

Abstract
BACKGROUND AND PURPOSE
Glutamate transporters play a major role in maintaining brain homeostasis and the astrocytic transporters, EAAT1 and EAAT2, are functionally dominant. Astrocytic excitatory amino acid transporters (EAATs) play important roles in various neuropathologies wherein astrocytes undergo cytoskeletal changes. Astrocytic plasticity is well documented, but the interface between EAAT function, actin and the astrocytic cytoskeleton is poorly understood. Because Rho kinase (ROCK) is a key determinant of actin polymerization, we investigated the effects of ROCK inhibitors on EAAT activity and astrocytic morphology.
EXPERIMENTAL APPROACH
The functional activity of glutamate transport was determined in murine cultured astrocytes after exposure to the ROCK inhibitors Fasudil (HA-1077) and Y27632 using biochemical, molecular and morphological approaches. Cytochemical analyses assessed changes in astrocytic morphology, F-/G-actin, and localizations of EAAT1/2.
RESULTS
Fasudil and Y27632 increased [3H]-d-aspartate (d-Asp) uptake into astrocytes, and the action of Fasudil was time-dependent and concentration-related. The rapid stellation of astrocytes (glial fibrillary acidic protein immunocytochemistry) induced by Fasudil was accompanied by reduced phalloidin staining of F-actin and increased Vmax for [3H]-d-Asp uptake. Immunoblotting after biotinylation demonstrated that Fasudil increased the expression of EAAT1 and EAAT2 on the cell surface. Immunocytochemistry indicated that Fasudil induced prominent labelling of astrocytic processes by EAAT1/2.
CONCLUSION AND IMPLICATIONS
These data show for the first time that ROCK plays a major role in determining the cell surface expression of EAAT1/2, providing new evidence for an association between transporter function and astrocytic phenotype. ROCK inhibitors, via the actin cytoskeleton, effect a consequent elevation of glutamate transporter function – this activity profile may contribute to their beneficial actions in neuropathologies.
Keywords: glutamate transporters, astrocytic phenotype, Rho kinase, Fasudil, glial scarring, actin, cell surface expression
Introduction
Active transport via sodium-dependent excitatory amino acid transporters (EAATs) is the primary mechanism maintaining homeostasis of extracellular l-glutamate (Glu), thereby preventing cell death due to excitotoxicity. EAAT1 [human equivalent glutamate-aspartate transporter (GLAST)] and EAAT2 [glutamate transporter 1 (GLT1)] in glial cells, particularly astrocytes, are responsible for the bulk of Glu uptake in the central nervous system (CNS) (Danbolt, 2001; Beart and O'Shea, 2007). Various manipulations of EAATs are beneficial under pathological conditions where EAAT function is impaired (Beart and O'Shea, 2007; Sheldon and Robinson, 2007). Mechanisms modulating the abundance and function of astrocytic EAATs are not well understood, yet a number of experimental treatments directed at cell surface and intracellular mechanisms that result in phenotypic changes resembling astrocytic activation have been shown to increase the activity or abundance of EAATs (Danbolt, 2001; Beart and O'Shea, 2007; Sheldon and Robinson, 2007). Astrocytes, well recognized as extremely plastic cells, play key roles in maintaining brain function via energetics, antioxidant activity, trophic factor synthesis, neurovascular coupling and Glu homeostasis (Ridet et al., 1997; Maragakis and Rothstein, 2006). Transport of Glu into astrocytes is another of their functions with EAAT2 (∼1% of proteome) considered an astrocyte-specific protein like the cytoskeletal marker glial fibrillary acidic protein (GFAP) (Cahoy et al., 2008) – additionally both proteins co-localize and GFAP modulates EAAT function (Hughes et al., 2004; Zhou and Sutherland, 2004; Shobha et al., 2007). Indeed, GFAP expression increases in astrocytes when they become reactive (termed ‘astrogliosis’) (Ridet et al., 1997; Maragakis and Rothstein, 2006) with probable alterations in EAAT biology, as we have shown an interdependence between astrocytic phenotype and EAAT function that involves altered transporter activity and/or localization (O'Shea et al., 2006; Zagami et al., 2009; Lau et al., 2010). Astrogliosis is now documented to be a graded event with both pro-survival and destructive components (Sofroniew and Vinters, 2010), and it is apparent that maintenance of EAAT function during brain perturbation (i.e. drug treatment, trauma or neurodegeneration) when astrogliosis occurs is of fundamental importance.
Actin is central to morphological changes of cells and exists in two forms: a filamentous polymer called F-actin and a globular monomer, known as G-actin. F-actin represents the polymerized form of actin, displaying stress fibres and focal adhesions, while G-actin is the depolymerized form of actin, recognizable by disappearance of these stress fibres and focal adhesions. In astrocytes, like all cells, the actin cytoskeleton is known to be regulated tightly by a family of small G-proteins called Rho GTPases (Rho, Rac, Cdc42) (Hall, 1998; Luo, 2002), which have been linked to distinct sets of actin-rich structures regulating dynamic surface protrusions, such as lamellipodia and filopodia, fundamental determinants of motility and migratory potential (Le Clainche and Carlier, 2008; Mattila and Lappalainen, 2008). Astrocytic morphology thereby is affected by the regulation of Rho-GTPases through their involvement in actin dynamics, by influencing the existing equilibrium between G-actin and F-actin. The three best-characterized Rho-GTPases are RhoA, Rac and Cdc42, which regulate the depolymerization/polymerization of actin via downstream, interactive signalling cascades (Etienne-Manneville and Hall, 2002; Luo, 2002; Raftopoulou and Hall, 2004). Active RhoA increases the stability of F-actin and acts via Rho associated coiled-coil containing kinase (ROCK) – not only is the ROCK/Rho system an important regulator via the actin cytoskeleton of cellular migration, but it also has determinant roles in cellular proliferation and survival (Riento and Ridley, 2003). Inhibitors of ROCK have shown potential as drugs that provide benefit for the management of various neurological disorders, including multiple sclerosis, spinal and hypoxic/ischaemic injury (Mueller et al., 2005; 2009;), probably by the inhibition of inflammatory events related to glial scarring (Mueller et al., 2005; Ding et al., 2010; Yu et al., 2010). Glial scar formation is part of the spectrum of adaptive changes in reactive astrogliosis and represents a physical barrier to regenerative events that follow a focal brain injury – in this milieu astrocytes becoming hypertrophic and proliferate (Sofroniew and Vinters, 2010).
Studies have shown that astrocytic process formation is accompanied by a shift from F-actin to G-actin; that is depolymerization of actin filaments (Goldman and Chiu, 1984; Goldman and Abramson, 1990; Baorto et al., 1992). Astrocytic morphology can be altered by molecules acting on the actin cytoskeletal signalling pathways, and EAAT expression and activity changes are often associated with altered astrocytic morphology. Such changes have been well documented for dibutyryl cAMP (Schlag et al., 1998) and by extensive work in our own laboratory (O'Shea et al., 2006; Zagami et al., 2009; Lau et al., 2010). Major cellular mechanisms considered to be important in the rearrangement of the actin cytoskeleton include cAMP, intracellular calcium and small Rho-GTPases (Kuhn et al., 2000). Of particular interest here is the recent description by Abe and Misawa (2003) of Rho kinase inhibitors producing rapid stellation of astrocytes. Thus, in the current study we sought to determine how the inhibition of ROCK activity affects the relationship between EAAT activity and astrocytic morphology. Using primary astrocytes in culture we describe for the first time that ROCK inhibition via the actin cytoskeleton produces rapid stellation with increased cell surface expression of EAATs and elevated transporter activity – a profile of activity that may contribute to beneficial actions of ROCK inhibitors in neuropathologies (Mueller et al., 2009). Preliminary accounts of this work have been presented (Lau et al., 2008; O'Shea et al., 2009).
Methods
The investigation received appropriate institutional ethical approval and was undertaken according to the guidelines of the National Health and Medical Research Council (Australia). The nomenclature used here for Glu transporters conforms to the BJP Guide for Receptors and Channels (Alexander et al., 2009).
