

Abstract
BACKGROUND AND PURPOSE
The peroxisome proliferator-activated receptor (PPAR)δ has been considered a therapeutic target for diabetes and obesity through enhancement of fatty acid oxidation. The present study aimed to characterize the effects of PPARδ agonists during insulin resistance of the whole body, muscle and liver.
EXPERIMENTAL APPROACH
Wistar rats and C57BL/J6 mice were fed a high fat diet (HF) and then treated with PPARδ agonists NNC61-5920 and GW501516. The effects on insulin resistance were evaluated by hyperinsulinaemic clamp or glucose tolerance tests combined with glucose tracers.
KEY RESULTS
In HF rats, 3 weeks of treatment with NNC61-5920 reduced the glucose infusion rate (by 14%, P < 0.05) and glucose disposal into muscle (by 20–30%, P < 0.01) during hyperinsulinaemic clamp. Despite increased mRNA expression of carnitine palmitoyltransferase-1, pyruvate dehydrogenase kinase 4 and uncoupling protein 3 in muscle, plasma and muscle triglyceride levels were raised (P < 0.01). Similar metabolic effects were observed after extended treatment with NNC61-5920 and GW501516 to 6 weeks. However, HF mice treated with NNC61-5920 improved their plasma lipid profile, glucose tolerance and insulin action in muscle. In both HF rats and mice, NNC61-5920 treatment attenuated hepatic insulin resistance and decreased expression of stearoyl-CoA desaturase 1, fatty acid translocase protein CD36 and lipoprotein lipase in liver.
CONCLUSIONS AND IMPLICATIONS
PPARδ agonists exacerbated insulin resistance in HF rats in contrast to their beneficial effects on metabolic syndrome in HF mice. These opposing metabolic consequences result from their different effects on lipid metabolism and insulin sensitivity in skeletal muscle of these two species.
Keywords: PPARδ, insulin sensitivity, lipid metabolism genes, rats, mice
Introduction
Insulin resistance is a major metabolic feature of type 2 diabetes and it is closely associated with dyslipidaemia and obesity. Although the mechanisms of insulin resistance are complex and may involve many factors, an excess accumulation of lipids in muscle and liver is an important and commonly observed contributing factor. As such elimination of this ectopic lipid accumulation often improves insulin sensitivity and glycaemia control. One such approach is to enhance fatty acid (FA) oxidation and catabolism in muscle, the largest organ for FA oxidation, which eventually reduces ectopic lipid accumulation to improve insulin sensitivity.
The peroxisome proliferator-activated receptor δ (PPARδ; receptor nomenclature follows Alexander et al., 2009) is a transcription factor ubiquitously expressed in the whole body and it is thought to mediate the expression of a number of genes to increase FA oxidation and mitochondrial biogenesis in muscle. Activation of PPARδ up-regulates carnitine palmitoyltransferase-1 (CPT1), uncoupling protein 3 (UCP3), pyruvate dehydrogenase kinase 4 (PDK4) and peroxisome proliferator-activated receptor γ coactivator -1α (PGC1α) and increases palmitate oxidation in muscle cells (Tanaka et al., 2003; Wang et al., 2003; Dimopoulos et al., 2007). Several studies in mice have shown improved glucose tolerance and muscle insulin sensitivity in both genetic and dietary models of insulin resistance after treatment with PPARδ agonists (Tanaka et al., 2003; Wang et al., 2003; Lee et al., 2006). Recently, the PPARδ agonist GW501516 has also been shown to reverse multiple metabolic abnormalities including reduction in plasma triglyceride, liver triglyceride content and insulin resistance (HOMA-IR) in moderately obese monkeys and men (Oliver et al., 2001; Riserus et al., 2008). These findings suggest that PPARδ may be a potential target for the treatment of insulin resistance, obesity and dyslipidaemia. However, the in vivo effects of PPARδ agonists on insulin resistance, particularly in muscle and liver, have not been well characterized.
Therefore, the initial aim of this study was to evaluate the therapeutic efficacy of NNC61-5920, a novel selective PPARδ agonist, on insulin resistance and lipid metabolism in the high fat-fed (HF) rats and to characterize its effects on insulin sensitivity and associated lipid metabolism in muscle and liver. Our findings show an unexpected worsening of muscle insulin resistance and triglyceride accumulation in HF rats after treatment with PPARδ agonists. Intriguingly, insulin resistance in HF mice was ameliorated in both muscle and liver along with correction of dyslipidaemia following NNC61-5920 administration, as previously reported for GW501516 (Tanaka et al., 2003; Wang et al., 2003; Lee et al., 2006). Our findings reveal distinct metabolic responses to PPARδ agonists between tissues and species and indicate a need for careful selection of preclinical species for characterization of the endpoint metabolic effects of PPARδ agonists intended for treatment of patients with the metabolic syndrome.
Methods
All animal care and experimental procedures were carried out with the approval of the Garvan Institute/St. Vincent's Hospital Animal Experimentation Ethics Committee, following guidelines issued by the National Health and Medical Research Council of Australia. Male Wistar rats and C57BL6/J mice were purchased from the Animal Resources Centre (Perth, Australia). The animals were kept in a temperature-controlled room (22 ± 1°C) on a 12 h light/dark cycle with free access to water. Mice and rats were fed ad libitum for 1 week on a standard chow diet (8% calories from fat, 21% calories from protein, 71% calories from carbohydrate, Gordon's Specialty Stock Feeds, Yanderra, New South Wales, Australia) and were then randomly allocated to remain on the chow (Ch) or to receive a HF diet. The HF diet consisted of ∼60% of fat as calories, as previously reported in detail (Kraegen et al., 1991).
Assessment of the metabolic effects of PPARδ activation in rats on a HF diet (HF rats).
Earlier studies in our laboratory showed a development of insulin resistance in liver and muscle in rats after 1 week of high-fat feeding (Kraegen et al., 1991). In the first study, the novel PPARδ agonist NNC61-5920 (Winzell et al., 2010) was administered as a food additive at 30 mg·kg−1·day−1 for 3 weeks starting 1 week after the rats (8 week of age, ∼200–220 g) were placed on the HF diet. One group of HF rats were treated with rosiglitazone (15 mg·kg−1·day−1) for 2 weeks based on our previous studies (Ye et al., 2003) as positive controls. In the second study, rats were fed a HF diet for 10 weeks and NNC61-5920 (15 mg·kg−1·day−1) was orally gavaged in 0.2 mL of 0.5% methylcellulose for the last 6 weeks of fat-feeding. In this extended study, rats were started on the HF diet from 4 weeks of age (approx 50 g) to minimize the influence of ageing or overweight on insulin sensitivity. One group of rats were administered GW501516 at 5 mg·kg−1·day−1 based on earlier studies in mice (Tanaka et al., 2003; Lee et al., 2006).
