currents are expressed in three subpopulations of putative nociceptive and two subpopulations of putative mechanosensory non-nociceptive neurons.
++++, very high; +++, high; ++, moderate; +, low; n.a., not applicable; ITCa2+, T-type Ca2+ currents; IHVACa2+, high-voltage-activated Ca2+ currents; INa+TTX-R, tetrodotoxin-resistant voltage-gated Na+ currents; ITRPV1, transient receptor potential vanilloid 1 (capsaicin-gated) currents; IB4, isolectin B4; DRG, dorsal root ganglion.
Studies using in situ hybridization have demonstrated that mRNA for CaV3.2 (α1H) is the most abundant isoform of T-channels in peripheral sensory neurons of small- and medium-size neurons (Talley et al., 1999). Patch-clamp recordings from small, acutely dissociated sensory neurons in mice lacking the CaV3.2 gene essentially showed a complete loss of T-currents (Chen et al., 2003; Nelson et al., 2007b). Consistent with this, CaV3.2 knockout mice phenotype is characterized with profound attenuation of acute pain responses (Choi et al., 2007). Bourinet et al. (2005) recently examined pain thresholds to both thermal and mechanical hyperalgesia in adult rats with molecular knockdown of CaV3.2 T-channels in DRG neurons. Intrathecally administered anti-CaV3.2 antisense oligonucleotides led to significant depression of pain responses in both acute thermal and mechanical pain testing. Importantly, this resulted in a concomitant decrease in T-current density in corresponding lumbar DRG neurons, diminished expression T-channel immunoreactivity in DRG tissue homogenates, and decreased CaV3.2 mRNA transcript expression in DRGs. Thus, this study confirmed that the major function of T-channels in DRG sensory neurons is to support acute nociceptive signals.
T-channels in dorsal horn of the spinal cord
The spinal dorsal horn (DH) contains the central terminals of peripheral afferent fibres, projection neurons of the spino-thalamic and other ascending tracts, and local inhibitory GABAergic neurons (interneurons). Little is known about the function of T-channels in the complex spinal DH circuitry. In one study, ethosuximide depressed excitability to both low- and high-intensity mechanically and thermally evoked neuronal responses recorded extracellularly from an anaesthetized rat with a spinal nerve injury (Matthews and Dickenson, 2001). However, the required concentrations were in a range up to 100 mM, at which ethosuximide was likely to affect various voltage-gated currents (e.g. Na+ currents) (Leresche et al., 1998). In a behavioural study, Dogrul et al. (2001) intrathecally injected mibefradil, which enhanced opioid-induced analgesia. Interestingly, mibefradil applied alone did not change baseline pain transmission.
T-channels are present in moderate density in DH neurons located in superficial lamina (Ryu and Randic, 1990). Ikeda et al. (2003), using acute spinal cord slice preparations, showed that nickel-sensitive T-currents in projection neurons of lamina I of the spinal DH work synergistically with neurokinin-1 (substance P) and NMDA receptors to enhance excitatory synaptic strength after brief (1 s) but high-frequency stimulation (100 Hz) of sensory afferents. This is important because lamina 1 of the DH receives synaptic input from small-diameter fibres that conduct pain signals from peripheral nerves. This study demonstrated that T-channels are crucial in raising intracellular calcium levels to the critical threshold necessary for activity- and calcium-dependent long-term potentiation (LTP) of synaptic activity resulting from stimulation of the peripheral nociceptors. However, high-frequency stimulation of afferent neurons may not be a physiologically relevant stimulus. Thus, Ikeda et al. (2006) followed with a demonstration that low-frequency stimulation of peripheral nociceptors such as that caused by inflammation can induce lasting synaptic plasticity in the form of LTP in another group of projection neurons in the spinal DH; these neurons also critically depended on T-channels. Furthermore, these investigators identified a whole signalling cascade that, in addition to T-channels, includes NMDA receptors, substance P (neurokinin) receptors, PKC, Ca2+ calmodulin-dependent kinase, intracellular calcium stores and nitric oxide.
Bao et al. (1998) showed, using intact spinal-cord slice preparations, that T-channels could be important in the presynaptic regulation of the spontaneous release of miniature excitatory synaptic currents (mEPSCs) in superficial spinal lamina, which receive their main sensory input from peripheral nociceptors. The frequency, but not the amplitude, of mEPSCs was diminished by mibefradil and nickel, but not by classical blockers of HVA-type Ca2+ currents. However, the major caveat of this study is that both mibefradil and nickel may, with similar potency, block R-type calcium channels (reviewed in Perez-Reyes, 2003). Thus, direct evidence of the function of T-channels in DH neurons is lacking. Attempts to obtain such evidence should be undertaken using newer, more selective drugs and/or knockout animals.
T-channels located both presynaptically and postsynaptically in DH neurons may have important functions in neuronal plasticity. This possibility is supported by the increase in synaptic strength, cellular excitability, and consequent susceptibility to sensitization of DH neurons, particularly in superficial laminas. These neurons could be especially important in mediation of the abnormal sensitivity to pain that occurs during inflammatory processes or as a result of peripheral nerve injury. This possibility is supported by the expression of transcripts for CaV3.3, CaV3.2 and CaV3.1 subtypes of T-channels in DH neurons (Talley et al., 1999), even though the function of the transcripts in these channels has not been clearly demonstrated. Isoform-specific antisense down-regulation of CaV3.2 and CaV3.3, but not CaV3.1 in spinal cord relieved mechanical allodynia and thermal hyperalgesia in rats with chronic compression of DRG (Wen et al., 2006). In contrast, mice lacking the CaV3.1 gene have attenuated pain responses in a neuropathic pain model resulting from L5 spinal nerve ligation (Shin et al., 2008). Additional studies will be needed to address the molecular basis of T-channel-mediated nociceptive processing in the DH of the spinal cord.
T-channels in the thalamus
Pain signals from the DH of the spinal cord traverse through the thalamus, the main sensory relay station in the CNS, before reaching higher CNS structures. T-currents in TC cells and the nucleus reticularis thalami have a critical function in the generation of low-amplitude oscillations involving reciprocally interconnected cortical and thalamic neurons. T-currents also are strongly implicated in the states of arousal and sleep, as well as absence seizures (Huguenard, 1996; Perez-Reyes, 2003). T-channel-dependent bursting activity and underlying low-threshold calcium spikes (LTS), which normally are associated with sleep, are accompanied by slow-frequency and high-amplitude electroencephalographic patterns that rarely occur during awake states.