Cell culture
The establishment of astrocytic cultures from the brains of C57BL/6 mice (post-natal day 1.5) was performed as described previously (O'Shea et al., 2006). Briefly, forebrains were dissected in ice-cold solution (Hanks balanced salt salution: 137 mM NaCl, 5.37 mM KCl, 4.1 mM NaHCO3, 0.44 mM KH2PO4, 0.13 mM Na2HPO4, 10 mM HEPES, 1 mM sodium pyruvate, 13 mM d(+)glucose, 0.01 g·L−1 phenol red), containing 3 mg·mL−1 BSA and 1.2 mM MgSO4, pH 7.4). Cells were chemically and mechanically dissociated, centrifuged, and the pellet resuspended in astrocytic medium [AM: DMEM, Dulbecco's modified eagle medium, 10% FBS, 100 U·mL−1 penicillin/streptomycin, 0.25% (v·v−1) Fungizone], preheated to 36.5°C at a volume of 5 mL per brain and plated at 10 mL per 75 cm2 flask. Cells were maintained in a humidified incubator supplied with 5% CO2 at 36.5°C and complete medium changes were carried out twice weekly. When a confluent layer had formed (10 Division), the cells were shaken overnight (180 r.p.m.) and rinsed in fresh medium to remove non-astrocytic cells. Astrocytes were subsequently detached using 5 mM EDTA (10 min at 37°C) and plated into 24-well plates or onto coverslips at 1 × 104 cells·cm−2, and incubated in a humidified atmosphere at 36.5°C with 5% CO2 overnight. A full medium change was performed to remove non-adherent cells and medium was subsequently changed every 3–4 days thereafter until cells were ready for use.
Cytochemistry and confocal microscopy
The procedures used have been described previously (O'Shea et al., 2006; Lau et al., 2010). Astrocytes on coverslips were washed with phosphate buffered saline (PBS: 137 mM NaCl, 0.5 M Na2HPO4, 0.5 M NaH2PO4, pH 7.4) and fixed in 4% paraformaldehyde in PBS for 10 min, followed by three washes with Tris buffered saline (TBS: 50 mM Tris–HCl, 1.5% NaCl, pH 7.6). Non-specific binding was blocked with 10% normal goat serum/normal donkey serum (NGS/NDS) in TBS containing 0.3% Triton X-100. Following three further washes, astrocytes were incubated with primary antibodies {[EAAT1: GLAST anti-A522 (ID: Ab 141), 1:1500; EAAT2: GLT1 anti-B12 (ID: Ab 150), (1:1500) (Danbolt et al., 1998)]; GFAP 1:3000} at 4°C overnight on a rocker. Astrocytes were then washed and incubated with secondary antibodies (Alexa Fluor 488 for GFAP, Alexa Fluor 647 for EAATs) and rhodamine-conjugated phalloidin (1:3333) for 3 h at room temperature. Astrocytes were then washed with TBS and observed using an Olympus FV1000 confocal microscope using a 100× oil immersion objective lens. Nuclear staining: Hoechst 33 342 (1:500) was included in the incubation with secondary antibodies, diluted in 2% (v·v−1) NGS or/and NDS in PBS containing 0.3% (v·v−1) Triton X-100, and incubated for 3 h at room temperature. Labelling of F-/G-actin concurrently was essentially as described with the following modifications: after the initial wash, astrocytes were incubated for 1 min in stabilizing solution [10 mM Tris + 0.15 M NaCl, 0.01% Triton X-100, 2 mM MgCl2, 0.2 mM DTT, 10% glycerol (v·v−1)] at 4°C. After fixation, cells were permeabilized with 0.5% Triton X-100 in PBS for 5 min followed by 15 min incubation with blocking solution (2% BSA, 0.1% Triton X-100 in PBS). Cells were again washed with PBS twice. Staining for F-actin and G-actin was carried out using rhodamine-conjugated phalloidin (1:3333) and Alexa Fluor 488-conjugated deoxyribonuclease I (DNAseI, 1:250), respectively, in blocking solution for 30 min in the dark.
Image analysis
Analysis of staining for F-Actin and G-actin was performed using ImageJ software (obtained from the NIH web site: http://rsb.info.nih.gov/ij). Images were converted to grayscale and the area above threshold was determined for each marker. The same threshold brightness level was used for all images stained for the same marker. Sixteen images per group, spread across two independent experiments, were analysed using Student's t-test.
Advanced analysis of astrocytic morphology was performed using an updated version of CSIRO's HCA-Vision software (Vallotton et al., 2007) adapted for cultured astrocytes. In order to make measurements on a per-cell basis, a surrogate for cell extent was used, because cells overlap and immunocytochemistry for GFAP does not label fine processes. We used a surrogate for cell extent commonly used in high throughput screening, that is, a doughnut or ring around the nucleus (labelled with Hoechst 33 342). If cells are closely packed, the pseudo-cellular regions from neighbouring nuclei deform to the mid-point between the two nuclei. This gives rise to pseudo-cellular regions approximating the actual cell shape. Within these pseudo-cellular regions, linear structures of GFAP were then identified using a sophisticated linear feature detector (Sun and Vallotton, 2009), capable of detecting lines that vary in both brightness and width even in the presence of noise and varying background brightness. These linear structures were then quantified by their line density and line angle variance. Line density is the proportion of each pseudo-cellular region covered by lines, while line angle variance is the variance of the orientation angles of these lines within the pseudo-cellular region. For a circular variable like the orientation angle of a line, the angle variance is defined as follows. Let Ai be the angle relative to the horizontal for lines numbered i = 1, 2, …, N, where Ai lies between −90° and +90°. Angle variance = 1 −✓(Σsin(2*π*Ai/180)2+Σcos(2*π*Ai/180)2)/N. Statistical significance was determined using Student's t-test.
Quantification of cellular viability and death
Cellular viability was assessed using a 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT; an index of mitochondrial function) reduction assay (Cheung et al., 1998). After treatment, MTT was added to the wells give a final concentration of 0.5 mg·mL−1, incubated with the cells at 36.5°C with 5% CO2 for 30 min. Media were aspirated and 300 µL of dimethyl sulphoxide was added into each well to dissolve the formazan product. The absorbance was subsequently measured at 570 nm using a Bio-Rad Benchmark Plus microplate spectrophometer. Lactate dehydrogenase assay was carried out using a commercially available kit (Roche). Medium (50 µL) was collected from each well and placed in a 96-well plate. The samples were incubated with the reaction mixture (Cytotoxicity Detection Kit from Roche, Sydney, Australia) according to the manufacturer's protocol and left in the dark for 30 min. The absorbance of the sample mixture was determined at 490 nm using a Bio-Rad Benchmark Plus microplate spectrophometer. Data were subjected to two-tailed, unpaired t-test using GraphPad Prism v.4.0 (GraphPad, San Diego, CA, USA).
[3H]-d -aspartate uptake
Excitatory amino acid transporter activity was determined using [3H]-d-aspartate ([3H]-d-Asp) uptake (Aprico et al., 2004). Cells were pre-incubated at 37°C for 5 min and then incubated with [3H]-d-Asp (50 nM, 5 min) with or without unlabelled d-Asp (0.1 µM–1 mM) or with 1 mM Glu (non-specific uptake) in uptake buffer (135 mM NaCl, 5 mM KCl, 0.6 mM MgSO4, 1 mM CaCl2, 6 mM d-glucose, 10 mM HEPES, pH 7.5). Uptake was terminated by washing at 4°C, and accumulated radioactivity determined by scintillation spectrometry (O'Shea et al., 2006). Analysis of data from studies of [3H]-d-Asp uptake was performed using GraphPad Prism v.4.0 (GraphPad, San Diego, CA, USA), as described in Results.
Biotinylation of cell surface proteins and Western blot analysis
This protocol was performed as described previously (O'Shea et al., 2006; Lau et al., 2010). Samples were pooled from six wells (n = 6 replicates), where total cell protein concentration was determined with the Bio-Rad Dc Assay Kit (Sydney, Australia) according to the manufacturer's specifications. Standard Western blot protocols were carried out with equal volumes of three fractions were loaded onto gels and membranes were incubated with primary antibodies [GLAST anti-A522, 1:15 000 (Danbolt et al., 1998); GLT1 1:10 000 (Williams et al., 2005)] overnight at 4°C. Following washing, membranes were incubated with horseradish-peroxidase-conjugated secondary antibodies (goat anti-rabbit IgG, 1:1000 or goat anti-mouse IgG, 1:1000) for 3 h at room temperature. Proteins were then visualized using enhanced chemiluminescence. As a control for protein loading, blots were subsequently probed for β-actin (primary antibody 1:10 000) using the same procedures. Densitometric analysis of Western blots was performed using ImageJ software (http://rsb.info.nih.gov.ij/ National Institutes of Health, Bethesda, MD, USA) to measure the area and density of proteins bands after subtracting the background of the autoradiographic film (Cimarosti et al., 2005). Because EAATs are believed to function as trimers (Yernool et al., 2004) and might be broken down to monomers and dimers during processing of protein samples, densitometric analyses determined the combined optical density of all three molecular species. Data were subjected to two-tailed, unpaired t-test using GraphPad Prism v.4.0 (GraphPad, San Diego, CA, USA).
