

Abstract
BACKGROUND AND PURPOSE
2,5-diaziridinyl-3-(hydroxymethyl)-6-methyl-1,4-benzoquinone (RH1) is a bioreductive agent that is activated by the two-electron reductase NAD(P)H quinone oxidoreductase 1 (NQO1). Although the cytotoxic efficacy of RH1 against tumours has been studied extensively, the molecular mechanisms underlying this anti-cancer activity have not yet been fully elucidated.
EXPERIMENTAL APPROACH
2,5-diaziridinyl-3-(hydroxymethyl)-6-methyl-1,4-benzoquinone-induced apoptosis and related signalling pathways in NQO1-negative and NQO1-overexpressing cells were evaluated. The role of p53 in RH1-induced cell death was investigated using parental and p53-deficient RKO human colorectal cancer cells by assaying clonogenic cell survival. Specific inhibitors and siRNAs targeting factors involved in RH1-induced apoptosis were used to clarify the roles played by such factors in RH1-activated apoptotic signalling pathways.
KEY RESULTS
2,5-diaziridinyl-3-(hydroxymethyl)-6-methyl-1,4-benzoquinone induced apoptosis and clonogenic death, dependent on NQO1 and p53. Treatment of NQO1-overexpressing cells with RH1 caused rapid disruption of mitochondrial membrane potential, nuclear translocation of apoptosis-inducing factor (AIF) and endonuclease G (Endo G) from mitochondria, and subsequent caspase-independent apoptotic cell death. siRNA targeting AIF and Endo G effectively attenuated RH1-induced apoptotic cell death. Moreover, RH1 induced cleavage of Bax, which targets mitochondria. RH1 significantly activated the c-Jun N-terminal kinase (JNK) pathway, and inhibition of this pathway suppressed RH1-induced mitochondria-mediated apoptosis. RH1-induced generation and mitochondrial translocation of cleaved Bax were blocked by the JNK inhibitor, SP600125. Inhibition of JNK with SP600125 attenuated the mitochondrial translocation of JNK.
CONCLUSIONS AND IMPLICATIONS
2,5-diaziridinyl-3-(hydroxymethyl)-6-methyl-1,4-benzoquinone activated JNK, resulting in mitochondria-mediated apoptotic cell death that was NQO1-dependent.
Keywords: RH1, mitochondria, JNK, Bax, AIF, Endo G
Introduction
The pharmacological properties of aziridinyl benzoquinones have been studied for several decades (Danson et al., 2004). Among these compounds, 2,5-diaziridinyl-3-(hydroxymethyl)-6-methyl-1,4-benzoquinone (RH1) is an especially potent bioreductive agent that has been shown to exert a profound anti-cancer activity in vivo and in vitro (Danson et al., 2007; Hussein et al., 2009). The anti-cancer activity of RH1 has been attributed to two-electron reduction by NAD(P)H quinone oxidoreductase 1 (NQO1, DT-diaphorase), an enzyme abundantly expressed in a variety of human solid tumours, including those of the breast, pancreas, lung and colon (Cummings et al., 2003; Kim et al., 2004a). NQO1-mediated reduction of RH1 results in activation of the aziridine rings, in turn inducing DNA alkylation and cross-linking (Cummings et al., 2003; Kim et al., 2004a; Kim et al., 2004b; Ward et al., 2005; Hussein et al., 2009). Several lines of evidence indicate that NQO1 positively regulates p53 stability by inhibiting p53 degradation (Asher et al., 2001; Nioi and Hayes, 2004). These findings further indicate that NQO1 plays a role in protecting against tumour development. p53 has been reported to enhance the therapeutic efficacy of various anti-cancer drugs (Nicholson, 2000). p53 not only modulates the transcriptional expression of various genes involved in cell cycle arrest and apoptosis (by functioning as a transcription factor), but is also involved in transcription-independent apoptosis (Haupt et al., 2003; Mihara et al., 2003). Thus, the development of anti-cancer drugs that can activate p53 in a manner that does not focus on p53 transcriptional activity is important in the establishment of feasible therapeutic strategies.
It has been suggested that the mitochondrial cell death pathway is regulated by the ratio of pro- to anti-apoptotic proteins, including members of the Bcl-2 family. Among these family members, Bax or Bak plays a critical role in loss of mitochondrial transmembrane potential (Huang and Strasser, 2000). Upon delivery of an apoptotic stimulus, cytosolic Bax translocates to the outer mitochondrial membrane, where the protein oligomerizes to form homodimers and creates pores facilitating release of cytochrome c, apoptosis-inducing factor (AIF) and endonuclease G (Endo G), from the intermembrane space of the mitochondrion to the cytosol. Cytosolic cytochrome c binds to apoptotic protease-activating factor 1, in a ternary complex with caspase-9, resulting in caspase-9 activation and caspase-9 in turn activates caspase-3 (Park et al., 2005). Cleavage of the inhibitor of caspase-activated DNase by caspase-3 results in activation of caspase-activated DNase, which fragments DNA into the characteristic oligonucleosomal-length fragments (Li et al., 1997; Sakahira et al., 1998; Daugas et al., 2000; Joza et al., 2001; Park et al., 2005). On the other hand, AIF and Endo G released from mitochondria are translocated to the nucleus, where these molecules trigger large-scale DNA fragmentation and condense chromatin, resulting in apoptotic cell death caused by a caspase-independent pathway (Daugas et al., 2000).
A number of chemotherapeutic agents have been known to cause proteolytic truncation of Bax, and trigger the mitochondrial cell death pathway (Yanase et al., 2000; Yeo et al., 2002; Cao et al., 2003). Bax is truncated at aspartate 33 by calpain, resulting in the formation of an 18 kDa cleavage product that is more potent than native Bax in terms of stimulating mitochondria-mediated apoptotic cell death (Toyota et al., 2003; Ariyama et al., 2006).
Mitogen-activated protein kinases (MAPKs), a family of serine/threonine kinases, are involved in intracellular signalling in response to a variety of stimuli. On the basis of structural differences, MAPKs can be divided into three major families: extracellular signal-regulated kinases (ERKs), c-Jun N-terminal kinases (JNKs) and p38 MAPKs. The ERK pathway activated by mitogenic stimuli is critical for regulation of cell growth, survival and differentiation (Xia et al., 1995; Klekotka et al., 2001; Park et al., 2003; Wada and Penninger, 2004). ERK is commonly thought to mediate survival when apoptotic stimuli are applied (Xia et al., 1995; Klekotka et al., 2001; Park et al., 2003; Wada and Penninger, 2004). However, several studies have shown that ERK may play an important role in apoptotic cell death (Wang et al., 2000; Bacus et al., 2001; Xiao and Singh, 2002; Choi et al., 2003). In contrast to ERKs, JNK and p38 MAPK respond strongly to a variety of stress signals, including those mediated by cytokines, hyperosmotic stress, ionizing radiation, ultraviolet irradiation and chemotherapeutic agents, and cause apoptosis (Xia et al., 1995; Klekotka et al., 2001; Park et al., 2003; Wada and Penninger, 2004).
Recent studies have shown that JNK can directly or indirectly modulate expression of certain members of the Bcl-2 family and can positively influence apoptotic cell death (Fan et al., 2000; Schroeter et al., 2003). Specifically, JNK has been shown to cause apoptosis via induction of mitochondrial translocation of Bax, or Bcl-2 phosphorylation (Fan et al., 2000; Schroeter et al., 2003; Kang and Lee, 2008). Additionally, JNK has been reported to move to mitochondria and to phosphorylate Bcl-xL in mitochondrial membranes (Fan et al., 2000; Kharbanda et al., 2000). Moreover, translocation of JNK to mitochondria can induce release of apoptogenic molecules from these organelles, thereby causing apoptotic cell death (Aoki et al., 2002; Chauhan et al., 2003).
In the present study, we investigated the signalling pathways leading to apoptotic cell death in response to RH1. We have shown that JNK activation contributed significantly to mitochondria-mediated apoptosis caused by RH1. Further, we present evidence that JNK activity was required for generation and mitochondrial translocation of cleaved Bax, and that JNK itself translocated to mitochondria in response to treatment with RH1. Detailed insights into molecular signalling pathways involved in RH1-induced apoptotic cell death may prove useful in the development of chemotherapeutic approaches and strategies for NQO1-directed tumour targeting.
Methods
Cell culture
Parental NQO1–-MDA-MB-231 human breast cancer cells, which are NQO1-deficient, and NQO1+-MDA-MB-231 human breast cancer cells, which are stably transfected with NQO1, were obtained from Dr David Boothman, University of Texas Southwestern Medical Center, Dallas, TX. Cells were cultured in RPMI 1640 medium (Gibco BRL, Grand Island, NY, USA) supplemented with 10% (v/v) bovine calf serum (Gibco BRL), penicillin (50 units·mL−1) and streptomycin (50 µg·mL−1), in a 37°C incubator under a mixture of 95% air and 5% CO2 (both v/v). RKO human colorectal cancer cells carrying wild-type p53 and a subline of RC10.1 cells deficient in p53 were maintained in DMEM medium supplemented with 10% (v/v) bovine calf serum, penicillin (50 units·mL−1) and streptomycin (50 µg·mL−1), in a 37°C incubator under a mixture of 95% air and 5% CO2 (both v/v).
Measurement of NQO1 activity
NAD(P)H quinone oxidoreductase 1 activity was measured by assessing of the dicoumarol-inhibited proportion of 2,6-dichlorophenolindophenol (DCPIP) reduction in S9 supernatants isolated from NQO1-- and NQO1+-MDA-MB-231 cells (Benson et al., 1980). Cells were trypsinized and resuspended in cold TE buffer (20 mM Tris-HCl, pH 7.4 and 2 mM EDTA), washed twice with ice-cold phosphate-buffered saline (PBS), and then resuspended in PBS containing 10 µg·µL−1 aprotinin. Cell suspensions were sonicated four times using 10 s pulses on ice, and S9 supernatants were harvested by centrifugation at 14 000×g for 20 min. Each 1 mL reaction mixture contained 25 mM Tris-HCl (pH 7.4), 180 µM NADPH, bovine serum albumin (BSA; 0.2 mg·mL−1), Tween-20 (0.01%, v/v), 0 or 20 µM dicoumarol and an appropriate volume of cell lysate. Reaction was initiated by addition of 40 µM DCPIP. Reduction of DCPIP was measured at room temperature at 600 nm (=21 × 103 M−1·cm−1) with or without 20 µM dicoumarol. Activities are presented as levels of reduced DCPIP (nM)·min−1·mg−1 protein.
Establishment of cell lines containing inducible NQO1 short hairpin RNA (shRNA)
A double-stranded oligonucleotide, of which one strand was 5′-TCG AGG CAG TAC ACA GAT ACC TTG ATT CAA GAG ATC AAG GTA TCT GTG TAC TGT TTT TTA CGC GTA-3′, was cloned into the pSingle-tTS-shRNA vector (Clontech Laboratories, Inc., Mountain View, CA, USA). RKO and RC10.1 cells were transfected with the plasmid or control empty vector, using Lipofectamine 2000 (Invitrogen) in accordance with the manufacturer's recommendations. Inducible NQO1 shRNA-containing stable clones were selected using 1 mg·mL−1 G418 for 7 days. Stable clones were isolated and treated with 1 µg·mL−1 doxycycline (Sigma), a tetracycline analogue, for 72 h, and endogenous NQO1 knock-down was determined by Western blot analysis using anti-NQO1 antibody.
Small interfering RNA (siRNA) transfection
RNA interference mediated by siRNAs was achieved using double-stranded RNA molecules. AIF (5′-GCA AGU UAC UUA UCA AGC UTT-3′) and JNK2 (5′-CUG UAA CUG UUG AGA UGU ATT-3′) siRNAs were purchased from Bioneer Corporation (Daejeon, Korea). Endo G (5′-GGA ACA ACC UGG AGA AAU ATT-3′) and JNK1 (5′-GAC CUA AAU AUG CUG GAU ATT-3′) siRNAs were the products of Samchully Pharm (Seoul, Korea). An unrelated control siRNA (5′-CCA CTA CCT GAG CAC CCA G-3′) that targeted green fluorescent protein DNA sequence was used as a control. For transfection, cells were seeded on 60 mm dishes and transfected at 30% confluence with the siRNA duplexes (100 nM), using Lipofectamine 2000 (Invitrogen) in accordance with the manufacturer's instructions. Assays were performed 24 h after transfection.