Food intake and body weight gain were monitored daily in the first 3 days of treatment and then twice a week. The effect of chronic PPARδ activation on insulin sensitivity was assessed in the conscious state 5–7 h after the removal of food and 15 h after the last oral gavage. The hyperinsulinaemic clamp was performed at an insulin infusion rate of 0.25 U·kg−1·h−1 to elevate circulating insulin levels to a half-maximal physiological concentration (Kraegen et al., 1991; Ye et al., 2003; Ye et al., 2006) with glucose infused at variable rates (GIR) to maintain euglycaemia. After plasma glucose levels reached the designated steady state, a bolus of 2-deoxy-D-[2,6-3H] glucose (2DG) and D-[U-14C] glucose was injected (i.v.) to determine glucose disappearance rate (Road) and hepatic glucose output rate (HGO). Under the euglycaemic conditions, HGO was calculated as Road – GIR where Road was determined by counting [14C] of the plasma collected at 2, 5, 10, 15, 20, 30 and 45 min after the tracer injection, as validated by this laboratory (James et al., 1986). The insulin sensitivity in muscle and adipose tissues was assessed by measuring glucose uptake (Rg') from these tissues freeze-clamped at the end of the hyperinsulinaemic clamp. Representative subcutaneous (in the right lower abdomen) and epididymal fat depot and liver were weighed before being frozen in liquid nitrogen. Tissues were also collected at the basal state for the measurement of metabolites and expression of genes regulated by PPARδ.
Assessment of the metabolic effects of PPARδ activation in mice
Male C57BL/6 mice aged 10 weeks were fed a HF diet for 6 weeks. After 2 weeks of high-fat feeding, mice were assigned to receive the PPARδ agonist NNC61-5920 (30 mg·kg−1·day−1) or the PPARγ agonist RSG (16 mg·kg−1·day−1) as an additive in the diet. An oral administration of NNC61-5920 at 30 mg·kg−1 has been shown to produce a maximal plasma concentration of ∼130 µg·ml−1 in db/db mice, ∼40 times of that (∼3 µg·ml−1) in rats produced at a dose of 2.27 mg·kg−1 (Winzell et al., 2010). We estimated that plasma concentrations of NNC61-5920 in mice at 30 mg·kg−1 would be within the range achieved in rats at the doses between 15 and 30 mg·kg−1·day−1. The effect on insulin action was assessed using a glucose tolerance test (GTT) after 5–7 h of fasting. Briefly, after basal blood samples, a 50% glucose solution was injected (i.p.) at a dose of 2 g·kg−1 body weight. Blood samples were obtained from the tail tip at the indicated times and glucose levels were measured using a glucometer (Accu-Check, Roche, New South Wales, Australia). During the GTT, [3H]-2DG and [14C]-glucose tracers were included in the glucose injection to allow the determination of glucose uptake and incorporation into lipids in tissues of interest based on previous studies from our laboratory (Cooney et al., 2004). Blood samples were taken at 0, 30, 60 and 90 min and diluted in 20 µL of 0.9% normal saline to determine plasma insulin levels and plasma radioactivity during the GTT. At the end of experiments, epididymal fat was dissected and weighed to assess the visceral adiposity.
Metabolite measurements
As described in our previous publications (Turner et al., 2009), glucose concentrations during the hyperinsulinaemic clamp in rats were determined using a glucose analyser (YSI 2300, YSI Inc., Yellow Springs, OH, USA). Plasma free FAs were determined using enzymatic colorimetric methods by commercial kits (Sigma-Aldrich, Sydney, Australia). Plasma, muscle and liver triglyceride contents were determined using a colorimetric assay kit (Triglycerides GPO-PAP; Roche Diagnostics, Indianapolis, IN, USA). Plasma insulin was determined by radioimmunoassay using a rat-specific kit (Linco Research, Inc., St. Charles, MO, USA).
qPCR determination of mRNA expression influenced by PPARδ
Total RNA was extracted from the red quadriceps muscle and liver of HF rats and HF mice, treated with NNC61-5920, using TRI-reagent (Sigma, St. Louis, MO, USA) followed by phenol–chloroform extraction, as described (Li et al., 2008). Briefly, RNA was purified with an RNeasy Mini Kit (Qiagen, GmbH, Germany) and digested with RNase-free DNase I (Ambion, Austin, TX, USA) during RNA purification. RNA was quantified using SYBR II by a spectrophotometer and the integrity was verified by agarose gel electrophoresis and by OD260/OD280 nm absorption ratio >1.90. cDNA was synthesized using Omniscript RT kit (QIAGEN, GmbH, Germany) according to the manufacturer's instruction. qPCR was performed using ABI Sequence Detection System (ABI 7900HT; ABI Prism, Foster City, CA, USA) or Rotor-Gene (RG-3000; Corbett Research, San Francisco, CA, USA). The species and tissue specific primers for CPT1, PDK4, UCP3, FA synthase (FAS), stearoyl-CoA desaturase-1 (SCD1), CD36 and lipoprotein lipase (LPL) were used to measure expression levels in the liver and muscle from rats and mice (Supporting Information Table S1). RNA samples were normalized for comparison by determining the ribosomal protein 36B4 as housekeeping gene as recommended for gene expression analysis across different tissues and animal species including rats and mice (Akamine et al., 2007).
Western blotting and determination of oxidative enzyme activity
As described previously (Turner et al., 2009), powdered muscle samples were suspended in a Western blotting buffer and solubilised for 2 h at 4°C. Equal amounts of tissue lysates (10–20 µg protein) were resolved by SDS-PAGE and mitochondrial metabolic capacity was assessed by specific antibodies against PGC-1α (Chemicon International, Temecula, CA, USA); muscle CPT-1 (Alpha Diagnostic International, San Antonio, TX, USA); UCP3 (Affinity Bioreagents, Golden, CO, USA) and an antibody cocktail that recognises several subunits of the mitochondrial respiratory chain (MS601) (Mitosciences, Eugene, OR, USA). Immunolabeled bands were quantitated by densitometry. Citrate synthase (CS) and β-hydroxyacyl-CoA dehydrogenase (β-HAD) were determined in muscle homogenates by spectrophotometric methods.
Statistical analyses
All results are presented as means ± SE. A one-way analysis of variance (anova) was used to assess the statistical significance across all groups. When tested as significant, a post hoc (Fisher-paired least significant difference) test was used to establish differences between groups. All data were processed in Excel 5.0, and statistical analyses were performed using the Statview SE + Graphic Program (Abacus Concepts-Brain Power, Farmington Hills, MI, USA).
Materials
NNC61-5920, rosiglitazone and GW501516 were from Novo Nordisk, Denmark. 2-Deoxy-D-[2,6-3H] glucose; 8.5–13.7 GBq·mmol−1 (230–370 mCi·mmol−1) and D-[U-14C] glucose: 370–740 GBq·mmol−1 (10–20 Ci·mmol−1) were from Amersham, Buckinghamshire, UK.
Results
Effects of 3 weeks of administration of NNC61-5920 in HF rats
As PPARδ agonists have been reported to reduce obesity and fat mass (Tanaka et al., 2003), we first examined the changes in body weight, fat mass and basal plasma parameters. Compared with chow-fed rats (Ch-Con), HF rats (HF-Con) displayed significantly (P < 0.05) greater body weight gain (24%), more central (80%) and subcutaneous fat mass (38%), higher plasma glucose (14%) and insulin (2.7-fold), and accumulation of triglyceride in the liver (threefold) and muscle (∼80%) (Table 1). Consistent with our previous studies (Ye et al., 2003), treatment of HF rats with rosiglitazone (HF-RSG) led to further increases in body weight gain and subcutaneous fat mass and decreased plasma insulin and liver triglyceride content. However, compared with the HF-Con group, treatment of HF rats with NNC61-5920 (HF-NNC) did not have any significant effects on food intake (data not shown), body weight gain, fat mass or plasma levels of glucose and insulin. Unexpectedly, triglyceride levels were significantly further elevated from HF-Con rats in both plasma (27%, P < 0.05) and particularly in muscle (70%, P < 0.01) in the HF-NNC group.
