

Abstract
BACKGROUND AND PURPOSE
Renal fibrosis acts as the common pathway leading to the development of end-stage renal disease. The present study investigated, in vivo and in vitro, the anti-fibrotic and anti-inflammatory effects, particularly on the epithelial to mesenchymal transition of renal tubular cells, exerted by honokiol, a phytochemical used in traditional medicine, and mechanisms underlying these effects.
EXPERIMENTAL APPROACH
Anti-fibrotic effects in vivo were assayed in a rat model of renal fibrosis [the unilateral ureteral obstruction (UUO) model]. A rat tubular epithelial cell line (NRK-52E) was stimulated by transforming growth factor-β1 (TGF-β1) and treated with honokiol to explore possible mechanisms of these anti-fibrotic effects. Gene or protein expression was analysed by Northern or Western blotting. Transcriptional regulation was investigated using luciferase activity driven by a connective tissue growth factor (CTGF) promoter.
KEY RESULTS
Honokiol slowed development of renal fibrosis both in vivo and in vitro. Honokiol treatment attenuated tubulointerstitial fibrosis and expression of pro-fibrotic factors in the UUO model. Honokiol also decreased expression of the mRNA for the chemokine CCL2 and for the intracellular adhesion molecule-1, as well as accumulation of type I (α1) collagen and fibronectin in UUO kidneys. Phosphorylation of Smad-2/3 induced by TGF-β1 and CTGF luciferase activity in renal tubular cells were also inhibited by honokiol.
CONCLUSIONS AND IMPLICATIONS
Honokiol suppressed expression of pro-fibrotic and pro-inflammatory factors and of extracellular matrix proteins. Honokiol may become a therapeutic agent to prevent renal fibrosis.
Keywords: honokiol, unilateral ureteral obstruction, chronic kidney disease, renal fibrosis, transforming growth factor-β1, inflammation
Introduction
Honokiol {[4-allyl-2-(3-allyl-4-hydroxy-phenyl)-phenol]}, a major bioactive compound isolated from the bark of ‘Houpo’ (Magnolia officinalis), has been used in traditional Asian medicine for many years. We and others have demonstrated that honokiol exerts anti-inflammatory effects and protects endothelial cells by inhibiting NF-κB signalling (Lee et al., 2005; Sheu et al., 2008). Honokiol also exhibits a number of pharmacological actions, including anti-oxidative, anti-microbial, anxiolytic, anti-neoplastic and anti-platelet effects. Several reports also suggest anti-inflammatory and anti-fibrotic roles of honokiol in studies on neutrophils and hepatic fibrosis (Son et al., 2000; Liou et al., 2003; Park et al., 2005). Our recent work also demonstrated that honokiol attenuated experimental anti-Thy1 nephritis and suppressed endothelial cell apoptosis induced by high glucose concentrations, by inhibiting oxidative stress (Chiang et al., 2006; Sheu et al., 2008).
Chronic kidney disease (CKD) is a worldwide public health problem. There is an increasing incidence and prevalence of patients with kidney failure requiring replacement therapy, with poor outcomes and high cost (Eknoyan et al., 2004; Castro et al., 2009). Tubulointerstitial fibrosis, regardless of the initial renal insults, is a common pathological presentation of CKD. Tubulointerstitial fibrosis, characterized by tubular atrophy, interstitial matrix deposition and mononuclear cell infiltration, is a more consistent predictor of functional impairment than glomerular damage (Risdon et al., 1968; Hewitson, 2009). During disease progression, a variety of adhesion molecules, chemokines and growth factors, such as intracellular adhesion molecule-1 (ICAM-1), the chemokine CCL2 (also known as monocyte chemoattractant protein-1), plasminogen activator inhibitor 1 (PAI-1), and transforming growth factor-β1 (TGF-β1), can further drive these pathological changes (Liu, 2006). Smad-connective tissue growth factor (CTGF) pathways transmit TGF-β1 receptor signalling to regulate the expression of pro-fibrotic genes and the epithelial-to-mesenchymal transition in tubular cells (Liu, 2010). Currently, pharmacological intervention with angiotensin converting enzyme inhibitors and angiotensin II receptor antagonists provide only partial renoprotection in diabetes and non-diabetes CKD by reducing glomerular and tubulointerstitial damage (mesangial activation, podocyte injury, tubulointerstitial injury and inflammatory cell infiltration) (Brenner et al., 2001; Izuhara et al., 2005; Hou et al., 2006). Efforts to explore other potential agents to achieve more comprehensive renoprotection are essential.
We hypothesized that honokiol may have the potential to prevent renal fibrosis. To test this hypothesis, we used unilateral ureteral obstruction (UUO) in rats to induce renal fibrosis, characterized by a normotensive, non-proteinuric and non-hyperlipidemic state, and without any immune or toxic renal insult (Klahr et al., 2002). We investigated the in vivo and in vitro efficacy of honokiol on UUO-induced and TGF-β-induced expression of pro-fibrotic genes, depositionof extracellular matrix (ECM) proteins, Smad signaling and CTGF luciferase activity. Our findings provide the first evidence that honokiol has the potential to be developed as a therapeutic agent to prevent renal fibrosis.
Methods
Experimental animals
All animal care and experimental procedures were approved by the boards of the Experimental Animal Center, College of Medicine, National Taiwan University, Taipei, Taiwan. Male Wistar rats (Taipei, Taiwan) weighing 150–200 g were housed in temperature-controlled conditions under a light/dark photo cycle with food and water supplied ad libitum. Following anaesthesia with pentobarbital sodium (50 mg·kg−1, i.p.), the left ureter was ligated at the ureteropelvic junction with 4-0 silk through a left flank incision (UUO model). A control group of rats were subjected to sham operations that were identical to those for the rats with UUO, except that the ureters were not ligated.
Experimental protocols for UUO
Four groups of rats (n = 24) were used. UUO rats were treated with either PBS (UUO group, n = 6) or honokiol (Wako Pure Chemical Industries, Ltd., Osaka, Japan) (2.5 mg·kg−1, twice per day; UUO/HK group, n = 6) by oral-gastric syringe feeding just after the animal had recovered from general anesthesia and through day 0 to day 13, with a total of with 28 doses of honokiol. Sham operated rats (SHM and SHM/HK groups) consisted of six age-matched rats in each group. At day 14, a multiple physiological recorder (TA240S; Gould, Valley View, OH, USA) was applied to record arterial blood pressure and heartbeat rate through the carotid artery, and then blood samples were taken and the rats killed. UUO and SHM kidneys were divided coronally into two parts. The first part was fixed in 10% neutral-buffered formalin for pathological examnination and the second part was quickly frozen in liquid nitrogen and stored at −70°C for protein and RNA extraction.
Semiquantitative assessment of renal fibrosis
Tubulointerstitial damage was graded in periodic acid-Schiff (PAS)-stained sections on a scale from 0 to 4 (0, no changes; 1, changes affecting <25%; 2, changes affecting 25 to 50%; 3, changes affecting 50 to 75%; 4, changes affecting 75 to 100% of the section) (Remuzzi et al., 1999). For further analysis of the degree of interstitial collagen deposition, Masson trichrome-stained sections were graded (0, no staining; 1, <25% staining; 2, 25 to 50% staining; 3, 50 to 75% staining; 4, 75 to 100% staining of the section) (Remuzzi et al., 1999). Tubulointerstitial damage was assessed in each rat and averaged for rats of each group. The same investigator who was unaware of the nature of the experimental groups analysed all renal pathology samples.
Electrophoresis and immunoblotting
Whole cell lysates were prepared as described previously (Chiang et al., 2006; Sheu et al., 2007). Proteins were separated by pre-cast 10% SDS-polyacrylamide gel electrophoresis, and then electrophoretically transferred from the gel onto polyvinylidene difluoride membranes. After blocking, blots were incubated with, anti-CTGF, anti-type I (α1) collagen, anti-fibronectin (BD Biosciences, Woburn, MA, USA), anti-PAI-1 and anti-TGF-β1 (Santa Cruz Biotechnology, Santa Cruz, CA, USA), anti-α-smooth muscle actin (α-SMA) and anti-β-actin and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (Sigma, St. Louis, MO, USA) antibodies in PBS within 0.1% Tween 20 for 1 h followed by three 10 min washes in PBS within 0.1% Tween 20. The membranes were then incubated with horseradish peroxidase-conjugated secondary antibodies for 60 min. Detection was performed with Western blotting reagent ECL (Amersham, Piscataway, NJ, USA), and the Kodak X-Omat films (Kodak, Rochester, NY, USA) exposed chemiluminescence. We used Scion Image (Scion, Frederick, MD, USA) for computer analysis of band pixel intensities.