Given the major function of the thalamus in sensory processing and the abundance of T-channel variants in different thalamic nuclei, is it possible that the alteration of their function leads to changes in nociception? Extracellular recordings from the medial thalamus of patients with neurogenic pain have shown abnormalities of LTS-mediated bursts that could at least contribute to persistent pain (Jeanmonod et al., 1996). However, slow-frequency oscillations in the lateral thalamus of patients with various chronic pain syndromes were found not to differ from those in the lateral thalamus of healthy patients (Radhakrishnan et al., 1999). These results suggest that the bursting activity of thalamic neurons, possibly mediated by distinct isoforms of T-channels, exhibits different patterns in a variety of chronic pain syndromes.
Through a combination of pharmacological, genetic and electrophysiological studies, it has been shown (Kim et al., 2003) that T-channels in ventroposterolateral thalamic relay neurons have an important function in visceral pain. Moreover, mice lacking the CaV3.1 gene did not exhibit low-threshold bursting in the thalamus. These mice also exhibited hyperalgesia to visceral pain induced by intraperitoneal injections of acidic solutions, a finding that was reproduced by the intra-thalamic infusion of mibefradil. Further studies have shown that both CaV3.1 T-type and L-type calcium channels in thalamic neurons, respectively, regulate burst and tonic firing mode of these cells, in PKC-dependent pathways (Cheong et al., 2008). Thus, in contrast to the pronociceptive function of the peripheral CaV3.2 T-channels in DRG and the spinal DH, activation of thalamic CaV3.1 T-channels has an anti-nociceptive effect.
Recent evidence also supports pro-nociceptive roles of CaV3.2 isoforms of T-channels in the thalamus during the development of acid-induced chronic muscle pain. Chen et al. (2010) found that that intramuscular injection of acid induces chronic mechanical hyperalgesia in wild-type and CaV3.1-deficient mice, but not in CaV3.2-deficient mice. Furthermore, administration of the T-channel blocker ethosuximide either intraperitoneally or intracerebroventricularly, but not intramuscularly or intrathecally, abolished mechanical hyperalgesia in wild-type mice. The development of mechanical hyperalgesia in wild-type mice requires a signalling cascade involving CaV3.2-dependent activation of extracellular signal-regulated kinase in the anterior nucleus of the paraventricular thalamus.
The function of T-type channels in animal models of chronic neuropathic pain
When nociceptors are in an increased state of responsiveness, they often respond to normal sensory stimuli as though they were painful (allodynia) and to mildly painful stimuli as though they were acutely painful (hyperalgesia). The electrophysiological correlates of these altered pain responses, collectively termed ‘peripheral sensitization’, include lower thresholds of activation, increased frequency of firing in response to suprathreshold stimuli and spontaneous firing (Coderre et al., 1993; Bhave and Gereau, 2004). Although sensitization of peripheral nociceptors contributes to various pain pathologies, the precise cellular mechanisms underlying sensitization are unclear. However, recent studies of different animal models of neuropathic pain support the importance of T-channels in initiating and maintaining chronic neuropathic pain. Thus, blockers of T-channels may provide much-needed new treatments for intractable neuropathic pain symptoms such as hyperalgesia and allodynia.
Chronic constrictive injury to the sciatic nerve
Jagodic et al. (2008) has shown that T-current density is significantly increased in small DRG cells in rats with CCI. Redox agents, locally injected, modulate thermal hyperalgesia by similar means, though to different degrees, in healthy rats and rats with CCI (Todorovic et al., 2004b). For example, L-cysteine given to rats with CCI induced thermal hyperalgesia of significantly smaller magnitude and shorter duration than the same doses in intact or sham-operated rats did. However, an oxidizing agent, 5,5′-dithio-bis-(2-nitrobenzoic acid), transiently abolished thermal hyperalgesia in neuropathic pain (NPP) rats and caused a higher degree of analgesia than it did in intact or sham-operated animals. These results suggest that the putative redox-sensitive modulatory site on T-channels in peripheral nociceptors may be in a reduced state after peripheral nerve injury. Similarly, Dogrul et al. (2003) found that local intraplantar injections of mibefradil abolished the progression and reversed CCI-induced mechanical and thermal hyperalgesia. In this study, mibefradil exerted similar effects when given systemically, but not intrathecally. Since mibefradil does not cross the blood-brain barrier, this argues that the major site of its antihyperalgesic effect in the CCI model is peripheral. Dogrul and colleagues found, in the same study, that systemic injections of ethosuximide reversed NPP in the rat CCI model.
Pathirathna et al. (2005b) reported that ECN, a neuroactive steroid and T-channel blocker, was about fivefold more potent in alleviating thermal hyperalgesia in neuropathic pain in rats with CCI than it was in sham-operated rats. Specific molecular knockdown of CaV3.2 T-currents in rat nociceptors in the CCI model provided complete relief from thermal and mechanical hyperalgesia and allodynia for up to 7–8 days (Bourinet et al., 2005). Conversely, up-regulation of CaV3.2 T-channels by endogenous H2S contributes to the maintenance of neuropathic pain in a spinal nerve injury model (Takahashi et al., 2010).
Chemotherapy-induced peripheral neuropathy
Many drugs that are effective in suppressing the growth of tumour cells also cause painful peripheral neuropathies that limit their use in cancer therapy. Flatters and Bennett (2004) found that systemic administration of ethosuximide alleviated mechanical and cold allodynia and hyperalgesia in rats with paclitaxel- and vincristine-induced painful peripheral neuropathy. Furthermore, chronic use of ethosuximide did not cause tolerance to analgesic effects. These authors reported that the administration of morphine and/or 5S,10R)-(+)-5-Methyl-10,11-dihydro-5H-dibenzo[a,d]cyclohepten-5,10-imine maleate, an NMDA blocker, had little effect on paclitaxel-induced neuropathic pain.
Diabetic neuropathy
Little is known about the function of VGCCs in the development of abnormal excitability of sensory neurons and NPP symptoms in animal models of diabetic neuropathy. In diabetic BB/W rats, a model for autoimmune-mediated type I diabetes, up-regulation of HVA Ca2+ current amplitude by about twofold occurred in small nociceptive DRG cells, consequently decreasing opioid-induced inhibition of HVA currents (Hall et al., 1996; 2001;). BB/W rats, after 8 months in a diabetic state, similarly up-regulated T-channels in small DRG cells (Hall et al., 1995). Similar up-regulation of HVA Ca2+ channels in small DRG cells has been reported in rats with streptozotocin (STZ)-induced diabetic neuropathy, a model of type I diabetes (Hall et al., 2001). In contrast, another study with STZ-induced diabetic neuropathy, which used electrophysiological recordings from acutely isolated IB4-positive medium-size DRG cells, found that T-current amplitude was up-regulated about twofold, while HVA current was not significantly altered (Jagodic et al., 2007; Messinger et al., 2009).