Data analysis
Details of statistical analyses are given above. All values are mean ± SEM and are from replicate observations (typically 3–6) from each of multiple independent experiments. Morphological observations employing confocal microscopy are from replicate culture wells with at least six photomicrographs per treatment from two or more independent experiments.
Drugs, chemical reagents and other materials
Hanks balanced salt salution, FBS, DMEM, Fungizone and penicillin/streptomycin were obtained from Gibco Invitrogen Corporation (Melbourne, Australia). Cells were grown in NUNC (Copenhagen, Denmark) culture flasks and Costar (Roskilde, Denmark) plates. Rhodamine–phalloidin, mouse anti-β-actin primary antibody and other chemicals not specified were obtained from Sigma-Aldrich (Sydney, Australia). Complete protease inhibitor cocktail tablets and Lumilight fluorescence solution were obtained from Roche Diagnostics (Castle Hill, Australia). EZ-linkTM Sulfo-NHS-Biotin and ImmunoPure Immobilized Avidin were from Pierce (Progen Biosciences, Queensland, Australia). [3H]-d-Asp and Packard Microscint PS scintillant were obtained from Perkin-Elmer Life Sciences Inc. (Boston, USA). Alexa Fluor 488-conjugated DNaseI and Alexa Fluor secondary antibodies were purchased from Molecular Probes (Melbourne, Australia). Fasudil, Y27632, NDS, NGS and mouse anti-GFAP antibody were obtained from Chemicon (Melbourne, Australia).
Results
Fasudil alters the organization of the actin cytoskeleton and GFAP, causing changes in astrocytic morphology
Under control conditions, murine astrocytes in culture grew as flattened, polygonal cells, most of which were GFAP-positive. Astrocytic cultures were treated with Fasudil at the concentration previously employed by Abe and Misawa (100 µM; Abe and Misawa, 2003). As visualized by phase contrast microscopy, treatment with Fasudil caused a dramatic change in astrocytic morphology, from a flattened polygonal shape to a stellate shape with spindly, elaborate processes within 24 h (Figure 1). This change occurred by retraction of cytoplasm, and became apparent within 30 min. Immunocytochemical staining for the astrocytic intermediate filament protein GFAP further demonstrated this change. The GFAP immunoreactivity was changed from a diffuse, cobblestone morphology (a typical epithelial cell-like appearance) around the nucleus to an elaborate, process-bearing morphology with condensed GFAP-positive fibrils. The effects of Fasudil on the actin-based cytoskeleton were examined by staining astrocytes with a high-affinity probe for F-actin, rhodamine-conjugated phalloidin. Control astrocytes displayed prominent well-organized actin fibres with densely packed stress fibres. In contrast, treatment with Fasudil caused dissipation of actin stress fibres, a change that was observed as early as 15 min following treatment (Figure 1) and there was subsequent thickening of the cytoplasmic ring as shown by the pattern of phalloidin labelling. In addition, the labelling of F-actin appeared to decrease in a time-dependent manner following treatment with Fasudil.
Figure 1.
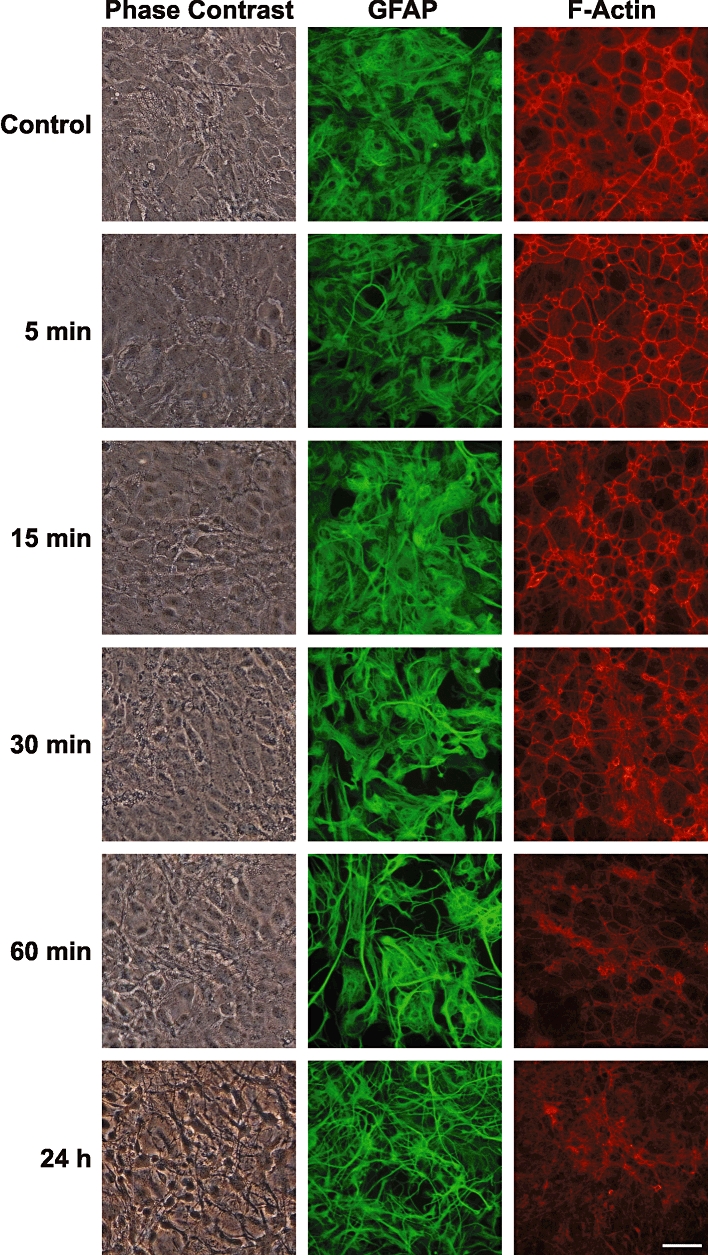
Time course of effects of Fasudil on astrocytic morphology and F-actin organization. Primary cultures of mouse astrocytes were treated with Fasudil (100 µM) and examined at various times using phase contrast microscopy (left column), GFAP immunocytochemistry (green) or labelling of F-actin with rhodamine-phalloidin (red). Scale bar = 50 µm.
Treatment with Fasudil (100 µM, 24 h) did not result in changes in astrocytic viability: significant changes were not observed in assays for cell death (lactate dehydrogenase assay) or cellular viability (MTT assay; data not shown).
More detailed analyses of the effects of Fasudil on the actin cytoskeleton were undertaken using fluorescent labelling of both F- and G-actin, using rhodamine-phalloidin and Alexa Fluor 488-conjugated DNaseI respectively (Figure 2). Image analysis revealed that Fasudil (100 µM, 24 h) decreased the abundance of F-actin (area above threshold decreased to 24 ± 5% of control) while increasing the labelling of G-actin (area above threshold increased to 480 ± 180% of control; both P < 0.05).
Figure 2.
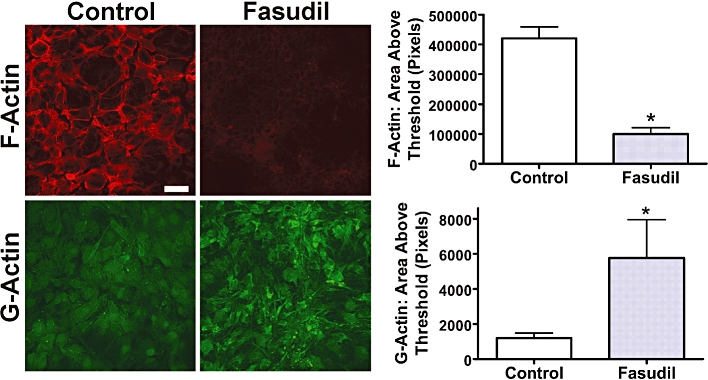
Effects of Fasudil on expression of F-actin and G-actin. Primary cultures of mouse astrocytes were treated with Fasudil (100 µM, 24 h), stained to identify F-actin (rhodamine-phalloidin; red) or G-actin (Alexa Fluor 488-conjugated DNaseI; green). Scale bar = 50 µm. Graphs demonstrate image analysis of area above threshold for F-actin and G-actin. *Significantly different from control, P < 0.05, Student's t-test. Data represent mean ± SEM (n = 16).