RNA extraction and Northern blot analyses
Total RNA isolated for Northern blot analysis was performed as described previously (Chiang et al., 2006). Human ICAM-1, CCL2 and GAPDH RNA probes were synthesized as described previously (Chiang et al., 2006).
Miscellaneous measurements
Blood samples were drawn from the carotid artery after blood pressure measurement. Serum blood urea nitrogen (BUN), creatinine (Cr), glutamic pyruvic transaminase (GPT) and lactate dehydrogenase (LDH) were analysed with the automatic analyser Integra 800 (Roche, Mannheim, Germany). The Department of Laboratory Medicine, National Taiwan University Hospital, performed all analyses.
Normal rat kidney (NRK)-52E cell culture
NRK-52E cells were used to study the direct effects of honokiol on the induction of pro-fibrotic factors by TGF-β1, which has been validated as an in vitro system to evaluate the efficiency of therapeutic agents on fibrosis. NRK-52E cells were purchased from the Food Industry Research and Development Institute (Hsinchu, Taiwan). They were cultured at 37°C in 5% CO2 in DMEM that was supplemented with 10% fetal bovine serum (FBS), penicillin G (100 U·mL−1), and streptomycin (100 µL·mL−1). The NRK-52E cells were seeded on 10 cm dishes with complete medium that contained 10% FBS (Life Technologies BRL, Rockville, MD, USA) till sub-confluence. Thereafter, the cells were kept in 0.5% FBS medium for 24 h and were pre-treated with honokiol (1, 3 or 10 µM) for 60 mins, then stimulated with recombinant human TGF-β1 (R&D Systems, Minneapolis, MN, USA) at a concentration of 5 ng·mL−1 in 0.5% FBS-added medium, for the periods indicated.
Cell viability
Viability of renal cells after treatment with honokiol was assessed by the MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide, Sigma] assay, as described by Sheu et al. (2005). This assay is based on the cellular conversion of the colorimetric reagent MTT, in the presence of electron-coupling reagent phenazine methosulfate, into soluble formazan by dehydrogenase enzymes found only in metabolically active, living cells. Formazan formation was measured on the basis of increased absorbance at 490 nm.
Immunoblotting in TGF-β1–or TGF-β1 and HK–co-treated NRK-52E cells
Cells were harvested and immunoblotting using whole cell lysates was carried out as described previously (Chiang et al., 2006; Sheu et al., 2007). In addition to the primary antibodies, anti-Smad-2/3 (Cell Signaling, Leiden, the Netherlands) and anti-phospho-Smad-2/3 (Upstate, Lake Placid, NY, USA) antibodies were also used.
Construction of CTGF promoter-luciferase reporter plasmids
The CTGF promoter (−747/+214) luciferase construct (pGL3-CTGF-Luc) was provided by Dr M.-L. Kuo (National Taiwan University) (Yu et al., 2009). Briefly, a 962-base pair (bp) fragment representing the 5' upstream region of the CTGF gene (−747 to 214 bp), based on GenBank accession number AL354866, was generated by PCR. The transcription initiation site was defined as +1. This fragment was ligated into the firefly luciferase (Rluc) reporter vector, pGL3-basic (Promega, Madison, WI, USA), and was designated pGL3-CTGF, then verified by sequencing.
Transient transfections and luciferase assay
NRK-52E cells (1 × 105 cells per well) were seeded onto 6-well plates and cells were transfected the following day using TransfectinTM reagent (Bio-Rad) with 2 ug of CTGF-Luc, the CTGF promoter constructs (−747/+214). After overnight incubation with transfection mixtures, culture medium was replaced by 0.5% FBS medium. A range of concentrations of honokiol (1, 3, 10 µM) were added, together with TGF-β1 to NRK52E cells. After incubation, cells were harvested and luciferase activity measured in a luminometer (Berthold Technologies GmbH & Co. KG, Bad Wildbad, Germany), using a dual-luciferase reporter assay system (Promega). The inhibition of CTGF-luciferase activity by honokiol was calculated as (CTGF-luciferase activity of honokiol treatment with TGF-β1/CTGF (−747/+214) luciferase activity caused by TGF-β1 alone) × 100%.
Statistical analysis
Data are presented as mean ± SEM. All analyses were performed by analysis of variance followed by a Fisher's least significant. All statistics calculations were done with SPSS 13.0 for Windows (SPSS, Chicago, IL, USA).
Results
During this study, no adverse effects observed in the experimental groups. No obvious change in appearance or mortality was found in rats with sham or UUO operation, treated with either honokiol or vehicle. Honokiol-tretaed rats ahd a trend towards a lower blood pressure but this was not significant (Table 1). However, we cannot exclude some influence of this lower blood pressure on the degree of renal damage. Body weight, heart rate, serum BUN, Cr, GPT and LDH were similar in all groups at the end of experiment (data not shown).
Table 1.
Baseline body weight and final body weight, blood pressure and heart rate at the end of the experiment (14 days)
| Baseline | After treatment | |||
|---|---|---|---|---|
| Study groups | Body weight (g) | Body weight (g) | Blood pressure (mmHg) | Heart rate (bpm) |
| Sham | 179.0 ± 13.4 | 243.4 ± 13.0 | 105 ± 9 | 443 ± 23 |
| Sham/HK | 165.8 ± 9.3 | 219.0 ± 16.5 | 116 ± 4 | 445 ± 5 |
| UUO | 174.0 ± 10.8 | 237.4 ± 10.9 | 130 ± 19 | 448 ± 22 |
| UUO/HK | 163.6 ± 7.6 | 216.0 ± 16.6 | 114 ± 23 | 424 ± 18 |
No differences in any of the variables measured were observed between any of the experimental groups.
UUO, unilateral ureteral obstruction; HK, honokiol; bpm, beats per minute.
Honokiol treatment attenuated tubulointerstitial damage and progressive fibrosis in UUO kidneys
UUO-induced tubulointerstitial damage was characterized as severe tubular dilatation, tubular atrophy and widened interstitial space on the PAS-stained kidney sections (Figure 1A, UUO group) compared with the control (Figure 1A, SHM group). In contrast, tubulointerstitial damage was reduced in UUO kidneys taken from the honokiol-treated rats (Figure 1A, UUO/HK group), compared with the vehicle-treated UUO kidneys (Figure 1A, UUO group). This finding was confirmed by semi-quantitative assessment of the tubulointerstitial damage on the PAS-stained sections of renal tissue (Figure 1A). UUO also increased collagen deposition in rat kidneys, as disclosed by the Masson's trichrome-stained sections (Figure 1B, UUO group), compared with the control (Figure 1B, SHM group). Honokiol treatment of UUO rats led to less collagen deposition in their kidneys (Figure 1B, UUO/HK group) compared with the vehicle-treated UUO kidneys (Figure 1B, UUO group). Semiquantitative assessment of the fibrotic area on the Masson's trichrome-stained sections of renal tissue is shown in Figure 1B.
Figure 1.