Selective knock-down of DRG CaV3.2 currents in vivo has effectively reversed mechanical and thermal hyperalgesia in STZ-induced diabetic neuropathy in rats (Messinger et al., 2009). Furthermore, significant up-regulation of CaV3.2 T-channel transcripts in DRG tissue homogenates and concomitant up-regulation of CaV3.2 T-currents in small nociceptive DRG cells has been reported in another model of painful diabetic neuropathy, leptin-deficient ob/ob mice (Latham et al., 2009). In the same study, these authors found that selective pharmacological antagonism of T-channels by systemically administered ECN effectively reversed thermal and mechanical hyperalgesia in diabetic ob/ob mice, but was completely ineffective in diabetic CaV3.2 knock-out mice. Thus, by blocking CaV3.2 T-channels in DRG cells, it may be possible to alleviate intractable pain in patients with diabetic neuropathy.
Conclusions
Recent results strongly implicate T-channels in the processing of nociceptive signals in both the peripheral and CNS. Moreover, many drugs and natural substances at clinically relevant concentrations modulate the function these channels. The CaV3.2 isoform of these channels expressed in peripheral sensory neurons, spinal DH and thalamic neurons has a well-documented function in supporting cutaneous, muscular and visceral nociceptive transmission. CaV3.2 channels are also strongly implicated as contributing to the development of neuropathic pain in conditions following mechanical injury of peripheral nerves and peripheral diabetic neuropathy. In contrast, current evidence supports the idea that activation of thalamic CaV3.1 channels may suppress visceral pain transmission. The putative role of the CaV3.3 isoform of T-channels in pain processing has not yet been validated. Because current treatments for both acute and chronic pain are insufficient and often associated with serious side effects, it would be highly useful to develop drug therapies that selectively target the CaV3.2 isoform of T-channels in pain pathways. Of particular interest would be selective voltage-dependent T-type channel blockers that have no effect in 10- to 100-fold higher concentrations on other classes of voltage-gated ion channels (e.g. Na+, K+, HVA-type Ca2+ channels). Many previous functional studies were done using non-selective Ca2+ channel blockers such as ethosuximide and mibefradil, and these data should be critically re-examined and interpreted using newly available molecular (in vivo knock-down) and/or genetic (knockout mice) tools (e.g. Latham et al., 2009). Furthermore, more selective and potent blockers of T-channel are being developed (e.g. TTA compounds, neuroactive steroids) that can further help in deciphering the roles of T-channels in sensory transmission and nociception in particular. However, it is certainly true that T-channel isoforms are expressed in many areas of the nervous system including non-nociceptive peripheral sensory neurons (Table 1), as well as vascular and cardiac tissues that can all be affected by T-channel blockers in vivo. Currently, very few clinical and pre-clinical studies are available to assess possible side effects of systemic blockade of T-channels (e.g. sedation, motor weakness, cardiac arrhythmia). However, based on currently available data using rodents, it appears that peripherally acting, voltage-dependent, potent and selective blockers of CaV3.2 isoform could offer pain relief in acute and chronic pain conditions with minimal side effects.
Acknowledgments
Supported by GM 075299 (to SMT), American Diabetes Association 7-09-BS-190 (to SMT), DA 029342 (to SMT and VJT) and Dr Harold Carron endowment (to VJT).
Glossary
Abbreviations
- 3βOH
[(3β, 5β, 17β)-3-hydroxyandrostane-17-carbonitrile]
- ADP
after-depolarizing potential
- CCI
chronic constrictive injury
- CNS
central nervous system
- DH
dorsal horn
- DRG
dorsal root ganglia
- DTNB
(5,5′-dithio-bis-(2-nitrobenzoic acid)
- ECN
[(3β, 5α, 17β)-17-hydroxyestrane-3-carbonitrile]
- GABA
γ-amino-butyric acid
- HVA
high voltage-activated
- KO
knock out
- LTP
long-term potentiation
- LTS
low threshold calcium spike
- LVA
low voltage activated
- mEPSCs
miniature excitatory postsynaptic currents
- MK-801
5S,10R)-(+)-5-Methyl-10,11-dihydro-5H-dibenzo[a,d]cyclohepten-5,10-imine maleate
- NMDA
N-methyl-D-aspartate
- NPP
neuropathic pain
- nRT
nucleus reticularis thalami
- PKC
protein kinase C
- PNS
peripheral nervous system
- ROCK
Rho-associated kinase
- STZ
streptozotocin
- TC
thalamo-cortical
- T-channels
(T)-type Ca2+ channels
- TTA-A2
[2-(4-Cyclopropylphenyl)-N-((1R)-1-{5-[(2,2,2-trifluoroethyl)oxo]-pyridin-2-yl}ethyl)acetamide]
- TTA-P2
3,5-dichloro-N-[1-(2,2-dimethyl-tetrahydro-pyran-4-ylmethyl)-4-fluoro-piperidin-4-ylmethyl]-benzamide
- TTX
tetrodotoxin
- VGCCs
voltage-gated calcium channels
- WT
wild type
Conflict of interest
None.
Supporting Information
Teaching Materials; Fig 1 as PowerPoint slide.