Table 1.
Basal metabolic variables after 3 weeks of treatment with NNC61-5920 in high fat-fed (HF) rats
| Ch-Con | HF-Con | HF-RSG | HF-NNC | |
|---|---|---|---|---|
| Body weight gain (g) | 66 ± 2 | 82 ± 6* | 104 ± 7**†† | 70 ± 8 |
| Epi fat mass (% BW) | 1.0 ± 0.1 | 1.8 ± 0.1** | 2.2 ± 0.1** | 1.8 ± 0.2* |
| Sub fat mass (% BW) | 1.6 ± 0.1 | 2.2 ± 0.2** | 3.5 ± 0.2**†† | 2.4 ± 0.3* |
| Liver weight (% BW) | 3.5 ± 0.1 | 3.9 ± 0.1 | 3.4 ± 0.1 | 5.8 ± 0.1**†† |
| Plasma glucose (mM) | 7.2 ± 0.2 | 8.2 ± 0.3* | 7.3 ± 0.2 | 8.7 ± 0.3** |
| Plasma insulin (mU/l) | 24 ± 2 | 64 ± 6** | 30 ± 8†† | 53 ± 11* |
| Plasma TG (mM) | 0.8 ± 0.1 | 1.1 ± 0.1 | 0.9 ± 0.1 | 1.4 ± 0.2**† |
| Plasma FAs (mM) | 0.5 ± 0.1 | 0.5 ± 0.0 | 0.4 ± 0.0 | 0.4 ± 0.1 |
| Liver TG (µmol/g) | 4.8 ± 0.2 | 14.8 ± 1.3** | 6.8 ± 0.8*†† | 13.4 ± 0.2** |
| Muscle TG (µmol/g) | 1.9 ± 0.1 | 3.4 ± 0.2** | 2.7 ± 0.2 | 5.8 ± 0.4**†† |
P < 0.05
P < 0.01 versus Ch-Con
P < 0.05
P < 0.01 versus HF-Con.
Male Wistar rats (200–220 g) were fed a high-fat diet for 4 weeks. After 1 week of the high-fat feeding, the PPARδ agonist NNC61-5920 (30 mg·kg−1·day−1) was added in the diet. As a positive control, the PPARγ agonist rosiglitazone (RSG, 16 mg·kg−1·day−1) was added in the diet for the last 2 weeks. Before the induction of NNC61-5920, body weight was similar among groups [normal chow control (Ch-Con) 243 ± 3 g; untreated HF rats (HF-Con) 236 ± 4 g, HF rats + rosiglitazone (HF-RSG) 245 ± 2 g and HF rats + NNC61-5920 (HF-NNC) 243 ± 2 g, P > 0.05, n = 9–12/group]. Fat mass was measured from epididymal (Epi) or subcutaneous (Sub) samples.
TG, triglyceride; FA, fatty acid.
Figure 1 shows insulin sensitivity as assessed by the hyperinsulinaemic-clamp after the treatment of NNC and RSG. Untreated HF rats (HF-Con) displayed typical insulin resistance at in the whole-body [reduced requirement for glucose infusion rate (GIR) to maintain euglycaemia, P < 0.01 vs. Ch-Con], peripheral tissues (decreased Road) and the liver (elevated HGO). Glucose uptake (Rg') in muscle and adipose tissue were also reduced following HF feeding. Rosiglitazone treatment significantly attenuated whole body insulin resistance (50% increase GIR vs. HF-Con, P < 0.01) by improving Road (via increased muscle Rg') and the suppression of HGO. In contrast, whole-body insulin resistance was exacerbated in HF rats treated with NNC61-5920 (GIR reduced by 14% vs. HF-Con, P < 0.05) due to a substantial decrease in Road (by 28%, P < 0.01) despite an improvement in insulin action by suppressing HGO (P < 0.01). The exacerbated insulin resistance in peripheral tissues was due to a worsening of insulin action in muscle (Rg' reduced by 30%, P < 0.01) whereas glucose uptake in fat tissue was not affected in HF rats (P > 0.05 vs. HF-Con).
Figure 1.
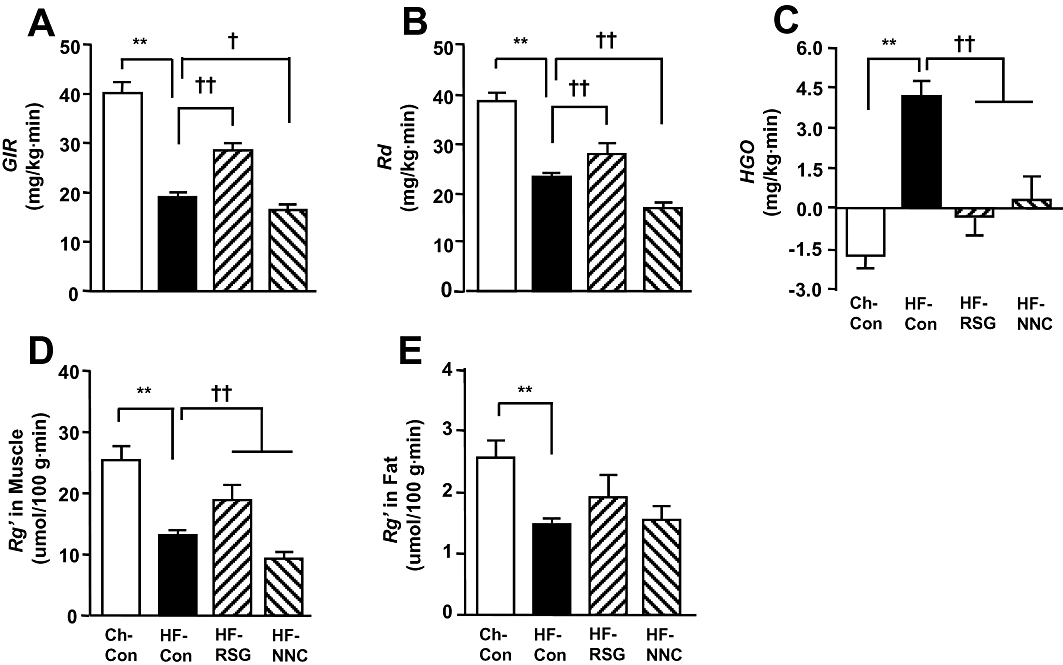
Effects of 3 weeks of treatment with the peroxisome proliferator-activated receptor δ (PPARδ) agonist NNC61-5920 on insulin action in high fat-fed (HF) rats. Hyperinsulinaemic clamp was performed at an insulin infusion rate of 0.25 U·kg−1·h−1 in the conscious state after 5–7 h of fasting. Plasma euglycaemia and hyperinsulinaemia were similar among all four groups during the clamp (data not shown). *P < 0.05, **P < 0.01 versus Ch-Con, †P < 0.05, ††P < 0.01 vs. HF-Con (n = 6–9). CH-Con, chow-fed rats; HF-Con, untreated HF rats; HF-RSG, HF-rats treated with rosiglitazone (16 mg·kg−1·day−1 in diet); HF-NNC, HF rats treated with NNC61-5920 (30 mg·kg−1·day−1 in diet).