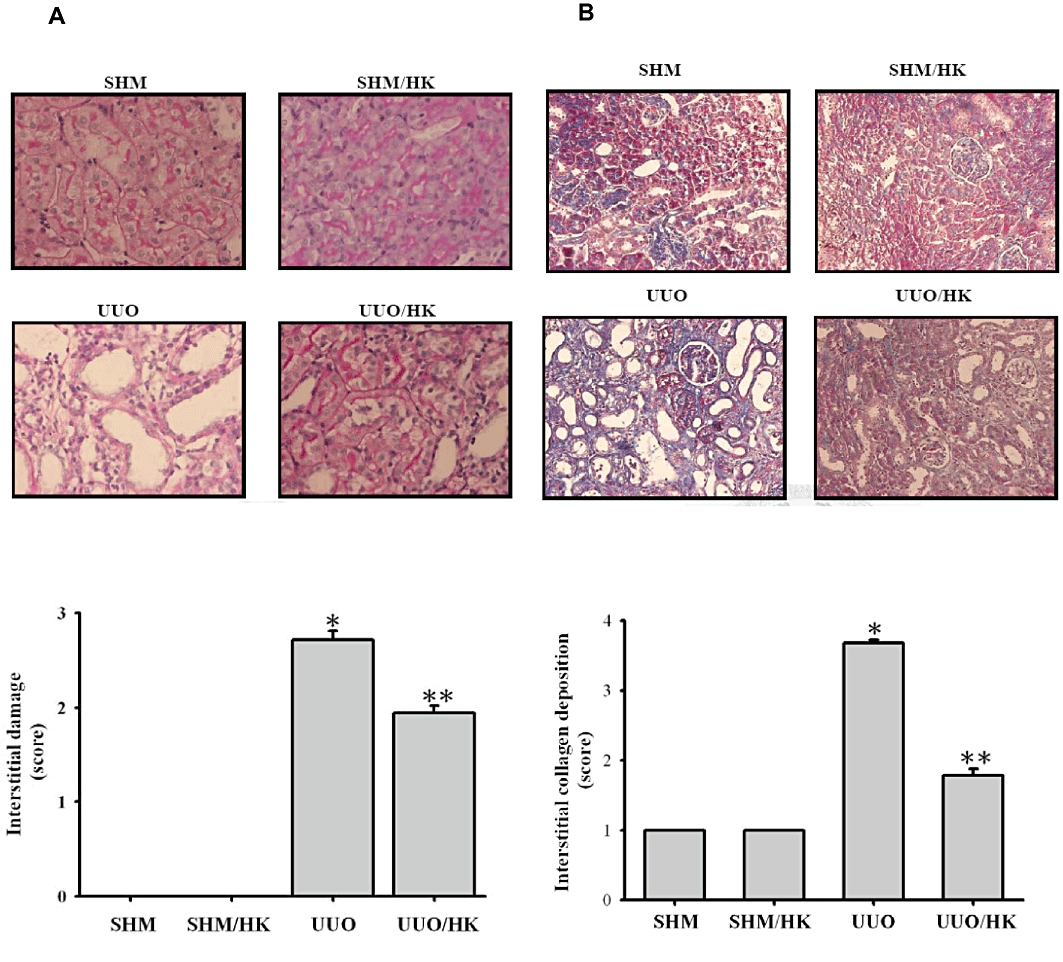
Honokiol attenuated renal interstitial damage and collagen deposition in UUO rats. Kidney tissue stained with (A) periodic acid-Schiff (PAS) and (B) Masson trichrome for sham-operated rats that were given vehicle (SHM) or honokiol (SHM/HK) and UUO rats that given vehicle or honokiol (UUO/HK) for 2 weeks after surgery. Magnification, ×200. In the lower part of this figure, the tubulointerstitial damage in PAS-stained sections and the degree of interstitial collagen deposition in Masson trichrome-stained sections of the kidney tissue was graded semi- quantitatively and averaged for rats of each group (n = 6 for each group). Data were expressed as mean ± SEM for each group. *P < 0.05 significantly different from SHM rats; **P < 0.05 significantly different from UUO rats.
Honokiol attenuated increase of proinflammatory mediators in UUO kidneys
Having demonstrated that honokiol attenuated renal pathology, we next examined the effect of honokiol on the expression of pro-inflammatory mediator genes, which induce leukocyte infiltration and activation at the place of injury. We examined the effects of honokiol treatment on ICAM-1 and CCL2 gene expression. The UUO kidneys showed up-regulation of the mRNA for ICAM-1, as well as that for CCL2 (Figure 2, UUO group) by 3.6- and 21.4-fold. Treatment with honokiol (Figure 2, UUO/HK group) diminished this augmented expression.
Figure 2.
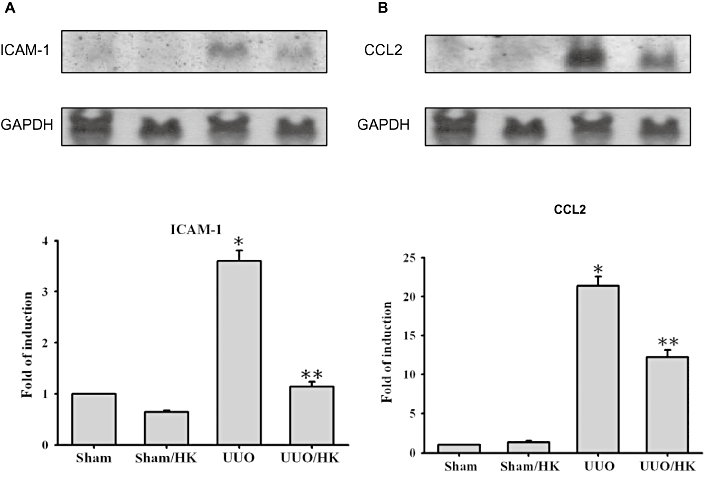
Honokiol reduced inflammatory response in UUO rats. UUO or sham groups were treated with or without honokiol (HK; 2.5 mg·kg−1·day−1, twice a day), for 14 days Tissue RNA was extracted and prepared for Northern blot analyses for CCL2, ICAM-1, and GAPDH. The UUO-induced expression of ICAM-1 (A) and CCL2 (B) RNA was reduced by honokiol. Quantification of the ICAM-1 and CCL2 RNA expression was performed by densitometric analysis (n = 6 for each group). Data were expressed as mean ± SEM for each group. *P < 0.05 significantly different from SHM rats; **P < 0.05 significantly different from UUO rats.
Honokiol attenuated increased expression of α-SMA and pro-fibrotic genes in UUO kidneys
UUO-induced tubulointerstitial damage would lead to activation of resident renal tubular cell and interstitial fibroblasts, as shown by induction of pro-fibrotic protein expression and accumulation of ECM components. As shown in Figure 3A, UUO increased TGF-β1 levels and this was prevented by treatment with honokiol. Furthermore, expression of α-SMA, a marker of tubular cell activation, was also increased by UUO, compared with the control (SHM group) (Figure 3B)and this up regulation of the α-SMA was inhibited by honokiol treatment (Figure 3B). This observation indicated that honokiol treatment inhibited the process of fibrosis in the obstructed kidneys, which is characterized by the gain of mesenchymal phenotype, and disruption of tubular basement membrane. To test our hypothesis further, we measured the effect of honokiol on the pro-fibrotic mediator CTGF, the levels of PAI-1 (which inhibits the degradation of ECM) and the accumulation of ECM proteins. As shown in Figure 4A and B, there was a significant increase in CTGF or PAI-1 protein expression in rat kidneys with UUO, which was reversed by honokiol treatment. This finding implies that honokiol can attenuate the genes upstream of ECM and modulates ECM accumulation. Next, we examined the effects of honokiol treatment on accumulation of ECM proteins in rats with UUO. The induction of UUO in rats led to the accumulation of interstitial type I (α1) collagen, as well as fibronectin (Figure 4C and D) by 245- and 15.3-fold. Treatment with honokiol of UUO-induced rats diminished the augmented expression of ECM proteins (Figure 4C and D).
Figure 3.