In order to study the changes in astrocytic morphology, the distribution of GFAP was analysed in images from confocal microscopy using more advanced algorithms. These analyses revealed significant changes in the arrangement of GFAP following treatment with Fasudil (Figure 3). Line angle variance (statistical mean for variance of the angles of ‘lines’ of GFAP staining within individual pseudo-cellular regions) was significantly reduced by Fasudil (to 81 ± 4% of control, P < 0.05), indicating a more aligned linear phenotype following treatment. In addition, the density of these lines within pseudo-cellular regions was also decreased (to 44 ± 5% of control, P < 0.05), demonstrating the decrease in the cellular area labelled by GFAP immunocytochemistry.
Figure 3.
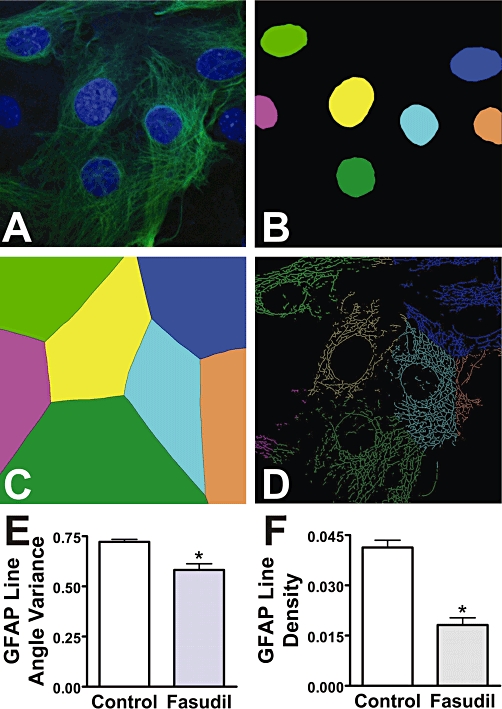
Morphological changes in astrocytes following treatment with Fasudil. Following treatment of mouse astrocytes with Fasudil (100 µM, 24 h), images of GFAP immunocytochemistry were subjected to advanced image analyses. (A) Original image showing staining of nuclei (blue) and GFAP (green). (B) Nuclei identified by the software and colour-coded. (C) Software-defined pseudo-cellular regions, colour-coded as per nuclei. (D) ‘Lines’ representing GFAP fibres, colour-coded as per nuclei. (E) Effects of Fasudil on cellular line angle variance for GFAP lines. (F) Effects of Fasudil on cellular line density for GFAP lines. *Significantly different from control, P < 0.05, Student's t-test. Data are from at least 43 images per condition across multiple independent cultures.
ROCK inhibitors up-regulate specific [3H]-d -aspartate uptake
To examine the specificity of the effects of Fasudil, primary astrocytic cultures were treated with Fasudil (100 µM) or another ROCK inhibitor Y-27632 (30 µM, 24 h). As demonstrated by labelling of F-actin (Figure 4), both compounds had similar effects on the astrocytic cytoskeleton, as described above. In addition, both ROCK inhibitors significantly increased the uptake of 3H]-d-Asp (Fasudil by 18 ± 4% and Y-27632 by 16 ± 3%; both P < 0.05).
Figure 4.
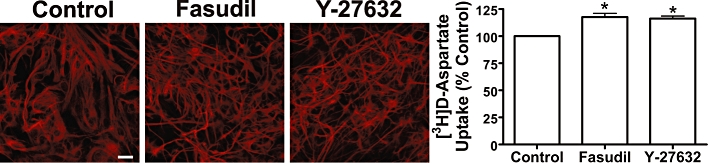
Effects of Rho kinase inhibition on astrocytic morphology and uptake of [3H]-d-aspartate mouse astrocytes. Treatment for 24 h with Fasudil (100 µM) or Y27632 (30 µM) produced similar changes in the pattern of GFAP expression. Scale bar = 50 µm. Both treatments significantly increased specific [3H]-d-aspartate uptake: one-way anova revealed a significant effect of treatment F[2,21] = 45.66, P < 0.01. *Significantly different from control, P < 0.05, Dunnett's multiple comparison post hoc test. Data represent mean ± SEM (n = 3–6).
The concentration dependence of the effects of Fasudil on [3H]-d-Asp uptake was examined employing a range of concentrations (1–100 µM) for 24 h. Results revealed a concentration-dependent increase in [3H]-d-Asp uptake, with a significant increase in uptake following treatment with 100 µM of Fasudil (142 ± 11% of control, P < 0.05; Figure 5A). The time-dependence of the effect of Fasudil on EAAT activity (1–72 h) was also investigated (Figure 5B), and a significant increase was observed at 24 h (144 ± 4% of control, P < 0.01), with further increases at 48 h (154 ± 5% of control, P < 0.01) and 72 h (187 ± 4% of control, P < 0.01).
Figure 5.
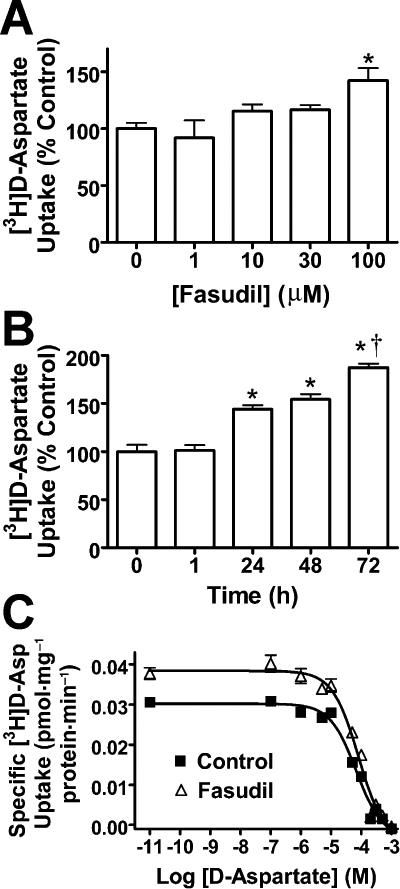
Detailed analyses of the effects of Fasudil on [3H]-d-aspartate uptake. (A) Concentration-related effects of Fasudil (24 h) on specific [3H]-d-Asp uptake. One-way anova revealed a significant effect of treatment F[4,15] = 4.268, P < 0.05. (B) Time-dependent effects of Fasudil (100 µM) on specific [3H]-d-aspartate uptake. One-way anova revealed a significant effect of treatment F[4,25] = 53.35, P < 0.0001. (C) Saturation analyses of [3H]-d-aspartate uptake into cultured mouse astrocytes. Data were fitted with a homologous competitive binding curve for one class of binding sites. Fasudil treatment produced a significant increase in Vmax, with no changes in KD. *Significantly different from control, P < 0.05; †significantly different from values at 24 and 48 h, P < 0.05, Dunnett's multiple comparison post hoc test. Data represent mean ± SEM (n = 9).
To examine how Fasudil affected EAAT activity, kinetic parameters of uptake (Vmax and KD) were determined by experiments examining the saturation analysis of [3H]-d-Asp uptake (Figure 5C). Fasudil treatment produced an increase in Vmax (control: 46 ± 5.4 pmol·mg−1·min−1; Fasudil: 71 ± 11 pmol·mg−1·min−1; P < 0.05) without significant change in KD[control: 68 (95% confidence interval 56–84) µM; Fasudil: 82 (63–110) µM, P > 0.05].
Cellular localization of EAAT1/2 by biotinylation and Western blotting
Western blotting for EAAT1 and EAAT2 in cultured mouse astrocytes revealed the presence of EAAT immunoreactivity in the biotinylated fraction (i.e. cell surface) as well as the cystolic fraction. While EAATs exist as functional trimers (Yernool et al., 2004) the disruptive nature of the procedures employed in biotinylation and cell lysis results in a complex mix of mono-, di- and trimeric species (Figure 6; cf. O'Shea et al., 2006) Following treatment with Fasudil (100 µM, 24 h), EAAT2 immunoreactivity was significantly increased in all fractions (Figure 6). While increases in cell surface expression of EAAT2 were in the order of ∼60%, greater increases were observed in cytosolic and total cellular fractions. In contrast to EAAT2, significant changes were not observed in either the total cellular EAAT1 or cytosolic compartments, yet significant increases (∼50%) were observed in the cell surface fraction (Figure 7). These changes could not be directly compared with cytosolic or total EAAT1 expression as samples from different fractions were processed independently. Western blotting for GFAP was also examined, and showed no changes following treatment (data not shown).