Quantification of clonogenic death
Various numbers of cells were plated on 60 mm dishes and treated with RH1 on the following day. Cells were then washed twice with PBS, and incubated in a 5% CO2 incubator at 37°C for 7–10 days. The colonies were fixed with methanol, stained with crystal violet (0.1% in methanol) and the number of colonies containing more than 50 cells was counted. The surviving cell fractions of treated groups were normalized against the plating efficiency of the untreated control cells.
Quantification of apoptosis
Propidium iodide (PI) staining
Cells were plated on 60 mm dishes at a cell density of 1 × 105 cells and treated with RH1 on the following day. At different time points, cells were trypsinized, washed in PBS and incubated with PI (45 µg·mL−1) for 5 min at room temperature, after which apoptosis was analysed using a flow cytometer (Becton Dickinson, Mountain View, CA, USA).
Annexin V/PI staining
Annexin V/PI staining was performed according to the instructions of the manufacturer (Calbiochem, San Diego, CA, USA). Briefly, 5 × 105 cells were washed with ice-cold PBS without Ca2+ and Mg2+ (Life Technologies, Carlsbad, CA, USA). Cells were then resuspended in 100 µL of binding buffer and incubated with 5 µL of PI and 2 µL of annexin V-fluorescein isothiocyanate (FITC) for 15 min in the dark at room temperature. Flow cytometric analysis was immediately performed.
Measurement of mitochondrial membrane potential
Cells (5 × 105 cells·mL−1) were exposed to 10 µM RH1 for various lengths of time, and then incubated for 30 min in 30 nM of 3,3′-dihexyloxacarbocyanine iodide [DiOC6(3), Molecular Probes, Eugene, OR, USA] at 37°C, harvested via trypsinization and washed three times with cold PBS. Mitochondrial membrane potential was determined by flow cytometry.
Western blotting
Cells were treated with lysis buffer [40 mM Tris-HCl (pH 8.0), 120 mM NaCl and 0.1% (v/v) NP40] supplemented with protease inhibitors, and centrifuged for 15 min at 12 000×g. Proteins were separated by SDS-PAGE and transferred to nitrocellulose membranes (Bio-Rad, Hercules, CA, USA). The membranes were blocked with 5% (w/v) non-fat dry milk in Tris-buffered saline and subsequently incubated for 1 h with primary antibodies, at room temperature. Specific reaction bands were detected using peroxidase-conjugated secondary antibodies, and proteins were visualized using the enhanced chemiluminescence system (Amersham Biosciences, Piscataway, NJ, USA) according to the manufacturer's recommendations.
RNA preparation and RT-PCR
Total RNA was extracted from NQO1+-MDA-MB-231 human breast cancer cells using the TRIzol reagent (Invitrogen), and 1 µg of isolated total RNA was reverse transcribed into cDNA. The primers used for PCR were: Bax forward primer (5′-GCT CTG AGC AGA TCA TGA AG-3′) and reverse primer (5′-CTT GAG CAC CAG TTT GCT GG-3′); AIF forward primer (5′-GAC CGT GTG CGT CCG AAG-3′) and reverse primer (5′-CTC CTG TTG CAA TCA AGC AC-3′); Endo G forward primer (5′-TTC TAC CTG AGC AAC GTC GC-3′) and reverse primer (5′-GTG CGT TGG GCA TCA CGT AG-3′); 18S rRNA (X03205) forward primer (5′-CGG CTA CCA CAT CCA AGG AA-3′) and reverse primer (5′-GCT GGA ATT ACC GCG GCT-3′). Amplified products were separated on 1% (w/v) ethidium bromide-stained agarose gels and expression levels were measured by luminescent image analysis.
Confocal microscopy
2,5-diaziridinyl-3-(hydroxymethyl)-6-methyl-1,4-benzoquinone-treated cells were washed twice in ice-cold PBS prior to fixation in ice-cold methanol. After blocking with 2% (w/v) BSA in PBS containing 0.2% (v/v) Triton X-100, cells were incubated for 1 h with primary antibodies against AIF and Endo G. Cells were then washed three times in blocking solution and incubated for an additional hour with secondary antibody conjugated to FITC (Molecular Probes). Nuclei were then stained with PI (Sigma) for 10 min. After three further washes with PBS, coverslips were mounted onto microscope slides using the ProLong Antifade mounting reagent (Molecular Probes). Slides were viewed using a confocal laser-scanning microscope (Nikon TE-2000E, Tokyo, Japan)
Preparation of mitochondrial and nuclear fractions
Cells were collected and washed twice in ice-cold PBS, resuspended in isotonic homogenization buffer [250 mM sucrose, 10 mM KCl, 1.5 mM MgCl2, 1 mM Na-EDTA, 1 mM dithiothreitol, 0.1 mM phenylmethylsulphonylfluoride, 10 mM Tris-HCl (pH 7.4)], incubated on ice for 20 min and homogenized using a Dounce glass homogenizer fitted with a loose pestle (Wheaton, Millville, NJ, USA) (70 strokes). Cell homogenates were centrifuged at 30×g to remove any unbroken cells. Supernatants were then re-centrifuged for 10 min at 750×g to separate nuclear and mitochondrial fractions. Each nuclear fraction (pellet) was washed three times in homogenization buffer, and resuspended in lysis buffer [50 mM Tris-HCl (pH 7.5), 150 mM NaCl, 1% (v/v) NP40, 0.5% (w/v) sodium deoxycholate] containing protease inhibitors, prior to Western blot analysis. After pelleting of the nuclear fraction, the supernatant was further subjected to 30 min of centrifugation at 14 000×g to pellet a mitochondria-rich fraction. Pellets were washed once in homogenization buffer, and then resuspended in lysis buffer [150 mM NaCl, 50 mM Tris-HCl (pH 7.5), 1% (v/v) NP40, 0.25% (w/v) sodium deoxycholate and 1 mM EGTA], with protease inhibitors, prior to Western blot analysis.
Statistical analysis
All Western blots and data presented are representative of at least three separate experiments. The optical density of all Western blots and RT-PCR data was quantified using Image J (NIH, Bethesda, MD, USA), and normalized to β-actin, Nucleoporin p62 (nuclear marker), mitochondrial HSP70 (mitochondrial marker), JNK, p38 MAPK, and ERK1/2 (Western blots) and 18S rRNA (RT-PCR data). Comparisons among groups were analysed using Student's t-test (SPSS Statistics version 17.0, Chicago, IL, USA). P-values < 0.05 were considered to be significant.
Materials
2,5-diaziridinyl-3-(hydroxymethyl)-6-methyl-1,4-benzoquinone was synthesized as previously described (Winski et al., 1998) and was dissolved in DMSO. Antibodies against Bax, AIF, Endo G, Bcl-2, p38 MAPK, JNK1, JNK2, JNK1/2, ERK1/2; caspases-8, -9 and -3; and Bid were purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA, USA). An antibody against mitochondrial heat shock protein 70 (HSP70) was the product of Affinity Bioreagents (Golden, CO, USA). Anti-β-actin, -rabbit IgG and -mouse IgG and chloramphenicol were purchased from Sigma (St. Louis, MO, USA). Antibodies against cytochrome c, Bcl-2, JNK1 and Bax were provided by BD Bioscience PharMingen (San Jose, CA, USA). Antibodies against phospho-p38 (Thr180/Tyr182), phospho-JNK1/2 (Thr183/Tyr185), phospho-ERK1/2(Thr202/Tyr204) and polyADP ribose polymerase (PARP) were obtained from Cell Signaling Technology (Beverly, MA, USA). Inhibitors specific for JNK (SP600125), MEK (PD98059), p38 MAPK (SB203580), cathepsin B (inhibitor II) and z-VAD-fmk (inhibitor of caspases) were purchased from Calbiochem (San Diego, CA, USA).
Results
NQO1-dependent apoptotic cell death in response to RH1
To investigate the kinetics of cell death induced by RH1, we treated parental NQO1--MDA-MB-231 cells deficient in NQO1, or NQO1+-MDA-MB-231 cells possessing abundant expression and activity of NQO1 (Figure 1A), with different doses of RH1 for various lengths of time, and analysed apoptotic cell death by PI staining. Figure 1B shows that a dose- and time-dependent increase in the proportion of cell death after RH1 treatment was observed, suggesting that NQO1+-MDA-MB-231 cells were more sensitive to RH1 than were the parental NQO1--MDA-MB-231 cells. To further assess whether the cytotoxic effect of RH1 was associated with induction of apoptosis, NQO1+-MDA-MB-231 cells were stained with Annexin V/PI after treatment with 10 µM RH1. Figure 1C shows that cells entered the initial stages of apoptosis soon after RH1 treatment, but the proportion of cells in early stage apoptosis began to decline from 12 h after RH1 treatment while the proportion of cells in late stage apoptosis progressively increased. Subsequently, necrosis was detected in a small cell population.
Figure 1.