References
- Bao J, Li JJ, Perl ER. Differences in Ca2+ channels governing generation of miniature and evoked excitatory synaptic currents in spinal laminae I and II. J Neurosci. 1998;18:8740–8750. doi: 10.1523/JNEUROSCI.18-21-08740.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbara G, Alloui A, Nargeot J, Lory P, Eschalier A, Bourinet E, et al. T-type calcium channel inhibition underlies the analgesic effects of the endogenous lipoamino acids. J Neurosci. 2009;29:13106–13114. doi: 10.1523/JNEUROSCI.2919-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartels P, Behnke K, Michels G, Groner F, Schneider T, Henry M, et al. Structural and biophysical determinants of single Ca(V)3.1 and Ca(V)3.2 T-type calcium channel inhibition by N(2)O. Cell Calcium. 2009;46:293–302. doi: 10.1016/j.ceca.2009.09.002. [DOI] [PubMed] [Google Scholar]
- Bessou P, Perl ER. Response of cutaneous sensory units with unmyelinated fibers to noxious stimuli. J Neurophysiol. 1969;32:1025–1043. doi: 10.1152/jn.1969.32.6.1025. [DOI] [PubMed] [Google Scholar]
- Bhave G, Gereau RW. Posttranslational mechanisms of peripheral sensitization. J Neurobiol. 2004;61:88–106. doi: 10.1002/neu.20083. [DOI] [PubMed] [Google Scholar]
- Bourinet E, Alloui A, Monteil A, Barrere C, Couette B, Poirot O, et al. Silencing of the Cav3.2 T-type calcium channel gene in sensory neurons demonstrates its major role in nociception. EMBO J. 2005;24:315–324. doi: 10.1038/sj.emboj.7600515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgess PR, Perl ER. Myelinated afferent fibres responding specifically to noxious stimulation of the skin. J Physiol. 1967;3:541–562. doi: 10.1113/jphysiol.1967.sp008227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardenas CG, Del Mar LP, Scroggs RS. Variation in serotonergic inhibition of calcium channel currents in four types of rat sensory neurons differentiated by membrane properties. J Neurophysiol. 1995;74:1870–1879. doi: 10.1152/jn.1995.74.5.1870. [DOI] [PubMed] [Google Scholar]
- Caterina M, Julius D. The vanilloid receptor: a molecular gateway to the pain pathway. Ann Rev Neurosci. 2001;24:487–517. doi: 10.1146/annurev.neuro.24.1.487. [DOI] [PubMed] [Google Scholar]
- Catterall WA. Structure and regulation of voltage-gated Ca2+ channels. Ann Rev Cell Dev Biol. 2000;16:521–555. doi: 10.1146/annurev.cellbio.16.1.521. [DOI] [PubMed] [Google Scholar]
- Chemin J, Monteil A, Perez-Reyes E, Nargeot J, Lory P. Direct inhibition of T-type calcium channels by the endogenous cannabinoid anandamide. EMBO J. 2001a;20:7033–7040. doi: 10.1093/emboj/20.24.7033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chemin J, Monteil A, Bourinet E, Nargeot J, Lory P. Alternatively spliced alpha(1G) (Ca(V)3.1) intracellular loops promote specific T-type Ca(2+) channel gating properties. Biophys J. 2001b;80:1238–1250. doi: 10.1016/S0006-3495(01)76100-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chemin J, Nargeot J, Lory P. Chemical determinants involved in anandamide-induced inhibition of T-type calcium channels. J Biol Chem. 2007a;282:2314–2323. doi: 10.1074/jbc.M610033200. [DOI] [PubMed] [Google Scholar]
- Chemin J, Mezghrani A, Bidaud I, Dupasquier S, Marger F, Barrère C, et al. Temperature-dependent modulation of CaV3 T-type calcium channels by protein kinases C and A in mammalian cells. J Biol Chem. 2007b;282:32710–32718. doi: 10.1074/jbc.M702746200. [DOI] [PubMed] [Google Scholar]
- Chen CC, Lamping KG, Nuno DW, Barresi R, Prouty SJ, Lavoie JL, et al. Abnormal coronary function in mice deficient in alpha1H T-type Ca2+ channels. Science. 2003;302:1416–1418. doi: 10.1126/science.1089268. [DOI] [PubMed] [Google Scholar]
- Chen WK, Liu IY, Chang YT, Chen YC, Chen CC, Yen CT, et al. Ca(v)3.2 T-type Ca2+ channel-dependent activation of ERK in paraventricular thalamus modulates acid-induced chronic muscle pain. J Neurosci. 2010;30:10360–10368. doi: 10.1523/JNEUROSCI.1041-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheong E, Lee S, Choi BJ, Sun M, Lee CJ, Shin HS. Tuning thalamic firing modes via simultaneous modulation of T- and L-type Ca2+ channels controls pain sensory gating in the thalamus. J Neurosci. 2008;28:13331–13340. doi: 10.1523/JNEUROSCI.3013-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi S, Na HS, Kim J, Lee J, Lee S, Kim D, et al. Attenuated pain responses in mice lacking CaV3.2 T-type channels. Genes Brain Behav. 2007;6:425–431. doi: 10.1111/j.1601-183X.2006.00268.x. [DOI] [PubMed] [Google Scholar]
- Chuang RSI, Jaffe H, Cribbs L, Perez-Reyess E, Swartz KJ. Inhibition of T-type voltage-gated calcium channels by a new scorpion toxin. Nat Neurosci. 1998;1:668–674. doi: 10.1038/3669. [DOI] [PubMed] [Google Scholar]
- Coderre TJ, Katz J, Vaccarino AL, Melzack R. Contribution of central neuroplasticity to pathological pain: review of clinical and experimental evidence. Pain. 1993;52:259–285. doi: 10.1016/0304-3959(93)90161-H. [DOI] [PubMed] [Google Scholar]
- Coste B, Crest M, Delmas P. Pharmacological dissection and distribution of NaN/Nav1.9, T-type Ca2+ currents, and mechanically activated cation currents in different populations of DRG neurons. J Gen Physiol. 2007;129:57–77. doi: 10.1085/jgp.200609665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coulter DA, Huguenard JR, Prince DA. Characterization of ethosuximide reduction of low-threshold calcium current in thalamic neurons. Ann Neurol. 1989;25:582–593. doi: 10.1002/ana.410250610. [DOI] [PubMed] [Google Scholar]
- Cribbs LL, Lee J, Yang J, Satin J, Zhang Y, Daud A, et al. Cloning and characterization of alpha1H from human heart, a member of the T-type Ca2+ channel gene family. Circ Res. 1998;83:103–109. doi: 10.1161/01.res.83.1.103. [DOI] [PubMed] [Google Scholar]