Figure 6.
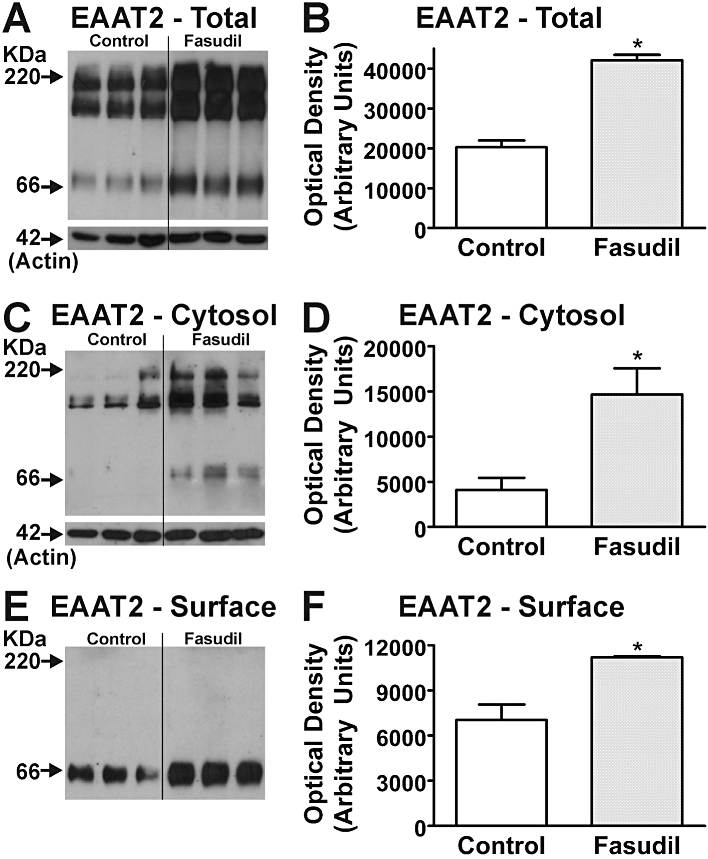
Effects of Fasudil on EAAT2 expression in mouse astrocytes. Western blots demonstrate EAAT2 expression in total cell (A), cytosolic (C) and cell surface (E) fractions of homogenates following treatment with Fasudil (100 µM, 24 h). Labelling of β-actin is included as a loading control. Each lane contains protein extracted from eight culture wells, with different lanes for each treatment being from independent cultures. Quantification of effects of Fasudil on abundance of EAAT2 protein in total cell (B), cytosolic (D) and cell surface (F) fractions of homogenates from cultured astrocytes. *Significantly different from control, P < 0.05, Student's t-test. Data in graphs represent mean ± SEM (n = 3).
Figure 7.
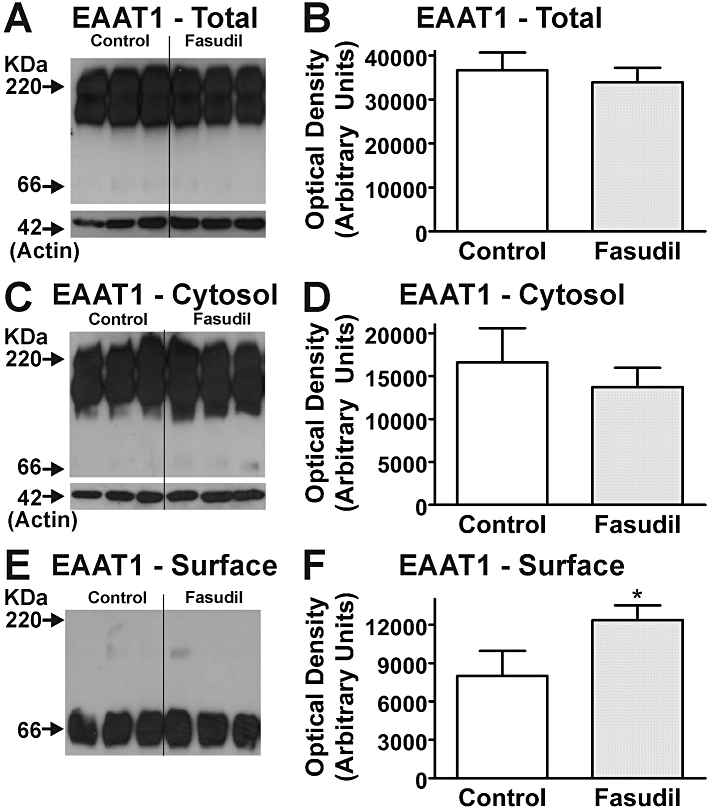
Effects of Fasudil on EAAT1 expression in mouse astrocytes. Western blots demonstrate EAAT1 expression in total cell (A), cytosolic (C) and cell surface (E) fractions of homogenates following treatment with Fasudil (100 µM, 24 h). Labelling of β-actin is included as a loading control. Each lane contains protein extracted from eight culture wells, with different lanes for each treatment being from independent cultures. Quantification of effects of Fasudil on abundance of EAAT1 protein in total cell (B), cytosolic (D) and cell surface (F) fractions of homogenates from cultured astrocytes. *Significantly different from control, P < 0.05, Student's t-test. Data in graphs represent mean ± SEM (n = 3).
GFAP, actin fibres and EAAT redistribution in astrocytes
Immunocytochemistry with confocal microscopy was used to investigate changes in cellular phenotype and the distribution of EAAT expression in murine cultured astrocytes, in concert with fluorescent labelling with rhodamine-conjugated phalloidin to detect F-actin (Figure 8). Stellation of astrocytes, demonstrated by GFAP was accompanied by a redistribution of EAAT2, where staining became more prominent, particularly at the margins (i.e. cell surface) of astrocytes. By comparison with EAAT2, the abundance of EAAT1 did not appear to increase in response to Fasudil treatment, although there was a net redistribution of EAAT1 resulting in a ∼50% increase at the cell surface. The redistribution of EAAT1 resulted in some clustering of immunoreactivity (see Figure 8).
Figure 8.
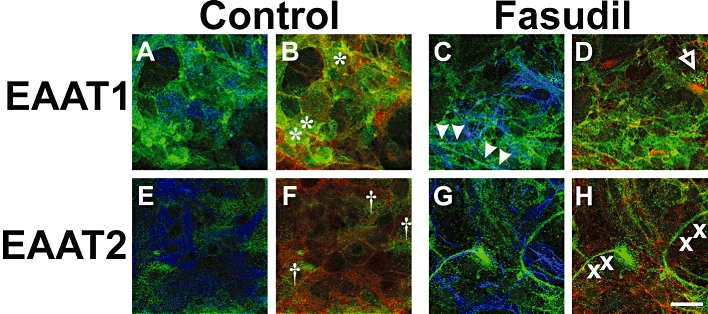
Effects of Fasudil on astrocytic morphology, F-actin organization and EAAT distribution. Control astrocytes (A, B, E, F) and cells treated with Fasudil (100 µM, 24 h; C, D, G, H) were stained for EAAT1 or EAAT2 (green) as well as the astrocytic protein GFAP (blue) or rhodamine-phalloidin to label F-Actin (red). In untreated astrocytes, EAAT1 tends to be dispersed over the cell body (* in panel B), while treatment with Fasudil results in a more punctate distribution of EAAT1 with some clustering (arrow in D) and alignment along astrocytic process (arrowheads in C). In contrast, EAAT2 is changed from a relatively uniform distribution over cell bodies († in F) to a more linear arrangement that does not coincide with processes or actin fibres (x in panel H). Scale bar = 30 µm.