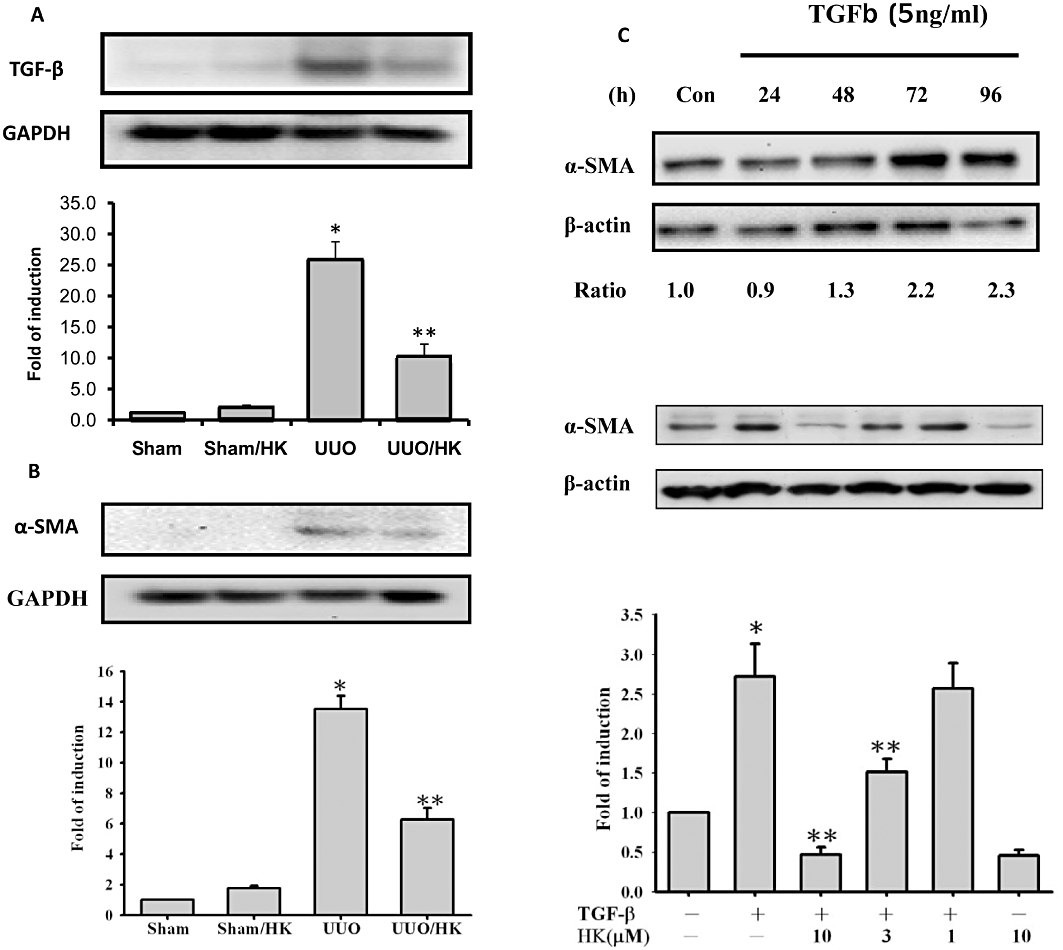
Honokiol blocked transforming growth factor-β1 (TGF-β1) and TGF-β1-induced α-SMA in UUO kidneys and cultured renal cells. In (A), levels of TGF-β1 were increased in extracts of UUO kidneys and this increase was blocked by honokiol. In (B), α-SMA protein expression, used as a marker of tubular cells activation, was increased in UUO kidneys and this increase was attenuated by honokiol (n = 6 for each group). In (C), results from NRK-52E cells treated with TGF-β1 (5 ng·mL−1) are shown. In the upper half, the time course of response of α-SMA levels to TGF-β1 is shown, over 96 h, with assay every 24 h. Levels of α-SMA protein were increased and peaked between 72 and 96 h. The numbers below the blot indicate the fold increase of α-SMA (control = 1). In the lower panel of (C), we show that the expression of α-SMA protein stimulated by TGF-β1 was inhibited by honokiol, concentration-dependently; assay after 72 h of TGF-β1 incubation (n = 4 for each group). Data are presented as mean ± SEM from three independent experiments. *P < 0.05 significantly different from control groups; **P < 0.05 significantly different from TGF-β1 groups.
Figure 4.
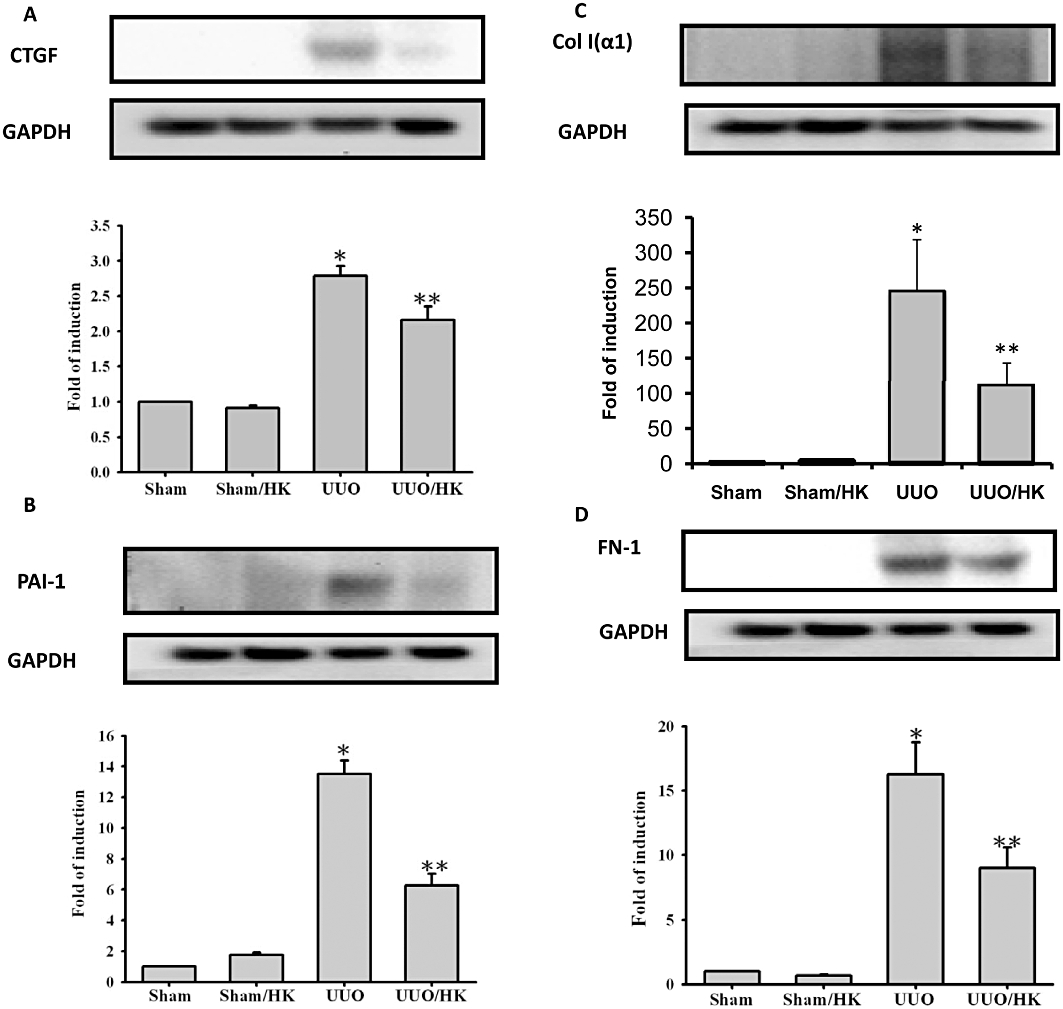
Honokiol attenuated connective tissue growth factor (CTGF), PAI-1, collagen-1 and fibronectin-1 in kidneys from UUO rats. UUO or sham groups were treated with or without honokiol (2.5 mg·kg−1·day−1, twice a day) for 14 days. Profibrotic factors (CTGF and PAI-1) and ECM proteins [type 1 collagen α (Col α1) and fibronectin-1 (FN-1)] were used as surrogate markers of fibrosis in UUO kidney. Tissue proteins were extracted and then were prepared for Western blot analyses, with specific antibodies. The UUO-induced Col α1, FN-1, CTGF and PAI-1 proteins were increased in UUO kidneys and these increases were attenuated by honokiol (n = 6 for each group). Data are presented as mean ± SEM from three independent experiments. *P < 0.05 significantly different from control groups; **P < 0.05 significantly different from transforming growth factor-β1 (TGF-β1) groups.
Honokiol blocked TGF-β1-induced profibrogenic factors and ECM accumulation in renal cells
We next tested the in vitro effects of honokiol on the viability and expression of pro-fibrotic factors in NRK-52E cells. Cell viability was not affected by honokiol over the concentration range used (1–10 µM; data not shown). Levels of α-SMA protein were increased after TGF-β1 stimulation of NRK-52E cells, peaking some time between 72 and 96 h (upper panel, Figure 3B). These increases in α-SMA protein were suppressed by honokiol, concentration-dependently (lower panel of Figure 3B).
Activated tubular cells act as a major source of ECM proteins. We therefore investigated whether honokiol could attenuate TGF-β1-stimulated expression of ECM in cultured tubular cells. As shown in Figure 5A and B, the fibrinogen and collagen α1 proteins, which are the major ECM components in interstitial fibrosis, were increased to 2.2- and 3.2-fold the control level in NRK-52E cells, incubated with TGF-β1, for up to 96 h (upper half of Figure 5A and B). Treatment of the cells with honokiol reduced this up-regulation of ECM proteins concentration-dependently (lower half of Figure 5A and B).
Figure 5.