- DePuy SD, Yao J, Hu C, McIntire W, Bidaud I, Lory P, et al. The molecular basis for T-type Ca2+ channel inhibition by G protein beta2gamma2 subunits. Proc Natl Acad Sci USA. 2006;103:14590–14595. doi: 10.1073/pnas.0603945103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dogrul A, Yesilyurt O, Isimer A, Guzeldemir ME. L-type and T-type calcium channel blockade potentiate the analgesic effects of morphine and selective mu opioid agonist, but not to selective delta and kappa agonist at the level of the spinal cord in mice. Pain. 2001;93:61–68. doi: 10.1016/S0304-3959(01)00293-7. [DOI] [PubMed] [Google Scholar]
- Dogrul A, Gardell LR, Ossipov MH, Tulunay FC, Lai J, Porecca F. Reversal of experimental neuropathic pain by T-type calcium channel blockers. Pain. 2003;105:159–168. doi: 10.1016/s0304-3959(03)00177-5. [DOI] [PubMed] [Google Scholar]
- Dubreuil A, Boukhaddaoui H, Desmadryl G, Martinez-Salgado C, Moshourab R, Lewin GR, et al. Role of T-type calcium current in identified d-hair mechanoreceptor neurons studied in vitro. J Neurosci. 2004;24:8480–8484. doi: 10.1523/JNEUROSCI.1598-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emerick MC, Stein R, Kunze R, McNulty MM, Regan MR, Hanck DA, et al. Profiling the array of Ca(v)3.1 variants from the human T-type calcium channel gene CACNA1G: alternative structures, developmental expression, and biophysical variations. Proteins. 2006;64:320–342. doi: 10.1002/prot.20877. [DOI] [PubMed] [Google Scholar]
- Ertel SI, Ertel EA, Clozel J. T-type Ca2+ channels and pharmacological blockade: potential pathophysiological relevance. Cardiovasc Drugs Ther. 1997;11:723–739. doi: 10.1023/a:1007706022381. [DOI] [PubMed] [Google Scholar]
- Flatters SJ, Bennett GJ. Ethosuximide reverses paclitaxel- and vincristine-induced painful peripheral neuropathy. Pain. 2004;109:150–161. doi: 10.1016/j.pain.2004.01.029. [DOI] [PubMed] [Google Scholar]
- Gruss M, Bushell TJ, Bright DP, Lieb WR, Mathie A, Franks NP. Two-pore-domain K+ channels are a novel target for the anesthetic gases xenon, nitrous oxide, and cyclopropane. Mol Pharmacol. 2004;65:443–452. doi: 10.1124/mol.65.2.443. [DOI] [PubMed] [Google Scholar]
- Hall KE, Sima AA, Wiley JW. Voltage-dependent calcium currents are enhanced in dorsal root ganglion neurones from the Bio Bred/Worchester diabetic rat. J Physiol. 1995;486:313–322. doi: 10.1113/jphysiol.1995.sp020814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall KE, Sima AA, Wiley JW. Opiate-mediated inhibition of calcium signaling is decreased in dorsal root ganglion neurons from the diabetic BB/W rat. J Clin Invest. 1996;97:1165–1172. doi: 10.1172/JCI118530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall KE, Liu J, Sima AA, Wiley JW. Impaired inhibitory G-protein function contributes to increased calcium currents in rats with diabetic neuropathy. J Neurophysiol. 2001;86:760–770. doi: 10.1152/jn.2001.86.2.760. [DOI] [PubMed] [Google Scholar]
- Herrington J, Lingle CJ. Kinetic and pharmacological properties of low voltage-activated Ca2+ current in rat clonal (GH3) pituitary cells. J Neurophysiol. 1992;68:213–232. doi: 10.1152/jn.1992.68.1.213. [DOI] [PubMed] [Google Scholar]
- Hildebrand ME, David LS, Hamid J, Mulatz K, Garcia E, Zamponi GW, et al. Selective inhibition of Cav3.3 T-type calcium channels by Galphaq/11-coupled muscarinic acetylcholine receptors. J Biol Chem. 2007;282:21043–21055. doi: 10.1074/jbc.M611809200. [DOI] [PubMed] [Google Scholar]
- Hu C, Depuy SD, Yao J, McIntire WE, Barrett PQ. Protein kinase A activity controls the regulation of T-type CaV3.2 channels by Gbetagamma dimers. J Biol Chem. 2009;284:7465–7473. doi: 10.1074/jbc.M808049200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang L, Keyser BM, Tagmose TM, Hansen JB, Taylor JT, Zhuang H, et al. NNC 55-0396 [(1S,2S)-2-(2-(N-[(3-benzimidazol-2-yl)propyl]-N-methylamino)ethyl)-6-fluoro-1,2,3,4-tetrahydro-1-isopropyl-2-naphtyl cyclopropanecarboxylate dihydrochloride]: a new selective inhibitor of T-type calcium channels. J Pharmacol Exp Ther. 2004;309:193–199. doi: 10.1124/jpet.103.060814. [DOI] [PubMed] [Google Scholar]
- Huguenard JR. Low-threshold calcium currents in central nervous system neurons. Annu Rev Physiol. 1996;58:329–348. doi: 10.1146/annurev.ph.58.030196.001553. [DOI] [PubMed] [Google Scholar]
- Iftinca M, Hamid J, Chen L, Varela D, Tadayonnejad R, Altier C, et al. Regulation of T-type calcium channels by Rho-associated kinase. Nat Neurosci. 2007;10:854–860. doi: 10.1038/nn1921. [DOI] [PubMed] [Google Scholar]
- Ikeda H, Heinke B, Ruscheweyh R, Sandkühler J. Synaptic plasticity in spinal lamina I projection neurons that mediate hyperalgesia. Science. 2003;299:1237–1240. doi: 10.1126/science.1080659. [DOI] [PubMed] [Google Scholar]
- Ikeda H, Stark J, Fischer H, Wagner M, Drdla R, Jäger T, et al. Synaptic amplifier of inflammatory pain in the spinal dorsal horn. Science. 2006;312:1659–1662. doi: 10.1126/science.1127233. [DOI] [PubMed] [Google Scholar]
- Jagodic MM, Pathirathna S, Nelson MT, Mancuso S, Joksovic PM, Rosenberg ER, et al. Cell-specific alterations of T-type calcium current in painful diabetic neuropathy enhance excitability of sensory neurons. J Neurosci. 2007;27:3305–3316. doi: 10.1523/JNEUROSCI.4866-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jagodic MM, Pathirathna S, Joksovic PM, Lee WY, Nelson MT, Su P, et al. Up-regulation of the T-type Ca2+ current in small rat sensory neurons after chronic constrictive injury of the sciatic nerve. J Neurophysiol. 2008;99:3151–3156. doi: 10.1152/jn.01031.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeanmonod D, Magnin M, Morel A. Low-threshold calcium spike bursts in the human thalamus. Common physiopathology for sensory, motor and limbic positive symptoms. Brain. 1996;119:363–375. doi: 10.1093/brain/119.2.363. [DOI] [PubMed] [Google Scholar]
- Jevtović-Todorović V, Todorović SM, Mennerick S, Powell S, Dikranian K, Benshoff N, et al. Nitrous oxide (laughing gas) is an NMDA antagonist, neuroprotectant and neurotoxin. Nat Med. 1998;4:460–463. doi: 10.1038/nm0498-460. [DOI] [PubMed] [Google Scholar]
- Jimenez C, Bourinet E, Leuranguer V, Richard S, Snutch TP, Nargeot J. Determinants of voltage-dependent inactivation affect Mibefradil block of calcium channels. Neuropharmacology. 2000;39:1–10. doi: 10.1016/s0028-3908(99)00153-7. [DOI] [PubMed] [Google Scholar]