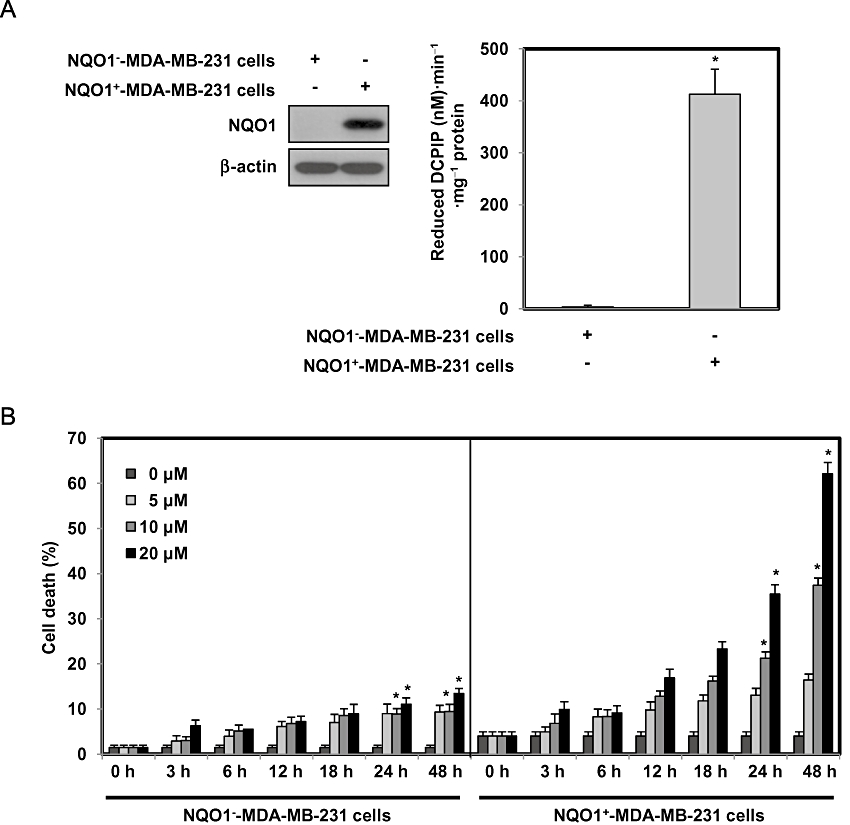
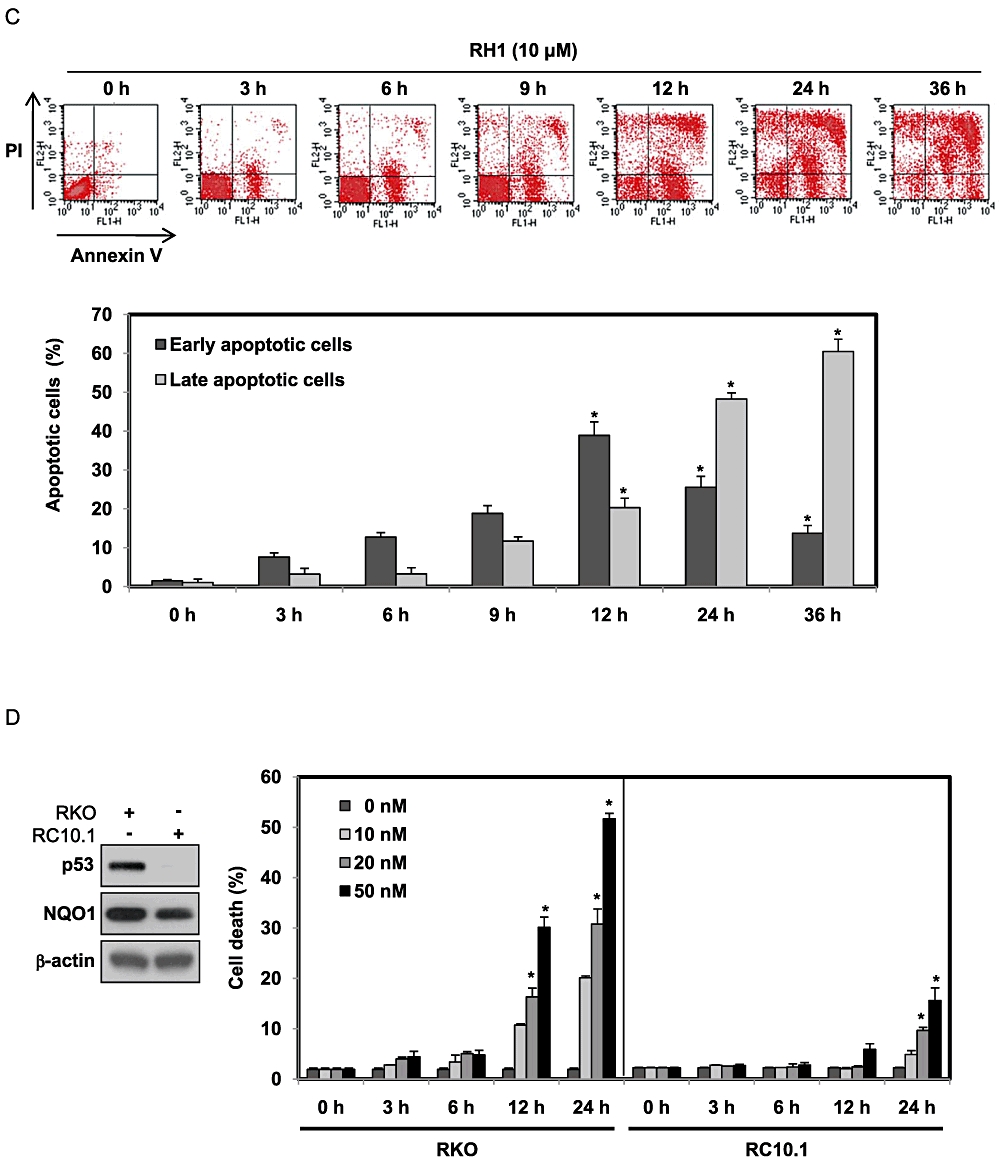
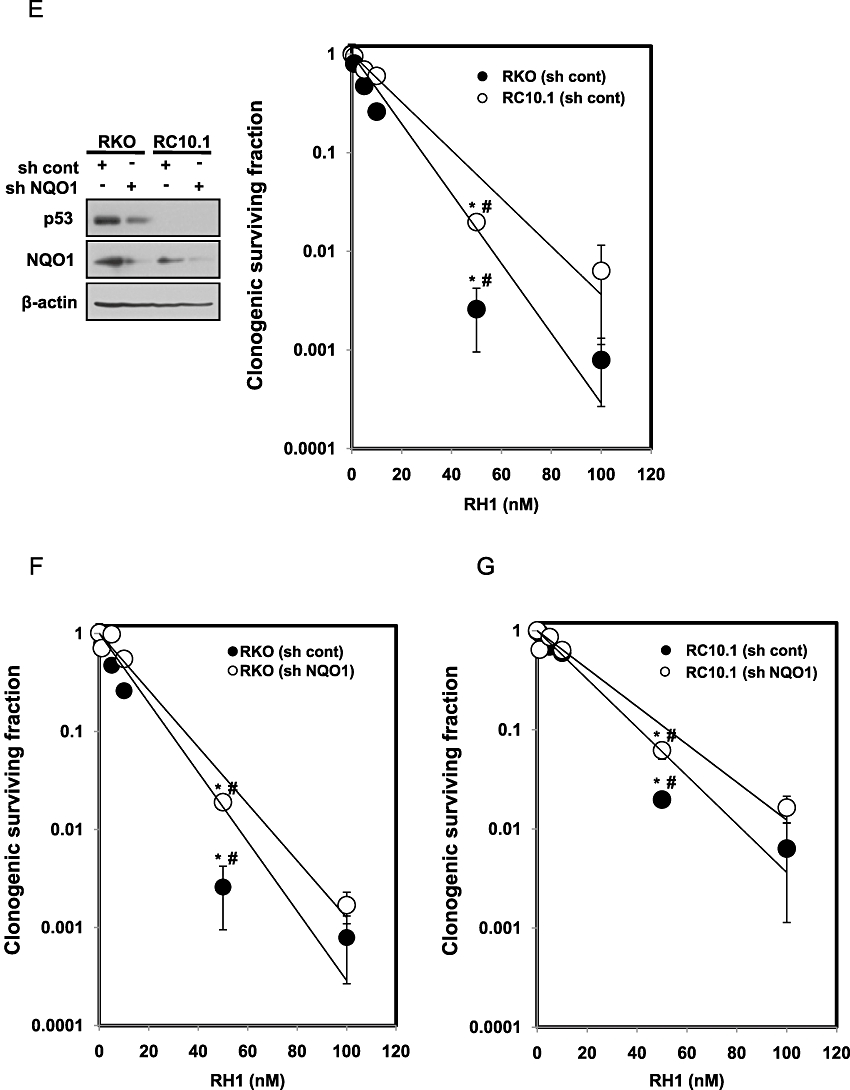
2,5-diaziridinyl-3-(hydroxymethyl)-6-methyl-1,4-benzoquinone (RH1) induces NAD(P)H quinone oxidoreductase 1 (NQO1)-dependent apoptotic cell death of MDA-MB-231 cells. (A) NQO1 activity levels in NQO1-- or NQO1+-MDA-MB-231 cells. NQO1 activity was measured as described in Methods. The data from three independent experiments are expressed as means ± SEM. *Significant difference between NQO1-- and NQO1+-MDA-MB-231 cells at P < 0.05. (B) NQO1-- or NQO1+-MDA-MB-231 cells were treated with various concentrations of RH1 for the indicated times. Cells were stained with propidium iodide (PI) and apoptotic cell numbers were measured by flow cytometry. Results from three independent experiments are expressed as means ± SEM. *Significant difference between control and RH1-treated cells at P < 0.05. (C) NQO1+-MDA-MB-231 cells were treated with 10 µM RH1 for the indicated times. Cells were stained with Annexin V/PI and apoptotic changes were measured by flow cytometry. Representative flow cytometric dot plots are shown: lower left quadrant, viable cells; lower right, early apoptotic cells; upper right, late apoptotic cells; upper left, necrotic cells. Apoptotic changes were quantitated as described below. Results from three independent experiments are expressed as means ± SEM. *Significant difference between RH1-untreated and -treated cells at P < 0.05. (D) RKO or RC10.1 cells were treated with various concentrations of RH1 for the indicated times. Cells were stained with PI, and apoptotic cell numbers measured using flow cytometry. Results from three independent experiments are expressed as means ± SEM. *Significant difference between RH1-untreated and -treated cells at P < 0.05.(E–G) Clonogenic survival of RKO cells and RC10.1 cells were compared. RC10.1 cells were resistant to RH1 treatment compared with parental RKO cells (E). F and G show that NQO1 shRNA effectively attenuated RH1-induced clonogenic cell death in both RKO and RC10.1 cells. Data for E–G are mean of three independent experiments ± SEM. *Significant difference between RH1-untreated and -treated cells; P < 0.05. #Significant difference between shRNA transfected cells in response to RH1 treatment; P < 0.05.
Previous reports indicated that NQO1 stabilizes p53 protein by interfering with 20S proteasome-mediated p53 degradation (Asher et al., 2001; Nioi and Hayes, 2004). Therefore, we determined whether p53 was required for RH1-induced apoptosis. Because MDA-MB-231 cells carry a mutant form of p53 (Olivier et al., 2002), we investigated the role of p53 in RH1-induced apoptotic cell death using human colon cancer RKO cells expressing wild-type p53, and RC10.1 cells deficient in p53 (p53-deficient RKO cells) (Figure 1D, left panel). Both cell lines expressed NQO1 protein although the expression levels in RC10.1 cells were slightly lower than that in RKO cells (Figure 1D, left panel). As shown in Figure 1D, at nanomolar concentrations, RH1 effectively induced apoptotic cell death in parental RKO cells in a dose- and time-dependent manner, but this was not observed in RC10.1 cells. We subsequently confirmed a role for p53 in RH1-induced cell death using a clonogenic cell survival assay. As shown in Figure 1E, RC10.1 cells were resistant to RH1 treatment, compared with parental RKO cells, suggesting that p53 was positively involved in RH1-induced clonogenic cell death. We next investigated whether NQO1 was also involved in this process, using both RKO and RC10.1 cells expressing NQO1 shRNA. Figure 1F and G show that NQO1 shRNA effectively attenuated RH1-induced clonogenic cell death in both RKO and RC10.1 cells. These results indicate that both p53 and NQO1 play key roles in RH1-induced apoptotic cell death as well as clonogenic cell death.
RH1 induces apoptotic cell death in a caspase-independent manner
As activation of caspases is an important mechanism whereby apoptotic cell death is induced (Park et al., 2003), we explored whether RH1 treatment activated caspases. Treatment of NQO1+-MDA-MB-231 cells with RH1 activated caspases-8, -9 and -3, and induced PARP cleavage, in a time-dependent manner (Figure 2A). Any requirement for caspase activity during RH1-induced apoptotic cell death was further investigated using a broad-spectrum caspase inhibitor, z-VAD-fmk. As shown in Figure 2B, pretreatment with z-VAD-fmk effectively blocked activation of caspases but did not attenuate RH1-induced apoptosis. We further confirmed the lack of effect of z-VAD-fmk on RH1-induced apoptosis by performing Annexin V/PI staining. Figure 2C shows that pretreatment with z-VAD-fmk did not inhibit apoptotic death caused by RH1. These findings indicate that RH1 inducesNQO1-dependent but caspase-independent apoptotic cell death in MDA-MB-231 cells.
Figure 2.

2,5-diaziridinyl-3-(hydroxymethyl)-6-methyl-1,4-benzoquinone (RH1) induces caspase-independent apoptotic cell death. (A) NAD(P)H quinone oxidoreductase 1 (NQO1)+-MDA-MB-231 cells were treated with 10 µM RH1 for the indicated times. Whole cell lysates were prepared and evaluated by Western blotting for expression of active caspases-8, -9 and -3; polyADP ribose polymerase (PARP); and β-actin. The relative density value of each band is shown below the Western blot. The data are representative of a typical experiment that was conducted three times (mean values, P < 0.05). (B) NQO1+-MDA-MB-231 cells were treated with 10 µM RH1 for 36 h in the presence or absence of 30 µM z-VAD-fmk. Whole cell extracts were directly examined by Western blotting using anti-active caspase-8, -active caspase-9, -active caspase-3, -PARP and -β-actin antibodies (left panel). The relative density value of each band is shown below the Western blot. The data are from a typical experiment that was conducted three times with similar results (mean values, P < 0.05). Cells were stained with propidium iodide (PI) and apoptotic cell numbers were measured by flow cytometry. Results of three independent experiments are expressed as means ± SEM. *Significant difference between control and RH1-treated cells at P < 0.05. **Significant difference between RH1-treated cells and z-VAD-fmk + RH1-treated cells at P < 0.05. (C) NQO1+-MDA-MB-231 cells were treated with 10 µM RH1 for 12 h in the presence or absence of 30 µM z-VAD-fmk. Cells were stained with Annexin V/PI and apoptotic changes were assessed by flow cytometry. Representative flow cytometric dot plots are shown: lower left quadrant, viable cells; lower right, early apoptotic cells; upper right, late apoptotic cells; upper left, necrotic cells. Apoptotic changes were quantitated as described below. Results from three independent experiments are expressed as means ± SEM. *Significant difference between control and RH1-treated cells; P < 0.05. **Significant difference between RH1-treated cells and z-VAD-fmk + RH1-treated cells; P < 0.05.