Discussion
The majority of synapses in the CNS are in close apposition with astrocytes, and astrocytic EAATs are responsible for the bulk of Glu transport in the CNS. Numerous in vitro and in vivo studies have demonstrated that transgenic ablation, antisense down-regulation or pharmacological inhibition of astrocytic EAATs result in increased extracellular Glu and neuronal death (reviewed in Danbolt, 2001; Beart and O'Shea, 2007; Sheldon and Robinson, 2007). Additionally, a body of evidence suggests impaired Glu transport results in alterations to Glu metabolism or astrocytic function contributing to neuronal damage (Rae et al., 2000; Had-Aissouni et al., 2002; Re et al., 2003). Conversely, elevation of astrocytic EAAT activity is neuroprotective in pathological models involving excitotoxicity (Beart and O'Shea, 2007; Sheldon and Robinson, 2007). Given that EAAT activity contributes to the determinant role of astrocytes in regulating synaptic function and the growing evidence for an interdependency between astrocytic phenotype and EAAT function (see Introduction), we speculated that there might be a yet undetermined role for the ROCK/Rho system in this multifaceted relationship. We confirmed the observations of Abe and Misawa (2003) of stellation of cultured astrocytes after Rho kinase inhibition by employing Fasudil and Y-27632. However, the key findings of the present study were that Fasudil induced a shift in the F-/G-actin ratio towards a predominance of monomeric G-actin, indicating that actin was central to the cytoskeletal changes, and that there was an appreciable up-regulation of both Glu transport and the cell surface expression of EAAT1 and EAAT2. Because ROCK inhibitors have emerged as neuro-regenerative drug candidates, possibly by their actions to reduce glial scarring (Mueller et al., 2009), our finding of their capacity to elevate Glu transport reveals an additional beneficial action likely to contribute to their therapeutic benefits in brain injury.
There is a considerable literature that astrocytes are extremely plastic cells that exhibit various biochemical and morphological changes in response to changes in their milieu induced by both physiological and pathological events. Such changes appear to occur across a broad continuum of events, being influenced in disease and trauma by the extent of multiple factors, which appear able to cause both short- and long-term responses (Ridet et al., 1997; Maragakis and Rothstein, 2006). Reactive astrogliosis is well documented in many neurological diseases and there is recent evidence that astrocytes display the capacity to strike remodelling within minutes (Haber et al., 2006). We recently demonstrated that changes in astrocytic morphology, particularly those governed by the actin cytoskeleton, may be linked to the activity and cell surface expression of EAATs (O'Shea et al., 2006; Zagami et al., 2009; Lau et al., 2010). There is a growing amount of data suggesting that EAAT redistribution is likely to involve lipid rafts, dynamin- and/or clathrin-dependent mechanisms (Beart and O'Shea, 2007) and that these events are linked to signalling cascades that influence cytoskeletal mechanisms. Rho family small GTPases may play a key role in regulating astrocytic actin organization (see Beart and O'Shea, 2007) and astrocytic process formation is accompanied by changes in actin polymerization (see Introduction). Increasing evidence indicates that EAATs are subject to at least two broad types of regulation; acutely by rapid trafficking and internalization, and in the longer-term by adaptive mechanisms involving transcriptional induction (Schlag et al., 1998; Duan et al., 1999; Figiel et al., 2003; Fournier et al., 2004; Rothstein et al., 2005; O'Shea et al., 2006; Lau et al., 2010). The present studies provide new insights into both of these events and were initiated to determine the effects of ROCK inhibition, which is known to cause cytoskeletal rearrangement (Abe and Misawa, 2003), on the regulation of EAAT expression and trafficking. A further aim of this study was to explore how EAAT function varied with changes in astrocytic morphology and, in particular, to explore its actin dependence. The rapid stellation as shown by GFAP immunolabelling was accompanied by dissipation of actin stress fibres. Moreover, the pronounced shift in the F-/G-actin ratio towards a predominance of G-actin indicated actin was central to the cytoskeletal changes. Indeed, these comparatively early changes in the actin cytoskeleton and astrocytic phenotype were followed by a slower elevation of Glu transport. This evidence of increased Glu transport over a much delayed time course than the morphological changes is suggestive of differential gene regulation affecting not only Rho-associated motility and migration, but also exerting diverse downstream functional effects (O'Shea et al., 2009), including on EAAT function
The ROCK inhibitors Fasudil and Y27632 both increased the specific uptake of [3H]-d-Asp. The effects of Fasudil were investigated in detail, and were found to be concentration-related and time-dependent and were attributed to a significant increase in Vmax, but no change in KM, indicative of increases in transporter abundance. Mechanistic insights were obtained by biotinylation and immunoblot analyses, and revealed an approximate doubling in total cellular EAAT2 protein expression, with parallel increases in cytosolic EAAT2 and a substantial (60%) enhancement in cell surface expression. These findings are consistent with the longer-term up-regulation of EAAT2 synthesis in concert with increased net trafficking to the cell surface. These changes in function and expression of EAAT2 were accompanied by parallel studies with phalloidin, a marker of F-actin. Treatment with Fasudil resulted in extensive changes in the patterns of phalloidin labelling and breakdown of actin stress fibres (cf. Abd-El-Basset and Fedoroff, 1997). Stellation of astrocytes was accompanied by a redistribution of EAAT2, particularly at the margins (i.e. cell surface) of astrocytes, where staining became more prominent, consistent with our evidence from biotinylation and immunoblotting. The overall abundance of EAAT1 did not appear to increase in response to Fasudil treatment, although there was an ∼50% increase of EAAT1 at the cell surface, with some clustering of EAAT1 noted in immunocytochemical studies – this action might reflect externalization of an ‘occluded’ EAAT1 pool closely associated with the plasma membrane (see Beart and O'Shea, 2007). Net increased uptake of [3H]-d-Asp was mediated apparently by both EAAT1 and EAAT2 based upon the Western analyses – uptake studies in the presence of the selective EAAT2 blocker dihydrokainate (Arriza et al., 1994) supported this interpretation (data not shown). Changes in the distribution of both EAAT2 and EAAT1 were strikingly similar to those previously observed in astrocytes in response to the endotoxin lipopolysaccharide (O'Shea et al., 2006), which also elevated Glu transport and cell surface expression of EAAT2 with similar effects on astrocytic morphology. The observed changes in EAAT abundance may result from alterations in synthesis and/or stability of these proteins combined with altered trafficking from the cytosolic pools, but further insights are needed to clarify the contribution of each of these mechanisms.
Rho associated coiled-coil containing kinase inhibitors have attracted considerable attention in the area of regenerative neurobiology (Mueller et al., 2005, 2009), initially for their actions in spinal traumatic injury, but subsequently for their possible application in both acute and chronic degenerative conditions. While they certainly have actions on glial scarring (Mueller et al., 2005), a component of reactive astrogliosis, recent work has reported other effects of Fasudil including on inflammation linked to T-cell migration (Yu et al., 2010), toxic aggregate formation (Bauer et al., 2009) and neurogenesis (Ding et al., 2010). Fasudil also has anti-vasospatic actions, being used to treat cerebral vasospasm (Satoh et al., 2001; Nishizawa and Laher, 2005), and also delays neuronal death in ischaemic injury (Satoh et al., 2001). Such actions almost certainly arise from the complex involvement of the Rho/ROCK system not only in cellular morphology, contractility and migration, but also in cellular proliferation and survival (Riento and Ridley, 2003; Pellegrin and Mellor, 2007), so multiple downstream effects on different cellular populations would be expected in the brain. Interestingly, while we found very rapid effects of ROCK inhibitors on astrocytic motility and cytoskeletal reorganization, these actions were maintained over the time course of experimentation and elevation of Glu transporter function was a relatively late event. Our ongoing work has examined alterations in the astrocyte transcriptome induced by Fasudil and revealed extensive changes to gene expression in major biological processes regulating cellular shape and motility viz. the actin cytoskeleton, axon guidance, transforming growth factor-β signalling and tight junctions (O'Shea et al., 2009). The numerous pro-survival responses exhibited therein in astrocytes complement our detailed description here of the ability of ROCK inhibitors to increase astrocytic cell surface expression of EAATs and to elevate Glu transport. Given the widespread involvement of excitotoxicity in acute and chronic neuropathologies (Beart and O'Shea, 2007), this action of ROCK inhibitors represents an unexpected beneficial mechanism likely to contribute to their therapeutic benefits in brain injury.
Acknowledgments
Supported by a Project Grant (#509319) from the NH&MRC (Australia). CLL and PMB acknowledge receipt of Dora Lush Postgraduate Scholarship and Research Fellowship, respectively, from the NH&MRC. Gifts of antibodies from Niels Danbolt (Oslo, Norway) and David Pow (Brisbane, Australia) are greatly appreciated. The authors acknowledge the advice received from Michael Robinson (Philadelphia, USA) with respect to the biotinylation procedures.