Effects of extended administration of PPARδ agonists NNC61-5920 and GW501516 in HF rats
To investigate whether the duration of treatment with NNC61-5920 was not long enough to show beneficial effects as reported in mice (Wang et al., 2003; Lee et al., 2006), we extended the administration of NNC61-5920 by oral gavage for 6 weeks. In this extended study, we also included the well-known PPARδ agonist GW501516 to compare its effects with NNC61-5920. Consistent with the results obtained from HF rats after 3 weeks of treatment with NNC61-5920, neither NNC61-5920 nor GW501516 was able to attenuate body weight gain, adiposity and triglyceride content in muscle and liver (Supporting Information Table S2). Compared with HF rats gavaged with vehicle, plasma triglyceride level in GW501516-treated rats was 30% higher (P < 0.05). During the hyperinsulinaemic-clamp, the GIRs, Rds and muscle Rg' were all significantly (P < 0.05) decreased (by 20–26%) in rats treated with NNC61-5920 and GW501516. Insulin-mediated suppression of HGO was significantly improved in NNC61-5920-treated rats (P < 0.05 vs. HF-Con) and a similar trend was also observed after treatment with GW501516 (P = 0.12).
Changes in markers indicative of PPARδ activation
It has been recently reported that activation of PPARδin vivo can lead to hepatomegaly (Lee et al., 2006; Faiola et al., 2008). Consistent with these earlier reports, we observed a significant increase in liver weight following 3 or 6 weeks of the treatment with NNC61-5920 (40–60%, P < 0.01 vs. HF-Con) (Table 1) and after 6 weeks of administration of GW501516 (∼16%, P < 0.01) (Table S2). We further examined expression of the genes responsive to the stimulation of the PPARδ agonist NNC61-5920 in muscle (Wang et al., 2003; Lee et al., 2006) and the liver (Qin et al., 2008). As shown in Figure 2, the mRNA from the PPARδ target genes, CPT1, PDK4 and UCP3, were elevated by 40, 90 and 30%, respectively (P < 0.05 for all) in muscle following 3 weeks of treatment with NNC61-5920, whereas mRNA from genes regulating lipid metabolism, such as SCD1, CD36 and LPL were significantly down-regulated in the liver. These findings were similar to the effects of other PPARδ agonists in the liver as previously reported (Wang et al., 2003; Lee et al., 2006; Qin et al., 2008) whereas the mRNA from the housekeeping gene 34B4 was not altered by NNC61-5920 in either HF rats (0.166 ± 0.033 vs. 0.121 ± 0.004 unit in HF rats, P = 0.211) or HF mice (0.174 ± 0.012 vs. 0.178 ± 0.010 unit in HF mice, P = 0.803). These data together suggest that PPARδ was activated in the target tissue of rats by the administration of NNC61-5920.
Figure 2.
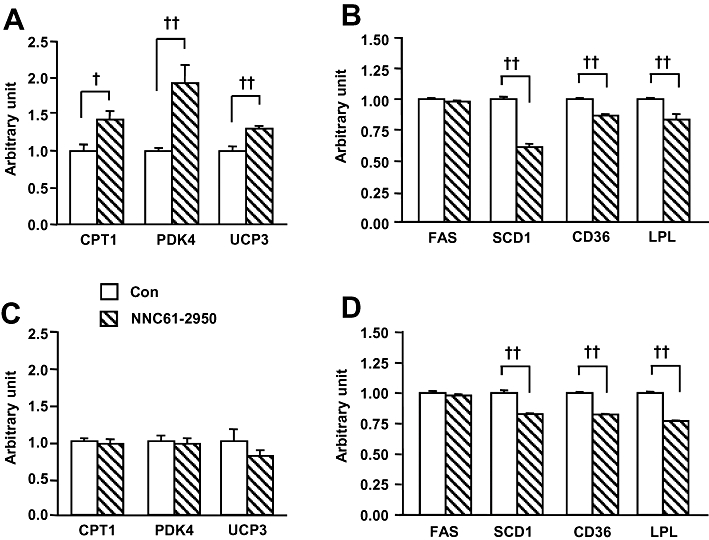
Expression of mRNA from peroxisome proliferator-activated receptor δ (PPARδ) responsive genes in rats as compared with mice after chronic administration of NNC61-5920. Muscle (A) and liver (B) were collected from high fat-fed rat after chronic administration of the PPARδ agonist NNC61-5920 as described in Figure 1. Muscle (C) and liver (D) were collected from high fat-fed (HF) mice after chronic administration of the PPARδ agonist NNC61-5920 as described in Figure 3. †P < 0.05, ††P < 0.01 (n = 5–6).
Effects of the PPARδ agonist NNC61-5920 in HF mice
As our findings in rats were opposite from the reported effects of the PPARδ agonist GW501516 in various mouse models (Tanaka et al., 2003; Wang et al., 2003; Lee et al., 2006), we decided to investigate whether or not there is species difference in their metabolic response to the PPARδ agonist NNC61-5920. In this study, C57BL/J6 mice at age of 8 weeks were fed a HF diet for 2 weeks to generate insulin resistance followed by the treatment of NNC61-5920 for 4 weeks. Body weight was matched before commencement of the treatment with NNC61-5920 and rosiglitazone (HF-Con, 23.8 ± 0.4 g; HF-RSG, 23.7 ± 0.5 g and HF-NNC, 23.8 ± 0.8 g, P > 0.05, n = 7–9/group). Food intake was not altered by NNC61-5920 (data not shown). Treatment with NNC61-5920 did not show any significant effect on the expression of CPT1, PDK4 and UCP3 in muscle (Figure 2C) whereas mRNA from the PPARδ target genes SCD1, CD36 and LPL, in the liver were significantly down-regulated (Figure 2D).
Interestingly, as illustrated in Figure 3 treatment with NNC61-5920 significantly decreased body weight gain (52% P < 0.01), epididymal fat mass (by 48%, P < 0.05) and plasma levels of triglyceride and FAs (50–60%, P < 0.01 vs. HF-Con). Consistent with earlier reports in obese and insulin resistant mice treated with GW501516 (Tanaka et al., 2003; Wang et al., 2003; Lee et al., 2006), treatment with NNC61-5920 significantly improved glucose tolerance without elevating plasma insulin levels (average of 30, 60 and 90 min during the GTT) (Figure 4A–C). When expressed as blood glucose AUC × plasma insulin (30–90 min average), whole body insulin resistance was abolished (Figure 4D) and this was attributed to the substantial reversal of insulin sensitivity in muscle (improved Rg' per unit plasma insulin) and the liver (glucose incorporation into lipids per unit plasma insulin (Figure 4E, F). The overall efficacy of NNC61-5920 in improving insulin action was comparable with that of rosiglitazone.
Figure 3.