RH1 induces apoptotic cell death via translocation of AIF and Endo G to the nucleus
Because AIF and Endo G are involved in a caspase-independent pathway of cell death (Daugas et al., 2000), we determined whether AIF and Endo G played roles in RH1-induced apoptotic cell death in NQO1+-MDA-MB-231 cells. When mitochondria receive apoptotic stimuli, AIF and Endo G are released from these organelles and are translocated to nuclei, where they induce DNA fragmentation (Daugas et al., 2000). As shown in Figure 3A and B, RH1 treatment induced translocation of AIF and Endo G to the nucleus, in a time-dependent manner, without causing any change in relevant gene expression. Confocal microscopy also clearly showed that RH1 treatment caused translocation of AIF and Endo G to nucleus, and induced nuclear condensation (Figure 3C). As shown in Figure 3D, siRNA targeting AIF or Endo G effectively attenuated RH1-induced apoptotic cell death. These results clearly indicate that translocation of AIF and Endo G from mitochondria to nuclei is essential for the caspase-independent cell death in NQO1+-MDA-MB-231 cells after RH1 treatment.
Figure 3.
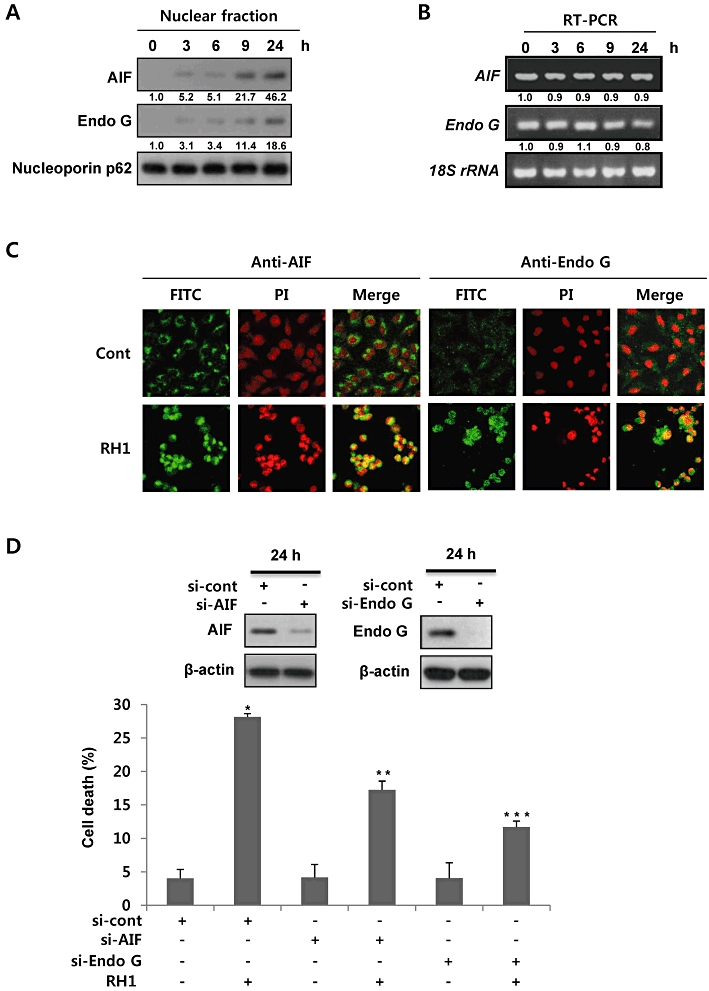
2,5-diaziridinyl-3-(hydroxymethyl)-6-methyl-1,4-benzoquinone (RH1) induces apoptotic cell death via nuclear translocation of apoptosis-inducing factor (AIF) and endonuclease G (Endo G). (A) Nuclear fractions were obtained from NAD(P)H quinone oxidoreductase 1 (NQO1)+-MDA-MB-231 cells treated with 10 µM RH1 for the indicated times, and were subjected to Western blot analysis using anti-AIF, -Endo G and -Nucleoporin p62 antibodies. The relative density value of each band is shown below the Western blot. The data are from a typical experiment that was conducted three times with similar results (mean values, P < 0.05). (B) NQO1+-MDA-MB-231 cells were treated with 10 µM RH1 for the indicated times, and RT-PCR products were then obtained using specific primers amplifying AIF mRNA, Endo G mRNA and 18S rRNA. The relative density value of each band is shown below the RT-PCR data. The data are from a typical experiment that was conducted three times with similar results (mean values, P < 0.05). (C) Representative confocal images showing translocation of AIF and Endo G to the nucleus, and nuclear condensation, at 24 h after treatment with 10 µM RH1. Nuclear translocation of AIF and Endo G is demonstrated by overlap of AIF or Endo G (green) and nuclear staining (red), resulting in a yellow colour. The data are from a typical experiment conducted three times. (D) NQO1+-MDA-MB-231 cells transfected with AIF or Endo G siRNA were treated with 10 µM RH1. After 36 h, cells were stained with propidium iodide (PI) and apoptotic cell numbers measured by flow cytometry. The results of three independent experiments are expressed as means ± SEM. *Significant difference between control and RH1-treated cells; P < 0.05. **Significant difference between si-cont RNA- and si-AIF RNA-transfected cells after RH1 treatment at P < 0.05. ***Significant difference between si-cont RNA- and si-Endo G RNA-transfected cells after RH1 treatment; P < 0.05.
RH1 induces generation and translocation of cleaved Bax to mitochondria
To determine the role played by the mitochondrial pathway in induction of apoptosis after RH1 treatment, we investigated in detail the changes in mitochondrial membrane potential, and the expression levels of Bcl-2 family proteins, in NQO1+-MDA-MB-231 cells after RH1 treatment. Figure 4A shows that RH1 treatment significantly disrupted mitochondrial membrane potential, in a time-dependent manner. Simultaneously, Bax level was markedly reduced, without any changes in the expression levels of Bcl-2 and Bid. The expression level of Bax gene was also not affected by RH1 treatment (Figure 4B and C). Bax is cleaved to an 18 kDa fragment from the 21 kDa native form in response to various stimuli (Toyota et al., 2003; Ariyama et al., 2006). As translocation of cleaved Bax from the cytosol to mitochondria induced a decline in mitochondrial membrane potential and subsequent release of proapoptotic proteins from mitochondria (Toyota et al., 2003; Ariyama et al., 2006), we determined whether RH1 treatment induced mitochondrial translocation of cleaved Bax. Interestingly, as shown in Figure 4D, RH1 treatment efficiently raised cleaved Bax levels within mitochondrial fractions for 24 h, coinciding with the observed changes in mitochondrial membrane potential. However, there were no indications of the 18-kDa cleaved form of Bax in whole cell lysates after RH1 treatment. As a cathepsin-like protease is known to be involved in the rapid degradation of the p18 kDa cleaved form of Bax in the cytosol (Cao et al., 2003), we pretreated cells with protease inhibitor MG132 before RH1 treatment and determined Bax in whole cell lysates. As shown in Figure 4E, when cells were treated with RH1 in the presence of MG132, cleaved Bax was detected in whole cell lysates, suggesting that cleaved Bax is readily degraded in the cytosol after RH1 treatment. Taken together, these results indicate that RH1-induced cell death involves an alteration in mitochondrial membrane potential mediated by intracellular redistribution of cleaved Bax.
Figure 4.
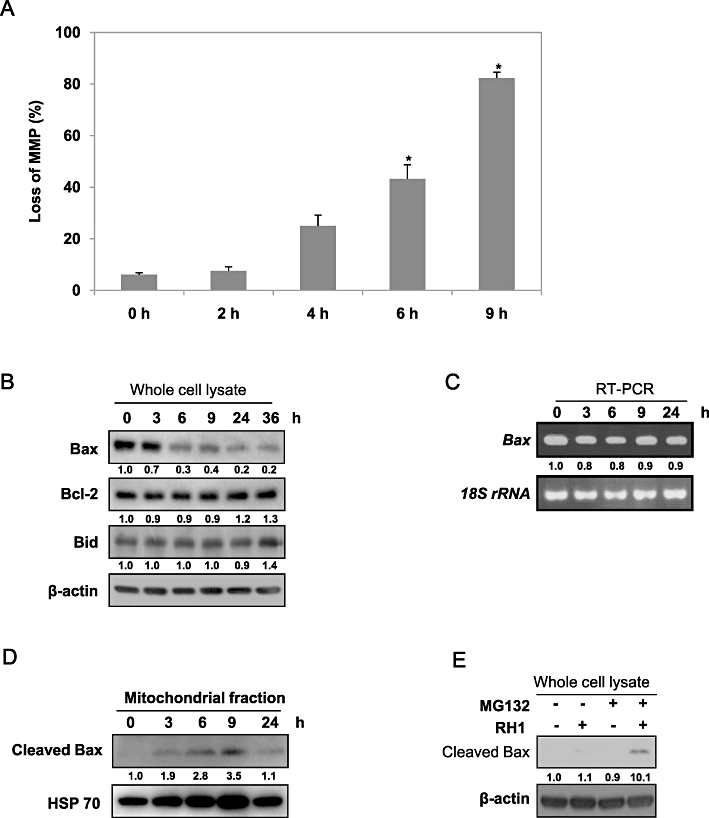
2,5-diaziridinyl-3-(hydroxymethyl)-6-methyl-1,4-benzoquinone (RH1) causes Bax cleavage and translocation of cleaved Bax to mitochondria. (A) Mitochondrial membrane potential (MMP) was determined by retention of the mitochondrial specific dye 3,3′-dihexyloxacarbocyanine iodide [DiOC6(3)]. After treatment with 10 µM RH1 for the indicated times, NAD(P)H quinone oxidoreductase 1 (NQO1)+-MDA-MB-231 cells were loaded with 30 nM DiOC6(3) for a further 30 min of RH1 treatment. After removal of medium, the concentration of retained DiOC6(3) was measured by flow cytometry. Results from three independent experiments are expressed as means ± SEM. *Significant difference between control and RH1-treated cells; P < 0.05. (B) NQO1+-MDA-MB-231 cells were treated with 10 µM RH1 for the times indicated. Whole cell lysates were prepared and Western blot analysis was conducted using anti-Bax, -Bcl2, -Bid and -β-actin antibodies. The relative density value of each band is shown below the Western blot. The data are from a typical experiment that was conducted three times with similar results (mean values, P < 0.05). (C) NQO1+-MDA-MB-231 cells were treated with 10 µM RH1 for the indicated times, and RT-PCR products were then obtained using specific primers recognizing Bax mRNA and 18S rRNA. The relative density value of each band is shown below the RT-PCR data. The data are from a typical experiment that was conducted three times with similar results (mean values, P < 0.05). (D) NQO1+-MDA-MB-231 cells were treated with 10 µM RH1 for the indicated times. Mitochondrial fractions were prepared and subjected to Western blot analysis using anti-Bax and -mitochondrial HSP70 antibodies. The relative density value of each band is shown below the Western blot. The data are from a typical experiment that was conducted three times with similar results (mean values, P < 0.05). (E) NQO1+-MDA-MB-231 cells were treated with 10 µM RH1 for 9 h in the presence or absence of 50 µM MG132. Whole cell lysates were prepared and Western blot analysis was conducted using anti-Bax and -β-actin antibodies. The relative density value of each band is shown below the Western blot. The data are from a typical experiment that was conducted three times with similar results (mean values, P < 0.05).
JNK activity is necessary for RH1-induced apoptotic cell death
As MAPKs have been implicated in the regulation of apoptosis in response to a variety of stimuli (Xia et al., 1995; Klekotka et al., 2001; Park et al., 2003; Wada and Penninger, 2004), we assessed the involvement of MAPKs in RH1-induced apoptotic cell death. We first measured the activities and protein levels of MAPKs following RH1 treatment in NQO1+-MDA-MB-231 cells. As shown in Figure 5A, RH1 treatment markedly increased the levels of the phosphorylated forms of JNK, p38 MAPK and ERK. Furthermore, JNK activation after RH1 treatment occurred earlier than did p38 MAPK or ERK activation, and no changes in the total cellular levels of JNK, p38 MAPK or ERK were evident. To identify the specific MAPK involved in RH1-induced apoptosis, we pretreated cells with SP600125, SB203580 and PD98059, inhibitors of JNK, p38 MAPK and MEK, respectively, prior to RH1 treatment. Interestingly, SP600125 selectively and effectively reduced RH1-induced apoptosis and disruption of mitochondrial membrane potential (Figure 5B and C). Pretreatment with PD98059 slightly attenuated RH1-mediated apoptosis and disruption of mitochondrial membrane potential, whereas pretreatment with SB203580 was ineffective in both respects.