Glossary
Abbreviations
- CNS
central nervous system
- DNaseI
deoxyribonuclease I
- EAAT2 (GLT1)
glutamate transporter 1
- EAATs
excitatory amino acid transporters
- GFAP
glial fibrillary acidic protein
- GLAST
glutamate-aspartate transporter
- Glu
l-glutamate
- MTT
3-(4,5-dimethylthiazol-2yl)-2,5-diphenyltetrazolium bromide
- NDS
normal donkey serum
- NGS
normal goat serum
- ROCK
Rho associated coiled-coil containing kinase
- Y27632
(R)-(+)-trans-N-(4-pyridyl)-4-(1-aminoethyl)-cyclohexane-carboxamide dihydrochloride
Conflict of interest
The authors state no conflict of interest.
Supporting Information
Teaching Materials; Figs 1–8 as PowerPoint slide.
References
- Abd-El-Basset EM, Fedoroff S. Upregulation of F-actin and alpha-actinin in reactive astrocytes. J Neurosci Res. 1997;49:608–616. doi: 10.1002/(SICI)1097-4547(19970901)49:5<608::AID-JNR11>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- Abe K, Misawa M. Astrocyte stellation induced by Rho kinase inhibitors in culture. Brain Res Dev Brain Res. 2003;143:99–104. doi: 10.1016/s0165-3806(03)00096-8. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Mathie A, Peters JA. Guide to receptors and channels (GRAC), 4th edition (2009) Br J Pharmacol. 2009;158(Suppl 1):S1–S254. doi: 10.1111/j.1476-5381.2009.00499.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aprico K, Beart PM, Crawford D, O'Shea RD. Binding and transport of [3H](2S,4R)-4-methylglutamate, a new ligand for glutamate transporters, demonstrate labeling of EAAT1 in cultured murine astrocytes. J Neurosci Res. 2004;75:751–759. doi: 10.1002/jnr.20013. [DOI] [PubMed] [Google Scholar]
- Arriza JL, Fairman WA, Wadiche JI, Murdoch GH, Kavanaugh MP, Amara SG. Functional comparisons of three glutamate transporter subtypes cloned from human motor cortex. J Neurosci. 1994;14:5559–5569. doi: 10.1523/JNEUROSCI.14-09-05559.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baorto DM, Mellado W, Shelanski ML. Astrocyte process growth induction by actin breakdown. J Cell Biol. 1992;117:357–367. doi: 10.1083/jcb.117.2.357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer PO, Wong HK, Oyama F, Goswami A, Okuno M, Kino Y, et al. Inhibition of Rho kinases enhances the degradation of mutant huntingtin. J Biol Chem. 2009;284:13153–13164. doi: 10.1074/jbc.M809229200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beart PM, O'Shea RD. Transporters for L-glutamate: an update on their molecular pharmacology and pathological involvement. Br J Pharmacol. 2007;150:5–17. doi: 10.1038/sj.bjp.0706949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cahoy JD, Emery B, Kaushal A, Foo LC, Zamanian JL, Christopherson KS, et al. A transcriptome database for astrocytes, neurons, and oligodendrocytes: a new resource for understanding brain development and function. J Neurosci. 2008;28:264–278. doi: 10.1523/JNEUROSCI.4178-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung NS, Pascoe CJ, Giardina SF, John CA, Beart PM. Micromolar L-glutamate induces extensive apoptosis in an apoptotic-necrotic continuum of insult-dependent, excitotoxic injury in cultured cortical neurones. Neuropharmacology. 1998;37:1419–1429. doi: 10.1016/s0028-3908(98)00123-3. [DOI] [PubMed] [Google Scholar]
- Cimarosti H, Jones NM, O'Shea RD, Pow DV, Salbego C, Beart PM. Hypoxic preconditioning in neonatal rat brain involves regulation of excitatory amino acid transporter 2 and estrogen receptor alpha. Neurosci Lett. 2005;385:52–57. doi: 10.1016/j.neulet.2005.05.006. [DOI] [PubMed] [Google Scholar]
- Danbolt NC. Glutamate uptake. Prog Neurobiol. 2001;65:1–105. doi: 10.1016/s0301-0082(00)00067-8. [DOI] [PubMed] [Google Scholar]
- Danbolt NC, Lehre KP, Dehnes Y, Chaudhry FA, Levy LM. Localization of transporters using transporter-specific antibodies. Methods Enzymol. 1998;296:388–407. doi: 10.1016/s0076-6879(98)96028-1. [DOI] [PubMed] [Google Scholar]
- Ding J, Li QY, Yu JZ, Wang X, Sun CH, Lu CZ, et al. Fasudil, a Rho kinase inhibitor, drives mobilization of adult neural stem cells after hypoxia/reoxygenation injury in mice. Mol Cell Neurosci. 2010;43:201–208. doi: 10.1016/j.mcn.2009.11.001. [DOI] [PubMed] [Google Scholar]
- Duan S, Anderson CM, Stein BA, Swanson RA. Glutamate induces rapid upregulation of astrocyte glutamate transport and cell-surface expression of GLAST. J Neurosci. 1999;19:10193–10200. doi: 10.1523/JNEUROSCI.19-23-10193.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Etienne-Manneville S, Hall A. Rho GTPases in cell biology. Nature. 2002;420:629–635. doi: 10.1038/nature01148. [DOI] [PubMed] [Google Scholar]
- Figiel M, Maucher T, Rozyczka J, Bayatti N, Engele J. Regulation of glial glutamate transporter expression by growth factors. Exp Neurol. 2003;183:124–135. doi: 10.1016/s0014-4886(03)00134-1. [DOI] [PubMed] [Google Scholar]
- Fournier KM, Gonzalez MI, Robinson MB. Rapid trafficking of the neuronal glutamate transporter, EAAC1: evidence for distinct trafficking pathways differentially regulated by protein kinase C and platelet-derived growth factor. J Biol Chem. 2004;279:34505–34513. doi: 10.1074/jbc.M404032200. [DOI] [PubMed] [Google Scholar]
- Goldman JE, Abramson B. Cyclic AMP-induced shape changes of astrocytes are accompanied by rapid depolymerization of actin. Brain Res. 1990;528:189–196. doi: 10.1016/0006-8993(90)91657-3. [DOI] [PubMed] [Google Scholar]
- Goldman JE, Chiu FC. Dibutyryl cyclic AMP causes intermediate filament accumulation and actin reorganization in astrocytes. Brain Res. 1984;306:85–95. doi: 10.1016/0006-8993(84)90358-5. [DOI] [PubMed] [Google Scholar]
- Haber M, Zhou L, Murai KK. Cooperative astrocyte and dendritic spine dynamics at hippocampal excitatory synapses. J Neurosci. 2006;26:8881–8891. doi: 10.1523/JNEUROSCI.1302-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Had-Aissouni L, Re DB, Nieoullon A, Kerkerian-Le Goff L. Importance of astrocytic inactivation of synaptically released glutamate for cell survival in the central nervous system – are astrocytes vulnerable to low intracellular glutamate concentrations? J Physiol Paris. 2002;96:317–322. doi: 10.1016/s0928-4257(02)00022-0. [DOI] [PubMed] [Google Scholar]
- Hall A. Rho GTPases and the actin cytoskeleton. Science. 1998;279:509–514. doi: 10.1126/science.279.5350.509. [DOI] [PubMed] [Google Scholar]
- Hughes EG, Maguire JL, McMinn MT, Scholz RE, Sutherland ML. Loss of glial fibrillary acidic protein results in decreased glutamate transport and inhibition of PKA-induced EAAT2 cell surface trafficking. Brain Res Mol Brain Res. 2004;124:114–123. doi: 10.1016/j.molbrainres.2004.02.021. [DOI] [PubMed] [Google Scholar]
- Kuhn TB, Meberg PJ, Brown MD, Bernstein BW, Minamide LS, Jensen JR, et al. Regulating actin dynamics in neuronal growth cones by ADF/cofilin and rho family GTPases. J Neurobiol. 2000;44:126–144. [PubMed] [Google Scholar]
- Lau CL, Beart PM, O'Shea RD. Inhibition of Rho-Kinase Alters Astrocytic Morphology and Stimulates Cell-Surface Expression of Glutamate Transporters. Washington, DC: Society for Neuroscience; 2008. Program No. 732.9. 2008 Neuroscience Meeting Planner. Online. [Google Scholar]