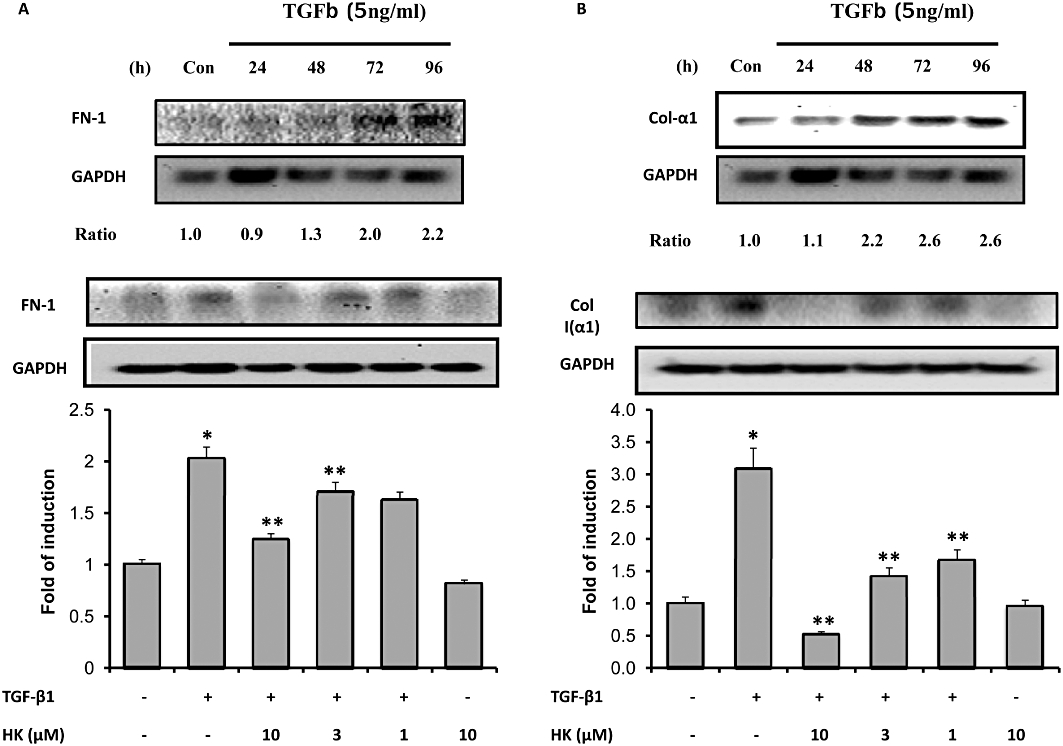
Honokiol treatment reduced fibronectin and collagen 1 expression in NRK-52E cells. NRK-52E cells were stimulated with transforming growth factor-β1 (TGF-β1; 5 ng·mL−1). In the upper half of (A), the time course of response of fibronectin (FN-1) is shown over 96 h; assay every 24 h. FN-1 protein levels were increased at all times and peaked at 96 h. The numbers below the blot indicate the fold increase of FN-1 (control = 1). In the lower panel, the TGF-β1-induced expression of FN-1 protein was reduced by honokiol in a concentration-dependent manner; assay at 96 h of TGF-β1 incubation (n = 4 for each group). In the upper half of (B), the time course of response of collagen 1 (Col 1α) is shown over 96 h; assay every 24 h. Col 1α protein levels were increased at all times and peaked after 72 h. The numbers below the blot indicate the fold increase of Col 1α (control = 1). In the lower panel, the TGF-β1-induced expression of Col 1α protein was reduced by honokiol in a concentration-dependent manner; assay at 72 h of TGF-β1 incubation (n = 4 for each group). Data are presented as mean ± SEM from three independent experiments. *P < 0.05 significantly different from control groups; **P < 0.05 significantly different from TGF-β1 groups.
To further examine possible mechanisms underlying attenuation of ECM accumulation by honokiol, we examined its effects on pro-fibrogenic factors, such as CTGF and PAI-1, in TGF-β1-stimulated cells. Exposure to TGF-β1 increased CTGF protein levels by 3.6-fold after 96 h in NRK-52E cells (Figure 6A, upper panels). Treatment with honokiol diminished this CTGF protein synthesis in a concentration -dependent manner (Figure 6A, lower panels). Another way to accumulate ECM proteins is to inhibit their normal degradation. We therefore measured levels of PAI-1, one of the most important endogenous inhibitors of ECM degradation, using Western blot analyses. In TGF-β1-stimulated NRK-52E cells, PAI-1 protein was increased 2.3-fold, compared with the control group, and honokiol attenuated this increase (Figure 6B, lower panel).
Figure 6.
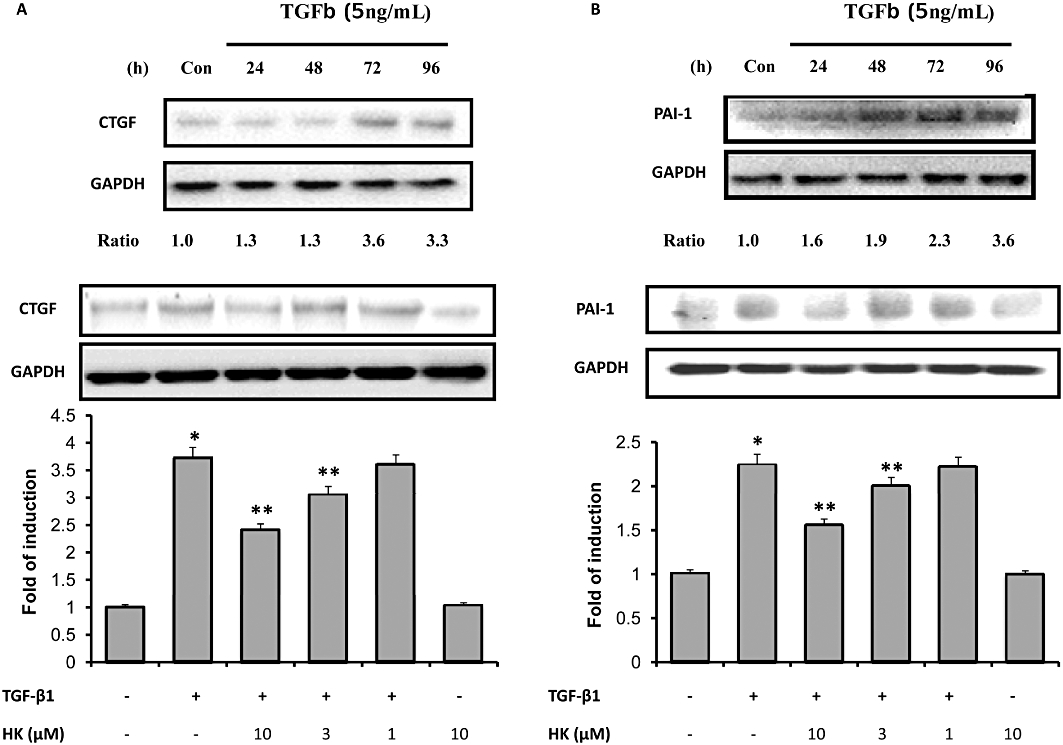
Honokiol treatment reduced connective tissue growth factor (CTGF), PAI-1 expression in NRK-52E cells. NRK-52E cells were stimulated with transforming growth factor-β1 (TGF-β1; 5 ng·mL−1). In the upper half of (A), the time course of response of CTGF is shown over 96 h; assayed every 24 h. CTGF protein levels were increased at all times and peaked between 72 and 96 h. The numbers below the blot indicate the fold increase of CTGF (control = 1). In the lower panel, the TGF-β1-induced expression of CTGF protein was reduced by honokiol in a concentration-dependent manner; assayed at 96 h of TGF-β1 incubation (n = 4 for each group). In the upper half of (B), the time course of response of PAI-1 is shown over 96 h; assayed every 24 h. PAI-1 protein levels were increased at all times and peaked at 96 h. The numbers below the blot indicate the fold increase of PAI-1 (control = 1). In the lower panel, the TGF-β1-induced expression of PAI-1 protein was reduced by honokiol in a concentration-dependent manner; assayed at 96 h of TGF-β1 incubation (n = 4 for each group). Data are presented as mean ± SEM from three independent experiments. *P < 0.05 significantly different from control groups; **P < 0.05 significantly different from TGF-β1 groups.