Figure 5.
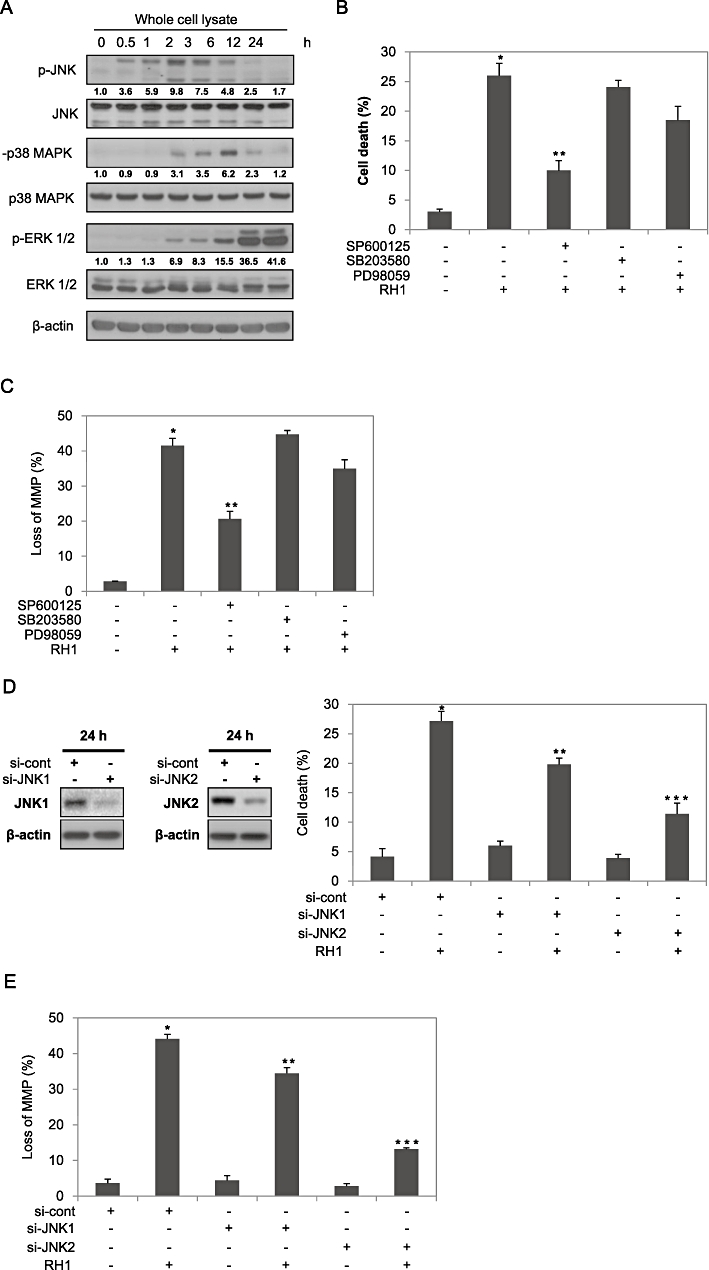
2,5-diaziridinyl-3-(hydroxymethyl)-6-methyl-1,4-benzoquinone (RH1) activates mitogen-activated protein kinases (MAPKs) in NAD(P)H quinone oxidoreductase 1 (NQO1)+-MDA-MB-231 cells. (A) NQO1+-MDA-MB-231 cells were treated with 10 µM RH1 for the times indicated. Whole cell lysates were subjected to Western blot analysis using anti-phospho-c-Jun N-terminal kinase (JNK)1/2, -JNK1/2, -phospho-p38 MAPK, -p38 MAPK, -phospho-extracellular signal-regulated kinase (ERK)1/2, -ERK1/2 and -β-actin antibodies. The relative density value of each band is shown below the Western blot. The data are from a typical experiment that was conducted three times with similar results (mean values, P < 0.05). (B) NQO1+-MDA-MB-231 cells were treated with 10 µM RH1 for 36 h in the presence or absence of 30 µM SP600125, SB203580 or PD98059. Cells were stained with PI and apoptotic cell numbers were measured by flow cytometry. Results from three independent experiments are expressed as means ± SEM. *Significant difference between control and RH1-treated cells; P < 0.05. **Significant difference between RH1- and SP600125 + RH1-treated cells; P < 0.05. (C) NQO1+-MDA-MB-231 cells were treated with 10 µM RH1 for 6 h in the presence or absence of 30 µM SP600125, SB203580 or PD98059. Cells were loaded with 30 nM of 3,3′-dihexyloxacarbocyanine iodide [DiOC6(3)] during the last 30 min of RH1 treatment. After removal of medium, the concentration of retained DiOC6(3) was measured by flow cytometry. The results from three independent experiments are expressed as means ± SEM. *Significant difference between control and RH1-treated cells; P < 0.05. **Significant difference between RH1- and SP600125 + RH1-treated cells; P < 0.05. (D) NQO1+-MDA-MB-231 cells were treated with 10 µM RH1 for 36 h in the presence or absence of siRNAs targeting JNK1 or JNK2. Cells were stained with PI and apoptotic cell numbers were analysed by flow cytometry. Results from three independent experiments are expressed as means ± SEM. *Significant difference between control and RH1-treated cells; P < 0.05. **Significant difference between si-cont RNA- and si-JNK1 RNA-transfected cells in response to RH1 treatment; P < 0.05. ***Significant difference between si-cont RNA- and si-JNK2 RNA-transfected cells after RH1 treatment; P < 0.05. (E) NQO1+-MDA-MB-231 cells were treated with 10 µM RH1 for 6 h in the presence or absence of siRNA targeting JNK1 or JNK2. Cells were loaded with 30 nM of DiOC6(3) during the last 30 min of RH1 treatment. After removal of medium, the concentration of retained DiOC6(3) was measured by flow cytometry. Results from three independent experiments are expressed as means ± SEM. *Significant difference between control and RH1-treated cells; P < 0.05. **Significant difference between si-cont RNA- and si-JNK1 RNA-transfected cells after RH1 treatment; P < 0.05. ***Significant difference between si-cont RNA- and si-JNK2 RNA-transfected cells after RH1 treatment; P < 0.05.
As it was apparent that JNK played a key role in RH1-induced mitochondrial apoptosis, we further determined which JNK isoform was required for the RH1-induced apoptosis using siRNA targeting JNK1 and JNK2. As shown in Figure 5D and E, siRNA targeting JNK2 significantly attenuated RH1-induced apoptosis and disruption of mitochondrial membrane potential, whereas siRNA targeting JNK1 only slightly reduced apoptosis and disrupted membrane potential. These results clearly indicate that JNK2 is an important mediator of RH1-induced mitochondrial apoptotic cell death in NQO1+-MDA-MB-231 cells.
JNK activity is critical for generation and mitochondrial translocation of cleaved Bax
We next investigated the effects of SP600125 and PD98059 on Bax cleavage and the nuclear translocation of AIF and Endo G by RH1 treatment. Figure 6A, B, C and D show that SP600125 was significantly more effective than PD98059 in blocking of all of RH1-induced Bax cleavage, mitochondrial translocation of cleaved Bax, and nuclear translocation of AIF and Endo G, without any change in the expression of Bax, AIF or Endo G genes. We confirmed SP600125 specificity using siRNA targeting JNK2. Transfection of cells with siRNA targeting JNK2 markedly decreased the mitochondrial translocation of cleaved Bax and inhibited the nuclear translocation of AIF and Endo G caused by RH1 treatment (Figure 6E and F). It has been reported that JNK positively regulates both the activity and expression of cathepsin B, a lysosomal protease involved in caspase-independent apoptosis (Foghsgaard et al., 2001; Tang et al., 2006; Werneburg et al., 2007; Bivik and Ollinger, 2008; Yamaguchi et al.,2008). We thus investigated the potential involvement of cathepsin B in RH1-induced apoptotic cell death, using a pharmacological inhibitor of the enzyme. As shown in Figure S1, pretreatment with the cathepsin B inhibitor did not attenuate apoptosis triggered by RH1. These results clearly show that the mitochondria-mediated apoptotic process requires induction of JNK activity after RH1 treatment.
Figure 6.
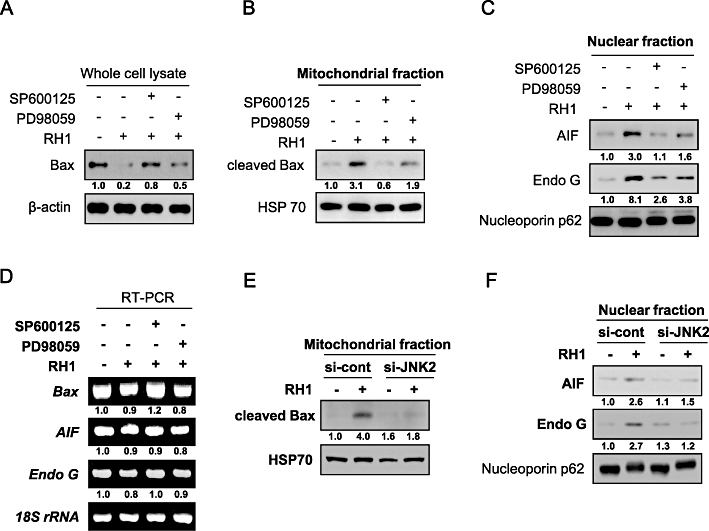
Activation of c-Jun N-terminal kinase (JNK) is required for mitochondrial translocation of cleaved Bax and nuclear translocation of apoptosis-inducing factor (AIF) and endonuclease G (Endo G) in response to 2,5-diaziridinyl-3-(hydroxymethyl)-6-methyl-1,4-benzoquinone (RH1) treatment. (A) NAD(P)H quinone oxidoreductase 1 (NQO1)+-MDA-MB-231 cells were treated with 10 µM RH1 for 9 h in the presence or absence of 30 µM SP600125 or PD98059. Whole cell lysates were subjected to Western blot analysis using anti-Bax and -β-actin antibodies. The relative density value of each band is shown below the Western blot. The data are from a typical experiment that was conducted three times with similar results (mean values, P < 0.05). (B) NQO1+-MDA-MB-231 cells were treated with 10 µM RH1 for 9 h in the presence or absence of 30 µM SP600125 or PD98059. Mitochondrial fractions were prepared and subjected to Western blot analysis using anti-Bax and -mitochondrial HSP70 antibodies. The relative density value of each band is shown below the Western blot. The data are from a typical experiment that was conducted three times with similar results (mean values, P < 0.05). (C) NQO1+-MDA-MB-231 cells were treated with 10 µM RH1 for 9 h in the presence or absence of 30 µM SP600125 or PD98059. Nuclear fractions were prepared and subjected to Western blot analysis using anti-AIF, -Endo G and -Nucleoporin p62 antibodies. The relative density value of each band is shown below the Western blot. The data are from a typical experiment that was conducted three times with similar results (mean values, P < 0.05). (D) NQO1+-MDA-MB-231 cells were treated with 10 µM RH1 for 9 h in the presence or absence of 30 µM SP600125 or PD98059, and RT-PCR products were obtained using specific primers recognizing Bax, AIF and Endo G mRNA, and 18S rRNA. The relative density value of each band is shown below the RT-PCR data. The data are from a typical experiment that was conducted three times with similar results (mean values, P < 0.05). (E) NQO1+-MDA-MB-231 cells were treated with 10 µM RH1 for 9 h in the presence or absence of siRNA targeting JNK2. Mitochondrial fractions were prepared and subjected to Western blot analysis using antibodies against Bax and mitochondrial HSP70. The relative density value of each band is shown below the Western blot. The data are from a typical experiment that was conducted three times with similar results (mean values, P < 0.05). (F) NQO1+-MDA-MB-231 cells were treated with 10 µM RH1 for 9 h in the presence or absence of siRNA targeting JNK2. Nuclear fractions were prepared and subjected to Western blotting using antibodies against AIF, Endo G and Nucleoporin p62. The relative density value of each band is shown below the Western blot. The data are from a typical experiment that was conducted three times with similar results (mean values, P < 0.05).