- Joksovic PM, Brimelow BC, Murbartian J, Perez-Reyes E, Todorovic SM. Contrasting anesthetic sensitivities of T-type Ca2+ channels of reticular thalamic neurons and recombinant Ca(v)3.3 channels. Br J Pharmacol. 2005;144:59–70. doi: 10.1038/sj.bjp.0706020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joksovic PM, Nelson MT, Jevtovic-Todorovic V, Patel MK, Perez-Reyes E, Campbell KP, et al. CaV3.2 is the major molecular substrate for redox regulation of T-type Ca2+ channels in the rat and mouse thalamus. J Physiol. 2006;574(Pt 2):415–430. doi: 10.1113/jphysiol.2006.110395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joksovic PM, Choe WJ, Nelson MT, Orestes P, Brimelow BC, Todorovic SM. Mechanisms of inhibition of T-type calcium current in the reticular thalamic neurons by 1-octanol: implication of the protein kinase C pathway. Mol Pharm. 2010;77:87–94. doi: 10.1124/mol.109.059931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang HW, Vitko I, Lee SS, Perez-Reyes E, Lee JH. Structural determinants of the high affinity extracellular zinc binding site on Cav3.2 T-type calcium channels. J Biol Chem. 2010;285:3271–3281. doi: 10.1074/jbc.M109.067660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawabata A, Ishiki T, Nagasawa K, Yoshida S, Maeda Y, Takahashi T, et al. Hydrogen sulfide as a novel nociceptive messenger. Pain. 2007;132:74–81. doi: 10.1016/j.pain.2007.01.026. [DOI] [PubMed] [Google Scholar]
- Kim D, Song I, Keum S, Lee T, Jeong M-J, Kim S-S, et al. Lack of the burst firing of thalamocortical relay neurons and resistance to absence seizures in mice lacking alpha(1G) T-type Ca(2+) channels. Neuron. 2001;31:35–45. doi: 10.1016/s0896-6273(01)00343-9. [DOI] [PubMed] [Google Scholar]
- Kim D, Park D, Choi S, Lee S, Sun M, Kim C, et al. Thalamic control of visceral nociception mediated by T-type Ca2+ channels. Science. 2003;302:117–119. doi: 10.1126/science.1088886. [DOI] [PubMed] [Google Scholar]
- Kraus RL, Li Y, Gregan Y, Gotter AL, Uebele VN, Fox SV, et al. In vitro characterization of T-type calcium channel antagonist TTA-A2 and in vivo effects on arousal in mice. J Pharmacol Exp Ther. 2010;335:409–417. doi: 10.1124/jpet.110.171058. [DOI] [PubMed] [Google Scholar]
- Latham JR, Pathirathna S, Jagodic MM, Choe WJ, Levin ME, Nelson MT, et al. Selective T-type calcium channel blockade alleviates hyperalgesia in ob/ob mice. Diabetes. 2009;58:2656–2665. doi: 10.2337/db08-1763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latour I, Louw DF, Beedle AM, Hamid J, Sutherland GR, Zamponi GW. Expression of T-type calcium channel splice variants in human glioma. Glia. 2004;48:112–119. doi: 10.1002/glia.20063. [DOI] [PubMed] [Google Scholar]
- Lee KH, Chung K, Chung JM, Coggeshall RE. Correlation of cell body size, axon size, and signal conduction velocity for individually labelled dorsal root ganglion cells in the cat. J Comp Neurol. 1986;15:335–346. doi: 10.1002/cne.902430305. [DOI] [PubMed] [Google Scholar]
- Lee JH, Daud AN, Cribbs LL, Lacerda AE, Pereverzev A, Klockner U, et al. Cloning and expression of a novel member of the low voltage-activated T-type calcium channel family. J Neurosci. 1999a;19:1912–1921. doi: 10.1523/JNEUROSCI.19-06-01912.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JH, Gomora JC, Cribbs LL, Perez-Reyes E. Nickel block of three cloned T-type calcium channels: low concentrations selectively block alpha1H. Biophys J. 1999b;77:3034–3042. doi: 10.1016/S0006-3495(99)77134-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leresche N, Parri HR, Erdemli G, Guyon A, Turner JP, Williams SR, et al. On the action of the anti-absence drug ethosuximide in the rat and cat thalamus. J Neurosci. 1998;18:4842–4853. doi: 10.1523/JNEUROSCI.18-13-04842.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llinas R. The intrinsic electrophysiological properties of mammalian neurons: insight into central nervous system function. Science. 1988;242:1654–1664. doi: 10.1126/science.3059497. [DOI] [PubMed] [Google Scholar]
- Lory P, Chemin J. Towards the discovery of novel T-type calcium channel blockers. Expert Opin Ther Targets. 2007;11:717–722. doi: 10.1517/14728222.11.5.717. [DOI] [PubMed] [Google Scholar]
- McDonough SI, Bean BP. Mibefradil inhibition of T-type calcium channels in cerebellar purkinje neurons. Mol Pharm. 1998;54:1080–1087. doi: 10.1124/mol.54.6.1080. [DOI] [PubMed] [Google Scholar]
- McGivern JG. Pharmacology and drug discovery for T-type calcium channels. CNS Neurol Disord Drug Targets. 2006;5:587–603. doi: 10.2174/187152706779025535. [DOI] [PubMed] [Google Scholar]
- McNulty MM, Hanck DA. State-dependent mibefradil block of Na+ channels. Mol Pharmacol. 2004;66:1652–1661. doi: 10.1124/mol.66.6.1652. [DOI] [PubMed] [Google Scholar]
- Maeda Y, Aoki Y, Sekiguchi F, Matsunami M, Takahashi T, Nishikawa H, et al. Hyperalgesia induced by spinal and peripheral hydrogen sulfide: evidence for involvement of Cav3.2 T-type calcium channels. Pain. 2009;142:127–132. doi: 10.1016/j.pain.2008.12.021. [DOI] [PubMed] [Google Scholar]
- Matsunami M, Tarui T, Mitani K, Nagasawa K, Fukushima O, Okubo K, et al. Luminal hydrogen sulfide plays a pronociceptive role in mouse colon. Gut. 2009;58:751–761. doi: 10.1136/gut.2007.144543. [DOI] [PubMed] [Google Scholar]
- Matthews EA, Dickenson AH. Effects of ethosuximide, a T-type Ca(2+) channel blocker, on dorsal horn neuronal responses in rats. Eur J Pharmacol. 2001;415:141–149. doi: 10.1016/s0014-2999(01)00812-3. [DOI] [PubMed] [Google Scholar]
- Messinger RB, Naik AK, Jagodic MM, Nelson MT, Lee WY, Choe WJ, et al. In-vivo silencing of the Cav3.2 T-type calcium channels in sensory neurons alleviates hyperalgesia in rats with streptozocin-induced diabetic neuropathy. Pain. 2009;145:184–195. doi: 10.1016/j.pain.2009.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller RJ. Presynaptic receptors. Annu Rev Pharmacol Toxicol. 1998;38:201–227. doi: 10.1146/annurev.pharmtox.38.1.201. [DOI] [PubMed] [Google Scholar]