Honokiol attenuated TGF-β1-induced up-regulation of the phosphorylation of Smad-2/3 in NRK-52E Cells
As a mechanistic experiment for the effects of HK, we examined whether the honokiol treatment affected Smad signalling, which has been known to play a major role in the process of TGF-β1-induced renal fibrosis (Li et al., 2002; Liu, 2004; 2006;). As shown in Figure 7, NRK-52E cells that were pre-treated with honokiol for 30 min followed by TGF-β1 treatment for an additional 30 min showed no change in the expression of total Smad-2/3 proteins, but the expression of phosphorylated Smad-2 and Smad-3 was increased in the TGF-β1-treated group. The subsequent pretreatment with honokiol attenuated both the basal and the TGF-β1-stimulated phosphorylation of Smad-2/3.
Figure 7.
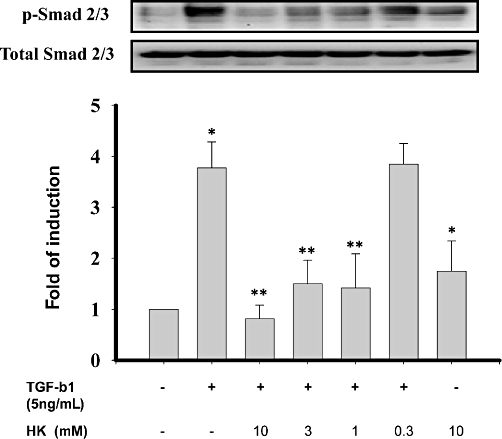
Honokiol inhibited basal and transforming growth factor-β1 (TGF-β1)-induced phosphorylation of Smad 2/3 in NRK-52E cells. NRK-52E cells were treated with 5 ng·mL−1 TGF-β1, with or without honokiol for 30 min. Smad 2/3 phosphorylation was assessed by immunoblotting using anti-phospho-Smad (p-Smad) 2/3 antibodies. The TGF-β1-induced Smad 2/3 phosphorylation in NRK-52E cells were reduced by honokiol, concentration-dependently. Results shown are representative of three independent experiments. Data are presented as mean ± SEM from three independent experiments. *P < 0.05 significantly different from control groups; **P < 0.05 significantly different from TGF-β1 groups.
Honokiol treatment attenuated TGF-β1-induced CTGF-luciferase activity in NRK-52E cells
To assay the effects of honokiol on TGF-β1-induced CTGF expression, NRK-52E cells were transiently transfected with CTGF-Luc plasmid and treated with TGF-β1 and the indicated concentrations of honokiol HK. As shown in Figure 8, TGF-β1 markedly increased CTGF promoter luciferase activity twofold, which was reduced by the addition of honokiol, in a concentration-dependent manner (Figure 8), from 17% inhibition at 1 µM to 59% inhibition at 10 µM.
Figure 8.
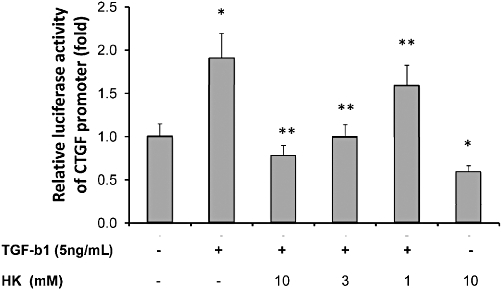
Honokiol inhibited transforming growth factor-β1 (TGF-β1)-induced connective tissue growth factor (CTGF) promoter activities in NRK-52E cells. NRK-52E cells were transfected with plasmids containing the wild-type CTGF promoter (−747/+214) luciferase construct (pGL3-CTGF-Luc). After overnight incubation with transfection mixtures, culture medium was replaced by 0.5% FBS medium. Honokiol (1, 3, 10 µM) was added together with TGF-β1 to NRK52E cells and luciferase activity measured. TGF-β1 markedly increased CTGF promoter luciferase activity, which was suppressed by honokiol, concentration -dependently. The inhibition by honokiol was 16 ± 0.3% at 1 µM, 48 ± 1% at 3 µM and 59 ± 1% at 10 µM. *P < 0.05; significantly different from TGF-β1-stimulated; #P < 0.05; significantly different from control group; n = 4.
Discussion
This study demonstrated that honokiol, a photochemical widely used in traditional medicine, attenuated renal interstitial fibrosis through its effects against pro-inflammatory and pro-fibrotic mediators in vitro and in vivo. These findings extended our previous study in applying honokiol not only in the treatment of glomerulonephritis but also for renal fibrosis (Chiang et al., 2006). This is the first evidence that honokiol has the potential to be developed as a therapeutic agent to prevent renal fibrosis. The wide range of biological effects exhibited by honokiol is thought to reflect its potent antioxidant properties, stemming from its polyphenol structure, which leads to anti-fibrotic and anti-inflammatory actions in renal fibrosis. There are several ways in which honokiol may prevent renal pathological insults in experimental tubulointerstitial fibrosis.
Firstly, TGF-β1 and its downstream signalling cascades play key roles in activating cellular pathological mechanisms in renal tubulointerstitial fibrosis, including induction of interstitial cell activation and of pro-fibrotic genes expression (Kaneto et al., 1993; Liu, 2006). Following ligand binding, the TGF-β receptor complex transiently interacts with receptor-activated Smads, which propagate TGF-β signalling and regulate the promoter activities of TGF-β target genes (Heldin et al., 1997; Derynck et al., 1998). TGF-β1 signalling may initiate pro-apoptotic effectors and/or pro-fibrotic activation of tubular epithelial cells, resulting in tubular degeneration and tubular atrophy. Induction of TGF-β1 may convert tubular epithelial cells and fibroblasts into activated myofibroblasts, which may be responsible for increased deposition of interstitial matrix in response to TGF-β/Smad signaling. Furthermore, considerable evidence indicates that CTGF plays a pivotal role in TGF-β-dependent tubulointerstitial fibrosis (Gupta et al., 2000; Burns et al., 2006). CTGF mediates the up-regulation of collagen type I by TGF-β1 in mesangial cells and fibroblasts; however, it mainly transduces myofibrotic activation to tubular epithelial cells (Gore-Hyer et al., 2002; Lin et al., 2005). Our study demonstrated that honokiol attenuated TGF-β1 induction in the UUO kidneys and also reduced CTGF expression in NRK-52E cell lines and in kidneys of UUO-treated rats. On this basis, honokiol may attenuate renal fibrosis through reducing CTGF expression. Previous studies showed that TGF-β1-induced TGF-β and CTGF expression can be blocked through Smad-dependent transcription (Lin et al., 2005) and Smad mediates TGF-β1 signaling leading to the activation of α-SMA transcription (Figure 3B and C) (Subramanian et al., 2004). Our present experiments showed that honokiol inhibited both basal and TGF-β1-mediated Smad-2/3 phosphorylation.
Next, the activation of interstitial cells, which are responsible for ECM deposition in tubulointerstitial fibrosis (Alpers et al., 1994; Remuzzi and Bertani, 1998; Garcia-Sanchez et al., 2010), may derived in part from the transition of tubular epithelial cells to myofibroblasts (Fan et al., 1999; Iwano et al., 2002) and activation of interstitial fibroblasts (Alpers et al., 1994; Grupp et al., 1997). During renal fibrosis, tubular epithelial cells acquire myofibroblast characteristics and interstitial fibroblast activation. In the present work, the TGF-β1-stimulated α-SMA over-expression in NRK-52E cells and the enhanced α-SMA expression in kidney of UUO rats were markedly reduced by honokiol. These observations suggest that TGF-β1-induced activation, observed in renal interstitial cells and UUO kidneys, could be effectively decreased by honokiol treatment.
Following UUO, kidneys showed clearly widened interstitial spaces, dilated renal tubules and increased collagen deposition. Collagen fibers were also increased in the interstitial space, forming a fine network. Treatment with honokiol blunted tubular dilatation, interstitial damage and the increase of collagen matrix induced by UUO. The abnormal ECM in fibrosis consists of an excess of normal components of ECM, such as fibronectin and collagen type IV, but also of an accumulation of pathological ECM components, such as collagen type Iα. (Fogo, 1999) These proteins characterize the scarring process and are usually irreversibly deposited in the fibrotic tissues. We found increased ECM deposition and synthesis in kidneys from UUO rats in vivo and in TGF-β1-induced cells in vitro, and this ECM deposition and synthesis was blocked by honokiol. On the other hand, the ECM is normally continually degraded and accumulation of ECM may also result from a deficit in ECM degradation. The plasminogen activation system is one of the most important degrading systems for the ECM (Eddy et al., 2006; Fogo, 2003) and several studies have shown that synthesis and deposition of the inhibitory protein PAI-1 are increased in the kidney during experimental and human nephropathies (Rerolle et al., 2000; Eddy et al., 2006; Seo et al., 2009). PAI-1 is also one of the targets regulated by TGF-β1 (Keeton et al., 1991). In the current study, we found that honokiol decreased UUO-induced PAI-1 expression in vivo and TGF-β1-induced PAI-1 expression in vitro. Combination of these data suggests that honokiol may attenuate renal fibrosis via not only by reduction of ECM synthesis but also by increasing ECM degradation.