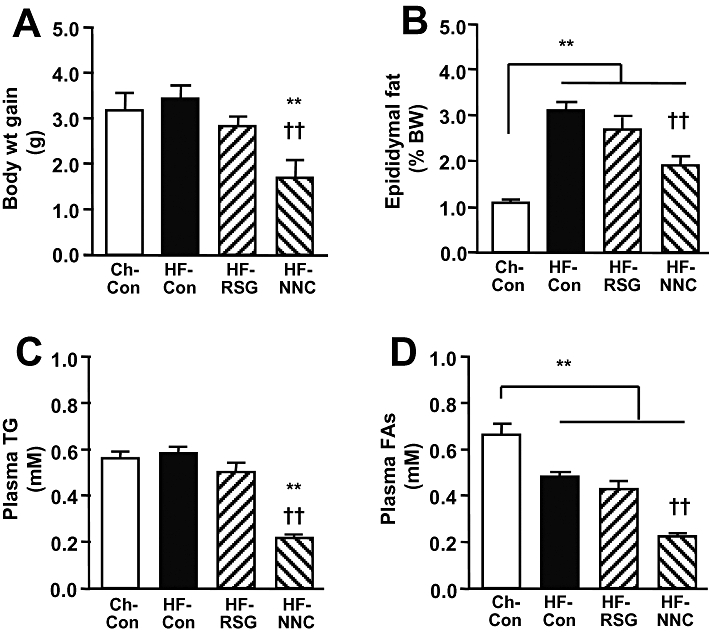
Effects of the peroxisome proliferator-activated receptor δ (PPARδ) agonist NNC61-5920 on body-weight gain, adiposity and plasma lipids in high fat-fed (HF) mice. Male C57 BL/J6 mice were fed a HF diet for 6 weeks and NNC61-5920 (30 mg·kg−1·day−1) or rosiglitazone (16 mg·kg−1·day−1) was administered for 4 weeks. (A) Body weight gain. (B) Epididymal fat mass. (C) Plasma triglyceride. (D) Plasma fatty acids. **P < 0.01 versus Ch-Con, ††P < 0.01 vs. HF-Con (n≥ 7).
Figure 4.
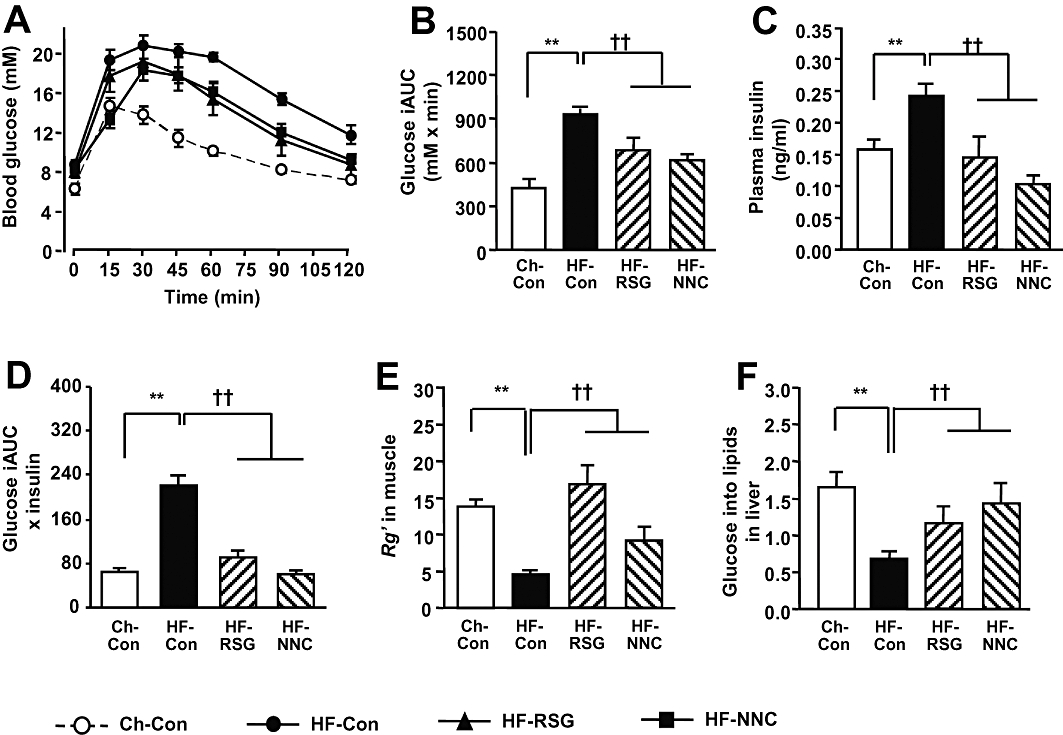
Effects of the peroxisome proliferator-activated receptor δ (PPARδ) agonist NNC61-5920 on insulin action in high fat-fed (HF) mice. Glucose tolerance test was carried out with i.p. injection of glucose solution (2 g glucose/kg BW) containing 3[H]-2DG and 14[C]-glucose tracers. (A) blood glucose during GTT; (B) increment area under the curve (AUC) of blood glucose; (C) plasma insulin levels during GTT (average of 30, 60 and 90 min); (D) whole body insulin resistance index; (E) Rg' index in quadriceps muscle during GTT [expressed as Rg' (mg·g−1·min−1) per mM blood glucose per ng/ml of plasma insulin]; (F) glucose incorporation into lipids in the liver during GTT (expressed in the same unit as for the Rg' index). **P < 0.01 vs. Ch-Con, ††P < 0.01 vs. HF-Con (n≥ 7).
Changes in key proteins and enzyme activity in muscle
As PPARδ agonists have been suggested to attenuate insulin resistance via increased muscle oxidative capacity in mice (Tanaka et al., 2003), we compared key mitochondrial proteins and enzyme activity in rats and mice. As shown in Figure 5, NNC61-5920 significantly increased UCP3 protein (by threefold, P < 0.01) and, unexpectedly, reduced CTP1 protein content (by 47%, P < 0.01). In HF mice, NNC61-5920 significantly increased the protein levels of Complex III (by 44%, P < 0.05) and UCP3 (by 2.1-fold, P < 0.01) with a similar effect on CTP1 (by 49%, P = 0.07). Although CS activity was not affected by the treatment of NNC61-2950, β-HAD activity was significantly increased in both rat (by 23%, P < 0.05) and mice (by 30%, P < 0.05).
Figure 5.
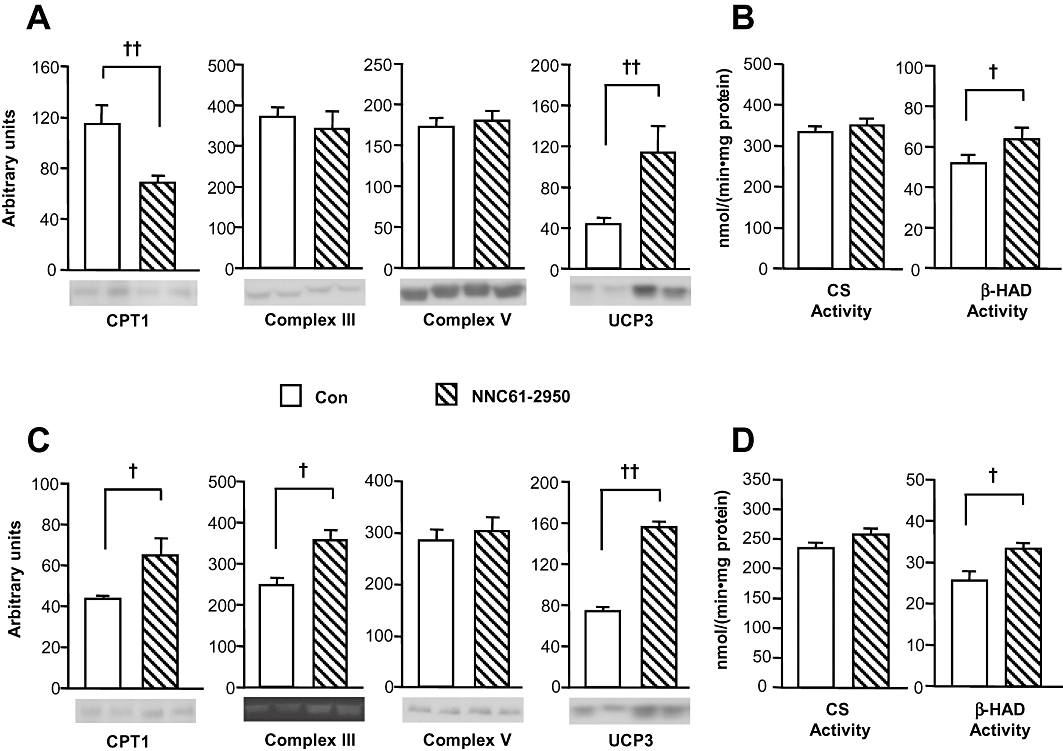
Effects of NNC61-2950 on mitochondrial markers and oxidative enzyme activity in muscle. Muscle samples were collected from HF rats (A, B) as described in Figure 1 or from HF mice (C,D) as described in Figure 3 after administration of NNC61-5920. Key mitochondrial protein levels were analysed by Western blot using specific antibodies (A, C) and the activity of citrate synthase (CS) and β-hydroxylacyl-CoA dehydrogenase (β-HAD) was determined by spectrophotometric methods (B, D). †P < 0.05, ††P < 0.01 (n = 5–6).