- Monteil A, Chemin J, Leuranguer V, Altier C, Mennessier G, Bourinet E, et al. Specific properties of T-type calcium channels generated by the human alpha 1I subunit. J Biol Chem. 2000;275:16530–16535. doi: 10.1074/jbc.C000090200. [DOI] [PubMed] [Google Scholar]
- Murbartián J, Arias JM, Lee JH, Gomora JC, Perez-Reyes E. Alternative splicing of the rat Ca(v)3.3 T-type calcium channel gene produces variants with distinct functional properties(1) FEBS Lett. 2002;528:272–278. doi: 10.1016/s0014-5793(02)03341-0. [DOI] [PubMed] [Google Scholar]
- Murbartián J, Arias JM, Perez-Reyes E. Functional impact of alternative splicing of human T-type Cav3.3 calcium channels. J Neurophysiol. 2004;92:3399–3407. doi: 10.1152/jn.00498.2004. [DOI] [PubMed] [Google Scholar]
- Nelson MT, Joksovic PM, Perez-Reyes E, Todorovic SM. The endogenous redox agent L-cysteine induces T-type Ca2+ channel-dependent sensitization of a novel subpopulation of rat peripheral nociceptors. J Neurosci. 2005;25:8766–8775. doi: 10.1523/JNEUROSCI.2527-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson MT, Joksovic PM, Su P, Kang HW, Van Deusen A, Baumgart JP, et al. Molecular mechanisms of subtype-specific inhibition of neuronal T-type calcium channels by ascorbate. J Neurosci. 2007a;27:12577–12583. doi: 10.1523/JNEUROSCI.2206-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson MT, Woo J, Kang HW, Vitko I, Barrett PQ, Perez-Reyes E, et al. Reducing agents sensitize C-type nociceptors by relieving high-affinity zinc inhibition of T-type calcium channels. J Neurosci. 2007b;27:8250–8260. doi: 10.1523/JNEUROSCI.1800-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson MT, Milescu LS, Todorovic SM, Scroggs RS. A modeling study of T-type Ca2+ channel gating and modulation by L-cysteine in rat nociceptors. Biophys J. 2010;98:197–206. doi: 10.1016/j.bpj.2009.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimura S, Fukushima O, Ishikura H, Takahashi T, Matsunami M, Tsujiuchi T, et al. Hydrogen sulfide as a novel mediator for pancreatic pain in rodents. Gut. 2009;58:762–770. doi: 10.1136/gut.2008.151910. [DOI] [PubMed] [Google Scholar]
- Orestes P, Bojadzic D, Lee J, Leach E, Salajegheh R, Digruccio MR, et al. Free radical signaling underlies inhibition of CaV3.2 T-type calcium channels by nitrous oxide in the pain pathway. J Physiol. 2010;589:135–148. doi: 10.1113/jphysiol.2010.196220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park JY, Kang HW, Moon HJ, Huh SU, Jeong SW, Soldatov NM, et al. Activation of protein kinase C augments T-type Ca2+ channel activity without changing channel surface density. J Physiol. 2006;577(Pt 2):513–523. doi: 10.1113/jphysiol.2006.117440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pathirathna S, Brimelow BC, Jagodic MM, Kathiresan K, Jiang X, Zorumski CF, et al. New evidence that both T-type Ca2+ channels and GABAA channels are responsible for the potent peripheral analgesic effects of 5alpha-reduced neuroactive steroids. Pain. 2005a;114:429–443. doi: 10.1016/j.pain.2005.01.009. [DOI] [PubMed] [Google Scholar]
- Pathirathna S, Todorovic SM, Covey DF, Jevtovic-Todorovic V. Novel 5alpha-reduced neuroactive steroids induce peripheral thermal anti-nociception in rats with neuropathic pain. Pain. 2005b;117:326–339. doi: 10.1016/j.pain.2005.06.019. [DOI] [PubMed] [Google Scholar]
- Perez-Reyes E. Molecular physiology of low-voltage-activated T-type calcium channels. Physiol Rev. 2003;83:117–161. doi: 10.1152/physrev.00018.2002. [DOI] [PubMed] [Google Scholar]
- Perez-Reyes E, Cribbs LL, Daud A, Lacerda AE, Barclay J, Williamson MP, et al. Molecular characterization of a neuronal low-voltage-activated T-type calcium channel. Nature. 1998;391:896–900. doi: 10.1038/36110. [DOI] [PubMed] [Google Scholar]
- Radhakrishnan V, Tsoukatos J, Davis KD, Tasker RR, Lozano AM, Dostrovsky JO. A comparison of the burst activity of lateral thalamic neurons in chronic pain and non-pain patients. Pain. 1999;80:567–575. doi: 10.1016/S0304-3959(98)00248-6. [DOI] [PubMed] [Google Scholar]
- Ryu PD, Randic M. Low- and high-voltage-activated calcium currents in rat spinal dorsal horn neurons. J Neurophysiol. 1990;63:273–285. doi: 10.1152/jn.1990.63.2.273. [DOI] [PubMed] [Google Scholar]
- Santi CM, Cayabyab FS, Sutton KG, McRory JE, Mezeyova J, Hamming KS, et al. Differential inhibition of T-type calcium channels by neuroleptics. J Neursoci. 2002;22:396–403. doi: 10.1523/JNEUROSCI.22-02-00396.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroeder JE, Fischbach PS, McCleskey EW. T-type calcium channels: heterogeneous expression in rat sensory neurons and selective modulation by phorbol esters. J Neurosci. 1990;10:947–951. doi: 10.1523/JNEUROSCI.10-03-00947.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scroggs RS, Fox AP. Calcium current variation between acutely isolated rat dorsal root ganglion neurons of different size. J Physiol. 1992;445:639–658. doi: 10.1113/jphysiol.1992.sp018944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin JB, Martinez-Salgado C, Heppenstall PA, Lewin GR. A T-type calcium channel required for normal function of a mammalian mechanoreceptor. Nat Neurosci. 2003;6:724–730. doi: 10.1038/nn1076. [DOI] [PubMed] [Google Scholar]
- Shin MC, Kim CJ, Min BI, Ogawa S, Tanaka E, Akaike N. A selective T-type Ca2+ channel blocker R(-) efonidipine. Naunyn Schmiedebergs Arch Pharmacol. 2008;377:411–421. doi: 10.1007/s00210-007-0239-6. [DOI] [PubMed] [Google Scholar]
- Shipe WD, Barrow JC, Yang ZQ, Lindsley CW, Yang FV, Schlegel KA, et al. Design, synthesis, and evaluation of a novel 4-aminomethyl-4-fluoropiperidine as a T-type Ca2+ channel antagonist. J Med Chem. 2008;51:3692–3695. doi: 10.1021/jm800419w. [DOI] [PubMed] [Google Scholar]