Finally, an influx of mononuclear cells takes place in the induction phase of renal fibrosis (Grande et al., 2010). Inflammatory cells migrate to the interstitium driven by the up-regulated expression of chemoattractant molecules (chemokines and adhesion molecules) produced by activated tubular cells, which further lead to local tissue injury (Noronha et al., 2002). In this work, we demonstrated that kidneys from UUO rats exhibited high levels of ICAM-1 and CCL2 mRNA during the injury, corresponding to the accumulation of macrophages in the interstitium. Because treatment with honokiol reduced the enhanced expression of both ICAM-1 and CCL2 mRNA, it is conceivable that by reducing the expression of these molecules, the recruitment of monocytes into the interstitium was diminished. These findings suggest that honokiol diminishes renal fibrosis, as least in part, through inhibiting mononuclear cell infiltration and expression of pro-inflammatory chemokines.
In summary, we have shown that honokiol, already used in traditional medicine, inhibited the progression of tubular dilatation, interstitial injury and renal fibrosis in the UUO model in rats, by decreasing pro-inflammatory factors, attenuating TGF-β1 and its downstream profibrotic factors, and thus decreasing ECM accumulation. Moreover, in cultures of renal tubular cells, honokiol directly attenuated TGF-β1-induced profibrotic signalling, most likely through inhibition of Smad-2/3 phosphorylation and subsequent promotor activation. These results suggested that one of the possible mechanisms of the renoprotective effect of honokiol is the inhibition of TGF-β1-induced renal fibrosis.
Acknowledgments
We thank Miss Yi-Wen Chang for their skillful technical support and the Second Core Laboratory of Department of Medical Research in National Taiwan University Hospital for equipment and facility support. This work was supported by National Science Council Grants (to S-H Liu; NSC 92-2320-B-002-095 and C-K Chiang; NSC 95-2314-B-002-307-MY2, NSC-97-2314-B-002-051-MY3) and Taiwan University Hospital (to C-K Chiang; NTUH-95-N02, NTUH-96-N-042 and NTUH -097 -001023).
Glossary
Abbreviations
- α-SMA
α-smooth muscle actin
- BUN
blood urea nitrogen
- CKD
chronic kidney disease
- Cr
creatinine
- CTGF
connective tissue growth factor
- ECM
extracellular matrix
- GPT
glutamic pyruvic transaminase
- ICAM-1
intracellular adhesion molecule-1
- LDH
lactic acid dehydrogenase
- MTT
3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide
- PAI-1
plasminogen activator inhibitor-1
- TGF-β1
transforming growth factor-β1
- UUO
unilateral ureteral obstruction
Conflicts of interest
The authors declare no conflict of interest.
Supporting Information
Teaching Materials; Figs 1–8 as PowerPoint slide.
References
- Alpers CE, Hudkins KL, Floege J, Johnson RJ. Human renal cortical interstitial cells with some features of smooth muscle cells participate in tubulointerstitial and crescentic glomerular injury. J Am Soc Nephrol. 1994;5:201–209. doi: 10.1681/ASN.V52201. [DOI] [PubMed] [Google Scholar]
- Brenner BM, Cooper ME, de Zeeuw D, Keane WF, Mitch WE, Parving HH, et al. Effects of losartan on renal and cardiovascular outcomes in patients with type 2 diabetes and nephropathy. N Engl J Med. 2001;345:861–869. doi: 10.1056/NEJMoa011161. [DOI] [PubMed] [Google Scholar]
- Burns WC, Twigg SM, Forbes JM, Pete J, Tikellis C, Thallas-Bonke V, et al. Connective tissue growth factor plays an important role in advanced glycation end product-induced tubular epithelial-to-mesenchymal transition: implications for diabetic renal disease. J Am Soc Nephrol. 2006;17:2484–2494. doi: 10.1681/ASN.2006050525. [DOI] [PubMed] [Google Scholar]
- Castro AF, Coresh J. CKD surveillance using laboratory data from the population-based National Health and Nutrition Examination Survey (NHANES) Am J Kidney Dis. 2009;53(Suppl 3):S46–S55. doi: 10.1053/j.ajkd.2008.07.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang CK, Sheu ML, Hung KY, Wu KD, Liu SH. Honokiol, a small molecular weight natural product, alleviates experimental mesangial proliferative glomerulonephritis. Kidney Int. 2006;70:682–689. doi: 10.1038/sj.ki.5001617. [DOI] [PubMed] [Google Scholar]
- Derynck R, Zhang Y, Feng XH. Smads: transcriptional activators of TGF-beta responses. Cell. 1998;95:737–740. doi: 10.1016/s0092-8674(00)81696-7. [DOI] [PubMed] [Google Scholar]
- Eddy AA, Fogo AB. Plasminogen activator inhibitor-1 in chronic kidney disease: evidence and mechanisms of action. J Am Soc Nephrol. 2006;17:2999–3012. doi: 10.1681/ASN.2006050503. [DOI] [PubMed] [Google Scholar]
- Eknoyan G, Lameire N, Barsoum R, Eckardt KU, Levin A, Levin N, et al. The burden of kidney disease: improving global outcomes. Kidney Int. 2004;66:1310–1314. doi: 10.1111/j.1523-1755.2004.00894.x. [DOI] [PubMed] [Google Scholar]
- Fan JM, Ng YY, Hill PA, Nikolic-Paterson DJ, Mu W, Atkins RC, et al. Transforming growth factor-beta regulates tubular epithelial-myofibroblast transdifferentiation in vitro. Kidney Int. 1999;56:1455–1467. doi: 10.1046/j.1523-1755.1999.00656.x. [DOI] [PubMed] [Google Scholar]
- Fogo AB. Mesangial matrix modulation and glomerulosclerosis. Exp Nephrol. 1999;7:147–159. doi: 10.1159/000020595. [DOI] [PubMed] [Google Scholar]
- Fogo AB. Renal fibrosis: not just PAI-1 in the sky. J Clin Invest. 2003;112:326–328. doi: 10.1172/JCI19375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Sanchez O, Lopez-Hernandez FJ, Lopez-Novoa JM. An integrative view on the role of TGF-beta in the progressive tubular deletion associated with chronic kidney disease. Kidney Int. 2010;77:950–955. doi: 10.1038/ki.2010.88. [DOI] [PubMed] [Google Scholar]
- Gore-Hyer E, Shegogue D, Markiewicz M, Lo S, Hazen-Martin D, Greene EL, et al. TGF-beta and CTGF have overlapping and distinct fibrogenic effects on human renal cells. Am J Physiol Renal Physiol. 2002;283:F707–F716. doi: 10.1152/ajprenal.00007.2002. [DOI] [PubMed] [Google Scholar]
- Grande MT, Perez-Barriocanal F, Lopez-Novoa JM. Role of inflammation in tubulo-interstitial damage associated to obstructive nephropathy. J Inflamm (Lond) 2010;7:19. doi: 10.1186/1476-9255-7-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grupp C, Lottermoser J, Cohen DI, Begher M, Franz HE, Muller GA. Transformation of rat inner medullary fibroblasts to myofibroblasts in vitro. Kidney Int. 1997;52:1279–1290. doi: 10.1038/ki.1997.453. [DOI] [PubMed] [Google Scholar]
- Gupta S, Clarkson MR, Duggan J, Brady HR. Connective tissue growth factor: potential role in glomerulosclerosis and tubulointerstitial fibrosis. Kidney Int. 2000;58:1389–1399. doi: 10.1046/j.1523-1755.2000.00301.x. [DOI] [PubMed] [Google Scholar]
- Heldin CH, Miyazono K, Dijke P. TGF-beta signalling from cell membrane to nucleus through SMAD proteins. Nature. 1997;390:465–471. doi: 10.1038/37284. [DOI] [PubMed] [Google Scholar]