Discussion
Muscle and liver are the two most important sites for insulin-mediated glucose homeostasis in the whole body and an excess lipid accumulation in these tissues has been causally linked to insulin resistance (Ruderman and Shulman, 2006). PPARδ is highly expressed in muscle and activation of this transcription factor has been suggested to up-regulate genes catalysing lipid metabolism to increase FA oxidation. Studies in both genetic and diet induced obese mice showed that PPARδ agonists such as GW501516 (Tanaka et al., 2003; Wang et al., 2003; Lee et al., 2006) exert beneficial efficacy in correcting insulin resistance and dyslipidaemia. Recently, pharmacological activation of PPARδ has been demonstrated to enhance running endurance in mice (Narkar et al., 2008). These findings suggest a therapeutic potential of PPARδ agonists for treatment of diabetes and obesity. However, treatment of rats with PPARδ agonists has been reported to inhibit glucose uptake and oxidation during contraction (Constantin et al., 2007; Constantin-Teodosiu et al., 2009). Therefore, we sought to characterize the effects of chronic administration of PPARδ agonists on insulin sensitivity in insulin resistant HF rats.
We first examined the efficacy of the novel PPARδ agonist NNC61-5920 (Sauerberg et al., 2007) using the hyperinsulinaemic clamp technique in HF rats following 3 weeks of administration, using a protocol reported previously for rosiglitazone (Ye et al., 2003), and the AMP-activated protein kinase (AMPK) activators, 5-aminoimidazole-4-carboxamide-1-β-D-ribofuranoside (Iglesias et al., 2002) and dihydroberberine (Turner et al., 2008). While rosiglitazone demonstrated a clear insulin-sensitizing efficacy as expected in this rat model (Ye et al., 2003), we were unable to detect an improvement of the whole-body insulin sensitivity after treatment with NNC61-5920. Instead, we found the whole-body insulin sensitivity was worsened due to exacerbated insulin resistance in muscle. These findings were opposite to our initial expectation based on previous studies in mice (Tanaka et al., 2003; Wang et al., 2003; Lee et al., 2006). However, several recent in vitro studies also failed to demonstrate beneficial effects of PPARδ agonists on glucose metabolism or insulin sensitivity in muscle from rats. For example, GW501516 has been shown to have no effect on glucose uptake with and without the presence of insulin in L6 myotubes derived from rats (Dimopoulos et al., 2007) or isolated epitrochlearis and soleus muscles from rats (Terada et al., 2006). To our knowledge, this is the first study showing a worsening of muscle insulin resistance in HF rats in vivo after treatment with PPARδ agonists. A recent study also showed impaired insulin action in muscle, when assessed ex vivo, from both normal diet and HF rats administered with GW501516 (Cresser et al., 2010). This suggests that the deleterious effect of PPARδ on insulin sensitivity in the muscle of rats observed in our study is unlikely to be due to the influence of high-fat feeding.
One argument for the lack of the beneficial effects might be that NNC61-5920 failed to activate PPARδ in the present study and the exacerbated muscle insulin resistance resulted from the off-target effects of this compound. However, rats treated with NNC61-5920 showed a significant enlargement of liver as has been noted in mice after administration of GW501516 (Lee et al., 2006) and GW0742 (Faiola et al., 2008). Importantly, NNC61-5920 significantly altered expression of PPARδ target genes including up-regulation of the mRNA from CPT1, UCP3 and PDK4 in muscle and down-regulation of that from FAS and SCD1 in the liver as reported for other PPARδ agonists (Wang et al., 2003; Lee et al., 2006; Qin et al., 2008). These data suggest that the exacerbation of insulin resistance in HF rats after treatment with NNC61-5920 was closely associated with changes in PPARδ responsive genes. To exclude the possibility of off-target effects of NNC61-5920, we included the well-known PPARδ agonist GW501516 and extended the treatment to 6 weeks and compared their effects. The results showed that GW501516 also worsened insulin resistance and elevated plasma triglycerides with enlarged liver similarly to NNC61-5920, indicating that exacerbation of insulin resistance is a common effect of PPARδ agonists in HF rats.
Because most reported beneficial effects of PPARδ agonists on the metabolic syndrome were obtained in mouse models including ob/ob (Wang et al., 2003), db/db (Lee et al., 2006; Winzell et al., 2010) and HF mice (Tanaka et al., 2003), we wondered whether NNC61-5920 was able to exert beneficial effects in mice as reported for other PPARδ agonists. Indeed, in HF mice treatment with NNC61-5920 significantly improved glucose tolerance, muscle insulin sensitivity and reduced body weight gain, visceral fat and plasma lipids as reported in various mouse models treated with PPARδ agonists. As observed in HF rats, NNC61-5920 also improved hepatic insulin action in HF mice (indicated by increased glucose incorporation into lipids during the GTT). Importantly, the pattern of inhibited expression of the PPARδ target genes, FAS, SCD1, CD36 and LPL, were almost identical in both species. PPARδ is known to inhibit expression of hepatic lipogenic genes FAS and SCD1 via insulin-induced gene-1 (Qin et al., 2008) and inhibition of hepatic lipogenesis has been shown to reverse insulin resistance (Dentin et al., 2006; Savage et al., 2006). These data together suggest that PPARδ agonists attenuated hepatic insulin resistance in HF rats and HF mice by suppressing lipogenic genes in the liver.