Mitochondrial translocation of JNK is involved in RH1-induced apoptosis
Several previous reports have demonstrated that translocation of JNK to mitochondria plays a role in apoptotic cell death in response to certain stimuli (Fan et al., 2000; Kharbanda et al., 2000; Aoki et al., 2002; Chauhan et al., 2003; Schroeter et al., 2003; Kang and Lee, 2008). To determine whether mitochondrial translocation of JNK was required for RH1-mediated apoptosis, we analysed the intracellular redistribution of JNK by Western blotting of both cytosolic and mitochondrial fractions. Figure 7A shows that RH1 treatment promoted translocation of both JNK and phospho-JNK to mitochondria, whereas the levels of both JNK and phospho-JNK in the cytosol initially increased and began to decrease 3 h after treatment with RH1. We then evaluated the effect of JNK inhibition on the mitochondrial translocation of JNK and phospho-JNK induced by RH1 treatment. As shown in Figure 7B, inhibition of JNK activity by pretreatment with SP600125 suppressed the mitochondrial translocation of both JNK and phospho-JNK. We then determined whether ERK activation was required for translocation of JNK to mitochondria, using PD98059, an ERK pathway inhibitor. Figure 7B shows that RH1-induced mitochondrial translocation of either JNK or phospho-JNK was not inhibited by PD98059. Additionally, we examined whether RH1 induces synthesis of JNK protein in mitochondria. As chloramphenicol has been reported to inhibit mitochondrial protein synthesis (Kroon, 1965; de Souza-Pinto et al., 2009), cells were treated with RH1 in the presence or absence of chloramphenicol, and Western blotting using an anti-JNK antibody was subsequently performed. As shown in Figure 7C, pretreatment with chloramphenicol in the presence of RH1 did not change the level of expression of JNK protein for 24 h in whole cell lysates. These data strongly suggest that activation and mitochondrial translocation of JNK are important for mitochondria-induced apoptotic cell death. These processes described above are upstream of nuclear translocation of AIF and Endo G in the RH1-induced apoptotic cascade.
Figure 7.
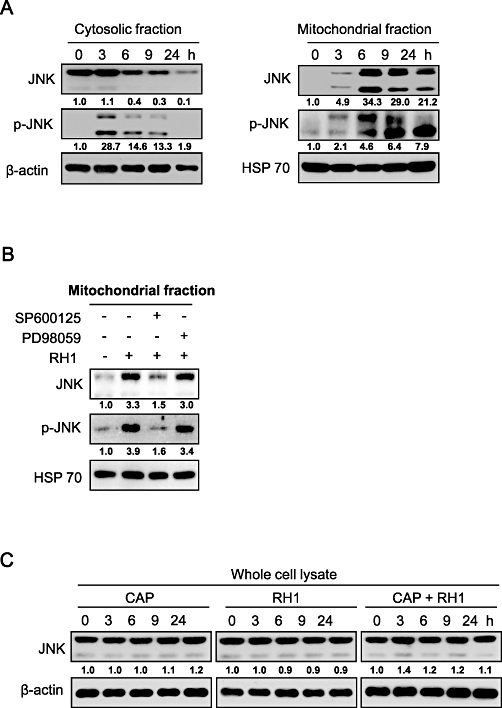
2,5-diaziridinyl-3-(hydroxymethyl)-6-methyl-1,4-benzoquinone (RH1) induces activation and mitochondrial translocation of c-Jun N-terminal kinase (JNK) in NAD(P)H quinone oxidoreductase 1 (NQO1)+-MDA-MB-231 cells. (A) NQO1+-MDA-MB-231 cells were treated with 10 µM RH1 for the indicated times. Cytosolic and mitochondrial fractions were prepared and subjected to Western blot analysis using anti-JNK1/2, -phospho-JNK1/2, -β-actin and -mitochondrial HSP70 antibodies. The relative density value of each band is shown below the Western blot. The data are from a typical experiment that was conducted three times with similar results (mean values, P < 0.05). (B) NQO1+-MDA-MB-231 cells were treated with 10 µM RH1 for 9 h in the presence or absence of 30 µM SP600125 or PD98059. Mitochondrial fractions were prepared and subjected to Western blotting using anti-JNK1/2, -phospho-JNK1/2 and -mitochondrial HSP70 antibodies. The relative density value of each band is shown below the Western blot. The data are from a typical experiment that was conducted three times with similar results (mean values, P < 0.05). (C) NQO1+-MDA-MB-231 cells were treated with 10 µM RH1 for the indicated times in the presence or absence of 50 µg·mL−1 chloramphenicol (CAP). Whole cell lysates were subjected to Western blot analysis using anti-JNK1/2 and β-actin antibodies. The relative density value of each band is shown below the Western blot. The data are from a typical experiment that was conducted three times with similar results (mean values, P < 0.05).
Discussion
2,5-diaziridinyl-3-(hydroxymethyl)-6-methyl-1,4-benzoquinone is a bioreductive agent that is reduced within cells by NQO1 (Cummings et al., 2003; Kim et al., 2004a). Reduction of RH1 activates aziridine residues, which subsequently induce DNA damage (Cummings et al., 2003; Kim et al., 2004a; Kim et al., 2004b; Ward et al., 2005; Hussein et al., 2009). Because NQO1 is abundantly expressed in a variety of cancer tissues, it has been suggested that such tissues can be selectively damaged by RH1. This enzyme-directed cancer-targeting agent is currently being tested in clinical trials (Cummings et al., 2003; Kim et al., 2004a; Kim et al., 2004b; Ward et al., 2005; Hussein et al., 2009). However, the molecular mechanisms underlying the therapeutic effects of the agent on solid tumours have not yet been clearly elucidated.
In the present study using NQO1+-MDA-MB-231 cells, we have demonstrated that translocation of AIF and Endo G from mitochondria to the nucleus was critical for RH1-induced caspase-independent cell death. We also showed that JNK2 activation was essential for mitochondria-mediated apoptotic cell death induced by RH1.
In the present work, RH1 preferentially induced apoptotic cell death in NQO1+-MDA-MB-231 cells, compared with NQO1--MDA-MB-231 cells. These results are consistent with previous reports on the role played by NOQ1 in mediation of RH1 toxicity (Cummings et al., 2003; Kim et al., 2004a). Caspase activation is known to be important in RH1-induced cell death (Dehn et al., 2005). Although we show here that RH1 induced caspase activation and PARP cleavage, the apoptotic cell death caused by RH1 appeared to be caspase-independent, because caspase inhibitors failed to attenuate the process.
Several anti-cancer drugs are known to exert cytotoxic effects via activation of p53-dependent apoptotic pathways (Lowe et al., 1993). NQO1 affects the sensitivity of cancer cells to a variety of anti-cancer drugs that induce apoptosis, by enhancing p53 stability. In the present study, we observed that RH1 is less effective in inducing apoptosis in NQO1+-MDA-MB-231 cells lacking p53 expression than in p53 expressing RKO cells. Therefore, it was likely that not only NQO1 status, but also p53 expression, affects the sensitivity of cells towards RH1. Furthermore, as MDA-MB-468 cells overexpressing NQO1 could be killed by nanomolar doses of RH1 (Dehn et al., 2005), unlike what was seen in NQO1+-MDA-MB-231 cells, RH1 cytotoxicity appears to vary in different cancer cell types.
Apoptosis-inducing factor and Endo G are known to function in a caspase-independent apoptosis pathway. Mitochondrial AIF and Endo G are translocated to the nucleus in response to death stimuli, where theses proteins initiate nuclear condensation and DNA fragmentation (Daugas et al., 2000). Consistent with these findings, we observed that AIF and Endo G were translocated to the nucleus after RH1 treatment, and that both AIF and Endo G activities were required for RH1-induced apoptosis.
Proteins of the Bcl-2 family play crucial roles in the regulation of mitochondria-mediated apoptosis (Huang and Strasser, 2000). In response to various stimuli, Bax is activated, translocated to the outer mitochondrial membrane and oligomerized, resulting in mitochondrial membrane permeabilization and release of mitochondrial apoptogenic molecules to the cytosol (Yanase et al., 2000; Yeo et al., 2002; Cao et al., 2003). In particular, proteolytic cleavage of native Bax (21 kDa) into an 18 kDa form by calpain has been shown to occur in cancer cells treated with a variety of chemotherapeutic agents (Toyota et al., 2003; Ariyama et al., 2006). This cleaved form of Bax is more potent than the native form in terms of disruption of mitochondrial membrane potential and induction of apoptotic cell death. Consistent with these reports, we found that RH1 treatment induced Bax cleavage without alterations in the protein levels of Bcl-2 or Bid. We also observed that the cleaved form of Bax was translocated to mitochondria upon treatment with RH1. Previous work showed that a truncated variety of Bid, induced by caspase-8 activation, triggered both mitochondrial dysfunction and release of apoptogenic molecules from mitochondria (Hengartner, 2000). Interestingly, we observed that the caspase-8 activation induced by RH1 treatment was not correlated with Bid cleavage, but that RH1 treatment nonetheless caused a reduction in mitochondrial membrane potential and led to nuclear translocation of both AIF and Endo G.
The MAPK family proteins are important mediators of apoptotic cell death in response to various stimuli (Xia et al., 1995; Klekotka et al., 2001; Park et al., 2003; Wada and Penninger, 2004). In particular, JNK and p38 MAPK have been implicated in activation of apoptotic cell death pathways in response to a variety of such stimuli, and after administration of chemotherapeutic agents (Xia et al., 1995; Klekotka et al., 2001; Park et al., 2003; Wada and Penninger, 2004). In agreement with these reports, we observed that RH1 treatment activated both JNK and p38 MAPK. Inhibition of JNK by a specific inhibitor or a targeted siRNA effectively attenuated RH1-induced apoptotic cell death, disruption of mitochondrial membrane potential, cleavage of Bax, mitochondrial translocation of cleaved Bax, and nuclear translocation of AIF and Endo G. However, inhibition of p38 MAPK did not affect RH1-induced apoptosis. These observations strongly indicate that activation of JNK is critically required for this process. Calpain has been shown to play a key role in the proteolytic cleavage of Bax (Toyota et al., 2003; Ariyama et al., 2006; Dadakhujaev et al., 2009; Su et al., 2010). The cited reports showed that calpain can be activated indirectly by JNK. As our results revealed that inhibition of JNK activity attenuated Bax cleavage induced by RH1, further studies are required to clarify the role of JNK in calpain-mediated Bax cleavage occurring in response to RH1. Nevertheless, our results clearly indicate that JNK activation is required for activity of the mitochondria-mediated apoptotic cell death pathway in response to RH1 treatment. Additionally, we found that inhibition of JNK2 was more effective than that of JNK1 in suppressing all of RH1-induced apoptosis, loss of mitochondrial membrane potential, mitochondrial translocation of cleaved Bax, and nuclear translocation of AIF and Endo G. These findings are consistent with the results of a previous study showing that JNK2 activity was required for mitochondria-mediated apoptosis in response to liver ischaemia (Theruvath et al., 2008).
Recently, it has been reported that DNA damage during mitosis induces JNK-mediated stress response leading to mitotic catastrophe, known to be another type of cell death (Ho and Li, 2010). In detail, this report suggested that the interaction between JNK and γH2AX during mitosis positively facilitate cell death signals. Furthermore, because RH1 has been already reported to induce DNA cross-linking, G2/M arrest and subsequent cell death in an NQO1-dependent manner (Dehn et al., 2005), RH1 is likely to cause mitotic catastrophe through the activation of JNK. Further investigation is in progress in our laboratory to shed light on the role of JNK in RH1-induced mitotic catastrophe.
Although ERK has often been implicated in cell survival and anti-apoptotic signalling pathways (Xia et al., 1995; Klekotka et al., 2001; Park et al., 2003; Wada and Penninger, 2004), some studies found that ERK played a positive role in activation of apoptotic signalling pathways (Wang et al., 2000; Bacus et al., 2001; Xiao and Singh, 2002; Choi et al., 2003). In the present work, we observed that ERK activation was weakly associated with the RH1-induced mitochondrial apoptotic cell death pathway. Although inhibition of ERK was less effective than was JNK inhibition in preventing RH1-induced apoptosis, inhibition of ERK did inhibit disruption of mitochondrial membrane potential, Bax cleavage, mitochondrial translocation of cleaved Bax, and nuclear translocation of AIF and Endo G induced by RH1 treatment. Several reports have indicated that ERK is also involved in activation of calpain (Glading et al., 2001; Glading et al., 2004), and it might thus be expected that activation of ERK in response to RH1 treatment should influence calpain-mediated cleavage of Bax, thus inducing mitochondrial apoptosis. However, additional studies are required to define precisely the role played by ERK in RH1 treatment-induced Bax cleavage.