- Hewitson TD. Renal tubulointerstitial fibrosis: common but never simple. Am J Physiol Renal Physiol. 2009;296:F1239–F1244. doi: 10.1152/ajprenal.90521.2008. [DOI] [PubMed] [Google Scholar]
- Hou FF, Zhang X, Zhang GH, Xie D, Chen PY, Zhang WR, et al. Efficacy and safety of benazepril for advanced chronic renal insufficiency. N Engl J Med. 2006;354:131–140. doi: 10.1056/NEJMoa053107. [DOI] [PubMed] [Google Scholar]
- Iwano M, Plieth D, Danoff TM, Xue C, Okada H, Neilson EG. Evidence that fibroblasts derive from epithelium during tissue fibrosis. J Clin Invest. 2002;110:341–350. doi: 10.1172/JCI15518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izuhara Y, Nangaku M, Inagi R, Tominaga N, Aizawa T, Kurokawa K, et al. Renoprotective properties of angiotensin receptor blockers beyond blood pressure lowering. J Am Soc Nephrol. 2005;16:3631–3641. doi: 10.1681/ASN.2005050522. [DOI] [PubMed] [Google Scholar]
- Kaneto H, Morrissey J, Klahr S. Increased expression of TGF-beta 1 mRNA in the obstructed kidney of rats with unilateral ureteral ligation. Kidney Int. 1993;44:313–321. doi: 10.1038/ki.1993.246. [DOI] [PubMed] [Google Scholar]
- Keeton MR, Curriden SA, van Zonneveld AJ, Loskutoff DJ. Identification of regulatory sequences in the type 1 plasminogen activator inhibitor gene responsive to transforming growth factor beta. J Biol Chem. 1991;266:23048–23052. [PubMed] [Google Scholar]
- Klahr S, Morrissey J. Obstructive nephropathy and renal fibrosis. Am J Physiol Renal Physiol. 2002;283:F861–F875. doi: 10.1152/ajprenal.00362.2001. [DOI] [PubMed] [Google Scholar]
- Lee J, Jung E, Park J, Jung K, Lee S, Hong S, et al. Anti-inflammatory effects of magnolol and honokiol are mediated through inhibition of the downstream pathway of MEKK-1 in NF-kappaB activation signaling. Planta Med. 2005;71:338–343. doi: 10.1055/s-2005-864100. [DOI] [PubMed] [Google Scholar]
- Li JH, Zhu HJ, Huang XR, Lai KN, Johnson RJ, Lan HY. Smad7 inhibits fibrotic effect of TGF-Beta on renal tubular epithelial cells by blocking Smad2 activation. J Am Soc Nephrol. 2002;13:1464–1472. doi: 10.1097/01.asn.0000014252.37680.e4. [DOI] [PubMed] [Google Scholar]
- Lin SL, Chen RH, Chen YM, Chiang WC, Lai CF, Wu KD, et al. Pentoxifylline attenuates tubulointerstitial fibrosis by blocking Smad3/4-activated transcription and profibrogenic effects of connective tissue growth factor. J Am Soc Nephrol. 2005;16:2702–2713. doi: 10.1681/ASN.2005040435. [DOI] [PubMed] [Google Scholar]
- Liou KT, Shen YC, Chen CF, Tsao CM, Tsai SK. The anti-inflammatory effect of honokiol on neutrophils: mechanisms in the inhibition of reactive oxygen species production. Eur J Pharmacol. 2003;475:19–27. doi: 10.1016/s0014-2999(03)02121-6. [DOI] [PubMed] [Google Scholar]
- Liu Y. Epithelial to mesenchymal transition in renal fibrogenesis: pathologic significance, molecular mechanism, and therapeutic intervention. J Am Soc Nephrol. 2004;15:1–12. doi: 10.1097/01.asn.0000106015.29070.e7. [DOI] [PubMed] [Google Scholar]
- Liu Y. Renal fibrosis: new insights into the pathogenesis and therapeutics. Kidney Int. 2006;69:213–217. doi: 10.1038/sj.ki.5000054. [DOI] [PubMed] [Google Scholar]
- Liu Y. New insights into epithelial-mesenchymal transition in kidney fibrosis. J Am Soc Nephrol. 2010;21:212–222. doi: 10.1681/ASN.2008121226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noronha IL, Fujihara CK, Zatz R. The inflammatory component in progressive renal disease – are interventions possible? Nephrol Dial Transplant. 2002;17:363–368. doi: 10.1093/ndt/17.3.363. [DOI] [PubMed] [Google Scholar]
- Park EJ, Zhao YZ, Kim YH, Lee BH, Sohn DH. Honokiol induces apoptosis via cytochrome c release and caspase activation in activated rat hepatic stellate cells in vitro. Planta Med. 2005;71:82–84. doi: 10.1055/s-2005-837757. [DOI] [PubMed] [Google Scholar]
- Remuzzi G, Bertani T. Pathophysiology of progressive nephropathies. N Engl J Med. 1998;339:1448–1456. doi: 10.1056/NEJM199811123392007. [DOI] [PubMed] [Google Scholar]
- Remuzzi G, Zoja C, Gagliardini E, Corna D, Abbate M, Benigni A. Combining an antiproteinuric approach with mycophenolate mofetil fully suppresses progressive nephropathy of experimental animals. J Am Soc Nephrol. 1999;10:1542–1549. doi: 10.1681/ASN.V1071542. [DOI] [PubMed] [Google Scholar]
- Rerolle JP, Hertig A, Nguyen G, Sraer JD, Rondeau EP. Plasminogen activator inhibitor type 1 is a potential target in renal fibrogenesis. Kidney Int. 2000;58:1841–1850. doi: 10.1111/j.1523-1755.2000.00355.x. [DOI] [PubMed] [Google Scholar]
- Risdon RA, Sloper JC, De Wardener HE. Relationship between renal function and histological changes found in renal-biopsy specimens from patients with persistent glomerular nephritis. Lancet. 1968;2:363–366. doi: 10.1016/s0140-6736(68)90589-8. [DOI] [PubMed] [Google Scholar]
- Seo JY, Park J, Yu MR, Kim YS, Ha H, Lee HB. Positive feedback loop between plasminogen activator inhibitor-1 and transforming growth factor-beta1 during renal fibrosis in diabetes. Am J Nephrol. 2009;30:481–490. doi: 10.1159/000242477. [DOI] [PubMed] [Google Scholar]
- Sheu ML, Ho FM, Yang RS, Chao KF, Lin WW, Lin-Shiau SY, et al. High glucose induces human endothelial cell apoptosis through a phosphoinositide 3-kinase-regulated cyclooxygenase-2 pathway. Arterioscler Thromb Vasc Biol. 2005;25:539–545. doi: 10.1161/01.ATV.0000155462.24263.e4. [DOI] [PubMed] [Google Scholar]
- Sheu ML, Liu SH, Lan KH. Honokiol induces calpain-mediated glucose-regulated protein-94 cleavage and apoptosis in human gastric cancer cells and reduces tumor growth. PLoS ONE. 2007;2:e1096. doi: 10.1371/journal.pone.0001096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheu ML, Chiang CK, Tsai KS, Ho FM, Weng TI, Wu HY, et al. Inhibition of NADPH oxidase-related oxidative stress-triggered signaling by honokiol suppresses high glucose-induced human endothelial cell apoptosis. Free Radic Biol Med. 2008;44:2043–2050. doi: 10.1016/j.freeradbiomed.2008.03.014. [DOI] [PubMed] [Google Scholar]
- Son HJ, Lee HJ, Yun-Choi HS, Ryu JH. Inhibitors of nitric oxide synthesis and TNF-alpha expression from Magnolia obovata in activated macrophages. Planta Med. 2000;66:469–471. doi: 10.1055/s-2000-8592. [DOI] [PubMed] [Google Scholar]
- Subramanian SV, Polikandriotis JA, Kelm RJ, Jr, David JJ, Orosz CG, Strauch AR. Induction of vascular smooth muscle alpha-actin gene transcription in transforming growth factor beta1-activated myofibroblasts mediated by dynamic interplay between the Pur repressor proteins and Sp1/Smad coactivators. Mol Biol Cell. 2004;15:4532–4543. doi: 10.1091/mbc.E04-04-0348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu CC, Hsu MJ, Kuo ML, Chen RF, Chen MC, Bai KJ, et al. Thrombin-induced connective tissue growth factor expression in human lung fibroblasts requires the ASK1/JNK/AP-1 pathway. J Immunol. 2009;182:7916–7927. doi: 10.4049/jimmunol.0801582. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.