While it is currently puzzling to us as to why rats and mice showed different metabolic consequence following treatment of PPARδ agonists, our data appear to point to muscle as a major site for this difference. Although muscle CPT1, PDK4 and UCP3 have been reported to be not regulated by other PPARδ agonists (Tanaka et al., 2003; Wang et al., 2003; Lee et al., 2006; Jucker et al., 2007; Narkar et al., 2008), in the present study NNC61-5920 significantly up-regulated CPT1, PDK4 and UCP3 in HF rats. As PPARδ agonists have been suggested to attenuate insulin resistance via increased muscle oxidative capacity in mice (Tanaka et al., 2003), we examined key mitochondrial proteins and the activity of CS and β-HAD to assess the changes in muscle oxidative capacity. Surprisingly, we observed a 47% reduction in CPT1 protein levels after treatment with NNC61-2950 despite increased UCP3 protein and β-HAD activity. As CPT1 is the major rate-limiting step for the β-oxidation of FAs in mitochondria, its reduced protein level alone could restrict muscle oxidative capacity for FAs. Interestingly, several recent studies have shown that PPARδ–induced expression of these FA oxidative genes is not coupled with enhanced mitochondrial function in rats (Constantin et al., 2007; Jucker et al., 2007; Constantin-Teodosiu et al., 2009). It is possible that in the absence of increased mitochondrial function, PPARδ–induced FA oxidation does not result in enhanced fuel utilization. Indeed, the unaltered body weight in HF rats treated with NNC61-5920 and GW501516 is suggestive of a lack of increase in fuel utilization. The lack of increased fuel utilization in HF rats treated with either NNC61-5920 or GW501516 is indicated by unaltered body weight gain, adiposity and food. Further studies are required to investigate whether the disparity between the mRNA and protein levels of CPT1 observed in HF rats results from PPARδ-mediated changes in post-transcription or protein degradation.
By comparison, administration of NNC61-5920 in HF mice appeared to increase metabolic rate (suggested by reductions in body weight gain and fat mass with unaltered food intake), as previously reported for GW501516 (Tanaka et al., 2003). In fact, NNC61-5920 induced increases in key oxidative proteins regulating FA oxidation such as mitochondrial CTP1, Complex III, UCP3 and the activity of β-HAD in a much more synchronised manner in mice, compared with rats. This may explain the findings of attenuated insulin resistance in HF mice after treatment with NNC61-5920. Interestingly, a recent study indicates the importance of physical activity for PPARδ–induced beneficial effects (Narkar et al., 2008). Further studies are required to investigate whether the beneficial effects of PPARδ agonists in mice are related to their higher physical activity than that of rats.
In summary, the present study investigated the proposed therapeutic effects of a novel and an established PPARδ agonist in two insulin-resistant rodent models induced by chronic high-fat feeding. Our data show a worsening of muscle insulin resistance in rats but improved insulin action in mice. While there is some evidence that PPARδ agonists may be of benefit in humans (Riserus et al., 2008), the present study along with other recent studies with the use of the PPARδ agonist GW610742X (Constantin-Teodosiu et al., 2009) indicates a need for careful selection of the species for preclinical characterization of the endpoint metabolic effects of PPARδ agonists intended for treatment of patients with the metabolic syndrome. Given the heterogenous effects of PPARδ agonists on insulin sensitivity in different rodent species, it will be necessary to characterize the effect of PPARδ agonists on insulin sensitivity in heterogenous populations of humans to determine their therapeutic efficacy.
Acknowledgments
This work was supported in part by the National Health and Medical Research Council (NHMRC) of Australia (JY and EWK) and Novo Nordisk (EWK, GC and JY). EWK and GJC are supported by Research Fellowship from NHMRC and NT by a Career Development Award. The excellent technical assistance of Miguel A. Iglesias is gratefully acknowledged. We also thank the Biological Testing Facility at the Garvan Institute for assistance with animal care.
Glossary
Abbreviations
- β-HAD
β-hydroxyacyl-CoA dehydrogenase
- 2DG
2-deoxy-D-[2,6-3H] glucose
- AUC
area under the curve (for blood glucose)
- CD36
fatty acid translocase protein CD36
- Ch
chow diet
- CPT-1
carnitine palmitoyltransferase-1
- CS
citrate synthase
- FA
fatty acid
- FAS
fatty acid synthase
- GIR
glucose infusion rate
- GTT
glucose tolerance test
- GW
- HF
high fat diet
- HGO
rate of hepatic glucose output
- HOMA-IR
insulin resistance index
- LPL
lipoprotein lipase
- NNC
NNC61-2950
- PDK4
pyruvate dehydrogenase kinase 4
- PGC1α
peroxisome proliferator-activated receptor γ coactivator-1α
- PPAR
peroxisome proliferator-activated receptor
- Road
rate of glucose disposal
- Rg'
rate of glucose uptake
- RSG
rosiglitazone
- SCD-1
stearoyl-CoA desaturase 1
- TG
triglyceride
- UCP3
uncoupling protein 3
Conflicts of interest
None.
Supporting information
Additional Supporting Information may be found in the online version of this article:
Table S1 Species and tissue specific primer sequences
Table S2 Effects of extended treatment of PPARδ agonists NNC61-5920 and GW501516 in rats
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- Akamine R, Yamamoto T, Watanabe M, Yamazaki N, Kataoka M, Ishikawa M, et al. Usefulness of the 5′ region of the cDNA encoding acidic ribosomal phosphoprotein P0 conserved among rats, mice, and humans as a standard probe for gene expression analysis in different tissues and animal species. J Biochem Biophys Methods. 2007;70:481–486. doi: 10.1016/j.jbbm.2006.11.008. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC), 4th edn. Br J Pharmacol. 2009;158(Suppl. 1):S1–S254. doi: 10.1111/j.1476-5381.2009.00499.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Constantin D, Constantin-Teodosiu D, Layfield R, Tsintzas K, Bennett AJ, Greenhaff PL. PPARdelta agonism induces a change in fuel metabolism and activation of an atrophy programme, but does not impair mitochondrial function in rat skeletal muscle. J Physiol. 2007;583:381–390. doi: 10.1113/jphysiol.2007.135459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Constantin-Teodosiu D, Baker DJ, Constantin D, Greenhaff PL. PPARdelta agonism inhibits skeletal muscle PDC activity, mitochondrial ATP production and force generation during prolonged contraction. J Physiol. 2009;587:231–239. doi: 10.1113/jphysiol.2008.164210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooney GJ, Lyons RJ, Crew AJ, Jensen TE, Molero JC, Mitchell CJ, et al. Improved glucose homeostasis and enhanced insulin signalling in Grb14-deficient mice. EMBO J. 2004;23:582–593. doi: 10.1038/sj.emboj.7600082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cresser J, Bonen A, Chabowski A, Stefanyk LE, Gulli R, Ritchie I, et al. Oral administration of a PPAR-delta agonist to rodents worsens, not improves, maximal insulin-stimulated glucose transport in skeletal muscle of different fibers. Am J Physiol Regul Integr Comp Physiol. 2010;299:R470–R479. doi: 10.1152/ajpregu.00431.2009. [DOI] [PubMed] [Google Scholar]