Notably, RH1-induced activation of JNK lasted for 2 h and then subsequently decreased (Figure 5A) at the time ERK activity began to increase (Figure 5A). Previous studies showed that ERK has been reported to increase the expression and activity of MKP2 and MKP7 (Paumelle et al., 2000; Masuda et al., 2003; Katagiri et al., 2005), which suppress JNK activation (Paumelle et al., 2000). Therefore, it seemed that activation of ERK was responsible for the decrease in JNK activity from 2 h after RH1 treatment in NQO1+-MDA-MB-231 cells.
It has been reported that JNK directly regulates mitochondrial function and release of apoptogenic molecules from mitochondria (Fan et al., 2000; Schroeter et al., 2003; Kang and Lee, 2008). It has also been shown that purified JNK itself can directly affect mitochondrial membrane potential and bioenergetics (Fan et al., 2000; Kharbanda et al., 2000; Aoki et al., 2002; Chauhan et al., 2003). In agreement with these reports, RH1 induced a marked increase in mitochondrial translocation of both JNK and phospho-JNK (Figure 7), although JNK activity in whole cell lysates declined steadily, beginning 2 h after treatment, in our present study.
The above results suggest that mitochondrial phospho-JNK may be protected from phosphatases, including MKP2 or MKP7. Furthermore, inhibition of JNK activity by chemical pretreatment effectively attenuated RH1-induced mitochondrial translocation of both JNK and phospho-JNK, but inhibition of ERK activity in this manner did not (Figure 7), implying that the ERK pathway was not involved in RH1-induced mitochondrial translocation of either JNK or phospho-JNK. Phospho-JNK of the mitochondrial membrane appears to interact with mitochondrial proteins responsible for induction of apoptosis after RH1 treatment. However, the mitochondrial targets of JNK during RH1-induced apoptotic cell death remain to be defined.
In summary, our results implicate JNK activity as a critical requirement for the cleavage of Bax and mitochondrial translocation of both cleaved Bax and JNK itself. This may be associated with nuclear translocation of AIF and Endo G from mitochondria, and apoptotic cell death following RH1 treatment, in NQO1-overexpressing MDA-MB-231 cells. Our findings also reveal that ERK activation plays a role in RH1-induced mitochondria-mediated apoptosis (Figure 8). Knowledge of the mechanisms underlying the anti-cancer activity of the bioreductive agent RH1 is important to improve the therapeutic efficacy of NQO1-directed cancer therapies. Involvement of JNK in RH1-induced mitotic death remains to be elucidated.
Figure 8.
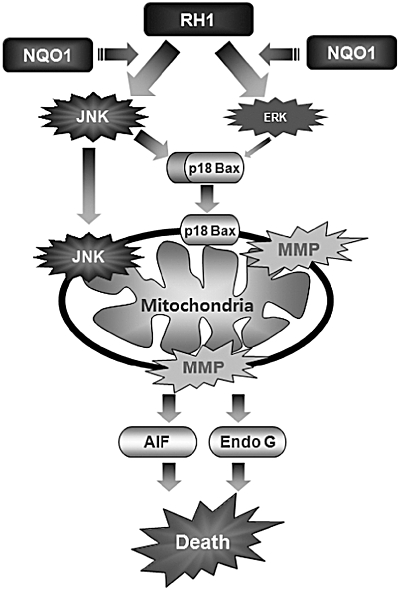
Schematic model of 2,5-diaziridinyl-3-(hydroxymethyl)-6-methyl-1,4-benzoquinone (RH1)-induced apoptotic cell death. RH1 treatment activates c-Jun N-terminal kinase (JNK) and extracellular signal-regulated kinase (ERK), both of which are involved in mitochondrial apoptotic cell death in NAD(P)H quinone oxidoreductase 1 (NQO1)+-MDA-MB-231 cells. Moreover, JNK activation induces cleavage of Bax and mitochondrial translocation of cleaved Bax, which causes loss of mitochondrial membrane potential, the consequent release of apoptosis-inducing factor (AIF) and endonuclease G (Endo G), and ultimately, apoptotic cell death. ERK activation plays a role in RH1-induced mitochondrial apoptosis. Notably, JNK itself translocates to mitochondria, where JNK appears to participate in phosphorylation of Bcl2 or Bcl-xL, resulting in apoptotic cell death. In the present study, elucidation of signalling pathways involved in RH1 function, may provide valuable insights into the design of NQO1-targeted cancer therapies.
Acknowledgments
This research was supported by the National Nuclear R&D Program through the National Research Foundation of Korea funded by the Ministry of Education, Science and Technology (2009-0078449), and Korea Health 21 R&D Project (A062254).
Glossary
Abbreviations
- AIF
apoptosis-inducing factor
- DiOC6(3)
3,3′-dihexyloxacarbocyanine iodide
- Endo
G, endonuclease G
- ERK
extracellular signal-regulated kinase
- JNK
c-Jun N-terminal kinase
- MAPK
mitogen-activated protein kinase
- PI
propidium iodide
- RH1
2,5-diaziridinyl-3-(hydroxymethyl)-6-methyl-1,4-benzoquinone
Conflict of interest
The authors hereby declare that there are no conflicts of interest.
Supporting information
Additional Supporting Information may be found in the online version of this article:
Figure S1 RH1 induces apoptotic cell death in a cathepsin B-independent manner. NQO1+-MDA-MB-231 cells were treated with 10 µM RH1 for 12 h in the presence or absence of 30 µM cathepsin B inhibitor. Cells were stained with Annexin V/PI and apoptotic changes were measured by flow cytometry. Representative flow cytometric dot plots are shown from: lower left quadrant, viable cells; lower right, early apoptotic cells; upper right, late apoptotic cells; upper left, necrotic cells. Apoptotic changes were quantitated as described above. Results from three independent experiments are expressed as means ± SEMs. *Significant difference between control and RH1-treated cells at P < 0.05. **Significant difference between RH1-treated cells and cathepsin B inhibitor + RH1-treated cells at P < 0.05.
References
- Aoki H, Kang PM, Hampe J, Yoshimura K, Noma T, Matsuzaki M, et al. Direct activation of mitochondrial apoptosis machinery by c-Jun N-terminal kinase in adult cardiac myocytes. J Biol Chem. 2002;277:10244–10250. doi: 10.1074/jbc.M112355200. [DOI] [PubMed] [Google Scholar]
- Ariyama H, Qin B, Baba E, Tanaka R, Mitsugi K, Harada M, et al. Gefitinib, a selective EGFR tyrosine kinase inhibitor, induces apoptosis through activation of Bax in human gallbladder adenocarcinoma cells. J Cell Biochem. 2006;97:724–734. doi: 10.1002/jcb.20678. [DOI] [PubMed] [Google Scholar]
- Asher G, Lotem J, Cohen B, Sachs L, Shaul Y. Regulation of p53 stability and p53-dependent apoptosis by NADH quinine oxidoreductase 1. Proc Natl Acad Sci USA. 2001;98:1188–1193. doi: 10.1073/pnas.021558898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bacus SS, Gudkov AV, Lowe M, Lyass L, Yung Y, Komarov AP, et al. Taxol-induced apoptosis depends on MAP kinase pathways (ERK and p38) and is independent of p53. Oncogene. 2001;202:147–155. doi: 10.1038/sj.onc.1204062. [DOI] [PubMed] [Google Scholar]
- Benson AM, Hunkeler MJ, Talalay P. Increase of NAD(P)H:quinone reductase by dietary antioxidants: possible role in protection against carcinogenesis and toxicity. Proc Natl Acad Sci USA. 1980;77:5216–5220. doi: 10.1073/pnas.77.9.5216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bivik C, Ollinger K. JNK mediates UVB-induced apoptosis upstream lysosomal membrane permeabilization and Bcl-2 family proteins. Apoptosis. 2008;13:1111–1120. doi: 10.1007/s10495-008-0240-7. [DOI] [PubMed] [Google Scholar]
- Cao X, Deng X, May WS. Cleavage of Bax to p18 Bax accelerates stress-induced apoptosis, and a cathepsin-like protease may rapidly degrade p18 Bax. Blood. 2003;102:2605–2614. doi: 10.1182/blood-2003-01-0211. [DOI] [PubMed] [Google Scholar]
- Chauhan D, Li G, Hideshima T, Podar K, Mitsiades C, Mitsiades N, et al. JNK-dependent Release of Mitochondrial Protein, Smac, during Apoptosis in Multiple Myeloma (MM) Cells. J Biol Chem. 2003;278:17593–17596. doi: 10.1074/jbc.C300076200. [DOI] [PubMed] [Google Scholar]
- Choi YJ, Lim SY, Woo JH, Kim YH, Kwon YK, Suh SI, et al. Sodium orthovanadate potentiates EGCG-induced apoptosis that is dependent on the ERK pathway. Biochem Biophys Res Commun. 2003;305:176–185. doi: 10.1016/s0006-291x(03)00719-8. [DOI] [PubMed] [Google Scholar]
- Cummings J, Ritchie A, Butler J, Ward TH, Langdon S. Activity profile of the novel aziridinylbenzoquinones MeDZQ and RH1 in human tumor xenografts. Anticancer Res. 2003;23:3979–3983. [PubMed] [Google Scholar]
- Dadakhujaev S, Jung EJ, Noh HS, Hah YS, Kim CJ, Kim DR. Interplay between autophagy and apoptosis in TrkA-induced cell death. Autophagy. 2009;5:103–105. doi: 10.4161/auto.5.1.7276. [DOI] [PubMed] [Google Scholar]
- Danson S, Ward TH, Butler J, Ranson M. DT-diaphorase: a target for new anticancer drugs. Cancer Treat Rev. 2004;30:437–449. doi: 10.1016/j.ctrv.2004.01.002. [DOI] [PubMed] [Google Scholar]
- Danson S, Ranson M, Denneny O, Cummings J, Ward TH. Validation of the comet-X assay as a pharmacodynamic assay for measuring DNA cross-linking produced by the novel anticancer agent RH1 during a phase 1 clinical trial. Cancer Chemother Pharmacol. 2007;60:851–861. doi: 10.1007/s00280-007-0432-9. [DOI] [PubMed] [Google Scholar]
- Daugas E, Nochy D, Ravagnan L, Loeffler M, Susin SA, Zamzami N, et al. Apoptosis-inducing factor (AIF): a ubiquitous mitochondrial oxidoreductase involved in apoptosis. FEBS Lett. 2000;476:118–123. doi: 10.1016/s0014-5793(00)01731-2. [DOI] [PubMed] [Google Scholar]
- Dehn DL, Inayat-Hussain SH, Ross D. RH1 induces cellular damage in an NAD(P)H:quinone oxidoreductase 1-dependent manner: relationship between DNA cross-linking, cell cycle perturbations, and apoptosis. J Pharmacol Exp Ther. 2005;313:771–779. doi: 10.1124/jpet.104.081380. [DOI] [PubMed] [Google Scholar]
- Fan M, Goodwin M, Vu T, Brantley-Finley C, Gaarde WA, Chambers TC. Vinblastine-induced phosphorylation of Bcl-2 and Bcl-XL is mediated by JNK and occurs in parallel with inactivation of the Raf-1/MEK/ERK cascade. J Biol Chem. 2000;275:29980–29985. doi: 10.1074/jbc.M003776200. [DOI] [PubMed] [Google Scholar]
- Foghsgaard L, Wissing D, Mauch D, Lademann U, Bastholm L, Boes M, et al. Cathepsin B acts as a dominant execution protease in tumor cell apoptosis induced by tumor necrosis factor. J Cell Biol. 2001;153:999–1010. doi: 10.1083/jcb.153.5.999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glading A, Uberall F, Keyse SM, Lauffenburger DA, Wells A. Membrane proximal ERK signaling is required for M-calpain activation downstream of epidermal growth factor receptor signaling. J Biol Chem. 2001;276:23341–23348. doi: 10.1074/jbc.M008847200. [DOI] [PubMed] [Google Scholar]
- Glading A, Bodnar RJ, Reynolds IJ, Shiraha H, Satish L, Potter DA, et al. Epidermal growth factor activates m-calpain (calpain II), at least in part, by extracellular signal-regulated kinase-mediated phosphorylation. Mol Cell Biol. 2004;24:2499–2512. doi: 10.1128/MCB.24.6.2499-2512.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haupt S, Berger M, Goldberg Z, Haupt Y. Apoptosis – the p53 network. J Cell Sci. 2003;116:4077–4085. doi: 10.1242/jcs.00739. [DOI] [PubMed] [Google Scholar]