- Sidach SS, Mintz IM. Kurtoxin, a gating modifier of neuronal high- and low-threshold ca channels. J Neurosci. 2002;22:2023–2034. doi: 10.1523/JNEUROSCI.22-06-02023.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song I, Kim D, Choi S, Sun M, Kim Y, Shin HS. Role of the alpha1G T-type calcium channel in spontaneous absence seizures in mutant mice. J Neurosci. 2004;24:5249–5257. doi: 10.1523/JNEUROSCI.5546-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi T, Aoki Y, Okubo K, Maeda Y, Sekiguchi F, Mitani K, et al. Upregulation of Ca(v)3.2 T-type calcium channels targeted by endogenous hydrogen sulfide contributes to maintenance of neuropathic pain. Pain. 2010;150:183–191. doi: 10.1016/j.pain.2010.04.022. [DOI] [PubMed] [Google Scholar]
- Talavera K, Staes M, Janssens A, Droogmans G, Nilius B. Mechanism of arachidonic acid modulation of the T-type Ca2+ channel alpha1G. J Gen Physiol. 2004;124:225–238. doi: 10.1085/jgp.200409050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talley EM, Cribbs LL, Lee JH, Daud A, Perez-Reyes E, Bayliss DA. Differential distribution of three members of a gene family encoding low voltage-activated (T-type) calcium channels. J Neurosci. 1999;19:1895–1911. doi: 10.1523/JNEUROSCI.19-06-01895.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang CM, Presser F, Morad M. Amiloride selectively blocks the low threshold (T) calcium channel. Science Wash DC. 1988;240:213–215. doi: 10.1126/science.2451291. [DOI] [PubMed] [Google Scholar]
- Tao J, Hildebrand ME, Liao P, Liang MC, Tan G, Li S, et al. Activation of corticotropin-releasing factor receptor 1 selectively inhibits CaV3.2 T-type calcium channels. Mol Pharmacol. 2008;73:1596–1609. doi: 10.1124/mol.107.043612. [DOI] [PubMed] [Google Scholar]
- Todorovic SM, Lingle CJ. Pharmacological properties of T-type Ca2+ current in adult rat sensory neurons: effects of anticonvulsant and anesthetic agents. J Neurophysiol. 1998;79:240–252. doi: 10.1152/jn.1998.79.1.240. [DOI] [PubMed] [Google Scholar]
- Todorovic SM, Prakriya M, Nakashima YM, Nilsson KR, Han M, Zorumski CF, et al. Enantioselective blockade of t-type Ca2+ current in adult rat sensory neurons by a steroid that lacks γ-aminobutyric acid-modulatory activity. Mol Pharmacol. 1998;54:918–927. doi: 10.1124/mol.54.5.918. [DOI] [PubMed] [Google Scholar]
- Todorovic SM, Jevtovic-Todorovic V, Meyenburg A, Mennerick S, Perez-Reyes E, Romano C, et al. Redox modulation of T-type calcium channels in rat peripheral nociceptors. Neuron. 2001a;31:75–85. doi: 10.1016/s0896-6273(01)00338-5. [DOI] [PubMed] [Google Scholar]
- Todorovic SM, Jevtovic-Todorovic V, Mennerick S, Perez-Reyes E, Zorumski CF. Cav3.2 channels is a molecular substrate for inhibition of T-type calcium currents in rat sensory neurons by nitrous oxide. Mol Pharmacol. 2001b;60:603–610. [PubMed] [Google Scholar]
- Todorovic SM, Pathirathna S, Brimelow BC, Jagodic MM, Ko SH, Jiang X, et al. 5β-reduced neuroactive steroids are novel voltage-dependent blockers of T-type Ca2+ channels in rat sensory neurons in vitro and potent peripheral analgesics in vivo. Mol Pharmacol. 2004a;66:1223–1235. doi: 10.1124/mol.104.002402. [DOI] [PubMed] [Google Scholar]
- Todorovic SM, Meyenburg A, Jevtovic-Todorovic V. Redox modulation of T-type calcium channels in vivo: alteration of nerve injury-induced thermal hyperalgesia. Pain. 2004b;109:328–339. doi: 10.1016/j.pain.2004.01.026. [DOI] [PubMed] [Google Scholar]
- Traboulsie A, Chemin J, Chevalier M, Quignard JF, Nargeot J, Lory P. Subunit-specific modulation of T-type calcium channels by zinc. J Physiol. 2007;578(Pt 1):159–171. doi: 10.1113/jphysiol.2006.114496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viana F, Pena E, Belmonte C. Specificity of cold thermotransduction is determined by differential ionic channel expression. Nat Neurosci. 2002;5:254–260. doi: 10.1038/nn809. [DOI] [PubMed] [Google Scholar]
- Welsby PJ, Wang H, Wolfe JT, Colbran RJ, Johnson ML, Barrett PQ. A mechanism for the direct regulation of T-type calcium channels by Ca2+/calmodulin-dependent kinase II. J Neurosci. 2003;23:10116–10121. doi: 10.1523/JNEUROSCI.23-31-10116.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen XJ, Li ZJ, Chen ZX, Fang ZY, Yang CX, Li H, et al. Intrathecal administration of Cav3.2 and Cav3.3 antisense oligonucleotide reverses tactile allodynia and thermal hyperalgesia in rats following chronic compression of dorsal root of ganglion. Acta Pharmacol Sin. 2006;27:1547–1552. doi: 10.1111/j.1745-7254.2006.00461.x. [DOI] [PubMed] [Google Scholar]
- White G. GABAA-receptor-activated current in dorsal root ganglion neurons freshly isolated from adult rats. J Neurophysiol. 1990;64:57–63. doi: 10.1152/jn.1990.64.1.57. [DOI] [PubMed] [Google Scholar]
- Wolfe JT, Wang H, Perez-Reyes E, Barrett PQ. Stimulation of recombinant Ca(v)3.2, T-type, Ca(2+) channel currents by CaMKIIgamma(C) J Physiol. 2002;538(Pt 2):343–355. doi: 10.1113/jphysiol.2001.012839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfe JT, Wang H, Howard J, Garrison JC, Barrett PQ. T-type calcium channel regulation by specific G-protein betagamma subunits. Nature. 2003;424:209–213. doi: 10.1038/nature01772. [DOI] [PubMed] [Google Scholar]
- Wu ZZ, Chen SR, Pan HL. Differential sensitivity of N- and P/Q-type Ca2+ channel currents to a mu opioid in isolectin B4-positive and -negative dorsal root ganglion neurons. J Pharmacol Exp Ther. 2004;311:939–947. doi: 10.1124/jpet.104.073429. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Cribbs LL, Satin J. Arachidonic acid modulation of alpha1H, a cloned human T-type calcium channel. Am J Physiol Heart Circ Physiol. 2000;278:H184–H193. doi: 10.1152/ajpheart.2000.278.1.H184. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Vilaythong AP, Yoshor D, Noebels JL. Elevated thalamic low-voltage-activated currents precede the onset of absence epilepsy in the SNAP25-deficient mouse mutant coloboma. J Neurosci. 2004;24:5239–5248. doi: 10.1523/JNEUROSCI.0992-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong X, Liu JR, Kyle JW, Hanck DA, Agnew WS. A profile of alternative RNA splicing and transcript variation of CACNA1H, a human T-channel gene candidate for idiopathic generalized epilepsies. Hum Mol Genet. 2006;15:1497–1512. doi: 10.1093/hmg/ddl068. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.