- Dentin R, Benhamed F, Hainault I, Fauveau V, Foufelle F, Dyck JR, et al. Liver-specific inhibition of ChREBP improves hepatic steatosis and insulin resistance in ob/ob mice. Diabetes. 2006;55:2159–2170. doi: 10.2337/db06-0200. [DOI] [PubMed] [Google Scholar]
- Dimopoulos N, Watson M, Green C, Hundal HS. The PPARdelta agonist, GW501516, promotes fatty acid oxidation but has no direct effect on glucose utilisation or insulin sensitivity in rat L6 skeletal muscle cells. FEBS Lett. 2007;581:4743–4748. doi: 10.1016/j.febslet.2007.08.072. [DOI] [PubMed] [Google Scholar]
- Faiola B, Falls JG, Peterson RA, Bordelon NR, Brodie TA, Cummings CA, et al. PPAR alpha, more than PPAR delta, mediates the hepatic and skeletal muscle alterations induced by the PPAR agonist GW0742. Toxicol Sci. 2008;105:384–394. doi: 10.1093/toxsci/kfn130. [DOI] [PubMed] [Google Scholar]
- Iglesias MA, Ye JM, Frangioudakis G, Saha AK, Tomas E, Ruderman NB, et al. AICAR administration causes an apparent enhancement of muscle and liver insulin action in insulin-resistant high-fat-fed rats. Diabetes. 2002;51:2886–2894. doi: 10.2337/diabetes.51.10.2886. [DOI] [PubMed] [Google Scholar]
- James DE, Burleigh KM, Kraegen EW. In vivo glucose metabolism in individual tissues of the rat. Interaction between epinephrine and insulin. J Biol Chem. 1986;261:6366–6374. [PubMed] [Google Scholar]
- Jucker BM, Yang D, Casey WM, Olzinski AR, Williams C, Lenhard SC, et al. Selective PPARdelta agonist treatment increases skeletal muscle lipid metabolism without altering mitochondrial energy coupling: an in vivo magnetic resonance spectroscopy study. Am J Physiol Endocrinol Metab. 2007;293:E1256–E1264. doi: 10.1152/ajpendo.00218.2007. [DOI] [PubMed] [Google Scholar]
- Kraegen EW, Clark PW, Jenkins AB, Daley EA, Chisholm DJ, Storlien LH. Development of muscle insulin resistance after liver insulin resistance in high-fat-fed rats. Diabetes. 1991;40:1397–1403. doi: 10.2337/diab.40.11.1397. [DOI] [PubMed] [Google Scholar]
- Lee CH, Kang K, Mehl IR, Nofsinger R, Alaynick WA, Chong LW, et al. Peroxisome proliferator-activated receptor delta promotes very low-density lipoprotein-derived fatty acid catabolism in the macrophage. Proc Natl Acad Sci USA. 2006;103:2434–2439. doi: 10.1073/pnas.0510815103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Heilbronn LK, Hu D, Poynten AM, Blackburn MA, Shirkhedkar DP, et al. Islet-1: a potentially important role for an islet cell gene in visceral fat. Obesity (Silver Spring) 2008;16:356–362. doi: 10.1038/oby.2007.76. [DOI] [PubMed] [Google Scholar]
- Narkar VA, Downes M, Yu RT, Embler E, Wang YX, Banayo E, et al. AMPK and PPARdelta agonists are exercise mimetics. Cell. 2008;134:405–415. doi: 10.1016/j.cell.2008.06.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliver WR, Jr, Shenk JL, Snaith MR, Russell CS, Plunket KD, Bodkin NL, et al. A selective peroxisome proliferator-activated receptor delta agonist promotes reverse cholesterol transport. Proc Natl Acad Sci USA. 2001;98:5306–5311. doi: 10.1073/pnas.091021198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin X, Xie X, Fan Y, Tian J, Guan Y, Wang X, et al. Peroxisome proliferator-activated receptor-delta induces insulin-induced gene-1 and suppresses hepatic lipogenesis in obese diabetic mice. Hepatology. 2008;48:432–441. doi: 10.1002/hep.22334. [DOI] [PubMed] [Google Scholar]
- Riserus U, Sprecher D, Johnson T, Olson E, Hirschberg S, Liu A, et al. Activation of peroxisome proliferator-activated receptor (PPAR)delta promotes reversal of multiple metabolic abnormalities, reduces oxidative stress, and increases fatty acid oxidation in moderately obese men. Diabetes. 2008;57:332–339. doi: 10.2337/db07-1318. [DOI] [PubMed] [Google Scholar]
- Ruderman NB, Shulman GI. The metabolic syndrome. In: De Groot, Jameson, editors. Endocrinology. 5th edn. Philadelphia, PA: Elsevier; 2006. pp. 1149–1166. [Google Scholar]
- Sauerberg P, Olsen GS, Jeppesen L, Mogensen JP, Pettersson I, Jeppesen CB, et al. Identification and synthesis of a novel selective partial PPARdelta agonist with full efficacy on lipid metabolism in vitro and in vivo. J Med Chem. 2007;50:1495–1503. doi: 10.1021/jm061202u. [DOI] [PubMed] [Google Scholar]
- Savage DB, Choi CS, Samuel VT, Liu ZX, Zhang D, Wang A, et al. Reversal of diet-induced hepatic steatosis and hepatic insulin resistance by antisense oligonucleotide inhibitors of acetyl-CoA carboxylases 1 and 2. J Clin Invest. 2006;116:817–824. doi: 10.1172/JCI27300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka T, Yamamoto J, Iwasaki S, Asaba H, Hamura H, Ikeda Y, et al. Activation of peroxisome proliferator-activated receptor delta induces fatty acid beta-oxidation in skeletal muscle and attenuates metabolic syndrome. Proc Natl Acad Sci USA. 2003;100:15924–15929. doi: 10.1073/pnas.0306981100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terada S, Wicke S, Holloszy JO, Han DH. PPARdelta activator GW-501516 has no acute effect on glucose transport in skeletal muscle. Am J Physiol Endocrinol Metab. 2006;290:E607–E611. doi: 10.1152/ajpendo.00430.2005. [DOI] [PubMed] [Google Scholar]
- Turner N, Li JY, Gosby A, To SW, Cheng Z, Miyoshi H, et al. Berberine and its more biologically available derivative, dihydroberberine, inhibit mitochondrial respiratory complex I: a mechanism for the action of berberine to activate AMP-activated protein kinase and improve insulin action. Diabetes. 2008;57:1414–1418. doi: 10.2337/db07-1552. [DOI] [PubMed] [Google Scholar]
- Turner N, Hariharan K, TidAng J, Frangioudakis G, Beale SM, Wright LE, et al. Enhancement of muscle mitochondrial oxidative capacity and alterations in insulin action are lipid species dependent: potent tissue-specific effects of medium-chain fatty acids. Diabetes. 2009;58:2547–2554. doi: 10.2337/db09-0784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang YX, Lee CH, Tiep S, Yu RT, Ham J, Kang H, et al. Peroxisome-proliferator-activated receptor delta activates fat metabolism to prevent obesity. Cell. 2003;113:159–170. doi: 10.1016/s0092-8674(03)00269-1. [DOI] [PubMed] [Google Scholar]
- Winzell MS, Wulff EM, Olsen GS, Sauerberg P, Gotfredsen CF, Ahren B. Improved insulin sensitivity and islet function after PPARdelta activation in diabetic db/db mice. Eur J Pharmacol. 2010;626:297–305. doi: 10.1016/j.ejphar.2009.09.053. [DOI] [PubMed] [Google Scholar]
- Ye JM, Iglesias MA, Watson DG, Ellis B, Wood L, Jensen PB, et al. PPARalpha /gamma ragaglitazar eliminates fatty liver and enhances insulin action in fat-fed rats in the absence of hepatomegaly. Am J Physiol Endocrinol Metab. 2003;284:E531–E540. doi: 10.1152/ajpendo.00299.2002. [DOI] [PubMed] [Google Scholar]
- Ye JM, Dzamko N, Hoy AJ, Iglesias MA, Kemp B, Kraegen E. Rosiglitazone treatment enhances acute AMP-activated protein kinase-mediated muscle and adipose tissue glucose uptake in high-fat-fed rats. Diabetes. 2006;55:2797–2804. doi: 10.2337/db05-1315. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.