- Hengartner MO. The biochemistry of apoptosis. Nature. 2000;407:770–776. doi: 10.1038/35037710. [DOI] [PubMed] [Google Scholar]
- Ho CY, Li HY. DNA damage during mitosis invokes a JNK-mediated stress response that leads to cell death. J Cell Biochem. 2010;110:725–731. doi: 10.1002/jcb.22583. [DOI] [PubMed] [Google Scholar]
- Huang DC, Strasser A. BH3-Only proteins-essential initiators of apoptotic cell death. Cell. 2000;103:839–842. doi: 10.1016/s0092-8674(00)00187-2. [DOI] [PubMed] [Google Scholar]
- Hussein D, Holt SV, Brookes KE, Klymenko T, Adamski JK, Hogg A, et al. Preclinical efficacy of the bioreductive alkylating agent RH1 against paediatric tumours (2009) Br J Cancer. 2009;101:55–63. doi: 10.1038/sj.bjc.6605100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joza N, Susin SA, Daugas E, Stanford WL, Cho SK, Li CY, et al. Essential role of the mitochondrial apoptosis-inducing factor in programmed cell death. Nature. 2001;410:549–554. doi: 10.1038/35069004. [DOI] [PubMed] [Google Scholar]
- Kang YH, Lee SJ. The role of p38 MAPK and JNK in Arsenic trioxide-induced mitochondrial cell death in human cervical cancer cells. J Cell Physiol. 2008;217:23–33. doi: 10.1002/jcp.21470. [DOI] [PubMed] [Google Scholar]
- Katagiri C, Masuda K, Urano T, Yamashita K, Araki Y, Kikuchi K, et al. Phosphorylation of Ser-446 determines stability of MKP-7. J Biol Chem. 2005;280:14716–14722. doi: 10.1074/jbc.M500200200. [DOI] [PubMed] [Google Scholar]
- Kharbanda S, Saxena S, Yoshida K, Pandey P, Kaneki M, Wang Q, et al. Translocation of SAPK/JNK to Mitochondria and Interaction with Bcl-xL in Response to DNA Damage. J Biol Chem. 2000;275:322–327. doi: 10.1074/jbc.275.1.322. [DOI] [PubMed] [Google Scholar]
- Kim JY, Kim CH, Stratford IJ, Patterson AV, Hendry JH. The bioreductive agent RH1 and gamma-irradiation both cause G2/M cell cycle phase arrest and polyploidy in a p53-mutated human breast cancer cell line. Int J Radiat Oncol Biol Phys. 2004a;58:376–385. doi: 10.1016/j.ijrobp.2003.09.040. [DOI] [PubMed] [Google Scholar]
- Kim JY, Patterson AV, Stratford IJ, Hendry JH. The importance of DT-diaphorase and hypoxia in the cytotoxicity of RH1 in human breast and non-small cell lung cancer cell lines. Anticancer Drugs. 2004b;15:71–77. doi: 10.1097/00001813-200401000-00011. [DOI] [PubMed] [Google Scholar]
- Klekotka PA, Santoro SA, Wang H, Zutter MM. Specific residues within the alpha 2 integrin subunit cytoplasmic domain regulate migration and cell cycle progression via distinct MAPK pathways. J Biol Chem. 2001;276:32353–33261. doi: 10.1074/jbc.M101921200. [DOI] [PubMed] [Google Scholar]
- Kroon AM. Protein synthesis in mitochondria. 3. On the effects of inhibitors on the incorporation of amino acids into protein by intact mitochondria and digitonin fractions. Biochim Biophys Acta. 1965;108:275–284. doi: 10.1016/0005-2787(65)90012-2. [DOI] [PubMed] [Google Scholar]
- Li P, Nijhawan D, Budihardjo I, Srinivasula SM, Ahmad M, Alnemri ES, et al. Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell. 1997;91:479–489. doi: 10.1016/s0092-8674(00)80434-1. [DOI] [PubMed] [Google Scholar]
- Lowe SW, Ruley HE, Jacks T, Housman DE. p53-dependent apoptosis modulates the cytotoxicity of anticancer agents. Cell. 1993;74:957–967. doi: 10.1016/0092-8674(93)90719-7. [DOI] [PubMed] [Google Scholar]
- Masuda K, Shima H, Katagiri C, Kikuchi K. Activation of ERK induces phosphorylation of MAPK phosphatase-7, a JNK specific phosphatase, at Ser-446. J Biol Chem. 2003;278:32448–32456. doi: 10.1074/jbc.M213254200. [DOI] [PubMed] [Google Scholar]
- Mihara M, Erster S, Zaika A, Petrenko O, Chittenden T, Pancoska P, et al. p53 has a direct apoptogenic role at the mitochondria. Mol Cell. 2003;11:577–590. doi: 10.1016/s1097-2765(03)00050-9. [DOI] [PubMed] [Google Scholar]
- Nicholson DW. From bench to clinic with apoptosis-based therapeutic agents. Nature. 2000;407:810–816. doi: 10.1038/35037747. [DOI] [PubMed] [Google Scholar]
- Nioi P, Hayes JD. Contribution of NAD(P)H:quinone oxidoreductase 1 to protection against carcinogenesis, and regulation of its gene by the Nrf2 basic-region leucine zipper and the arylhydrocarbon receptor basic helix-loop-helix transcription factors. Mutat Res. 2004;555:149–171. doi: 10.1016/j.mrfmmm.2004.05.023. [DOI] [PubMed] [Google Scholar]
- Olivier M, Eeles R, Hollstein M, Khan MA, Harris CC, Hainaut P. The IARC TP53 database: new online mutation analysis and recommendations to users. Hum Mutat. 2002;19:607–614. doi: 10.1002/humu.10081. [DOI] [PubMed] [Google Scholar]
- Park MT, Choi JA, Kim MJ, Um HD, Bae S, Kang CM, et al. Suppression of extracellular signal-related kinase and activation of p38 MAPK are two critical events leading to caspase-8- and mitochondria-mediated cell death in phytosphingosine-treated human cancer cells. J Biol Chem. 2003;278:50624–50634. doi: 10.1074/jbc.M309011200. [DOI] [PubMed] [Google Scholar]
- Park MT, Kim MJ, Kang YH, Choi SY, Lee JH, Choi JA, et al. Phytosphingosine in combination with ionizing radiation enhances apoptotic cell death in radiation-resistant cancer cells through ROS-dependent and -independent AIF release. Blood. 2005;105:1724–1733. doi: 10.1182/blood-2004-07-2938. [DOI] [PubMed] [Google Scholar]
- Paumelle R, Tulasne D, Leroy C, Coll J, Vandenbunder B, Fafeur V. Sequential activation of ERK and repression of JNK by scatter factor/hepatocyte growth factor in madin-darby canine kidney epithelial cells. Mol Biol Cell. 2000;11:3751–3763. doi: 10.1091/mbc.11.11.3751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakahira H, Enari M, Nagata S. Cleavage of CAD inhibitor in CAD activation and DNA degradation during apoptosis. Nature. 1998;391:96–99. doi: 10.1038/34214. [DOI] [PubMed] [Google Scholar]
- Schroeter H, Boyd CS, Ahmed R, Spencer JP, Duncan RF, Rice-Evans C, et al. c-Jun N-terminal kinase (JNK)-mediated modulation of brain mitochondria function: new target proteins for JNK signalling in mitochondrion-dependent apoptosis. Biochem J. 2003;372:359–369. doi: 10.1042/BJ20030201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Souza-Pinto NC, Mason PA, Hashiguchi K, Weissman L, Tian J, Guay D, et al. Novel DNA mismatch-repair activity involving YB-1 in human mitochondria. DNA Repair (Amst) 2009;8:704–719. doi: 10.1016/j.dnarep.2009.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su LT, Chen HC, González-Pagán O, Overton JD, Xie J, Yue L, et al. TRPM7 activates m-calpain by stress-dependent stimulation of p38 MAPK and c-Jun N-terminal kinase. J Mol Biol. 2010;396:858–869. doi: 10.1016/j.jmb.2010.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang PS, Tsang ME, Lodyga M, Bai XH, Miller A, Han B, et al. Lipopolysaccharide accelerates caspase-independent but cathepsin B-dependent death of human lung epithelial cells. J Cell Physiol. 2006;209:457–467. doi: 10.1002/jcp.20751. [DOI] [PubMed] [Google Scholar]
- Theruvath TP, Snoddy MC, Zhong Z, Lemasters JJ. Mitochondrial permeability transition in liver ischemia and reperfusion: role of c-Jun N-terminal kinase 2. Transplantation. 2008;85:1500–1504. doi: 10.1097/TP.0b013e31816fefb5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toyota H, Yanase N, Yoshimoto T, Moriyama M, Sudo T, Mizuguchi J. Calpain-induced Bax-cleavage product is a more potent inducer of apoptotic cell death than wild-type Bax. Cancer Lett. 2003;189:221–230. doi: 10.1016/s0304-3835(02)00552-9. [DOI] [PubMed] [Google Scholar]
- Wada T, Penninger JM. Mitogen-activated protein kinases in apoptosis regulation. Oncogene. 2004;23:2838–2849. doi: 10.1038/sj.onc.1207556. [DOI] [PubMed] [Google Scholar]
- Wang X, Martindale JL, Holbrook NJ. Requirement for ERK activation in cisplatin-induced apoptosis. J Biol Chem. 2000;275:39435–39443. doi: 10.1074/jbc.M004583200. [DOI] [PubMed] [Google Scholar]
- Ward TH, Danson S, McGown AT, Ranson M, Coe NA, Jayson GC, et al. Preclinical evaluation of the pharmacodynamic properties of 2,5-diaziridinyl-3-hydroxymethyl-6-methyl-1,4-benzoquinone. Clin Cancer Res. 2005;11:2695–2701. doi: 10.1158/1078-0432.CCR-04-1751. [DOI] [PubMed] [Google Scholar]
- Werneburg NW, Guicciardi ME, Bronk SF, Kaufmann SH, Gores GJ. Tumor necrosis factor-related apoptosis-inducing ligand activates a lysosomal pathway of apoptosis that is regulated by Bcl-2 proteins. J Biol Chem. 2007;282:28960–28970. doi: 10.1074/jbc.M705671200. [DOI] [PubMed] [Google Scholar]
- Winski SL, Hargreaves RH, Butler J, Ross D. A new screening system for NAD(P)H:quinone oxidoreductase (NQO1)-directed antitumor quinones: identification of a new aziridinylbenzoquinone, RH1, as a NQO1-directed antitumor agent. Clin Cancer Res. 1998;4:3083–3088. [PubMed] [Google Scholar]
- Xia Z, Dickens M, Raingeaud J, Davis RJ, Greenberg ME. Opposing effects of ERK and JNK-p38 MAP kinases on apoptosis. Science. 1995;270:1326–1331. doi: 10.1126/science.270.5240.1326. [DOI] [PubMed] [Google Scholar]
- Xiao D, Singh SV. Phenethyl isothiocyanate-induced apoptosis in p53-deficient PC-3 human prostate cancer cell line is mediated by extracellular signal-regulated kinases. Cancer Res. 2002;62:3615–3619. [PubMed] [Google Scholar]
- Yamaguchi T, Naruishi K, Arai H, Nishimura F, Takashiba S. IL-6/sIL-6R enhances cathepsin B and L production via caveolin-1-mediated JNK-AP-1 pathway in human gingival fibroblasts. J Cell Physiol. 2008;217:423–432. doi: 10.1002/jcp.21517. [DOI] [PubMed] [Google Scholar]
- Yanase N, Ohshima K, Ikegami H, Mizuguchi J. Cytochrome c release, mitochondrial membrane depolarization, caspase-3 activation, and Bax-alpha cleavage during IFN-alpha-induced apoptosis in Daudi B lymphoma cells. J Interferon Cytokine Res. 2000;20:1121–1129. doi: 10.1089/107999000750053799. [DOI] [PubMed] [Google Scholar]
- Yeo JK, Cha SD, Cho CH, Kim SP, Cho JW, Baek WK, et al. Se-methylselenocysteine induces apoptosis through caspase activation and Bax cleavage mediated by calpain in SKOV-3 ovarian cancer cells. Cancer Lett. 2002;182:83–92. doi: 10.1016/s0304-3835(02)00075-7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.