- Lau CL, Beart PM, O'Shea RD. Transportable and non-transportable inhibitors of L-glutamate uptake produce astrocytic stellation and increase EAAT2 cell surface expression. Neurochem Res. 2010;35:735–742. doi: 10.1007/s11064-010-0130-6. [DOI] [PubMed] [Google Scholar]
- Le Clainche C, Carlier MF. Regulation of actin assembly associated with protrusion and adhesion in cell migration. Physiol Rev. 2008;88:489–513. doi: 10.1152/physrev.00021.2007. [DOI] [PubMed] [Google Scholar]
- Luo L. Actin cytoskeleton regulation in neuronal morphogenesis and structural plasticity. Annu Rev Cell Dev Biol. 2002;18:601–635. doi: 10.1146/annurev.cellbio.18.031802.150501. [DOI] [PubMed] [Google Scholar]
- Maragakis NJ, Rothstein JD. Mechanisms of Disease: astrocytes in neurodegenerative disease. Nat Clin Pract Neurol. 2006;2:679–689. doi: 10.1038/ncpneuro0355. [DOI] [PubMed] [Google Scholar]
- Mattila PK, Lappalainen P. Filopodia: molecular architecture and cellular functions. Nat Rev Mol Cell Biol. 2008;9:446–454. doi: 10.1038/nrm2406. [DOI] [PubMed] [Google Scholar]
- Mueller BK, Mack H, Teusch N. Rho kinase, a promising drug target for neurological disorders. Nat Rev Drug Discov. 2005;4:387–398. doi: 10.1038/nrd1719. [DOI] [PubMed] [Google Scholar]
- Mueller BK, Mueller R, Schoemaker H. Stimulating neuroregeneration as a therapeutic drug approach for traumatic brain injury. Br J Pharmacol. 2009;157:675–685. doi: 10.1111/j.1476-5381.2009.00220.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishizawa S, Laher I. Signaling mechanisms in cerebral vasospasm. Trends Cardiovasc Med. 2005;15:24–34. doi: 10.1016/j.tcm.2004.12.002. [DOI] [PubMed] [Google Scholar]
- O'Shea RD, Lau CL, Farso MC, Diwakarla S, Zagami CJ, Svendsen BB, et al. Effects of lipopolysaccharide on glial phenotype and activity of glutamate transporters: evidence for delayed up-regulation and redistribution of GLT-1. Neurochem Int. 2006;48:604–610. doi: 10.1016/j.neuint.2005.12.028. [DOI] [PubMed] [Google Scholar]
- O'Shea RD, Lau CL, Sheean RK, Svendsen BB, Bischof L, Beart PM. Astrocyte biology, phenotype and EAAT activity. J Neurochem. 2009;110(Suppl 2):85. [Google Scholar]
- Pellegrin S, Mellor H. Actin stress fibres. J Cell Sci. 2007;120:3491–3499. doi: 10.1242/jcs.018473. [DOI] [PubMed] [Google Scholar]
- Rae C, Lawrance ML, Dias LS, Provis T, Bubb WA, Balcar VJ. Strategies for studies of neurotoxic mechanisms involving deficient transport of L-glutamate: antisense knockout in rat brain in vivo and changes in the neurotransmitter metabolism following inhibition of glutamate transport in guinea pig brain slices. Brain Res Bull. 2000;53:373–381. doi: 10.1016/s0361-9230(00)00372-5. [DOI] [PubMed] [Google Scholar]
- Raftopoulou M, Hall A. Cell migration: Rho GTPases lead the way. Dev Biol. 2004;265:23–32. doi: 10.1016/j.ydbio.2003.06.003. [DOI] [PubMed] [Google Scholar]
- Re DB, Boucraut J, Samuel D, Birman S, Kerkerian-Le Goff L, Had-Aissouni L. Glutamate transport alteration triggers differentiation-state selective oxidative death of cultured astrocytes: a mechanism different from excitotoxicity depending on intracellular GSH contents. J Neurochem. 2003;85:1159–1170. doi: 10.1046/j.1471-4159.2003.01752.x. [DOI] [PubMed] [Google Scholar]
- Ridet JL, Malhotra SK, Privat A, Gage FH. Reactive astrocytes: cellular and molecular cues to biological function. Trends Neurosci. 1997;20:570–577. doi: 10.1016/s0166-2236(97)01139-9. [DOI] [PubMed] [Google Scholar]
- Riento K, Ridley AJ. Rocks: multifunctional kinases in cell behaviour. Nat Rev Mol Cell Biol. 2003;4:446–456. doi: 10.1038/nrm1128. [DOI] [PubMed] [Google Scholar]
- Rothstein JD, Patel S, Regan MR, Haenggeli C, Huang YH, Bergles DE, et al. Beta-lactam antibiotics offer neuroprotection by increasing glutamate transporter expression. Nature. 2005;433:73–77. doi: 10.1038/nature03180. [DOI] [PubMed] [Google Scholar]
- Satoh S, Utsunomiya T, Tsurui K, Kobayashi T, Ikegaki I, Sasaki Y, et al. Pharmacological profile of hydroxy fasudil as a selective rho kinase inhibitor on ischemic brain damage. Life Sci. 2001;69:1441–1453. doi: 10.1016/s0024-3205(01)01229-2. [DOI] [PubMed] [Google Scholar]
- Schlag BD, Vondrasek JR, Munir M, Kalandadze A, Zelenaia OA, Rothstein JD, et al. Regulation of the glial Na+-dependent glutamate transporters by cyclic AMP analogs and neurons. Mol Pharmacol. 1998;53:355–369. doi: 10.1124/mol.53.3.355. [DOI] [PubMed] [Google Scholar]
- Sheldon AL, Robinson MB. The role of glutamate transporters in neurodegenerative diseases and potential opportunities for intervention. Neurochem Int. 2007;51:333–355. doi: 10.1016/j.neuint.2007.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shobha K, Vijayalakshmi K, Alladi PA, Nalini A, Sathyaprabha TN, Raju TR. Altered in-vitro and in-vivo expression of glial glutamate transporter-1 following exposure to cerebrospinal fluid of amyotrophic lateral sclerosis patients. J Neurol Sci. 2007;254:9–16. doi: 10.1016/j.jns.2006.12.004. [DOI] [PubMed] [Google Scholar]
- Sofroniew MV, Vinters HV. Astrocytes: biology and pathology. Acta Neuropathol. 2010;119:7–35. doi: 10.1007/s00401-009-0619-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun C, Vallotton P. Fast linear feature detection using multiple directional non-maximum suppression. J Microsc. 2009;234:147–157. doi: 10.1111/j.1365-2818.2009.03156.x. [DOI] [PubMed] [Google Scholar]
- Vallotton P, Lagerstrom R, Sun C, Buckley M, Wang D, De Silva M, et al. Automated analysis of neurite branching in cultured cortical neurons using HCA-Vision. Cytometry A. 2007;71:889–895. doi: 10.1002/cyto.a.20462. [DOI] [PubMed] [Google Scholar]
- Williams SM, Sullivan RK, Scott HL, Finkelstein DI, Colditz PB, Lingwood BE, et al. Glial glutamate transporter expression patterns in brains from multiple mammalian species. Glia. 2005;49:520–541. doi: 10.1002/glia.20139. [DOI] [PubMed] [Google Scholar]
- Yernool D, Boudker O, Jin Y, Gouaux E. Structure of a glutamate transporter homologue from Pyrococcus horikoshii. Nature. 2004;431:811–818. doi: 10.1038/nature03018. [DOI] [PubMed] [Google Scholar]
- Yu JZ, Ding J, Ma CG, Sun CH, Sun YF, Lu CZ, et al. Therapeutic potential of experimental autoimmune encephalomyelitis by Fasudil, a Rho kinase inhibitor. J Neurosci Res. 2010;88:1664–1672. doi: 10.1002/jnr.22339. [DOI] [PubMed] [Google Scholar]
- Zagami CJ, Beart PM, Wallis N, Nagley P, O'Shea RD. Oxidative and excitotoxic insults exert differential effects on spinal motoneurons and astrocytic glutamate transporters: implications for the role of astrogliosis in amyotrophic lateral sclerosis. Glia. 2009;57:119–135. doi: 10.1002/glia.20739. [DOI] [PubMed] [Google Scholar]
- Zhou J, Sutherland ML. Glutamate transporter cluster formation in astrocytic processes regulates glutamate uptake activity. J Neurosci. 2004;24:6301–6306. doi: 10.1523/JNEUROSCI.1404-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.